Introduction
DNA methylation is a key epigenetic mechanism
whereby a methyl group is added to the 5′ position of a cytosine
pyrimidine ring at promoter regions of a gene (1). In humans, DNA methylation occurs
mainly in the dinucleotide CpG sites (2). Aberrant DNA methylation has been
identified as one of the important factors that contributes to
cancer due to silencing of the tumour suppressor genes (3). The mechanisms underlying the
silencing effects of DNA methylation are associated with chromatin
configuration (4). When the CpG
sites remain unmethylated, DNA binding proteins and transcription
factors can easily access to the promoter site and active gene
expression will occur (5,6). Methylation of CpG sites results in
changes in the chromatin structure that interfere with the binding
capacity of the standard machinery for transcription factors
resulting in gene silencing (7,8). In
general, there are two types of DNA methylation including promoter
hypermethylation and global hypomethylation.
Promoter hypermethylation is associated with low
expression or silencing of tumour suppressor genes, for example,
the AT-rich interactive domain 1A (ARID1A) gene in breast
cancer (9). Hypermethylation of
the ARID1A promoter has resulted in transcription
inactivation despite the changes in the gene copy number, mutations
and histone modifications (10).
Previous studies reported several hypermethylated genes as
potential DNA methylation markers for breast cancer including
TUSC5, DOK7, KLF11, SIM1, NT5E
and OTP (11–15). However, there is a need to further
explore the reliability, sensitivity and specificity of these
biomarkers.
Focal hypermethylation can affect not only discrete
genes but also a large region of the chromosome that leads to
long-range epigenetic silencing (LRES) (16). In LRES, methylation of a particular
gene may also suppress expression of neighboring unmethylated genes
(17). This phenomenon was
observed in two specific gene clusters that were related to breast
cancer (16,18). In one of these examples, aberrant
methylation in the CpG islands of HOXA1, HOXA7,
HOXA5 and HOXA9 in chromosome 7 caused silencing of
HOXA1 to HOXA10 genes. In the second report,
hypermethylation of CpG islands in protocadherin (PCDH) in
chromosome 5 resulted in low expression of the PCDH gene
cluster (16,18). Global hypomethylation commonly
occurs at repetitive elements such as satellite DNA sequences
(19). An example of this
mechanism was documented in a breast cancer study where there was
hypomethylation of the satellite sequence of SATR-1 gene
(20). A study using deep
sequencing identified long-range hypomethylation with focal
hypermethylation of nuclear-associated domains and these were
likely to cause gene silencing in colorectal cancer (21).
Recent publications have used multiple genomic
datasets to get a clearer picture of the genetic events in cancers
including breast cancer (22–24).
This is because a single dataset analysis gives limited information
and does not fully reflect the actual events in the cells. In the
present study, we performed an integrative analysis combining DNA
methylation and gene expression profiling datasets to identify the
significant genes and their biological pathways that can served as
potential therapeutic targets for breast cancer.
Materials and methods
Clinical samples
Approval for the study was obtained from the Ethics
Committee of Universiti Kebangsaan Malaysia (ref. UKM
1.5.3.5/244/SPP/UMBI-003-2012). Subject recruitment was carried out
at the Universiti Kebangsaan Malaysia Medical Centre and Hospital
Kuala Lumpur, Malaysia. Primary breast tumour and adjacent
non-cancerous breast tissue samples were collected from 87 female
patients after a written informed consent. The collected tissues
were kept in liquid nitrogen and stored at −80°C before further
analysis. All tissues were sectioned at 5–7 μm thickness using a
cryostat (Microtome Cryostat HM550; Microm International GmbH,
Walldorf, Germany) and stained with haematoxylin and eosin
(H&E) before being viewed and confirmed by a histopathologist.
Tissues that contained > 80% of malignant cells were included.
We only used non-cancerous tissues which were free from malignant
or inflammatory cells and contained mainly ductal and lobular
cells. Laser capture microdissection (Arcturus Engineering,
Mountain View, CA, USA) was used for tissues that contained <80%
of cancer or non-cancerous cells, and this followed the previously
described procedure (25).
DNA methylation profiling
Genomic DNA was isolated using two different kits,
the DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany) and the
QIAamp® DNA Micro kit (Qiagen), according to the
protocol of the manufacturers. DNA integrity was assessed by 1.0%
agarose gel electrophoresis under 60 V for 1 h. We quantified
concentration and purity of the extracted DNA using NanoDrop
(Thermo Fisher Scientific, Leicester, UK).
Bisulphite conversion of genomic DNA was carried out
using EZ DNA Methylation-Gold kit (Zymo Research, Irvine, CA, USA).
DNA methylation analysis of 76 tumours and 25 adjacent normal
breast samples was performed using the Illumina’s Infinium
HumanMethylation27 Beadchip kit (Illumina, Inc., San Diego, CA,
USA) based on the manufacturer’s protocol and followed the
previously described procedure (26). The data generated by GenomeStudio
was further analysed using Partek Genomics Suite 6.6 (Partek Inc.,
St. Louis, MO, USA). A 4-way analysis of variance (ANOVA) was used
to compare CpG loci methylation data across tumour and normal
groups. The different sources of variation in the entire data in
this ANOVA model included sample group (tumour/normal), batch
effect, status of oestrogen receptor (positive/negative) and
sources of tissue type (frozen section/laser capture
microdissection). Adjustment for the different sources of variation
such as batch effect, status of oestrogen receptor and sources of
tissue type was done. We further used the filtering characteristics
of fold-change -2 to 2 and a false discovery rate (FDR) at
P<0.05 to identify the differentially methylated genes.
Gene expression profiling
Total RNA was extracted using the RNeasy Plus Mini
kit (Qiagen) according to the manufacturer’s protocol. RNA was
quantified using NanoDrop (Thermo Fisher Scientific) and Agilent
RNA 6000 Nano kit (Agilent Technologies GmbH, Waldbronn, Germany).
RNA samples with OD 260/280 of 1.8–2.2 and RNA integrity number
≥6.5 were included in the present study. Gene expression profiling
of 15 tumours and 5 adjacent normal breast tissues was performed
using GeneChip® Human Gene 1.0 ST array (Affymetrix,
Santa Clara, CA, USA). Data were extracted using the
Affymetrix® Genotyping Console™ (Affymetrix) and were
further analysed using Partek Genomics Suite 6.6 (Partek Inc.).
Data were normalised using quantile normalisation and robust
multi-array analysis (RMA) background correction. Filtering
characteristics of fold-change -1.5 to 1.5 and a FDR at P<0.05
were used in identifying the differentially expressed genes.
Integrative genomic and epigenomic
analysis
In order to identify overlapping genes across
different datasets, gene symbols from each of the datasets were
used. Filtered datasets in either in CSV (comma separated values)
or tab delimited format with significant cut-off values from each
of the datasets were import into MySQL relational database for
downstream data analysis. Each of the datasets was compared in
pairwise (gene expression vs. methylation). Unique gene symbol
found between the overlapping comparisons were used as a gene list
for downstream analysis. These overlapping genes were then analysed
using KEGG pathway and DAVID v6.7 for functional annotation,
classification and enrichment analysis. Functional classification
and signalling pathway that showed P≤0.05 was considered
significantly enriched. Expression values and methylation values
were also extracted from the datasets for circular map
generation.
Methylation-specific multiplex
ligation-dependent probe amplification (MS-MLPA)
MS-MLPA was performed to confirm the methylation
microarray results. The SALSA MLPA Kit P200-A1 Reference-1
(MRC-Holland, Amsterdam, The Netherlands) was used to detect
aberrant methylation for genes GPX7, SPARC,
TIMP3, BTG4 and SFRP2 in 70 tumours and 23
normal samples. Briefly, 5 μl of 50 ng/μl DNA was denatured at 95°C
for 30 min followed by cooling down to 25°C and adding of the
probes mix. The sample was then incubated at 60°C for 18 h. The
sample was then divided into 2 tubes, one used as a control without
the HhaI enzyme while another tube contained the HhaI
enzyme (Promega, Madison, WI, USA) for the digestion. PCR was
performed on all samples using the thermal cycler (Applied
Biosystems, Foster City, CA, USA). The amplified products were
further subjected to fragment analysis using the ABI 3500 Genetic
Analyzer (Applied Biosystems). Data analysis was performed using
the Coffalyser version 1.0.0.43 (MRC-Holland). Quantification of
methylation status for each gene was obtained by comparing the
probes relative peak area ratio from the digested samples with
those obtained from the undigested samples. Digested samples with
probes of relative peak area ratio ≥0.25 were considered as
methylated.
Methylation-specific quantitative
polymerase chain reaction (MS-qPCR)
MS-qPCR was performed to further confirm the
methylation microarray results. Bisulphite conversion of genomic
DNA was carried out using BisulFlash DNA modification kit
(Epigentek Group Inc., New York, NY, USA) followed the
manufacturer’s protocol. Methylation specific qPCR Fast kit
(Epigentek Group Inc.) was used to detect aberrant methylation for
genes SFRP1 and NRG1 in 50 samples. Basically, there
were two types of primers in this assay, namely methylated primer
and unmethylated primer. The methylated primer sequences for
SFRP1 were 5′-GTTTTTAGTCGGATATCGGTTC-3′ (forward) and
3′-CACGTTATAACACAACCGCA-5′ (reverse) while the unmethylated primer
sequences were 5′-GTGAGTTTTTAGTTGGATATTGGTTT-3′ (forward) and
3′-CCCACATTATAACACAACCACA-5′ (reverse). For gene NRG1, the
methylated primer sequences were 5′-CGGATTGGGGTAAAATAAGTTC-3′
(forward) and 3′-ACAATAATAACAACAACGACAACGA-5′ (reverse) while the
unmethylated primer sequences were 5′-AGAGTTGGGTAGAGTTTGAATTGA-3′
(forward) and 3′-CAACAATAATAACAACAACAACAACAAC-5′ (reverse). We used
β-actin as the positive control in this assay. The MS-qPCR was
carried out using Applied Biosystem 7500 Fast Real-Time PCR system
(Applied Biosystems) followed the manufacturer’s protocol. Data
analysis for gene SFRP1 was based on 43 tumour and 7 normal
breast tissues. For gene NRG1, data analysis was performed
based on 47 tumour and 2 normal breast tissues as one sample had
been identified as an outlier and excluded from the data analysis.
Data generated by MS-qPCR were further analysed by using Microsoft
Excel. Ct value of each sample was normalised with the Ct value of
the β-actin. Percentage of methylation level for each sample was
calculated based on a previous study (27). Unpaired t-test was used to test the
significance of the results.
Results
Novel gene clusters are hypermethylated
in breast cancer
DNA methylation analysis was conducted on 87 breast
cancer patients with a mean age of 55.90±11.17 years. The
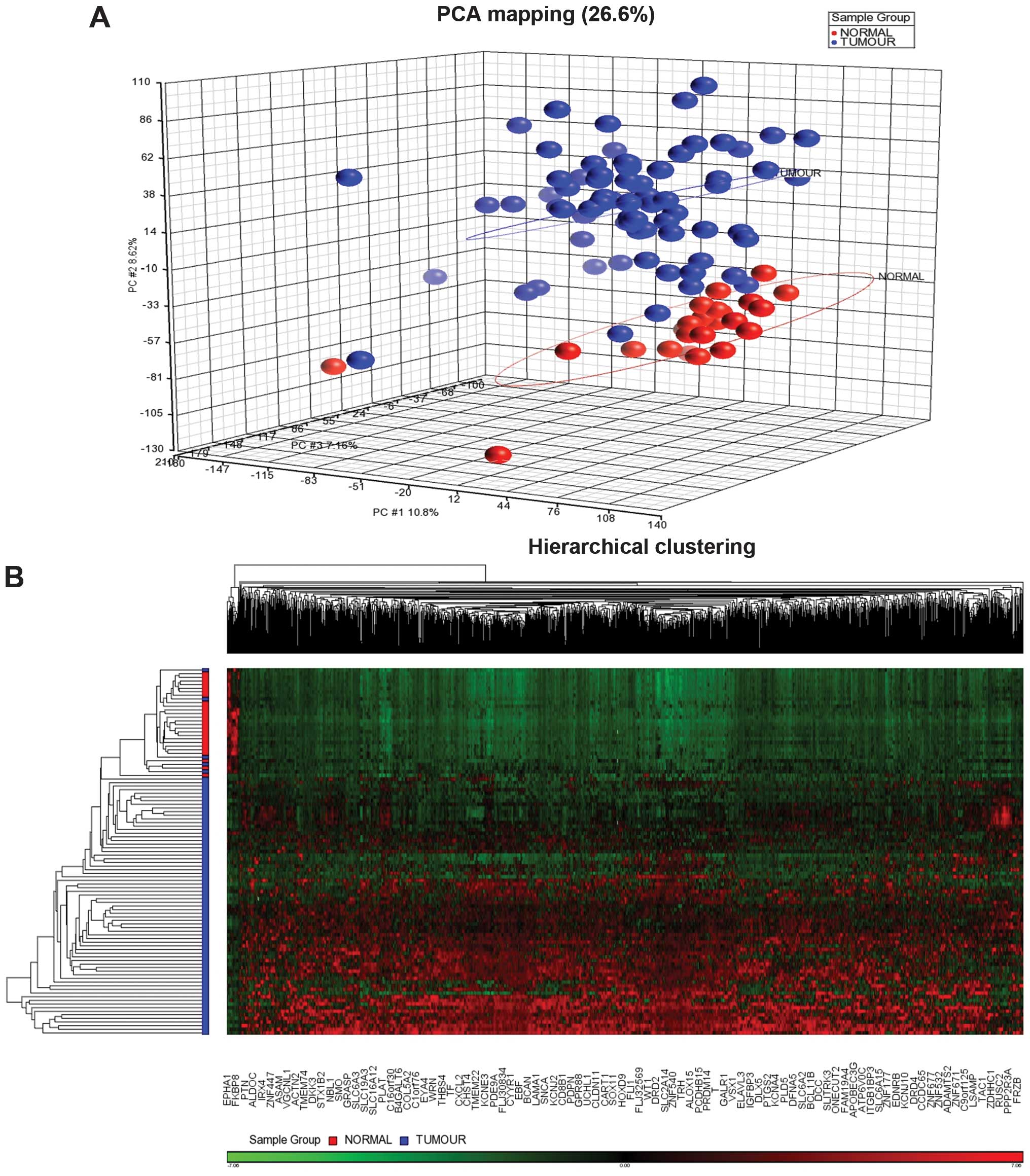
epidemiological data of the patients are shown in Table I. Principal component analysis
(PCA) showed the tumour and normal samples were clustered
distinctly (Fig. 1). Supervised
hierarchical clustering distinctly separated the tumour group from
the adjacent normal breast tissues. Filtering using a fold-change
−2 to 2 and a FDR P<0.05 generated 1,411 CpG sites which cover
1,049 genes. These significantly methylated CpG sites were further
grouped into 1,389 sites with high methylation level or
hypermethylation and 22 with low methylation level or
hypomethylation (Fig. 1). We
excluded 133 false CpG sites from the pathway analysis and 8 CpG
sites which were located on the X chromosome. There were 9 clusters
of genes that were hypermethylated including two which (HOXA
and PCDH) were previously reported in breast cancer
(Table II) (16,18).
 | Table IThe epidemiological data of the
patients. |
Table I
The epidemiological data of the
patients.
| Ages (years) | Mean | 55.90±11.17 |
| Range | | 32–78 |
| Tumour grade | I | 13.2% |
| II | 42.1% |
| III | 44.7% |
| Histological
type | IDC | 89.5% |
| Non-IDC | 10.5% |
| Oestrogen
receptor | Positive | 63.1% |
| Negative | 36.9% |
| Progesterone
receptor | Positive | 40.7% |
| Negative | 59.3% |
| HER2
amplification | Positive | 43.4% |
| Negative | 56.6% |
| Triple
negative | | 12 patients |
| Table IIGene clusters that were identified
from DNA methylation profiling. |
Table II
Gene clusters that were identified
from DNA methylation profiling.
| Cluster | Chromosome | Genes involved |
|---|
| 1 | 2 | HOXD8,
HOXD9, HOXD11, HOXD12, HOXD13 |
| 2 | 2 | IHH,
PTPRN, DES |
| 3 | 3 | ZNF660,
ZNF501, ZNF502 |
| 4 | 4 | CXCL1,
CXCL2, CXCL3, CXCL5, CXCL6 |
| 5 | 5 | PCDHAC1,
PCDHB2, PCDHB15, PCDHGA12, PCDHGB4,
PCDHGB7 |
| 6 | 5 | ZFP2,
ZNF454, GRM6, ZNF354C |
| 7 | 6 | HIST1H1A,
HIST1H2BB, HIST1H3C, HIST1H4F,
HIST1H3I, HIST1H3J, HIST1H4J,
HIST1H4K |
| 8 | 7 | HOXA1,
HOXA4, HOXA7, HOXA9, HOXA13 |
| 9 | 16 | HBA1,
HBA2, HBQ1 |
Gene expression pattern in breast cancer
through genome-wide expression microarray
Gene expression microarray analysis was performed on
15 tumour and 5 normal methylation-matched samples. Filtering using
a fold-change of −1.5 to 1.5 and a FDR P-value of <0.05
identified 867 differentially expressed genes. Principal component
analysis (PCA) showed the tumour and normal samples were clustered
distinctly (Fig. 2). The
supervised hierarchical clustering revealed 404 upregulated and 463
downregulated genes in cancer compared to non-cancerous samples
(Fig. 2). The top 10 upregulated
genes were CASC5, CENPF, KIF23, DTL,
MK167, TPX2, NUF2, KIF4A, NUSAP1
and BUB1B while the top 10 downregulated genes were
PAK3, B3GALT1, CX3CL1, EDN3,
KCNMB1, HOXA5, NRG1, KLHL13,
TSHZ2 and IL17RD. Gene Ontology enrichment analysis
revealed that most of the genes were enriched in cell
proliferation, viral reproduction, pigmentation, growth, rhythmic
process, cell killing and metabolic process under the biological
process. For the molecular function, most of the genes were
enriched in the chemoattractant, structural molecule, translation
regular, enzyme regulator, transporter and binding activity. For
the cellular component, most of the genes were active in
extracellular region and synapse. For the gene set enrichment
analysis (GSEA) with P<0.05, most of the genes are involved in
cell proliferation, spindle, M phase of mitotic cell cycle,
microtubule-based movement, microtubule motor activity, condensed
chromosome kinetochore and microtube.
Integrative analysis showed 64 driver
genes involved in breast cancer
The integrative analysis revealed 64 overlapping
significant genes between DNA methylation and gene expression
analysis. Notably, all of the overlapped genes were hypermethylated
with 51 showing negative association and 13 positive association
(Table III). The 64 overlapping
genes were further mapped to the KEGG database as shown in Table IV.
| Table IIIOverlapping significant genes between
DNA methylation and gene expression analysis. |
Table III
Overlapping significant genes between
DNA methylation and gene expression analysis.
| Genes | Status of
methylation | Gene
expression |
|---|
| ADAMTS5 |
Hypermethylation | Downregulated |
|
ADCYAP1R1 |
Hypermethylation | Downregulated |
| AGTR1 |
Hypermethylation | Downregulated |
| CCND2 |
Hypermethylation | Downregulated |
| CD200 |
Hypermethylation | Downregulated |
| CDH8 |
Hypermethylation | Downregulated |
| CHL1 |
Hypermethylation | Downregulated |
| CLDN11 |
Hypermethylation | Downregulated |
| CNN1 |
Hypermethylation | Downregulated |
| CNTN1 |
Hypermethylation | Downregulated |
| CNTNAP3 |
Hypermethylation | Downregulated |
| CTTNBP2 |
Hypermethylation | Downregulated |
| CXCL2 |
Hypermethylation | Downregulated |
| CYP24A1 |
Hypermethylation | Downregulated |
| CYYR1 |
Hypermethylation | Downregulated |
| D4S234E |
Hypermethylation | Downregulated |
| DAB2IP |
Hypermethylation | Downregulated |
| DKK3 |
Hypermethylation | Downregulated |
| EDN3 |
Hypermethylation | Downregulated |
| EFHA2 |
Hypermethylation | Downregulated |
| EPHB1 |
Hypermethylation | Downregulated |
| FGF2 |
Hypermethylation | Downregulated |
| GLP1R |
Hypermethylation | Downregulated |
| GRIA4 |
Hypermethylation | Downregulated |
| HOXA4 |
Hypermethylation | Downregulated |
| HOXA7 |
Hypermethylation | Downregulated |
| HOXA9 |
Hypermethylation | Downregulated |
| HPSE2 |
Hypermethylation | Downregulated |
| KCNJ2 |
Hypermethylation | Downregulated |
| STAC2 |
Hypermethylation | Downregulated |
| SYN2 |
Hypermethylation | Downregulated |
| ZNF667 |
Hypermethylation | Downregulated |
| KCTD14 |
Hypermethylation | Downregulated |
| KIT |
Hypermethylation | Downregulated |
| KLK10 |
Hypermethylation | Downregulated |
| LMOD1 |
Hypermethylation | Downregulated |
| MAMDC2 |
Hypermethylation | Downregulated |
| MT1E |
Hypermethylation | Downregulated |
| NDRG2 |
Hypermethylation | Downregulated |
| NRG1 |
Hypermethylation | Downregulated |
| OSR1 |
Hypermethylation | Downregulated |
| PAK7 |
Hypermethylation | Downregulated |
| PDE1C |
Hypermethylation | Downregulated |
| PDK4 |
Hypermethylation | Downregulated |
| PTN |
Hypermethylation | Downregulated |
| PTPRZ1 |
Hypermethylation | Downregulated |
| RELN |
Hypermethylation | Downregulated |
| SCARA5 |
Hypermethylation | Downregulated |
| SFRP1 |
Hypermethylation | Downregulated |
| SLC27A6 |
Hypermethylation | Downregulated |
| SLC34A2 |
Hypermethylation | Downregulated |
| AEBP1 |
Hypermethylation | Upregulated |
| C7orf11 |
Hypermethylation | Upregulated |
| CASC5 |
Hypermethylation | Upregulated |
| COL12A1 |
Hypermethylation | Upregulated |
| COL1A1 |
Hypermethylation | Upregulated |
| COL1A2 |
Hypermethylation | Upregulated |
| COL4A1 |
Hypermethylation | Upregulated |
| FBN1 |
Hypermethylation | Upregulated |
| H2AFY |
Hypermethylation | Upregulated |
| NID2 |
Hypermethylation | Upregulated |
| NOX4 |
Hypermethylation | Upregulated |
| THBS2 |
Hypermethylation | Upregulated |
| THY1 |
Hypermethylation | Upregulated |
| Table IVSignificant pathways identified from
mapping of 64 overlapping genes to KEGG database. |
Table IV
Significant pathways identified from
mapping of 64 overlapping genes to KEGG database.
| KEGG ID | Pathways | No. of genes | P-values | Genes | Enrichment
scores |
|---|
| hsa04510 | Focal adhesion | 7 | 2.62E-04 | PAK7,
COL4A1, CCND2, COL1A2, RELN,
COL1A1, THBS2 | 7.08 |
| hsa04512 | ECM-receptor
interaction | 5 | 5.72E-04 | COL4A1,
COL1A2, RELN, COL1A1, THBS2 | 12.11 |
For the pathway analysis, two of the enriched
pathways identified were the focal adhesion and extracellular
matrix-receptor interaction. Seven genes (PAK7,
COL4A1, CCND2, COL1A2, RELN,
COL1A1 and THBS2) are involved in the focal adhesion
while five genes (COL4A1, COL1A2, RELN,
COL1A1 and THBS2) are involved in extracellular
matrix-receptor interaction.
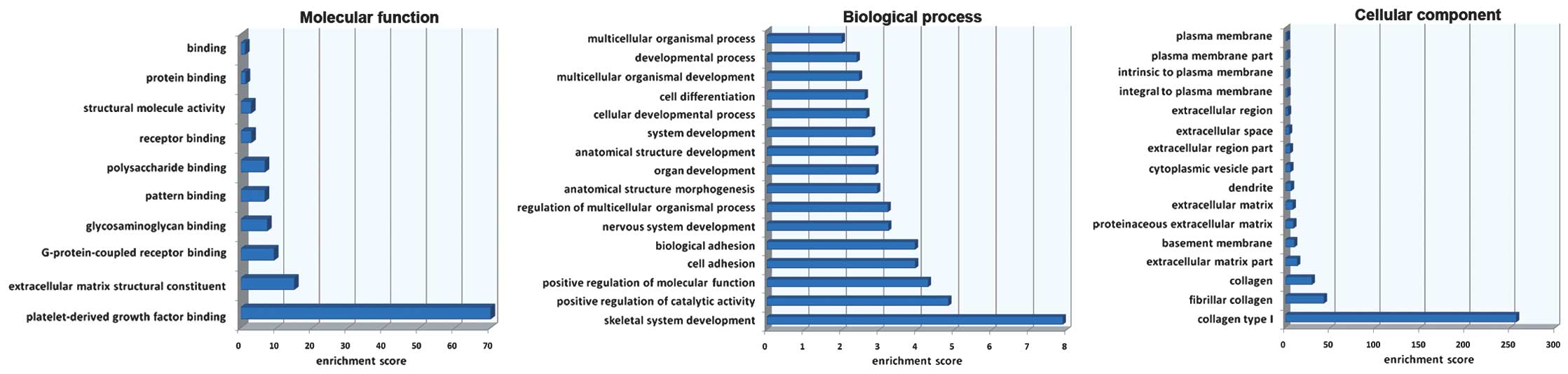
Gene Ontology (GO) enrichment analysis was carried
out on the overlapping genes. The enrichment analysis showed that
for the biological process, most of the genes were enriched in the
skeletal system development. For the molecular function, most of
the genes were enriched in the platelet-derived growth factor
binding while for the cellular component, most of the genes were
involved in collagen type I (Fig.
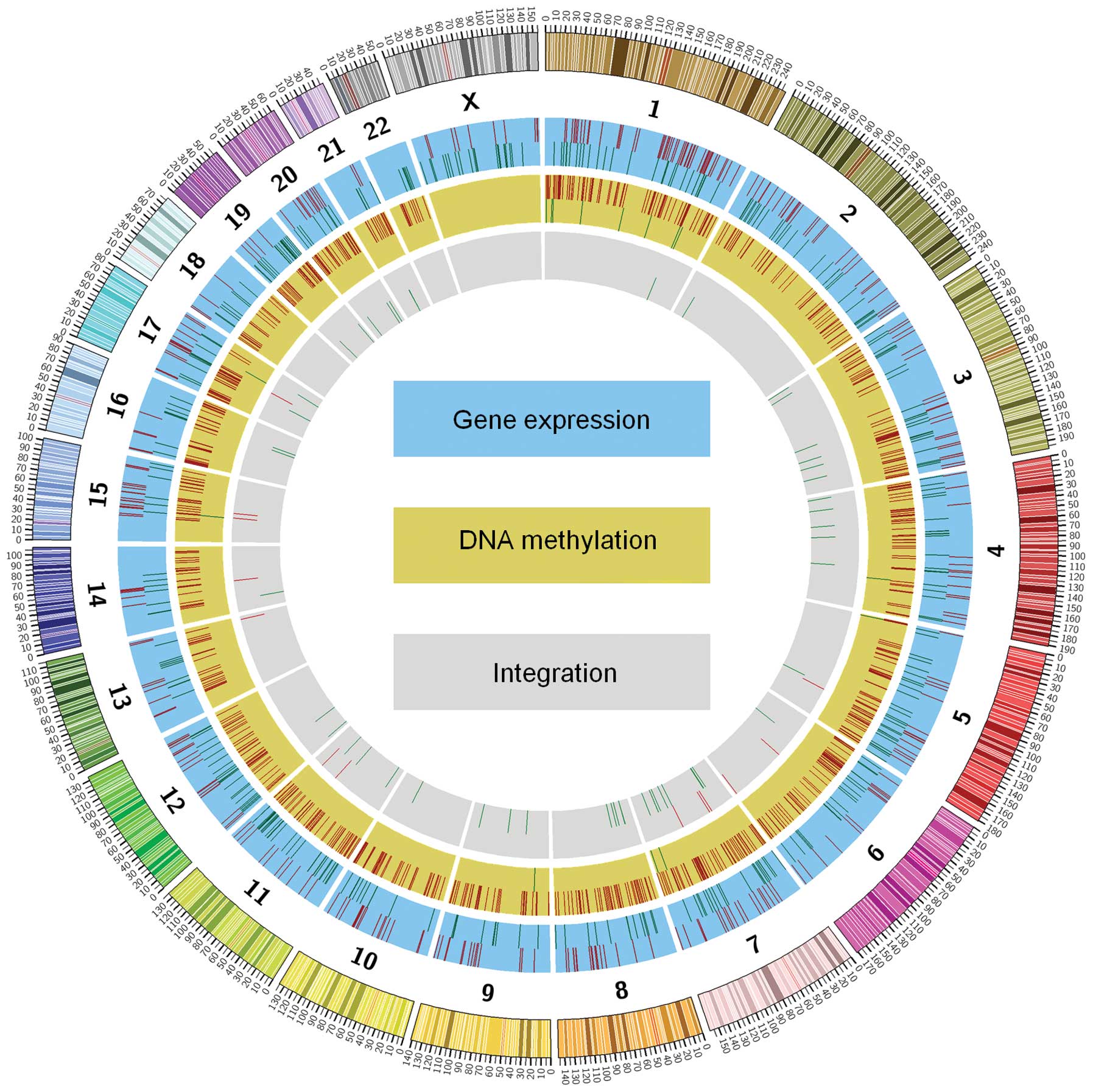
3). The circular map generated showed the overall visualisation
of overlapping genes according to chromosomes (Fig. 4).
MS-MLPA and MS-qPCR confirmed the
methylation profiling results
A total of 7 genes were selected for the validation
of methylation microarray results. The genes were selected based on
their association of breast cancer and the pathways of these genes
involved. Five genes included GPX7, SPARC,
TIMP3, BTG4 and SFRP2 were validated with
MS-MPLA while another two genes, NRG1 and SFRP1, were
validated using MS-qPCR. Both MS-MLPA and MS-qPRC results showed
that the breast cancer samples have higher percentages of
methylation compared to normal samples for all the selected genes
(Figs. 5 and 6). The highest percentage of methylation
was seen in TIMP3 followed by SPARC, SFRP2,
BTG4 and GPX7 in MS-MLPA while MS-qPCR showed that
the average methylation percentage in tumour samples for
NRG1 and SFRP1 were 53.5 and 27.2%, respectively,
with P-value <0.001.
Discussion
DNA methylation mechanism is a reversible process
(28). Reversibility of DNA
methylation can result in gene re-expression that leads to normal
gene regulation (29). Based on
this process, it can serve as a potential therapeutic target in
cancer. Up to date, numerous DNA methylation studies have been
carried out in breast cancer. However, most of the studies provided
results that were obtained from single dataset analysis and gave
limited information in terms of associating methylation to the
transcriptome datasets. We have performed an integrative genomic
analysis to identify the significant genes that can serve as
important prognostic indicators and potential therapeutic targets
for breast cancer. We found 9 gene clusters in breast cancer from
the DNA methylation profiling analysis. Two of the gene clusters
namely HOXA and PCDH were previously reported in
breast cancer (16,18). These two gene clusters were
involved in LRES which can cause multiple genes from the same
cluster to become silenced. It is highly possible that the same
mechanism could appear in the other 7 gene clusters that may lead
to silencing of their nearby genes.
Integrative analysis identified 64 hypermethylated
genes with 51 showing negative association and 13 with positive
association. Out of the 51 hypermethylated genes with low gene
expression, 14 genes were associated with breast cancer, including
DAB2IP, NDRG2, AGTR1, CXCL2,
CCND2, DKK3, FGF2, KLK10, NRG1,
PTN, PTPRZ1, SFRP1, RELN and KIT
(30–43). Five of these genes, DAB2IP,
DKK3, KLK10, NGR1 and SFRP1, were
tumour suppressor genes (41,44–47).
Similar findings were documented in previous breast cancer studies
that confirmed the reliability of our results (30,35,37,38,41,48–50).
DAB2IP is a Ras GTPase-activating tumour
suppressor protein which plays an important role in maintaining
cell homeostasis (51,52). Hypermethylation of this gene might
disrupt the cell homeostasis and lead to breast carcinogenesis. The
DKK3 encodes a protein that plays a role in inhibiting
planar cell polarity pathway which regulates cell adhesion,
motility and polarity (35,53).
Therefore, inactivation of this gene by hypermethylation might
activate planar cell polarity pathway and cause metastasis in
breast cancer. The SFRP1 is the modulator of Wnt signaling
pathway which plays a significant role in embryonic development,
cell differentiation and proliferation (41,54).
SFRP1 protein can inhibit the Wnt signalling pathway by
binding to WNT1 molecules (50).
It has been proposed that inactivation of SFRP1 is an early
event in breast cancer and downregulation of this gene has also
been shown to be associated with poor prognosis in breast cancer
(41,49).
Examples of non-tumour suppressor genes which were
hypermethylated with low expression and have crucial roles in
breast carcinogenesis included NDRG2, CCND2 and
FGF2. NDRG2 plays a role in stress responses, cell
proliferation and differentiation (55). A previous study showed that this
gene can regulate CD24 expression to decrease metastasis in breast
cancer (56). Thus,
hypermethylation that leads to silencing of NDRG2 might
contribute to metastasis of breast cancer cells. CCND2 is
involved in cell cycle regulation as it regulates the transition
from G1 to S phase during cell cycle (34). This gene was found to be
inactivated and hypermethylated in other studies (34,57).
In addition, FGF2 is a growth factor that regulates
epithelial cell proliferation, migration and angiogenesis (58). Loss of expression in FGF2
might probably lead to uncontrolled cell proliferation and
metastasis of breast cancer.
Our integrative analysis approach showed that
PTPRZ1, AGTR1, PTN and CXCL2 were
hypermethylated and showed low expression. This observation
contradicts with other studies on gene expression (32,33,39,40).
However, those studies have no information on the methylation
status of these genes.
We discovered 13 hypermethylated genes with high
expression. Three genes, namely COL1A2, FBN1 and
COL4A1, were previously reported in breast cancer (59–61).
Previous studies showed that COL1A2 and FBN1 were
hypermethylated and down regulated in the breast cancer cell lines
(61,62). However, the present study showed
that both of these genes were hypermethylated and upregulated in
breast tumour tissues. The difference between the results might be
due to the nature of the biological samples which gave distinct
gene expression patterns (63).
For gene COL4A1, its protein was reported to be elevated in
the serum of primary breast cancer patients (60). This finding somewhat supported our
results as COL4A1 was also upregulated. It has been
suggested that the positive association between hypermethylation
and gene expression could probably be explained by the presence of
long non-coding RNA (64).
We further compared our results with the genes
listed in the MammaPrint assay (Agendia, Amsterdam, The
Netherlands), Oncotype DX assay (Genomic Health, Redwood City, CA,
USA) and Ion Ampliseq™ Comprehensive Cancer Panel (Life
Technologies, Carlsbad, CA, USA) to check for overlapping genes.
There were only 5 genes (CASC5, CCND2, COL1A1,
EPHB1 and KIT) found to overlap with the gene list in
Ion Ampliseq™ Comprehensive Cancer Panel whereas none of our genes
were in the gene list of the MammaPrint and Oncotype DX assays.
For the pathway analysis, two of the enriched
pathways which have been identified are the focal adhesion and
extracellular matrix-receptor interaction. There were 7 genes
(PAK7, COL4A1, CCND2, COL1A2,
RELN, COL1A1 and THBS2) involved in focal
adhesion while 5 genes (COL4A1, COL1A2, RELN,
COL1A1 and THBS2) involved in extracellular
matrix-receptor interactions. PAK7 belongs to the PAK family
of Ser/Thr protein kinases which are known to regulate cytoskeleton
dynamics, proliferation and cell survival signalling (65) while RELN encodes a
glycoprotein that acts as a regulator for neuronal migration
(66). Previous study showed that
RELN was inactivated by hypermethylation and silencing of
RELN was associated with poor prognosis in breast cancer
(43). Finally, THBS2 is
involved in inhibiting angiogenesis (67). Aberrant methylation of these genes
might disrupt the pathways involved and further contributes to
breast cancer.
In conclusion, we have successfully performed the
integrative genomic analysis from DNA methylation and gene
expression profiling datasets and revealed a focused list of key
genes in breast cancer. This list will further be used to study in
more detail the pathogenesis of breast cancer using the interactive
genomics data.
Acknowledgements
The present study was funded by grant from the
Ministry of Science, Technology and Innovation
(07-05-MGI-GMB016).
References
|
1
|
Jones PA: Overview of cancer epigenetics.
Semin Hematol. 42:S3–S8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weber M, Hellmann I, Stadler MB, et al:
Distribution, silencing potential and evolutionary impact of
promoter DNA methylation in the human genome. Nat Genet.
39:457–466. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davis CD and Uthus EO: DNA methylation,
cancer susceptibility, and nutrient interactions. Exp Biol Med
(Maywood). 229:988–995. 2004.PubMed/NCBI
|
|
4
|
Jimenez-Useche I and Yuan C: The effect of
DNA CpG methylation on the dynamic conformation of a nucleosome.
Biophys J. 103:2502–2512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ito K, Barnes PJ and Adcock IM:
Glucocorticoid receptor recruitment of histone deacetylase 2
inhibits interleukin-1beta-induced histone H4 acetylation on
lysines 8 and 12. Mol Cell Biol. 20:6891–6903. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jovanovic J, Ronneberg JA, Tost J and
Kristensen V: The epigenetics of breast cancer. Mol Oncol.
4:242–254. 2010. View Article : Google Scholar
|
|
7
|
Beisel C and Paro R: Silencing chromatin:
comparing modes and mechanisms. Nat Rev Genet. 12:123–135. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang BH, Parkin DM, Cai L and Zhang ZF:
Cancer burden and trends in the Asian Pacific Rim region. Asian Pac
J Cancer Prev. 5:96–117. 2004.PubMed/NCBI
|
|
9
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar
|
|
10
|
Zhang X, Sun Q, Shan M, et al: Promoter
hypermethylation of ARID1A gene is responsible for its low mRNA
expression in many invasive breast cancers. PloS One. 8:e539312013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bubnov V, Moskalev E, Petrovskiy Y, Bauer
A, Hoheisel J and Zaporozhan V: Hypermethylation of TUSC5 genes in
breast cancer tissue. Exp Oncol. 34:370–372. 2012.PubMed/NCBI
|
|
12
|
Heyn H, Carmona FJ, Gomez A, et al: DNA
methylation profiling in breast cancer discordant identical twins
identifies DOK7 as novel epigenetic biomarker. Carcinogenesis.
34:102–108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Faryna M, Konermann C, Aulmann S, et al:
Genome-wide methylation screen in low-grade breast cancer
identifies novel epigenetically altered genes as potential
biomarkers for tumor diagnosis. FASEB J. 26:4937–4950. 2012.
View Article : Google Scholar
|
|
14
|
Lo Nigro C, Monteverde M, Lee S, et al:
NT5E CpG island methylation is a favourable breast cancer
biomarker. Br J Cancer. 107:75–83. 2012.PubMed/NCBI
|
|
15
|
Kim MS, Lee J, Oh T, et al: Genome-wide
identification of OTP gene as a novel methylation marker of
breast cancer. Oncol Rep. 27:1681–1688. 2012.PubMed/NCBI
|
|
16
|
Novak P, Jensen T, Oshiro MM, Watts GS,
Kim CJ and Futscher BW: Agglomerative epigenetic aberrations are a
common event in human breast cancer. Cancer Res. 68:8616–8625.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coolen MW, Stirzaker C, Song JZ, et al:
Consolidation of the cancer genome into domains of repressive
chromatin by longrange epigenetic silencing (LRES) reduces
transcriptional plasticity. Nat Cell Biol. 12:235–246.
2010.PubMed/NCBI
|
|
18
|
Novak P, Jensen T, Oshiro MM, et al:
Epigenetic inactivation of the HOXA gene cluster in breast
cancer. Cancer Res. 66:10664–10670. 2006.
|
|
19
|
Ehrlich M: DNA methylation in cancer: too
much, but also too little. Oncogene. 21:5400–5413. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Costa FF, Paixao VA, Cavalher FP, et al:
SATR-1 hypomethylation is a common and early event in breast
cancer. Cancer Genet Cytogenet. 165:135–143. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berman BP, Weisenberger DJ, Aman JF, et
al: Regions of focal DNA hypermethylation and long-range
hypomethylation in colorectal cancer coincide with nuclear
lamina-associated domains. Nat Genet. 44:40–46. 2012. View Article : Google Scholar
|
|
22
|
Li M, Balch C, Montgomery JS, et al:
Integrated analysis of DNA methylation and gene expression reveals
specific signaling pathways associated with platinum resistance in
ovarian cancer. BMC Med Genomics. 2:342009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cancer Genome Atlas Network. Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun Z, Asmann YW, Kalari KR, et al:
Integrated analysis of gene expression, CpG island methylation, and
gene copy number in breast cancer cells by deep sequencing. PloS
One. 6:e174902011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mokhtar NM, Ramzi NH, Yin-Ling W, Rose IM,
Hatta Mohd Dali AZ and Jamal R: Laser capture microdissection with
genome-wide expression profiling displayed gene expression
signatures in endometrioid endometrial cancer. Cancer Invest.
30:156–164. 2012. View Article : Google Scholar
|
|
26
|
Bibikova M, Le J, Barnes B, et al:
Genome-wide DNA methylation profiling using Infinium®
assay. Epigenomics. 1:177–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eads CA, Danenberg KD, Kawakami K, et al:
MethyLight: a high-throughput assay to measure DNA methylation.
Nucleic Acids Res. 28:E322000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mascolo M, Siano M, Ilardi G, et al:
Epigenetic disregulation in oral cancer. Int J Mol Sci.
13:2331–2353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qu Z, Fu J, Yan P, Hu J, Cheng SY and Xiao
G: Epigenetic repression of PDZ-LIM domain-containing protein 2:
implications for the biology and treatment of breast cancer. J Biol
Chem. 285:11786–11792. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yano M, Toyooka S, Tsukuda K, et al:
Aberrant promoter methylation of human DAB2 interactive protein
(hDAB2IP) gene in lung cancers. Int J Cancer. 113:59–66. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeschke J, Van Neste L, Glockner SC, et
al: Biomarkers for detection and prognosis of breast cancer
identified by a functional hypermethylome screen. Epigenetics.
7:701–709. 2012. View Article : Google Scholar
|
|
32
|
Rhodes DR, Ateeq B, Cao Q, et al: AGTR1
overexpression defines a subset of breast cancer and confers
sensitivity to losartan, an AGTR1 antagonist. Proc Natl Acad Sci
USA. 106:10284–10289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bieche I, Chavey C, Andrieu C, et al: CXC
chemokines located in the 4q21 region are up-regulated in breast
cancer. Endocr Relat Cancer. 14:1039–1052. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Evron E, Umbricht CB, Korz D, et al: Loss
of cyclin D2 expression in the majority of breast cancers is
associated with promoter hypermethylation. Cancer Res.
61:2782–2787. 2001.PubMed/NCBI
|
|
35
|
Veeck J, Bektas N, Hartmann A, et al: Wnt
signalling in human breast cancer: expression of the putative Wnt
inhibitor Dickkopf-3 (DKK3) is frequently suppressed by promoter
hypermethylation in mammary tumours. Breast Cancer Res.
R82:2008.PubMed/NCBI
|
|
36
|
Cao XC, Zhang WR, Cao WF, et al:
Aquaporin3 is required for FGF-2-induced migration of human breast
cancers. PloS One. 8:e567352013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sidiropoulos M, Pampalakis G, Sotiropoulou
G, Katsaros D and Diamandis EP: Downregulation of human kallikrein
10 (KLK10/NES1) by CpG island hypermethylation in breast, ovarian
and prostate cancers. Tumour Biol. 26:324–336. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fernandez SV, Snider KE, Wu YZ, Russo IH,
Plass C and Russo J: DNA methylation changes in a human cell model
of breast cancer progression. Mutat Res. 688:28–35. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Garver RI Jr, Radford DM, Donis-Keller H,
Wick MR and Milner PG: Midkine and pleiotrophin expression in
normal and malignant breast tissue. Cancer. 74:1584–1590. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Perez-Pinera P, Garcia-Suarez O,
Menendez-Rodriguez P, et al: The receptor protein tyrosine
phosphatase (RPTP)β/ζ is expressed in different subtypes of human
breast cancer. Biochem Biophys Res Commun. 362:5–10. 2007.
|
|
41
|
Lo PK, Mehrotra J, D’Costa A, et al:
Epigenetic suppression of secreted frizzled related protein 1
(SFRP1) expression in human breast cancer. Cancer Biol Ther.
5:281–286. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ko CD, Kim JS, Ko BG, et al: The meaning
of the c-kit proto-oncogene product in malignant transformation in
human mammary epithelium. Clin Exp Metastasis. 20:593–597. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stein T, Cosimo E, Yu X, et al: Loss of
reelin expression in breast cancer is epigenetically controlled and
associated with poor prognosis. Am J Pathol. 177:2323–2333. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dote H, Toyooka S, Tsukuda K, et al:
Aberrant promoter methylation in human DAB2 interactive protein
(hDAB2IP) gene in breast cancer. Clin Cancer Res.
10:2082–2089. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hoang BH, Kubo T, Healey JH, et al:
Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma
cells by modulating the Wnt-beta-catenin pathway. Cancer Res.
64:2734–2739. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Goyal J, Smith KM, Cowan JM, Wazer DE, Lee
SW and Band V: The role for NES1 serine protease as a novel tumor
suppressor. Cancer Res. 58:4782–4786. 1998.PubMed/NCBI
|
|
47
|
Huang HE, Chin SF, Ginestier C, et al: A
recurrent chromosome breakpoint in breast cancer at the
NRG1/neuregulin 1/heregulin gene. Cancer Res. 64:6840–6844. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kioulafa M, Kaklamanis L, Stathopoulos E,
Mavroudis D, Georgoulias V and Lianidou ES: Kallikrein 10 (KLK10)
methylation as a novel prognostic biomarker in early breast cancer.
Ann Oncol. 20:1020–1025. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Veeck J, Niederacher D, An H, et al:
Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer
is associated with unfavourable prognosis. Oncogene. 25:3479–3488.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dahl E, Veeck J, An H, et al: Epigenetic
inactivation of the WNT antagonist SFRP1 in breast cancer. Verh
Dtsch Ges Pathol. 89:169–177. 2005.(In German).
|
|
51
|
Xie D, Gore C, Zhou J, et al: DAB2IP
coordinates both PI3K-Akt and ASK1 pathways for cell survival and
apoptosis. Proc Natl Acad Sci USA. 106:19878–19883. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang X, Li N, Li X, et al: Low expression
of DAB2IP contributes to malignant development and poor prognosis
in hepatocellular carcinoma. J Gastroenterol Hepatol. 27:1117–1125.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Henderson DJ, Phillips HM and Chaudhry B:
Vang-like 2 and noncanonical Wnt signaling in outflow tract
development. Trends Cardiovasc Med. 16:38–45. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cadigan KM and Nusse R: Wnt signaling: a
common theme in animal development. Genes Dev. 11:3286–3305. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang J, Liu J, Li X, et al: The physical
and functional interaction of NDRG2 with MSP58 in cells. Biochem
Biophys Res Commun. 352:6–11. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zheng J, Liu Q, Li Y, et al: NDRG2
expression regulates CD24 and metastatic potential of breast cancer
cells. Asian Pac J Cancer Prev. 11:1817–1821. 2010.PubMed/NCBI
|
|
57
|
Euhus DM, Bu D, Milchgrub S, et al: DNA
methylation in benign breast epithelium in relation to age and
breast cancer risk. Cancer Epidemiol Biomarkers Prev. 17:1051–1059.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bikfalvi A, Klein S, Pintucci G and Rifkin
DB: Biological roles of fibroblast growth factor-2. Endocr Rev.
18:26–45. 1997.
|
|
59
|
Loss LA, Sadanandam A, Durinck S, et al:
Prediction of epigenetically regulated genes in breast cancer cell
lines. BMC Bioinformatics. 11:3052010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mazouni C, Arun B, Andre F, et al:
Collagen IV levels are elevated in the serum of patients with
primary breast cancer compared to healthy volunteers. Br J Cancer.
99:68–71. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Summers KM, Bokil NJ, Baisden JM, et al:
Experimental and bioinformatic characterisation of the promoter
region of the Marfan syndrome gene, FBN1. Genomics.
94:233–240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sengupta PK, Smith EM, Kim K, Murnane MJ
and Smith BD: DNA hypermethylation near the transcription start
site of collagen α2(I) gene occurs in both cancer cell lines and
primary colorectal cancers. Cancer Res. 63:1789–1797.
2003.PubMed/NCBI
|
|
63
|
Ross DT and Perou CM: A comparison of gene
expression signatures from breast tumors and breast tissue derived
cell lines. Dis Markers. 17:99–109. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Clark MB, Johnston RL, Inostroza-Ponta M,
et al: Genome-wide analysis of long noncoding RNA stability. Genome
Res. 22:885–898. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kumar R, Gururaj AE and Barnes CJ:
p21-activated kinases in cancer. Nat Rev Cancer. 6:459–471. 2006.
View Article : Google Scholar
|
|
66
|
Dohi O, Takada H, Wakabayashi N, et al:
Epigenetic silencing of RELN in gastric cancer. Int J Oncol.
36:85–92. 2010.
|
|
67
|
Bornstein P: Thrombospondins function as
regulators of angiogenesis. J Cell Commun Signal. 3:189–200. 2009.
View Article : Google Scholar
|