Introduction
Vitamin C (also known as an ascorbate or ascorbic
acid) is an essential micronutrient and considered to function as
an anti-cancer drug (1,2). Many studies have reported the
importance of vitamin C cytotoxicity. However, the use of vitamin C
as an anticancer therapeutic agent and its effectiveness remain
debatable (3–6). In previous reports, 0.3–20 mM of
vitamin C has been used for a pharmacological concentration in
various cancer cell lines (4,7).
This range of concentration is considered to effectively induce
cancer cell death in vitro and to inhibit tumor growth in
vivo. This can be achieved through intravenous (i.v.) or
intraperitoneal (i.p.) injection, but not through oral dosing
(4,8). Vitamin C is thought to modify the
expression levels of the proteins that are involved in many
different signaling pathways, which ultimately trigger cancer cells
to be more susceptible to vitamin C (9,10).
Most importantly, vitamin C has a significant advantage because it
can selectively kill cancer cells but not normal cells in
pharmacological concentrations. It is a crucial characteristic to
be an ideal anticancer drug (11,12).
Considering these facts, vitamin C can be regarded as an ideal
therapeutic agent. However, its action mechanism has not yet been
clearly defined. We therefore have attempted to elucidate the
physiological mechanism how vitamin C can act as an anticancer
agent. We previously showed that vitamin C stabilizes p53 by
inducing MDM2 polyubiquitination/degradation. Stabilized p53 in
turn aggravates intracellular oxidative stress and consequently
causes cancer cells to be more sensitive to vitamin C (13). In this process, p53-dependent
enhancement of vitamin C cytotoxicity is caused by increased ROS
(reactive oxygen species) generation via a differentially regulated
p53 transcriptional network (13).
In addition, we found that the p34SEI-1 protein level
was decreased in response to vitamin C treatment.
p34SEI-1 has been shown to have multiple biological
functions in cells (14–17). In particular, p34SEI-1
plays vital roles in cell cycle as a transcriptional co-factor and
in apoptosis as an anti-apoptotic oncoprotein (14,18,19).
We previously showed that it inhibits cancer cell death by
stabilizing XIAP (X-linked inhibitor of apoptosis protein), a
potent inhibitor of apoptosis and inhibiting ROS-induced cell death
through suppression of ASK1 (18,19).
Therefore, p34SEI-1 downregulation could be one of
efficient means of suppressing tumorigenesis. On the basis of these
observations, we hypothesized that decreased p34SEI-1
might be responsible for the p53-mediated vitamin C cytotoxicity in
cancer cells.
Considering the facts that vitamin C induces p53
activation and p34SEI-1 downregulation at the protein
level, we initially focused on the p53 function that controls the
stability of the target proteins by regulating ubiquitination and
subsequent proteasomal degradation among the p53-mediated many
other regulating systems. This process can be mediated by three
different types of enzymes (E1, E2 and E3) that are required for
the ubiquitination-proteasomal pathway (20–23).
Among them, the expression of SIAH1, E3 ubiquitin-protein ligase,
is strongly dependent on p53 (24,25).
It is a member of the SIAH family that belong to the ring domain
ubiquitin ligases (26). SIAH1 is
involved in numerous cellular processes including apoptosis, tumor
suppression, cell cycle, axon guidance, transcription regulation
and tumor necrosis factor signaling (27–30).
In particular, SIAH1 negatively affects cancer cell survival and
proliferation. Thus, deregulation of the SIAH1 expression is
strongly related to cancer progression (31–33).
Accordingly, SIAH1 activation could be an important factor to block
tumorigenesis (33).
In this report, we propose that vitamin C treatment
of cancer cells enhances p53 activity, which in turn induces the
SIAH1-mediated polyubiquitination/degradation of the
p34SEI-1 oncoprotein and ultimately increases cancer
cells cytotoxicity.
Materials and methods
Cell lines, cell culture and vitamin C
preparation
HCT116+/+ (p53 wild-type) and
HCT116−/− (p53 null-type) colon cancer cell lines were
provided by Dr Bert Vogelstein (Johns Hopkins University, USA) and
MCF7 was obtained from the ATCC (American Type Culture Collection,
USA). Each cell line was cultured in DMEM (Dulbecco’s modified
Eagle’s medium) medium (WelGENE, Korea) supplemented with 10% fetal
bovine serum and 1% antibiotic-antimycotic (both from Gibco BRL,
USA). Cells were cultured at 37°C in a humidified atmosphere
composed of 95% air and 5% CO2. Vitamin C was adjusted
to pH 7.0 using sodium hydroxide and it was prepared immediately
prior to use.
Western blotting
Western blot analysis was performed as previously
described (13). The antibodies
used in this study were purchased as follows: p53 (Santa Cruz
Biotechnology, sc-126, USA), phospho-p53 (Ser46) (Santa Cruz
Biotechnology, sc-101764), phospho-p53 (Ser15) (Cell Signaling
Technology, cat. no. 9284, USA), phosphor-p38MAPK (Thr180/Tyr182)
(Cell Signaling Technology, cat. no. 9211), p21 (Santa Cruz
Biotechnology, sc-397), Cdc25C (Santa Cruz Biotechnology, sc-6950),
XIAP (Santa Cruz Biotechnology, sc-8789), Caspase 9 (Cell Signaling
Technology, cat. no. 9508), Cleaved caspase 9 (Cell Signaling
Technology, cat. no. 9505), p34SEI-1 (Enzo Life
Sciences, cat. no. ALX-804-645, Australia), SIAH1 (Santa Cruz
Biotechnology, sc-5505), and γ-tubulin (Santa Cruz Biotechnology,
sc-7396).
Transfection
Transfection was performed after plating cells in 60
mm3 dish with 90% confluence of cells using
Lipofectamine 2000 (Invitrogen, USA). To induce p53 overexpression,
cells were transiently transfected with either a control vector
(pcDNA3.1) or a p53 overexpressing vector (pcDNA3.1/p53) for 48–72
h. To suppress endogenous p53 level, cells were transfected with a
control vector (pLKO.1) or a p53 shRNA silencing vector
(pLKO.1/p53-shRNA) for 48 h (13).
To induce overexpression of wild-type SIAH1 or mutant type
SIAH1C44S, cells were transfected with pCMV-SPORT6/SIAH1
or pCMV-SPORT6/SIAH1C44S, respectively. pCMV-SPORT6
plasmid was used as a control vector. pCMV-SPORT6/SIAH1 plasmid
(clone number hMU004814) was purchased from Korea Human Gene Bank
(Medical Genomics Research center, KRIBB, Korea).
pCMV-SPORT6/SIAH1C44S plasmid was constructed by
introducing SIAH1C44S mutation into wild-type SIAH1 gene
in pCMV-SPORT6/SIAH1 (34).
Mutagenesis was achieved by using site directed mutagenesis method
(Quick change site-directed mutagenesis kit, Stratagene, cat. no.
200519, USA) and the following primer pairs: SIAH1: forward,
(5′-GTCTTTTTGAGTGTCC AGTCAGCTTTGACTATGTGTTAC-3′); and reverse
(5′-GTAA CACATAGTCAAAGCTGACTGGACACTCAAAAAGAC-3′). To suppress the
endogenous SIAH1 expression, cells were transfected with shRNA
control vector (pLKO.1) or a SIAH1 silencing plasmid
(pLKO.1/SIAH1-shRNA) for 48 h. For modulating of
p34SEI-1 expression level, cells were transfected with a
control vector (pcDNA3.1), a p34SEI-1 overexpressing
vector (pcDNA3.1/p34SEI-1), scrambled siRNA (scRNA) or
p34SEI-1 small interfering RNA (si-p34SEI-1,
5′-CAGUGUGG CUGACAACUUACUGG-3′) for 48–72 h. The expression level
of each protein was examined by using western blot analysis.
Flow cytometric analysis
To detect the cell cycle arrest, vitamin C treated
cells were harvested and washed three times with PBS
(phosphate-buffered saline). Resulting cells were added with 70%
ethanol immediately and maintained at 4°C for 1–3 h to allow cell
fixation. Cells were washed with PBS and then suspended in 0.1
mg/ml propidium iodide solution at 4°C, followed by addition of 20
μl of 10 mg/ml RNase and incubation at 37°C for 30 min. Stained
samples were analyzed by using flow cytometry (BD Biosciences,
USA), in which 1×104 cells were recorded for each
sample. Apoptosis was assessed by Annexin V-binding and propidium
iodide staining. Cells were harvested and washed three times with
PBS and then incubated with 1 μl of Annexin V-fluorescein
isothiocynate (FITC) and 2.5 μl of propidium iodide (Enzo, cat. no.
ADI-ADK-700) for 15 min on ice in the dark and analyzed on a
FACSCanto machine (BD Biosciences).
Measurement of vitamin C
cytotoxicity
Cell viability was determined by employing the MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide,
Sigma-Aldrich, USA) assay as described previously (13). HCT116 cells in 96-well plates were
treated with 0.3 mM of vitamin C for 8 h. The resulting cells were
then incubated with MTT for 4 h at 37°C and the cell cytotoxicity
was measured by using Perkin-Elmer Wallac 1420 Victor2™ microplate
reader (Perkin-Elmer, USA). Cell death was measured by using trypan
blue exclusion assay.
Colony forming assay
Cells were seeded at a density of 2×102
per 60 mm3 dish and treated with 2 mM of vitamin C for
24 h after pretreatment with or without 2 mM of NAC
(N-acetylL-cysteine) and then cultured for 17 days. Colonies were
fixed with 3.7% formaldehyde and counted after staining with 0.05%
crystal violet solution.
Co-immunoprecipitation (co-IP) and
ubiquitination assay
Cell lysates were prepared by lysing cells in RIPA
buffer with protease inhibitors, followed by pre-clearing with
protein A/G Sepharose (Santa Cruz Biotechnology, sc-2020). The
pre-cleared lysates were incubated with either
anti-p34SEI-1 (Enzo Life Sciences, cat. no. ALX-804-645)
or anti-SIAH1 (Santa Cruz Biotechnology, sc-5505) antibodies for 16
h at 4°C with continuous agitation, in which the protein A/G
Sepharose was added. The resulting complex with antibody and
agarose A/G bead was centrifuged at 10,000 × g for 5 min and then
washed with RIPA buffer three times. The proteins were eluted from
the beads by boiling in an SDS sample buffer. They were then
analyzed using a western blotting with the corresponding
antibodies. For the p34SEI-1 ubiquitination experiments,
HCT116+/+ or HCT116−/− cells were transfected
with indicating plasmids. The cells were then treated with 20 μM of
MG132 proteasome inhibitor (A.G. Scientific, cat. no. M-1157, USA)
for 16 h and lysed with RIPA buffer containing protease inhibitors.
The lysates were centrifuged to obtain cytosolic proteins.
Ubiquitinated p34SEI-1 was immunoprecipitated with
anti-p34SEI-1 antibody, followed by immunoblotting with
anti-Ub antibody (Santa Cruz Biotechnology, sc-8017).
Results
Vitamin C cytotoxicity can be enhanced by
p53
To confirm the dependence of vitamin C induced
cytotoxicity on p53 (13), the
effect of p53 on vitamin C induced cytotoxicity was checked after
p53 wild-type HCT116+/+ and p53 null mutant-type
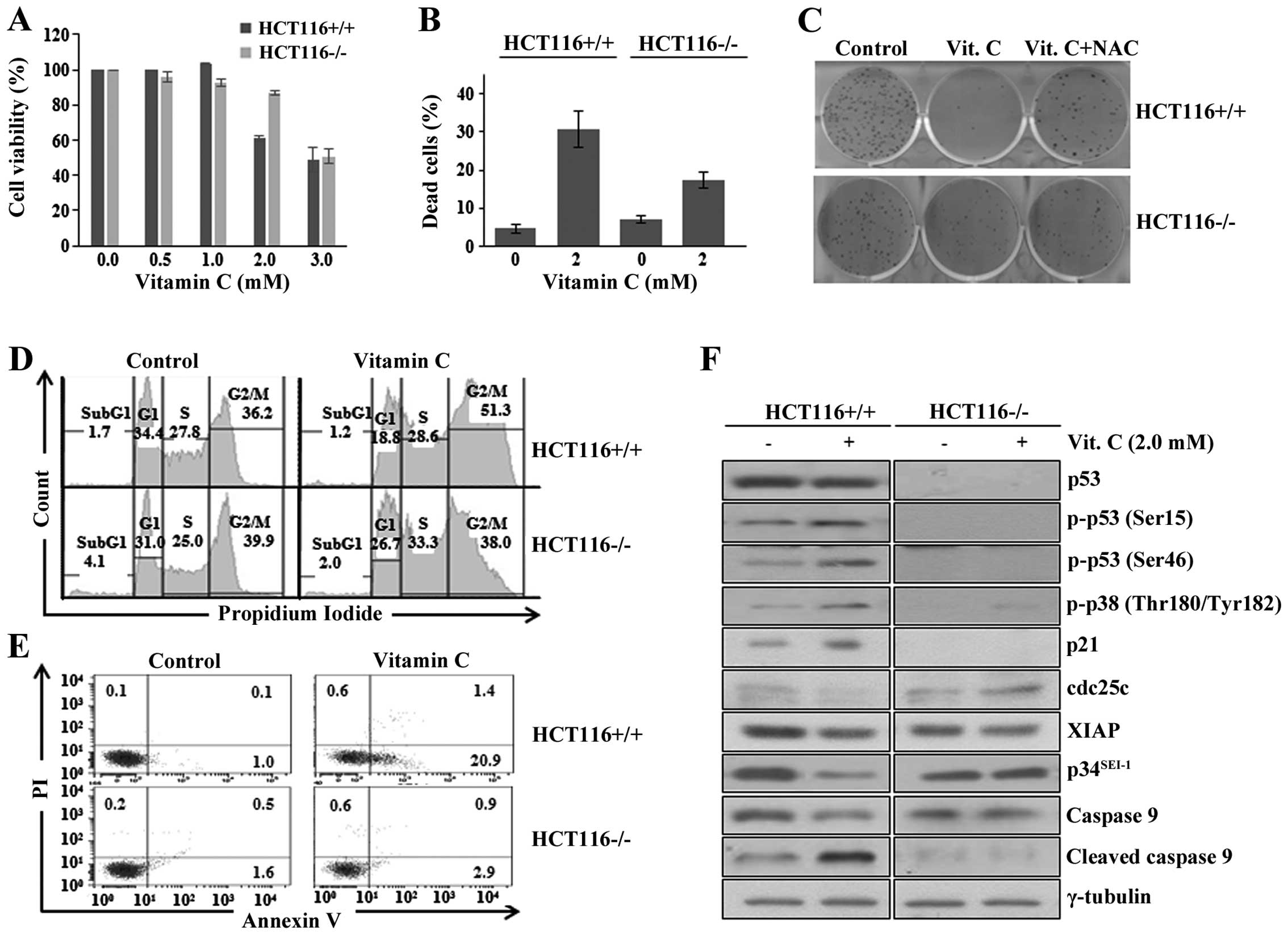
HCT116−/− colon cancer cells were treated with indicated
doses of vitamin C (Fig. 1A). Cell
viability was significantly decreased in HCT116+/+,
suggesting that p53 enhances vitamin C induced cytotoxicity. Cell
death of vitamin C-treated HCT116+/+ was approximately
two times higher than that of the HCT116−/− cells
(Fig. 1B). Colony forming assay
was employed to further examine the relationship between vitamin C
cytotoxicity and p53. Colony formation of HCT116+/+
cells was greatly decreased by vitamin C treatment compared to
those of control and HCT116−/− cells (Fig. 1C). The treatment of NAC antioxidant
alleviated vitamin C-mediated decrease in colony forming of
HCT116+/+, which is consistent with our previous results
(13). The data indicate that
vitamin C kills cancer cells by enhancing ROS production in the
presence of p53. Next question was how p53 can positively affect
vitamin C induced cytotoxicity in cancer cells. Previous reports
have shown that vitamin C affects various signaling pathways
related with cell cycle and apoptosis (12,35–37).
In our present study, vitamin C induced G2/M arrest at a higher
level in HCT116+/+ cells compared to that in
HCT116−/− cells (Fig.
1D). Furthermore, much higher level of apoptosis was also
induced in HCT116+/+ than that of HCT116−/−
cells in response to vitamin C treatment (Fig. 1E). These findings indicate that
both cell cycle arrest and apoptosis was much more sensitive to
vitamin C treatment in wild-type p53 expressing HCT116 cells. Based
on these results, western blot analysis was performed to examine
the changes in expression levels of genes that play vital roles in
cell cycle arrest and apoptosis in vitamin C-treated HCT116 cell
lines. Vitamin C treatment to HCT116+/+ resulted in
phosphorylation of p53 on serine 15 (p-p53 Ser15) and serine 46
(p-p53 Ser46) residues probably due to p38MAPK activation (Fig. 1F). This conclusion was supported by
treatment of p38 inhibitor, SB203580, in which phosphorylation
levels of both p38 and p53 (Ser46) were decreased (data not shown).
It is well known that the phosphorylation of p53 on the serine 15
residue leads to p53 stabilization and activation and the
phosphorylation of p53 on the serine 46 residue induces apoptosis
(38,39) by activating p38MAPK via
phosphorylation on two residues, threonine 180 and threonine 182
(Thr180/Tyr182) (13,40,41).
The effect of vitamin C on cell cycle was checked by examining the
expression levels of p21 and cdc25c, major cell cycle regulators.
Vitamin C treatment increased and decreased p21 and cdc25c protein
levels only in HCT116+/+ cells, respectively. It
suggests that vitamin C treatment induced G2/M cell cycle arrest in
a p53-dependent manner. Next, we checked the altered expression
levels of apoptosis relating proteins, XIAP, p34SEI-1,
and caspase 9. XIAP plays as a potent inhibitor of apoptosis by
inhibiting caspase 3, 7 and 9 activation and its overexpression
confers resistance to tumor chemotherapy (42,43).
Interestingly, XIAP expression is known to be negatively regulated
by p53 (44). Therefore, XIAP
protein level was checked to examine involvement of XIAP in vitamin
C-mediated apoptosis. XIAP expression was significantly decreased
in HCT116+/+ cells but not in HCT116−/− cells
in response to vitamin C. According to our previous result, XIAP
stability can be greatly increased by p34SEI-1 (19). Very interestingly,
p34SEI-1 protein level was decreased only in
HCT116+/+ cells but not in HCT116−/− cells.
It suggests that p53 is responsible for the downregulation of the
XIAP and p34SEI-1 proteins in response to the vitamin C
treatment. This conclusion was confirmed by checking caspase 9
activation, one of direct targets of XIAP (45,46).
Cleaved active form of caspase 9 was found to be increased in
vitamin C-treated HCT116+/+ but not in
HCT116−/− (Fig. 1F).
The data imply that vitamin C may initiate the apoptotic process
through downregulating XIAP and p34SEI-1 in p53
wild-type cells. Considering that p34SEI-1 plays
important roles in tumorigenesis as a vital regulator of cell cycle
and apoptosis, we hypothesized that p34SEI-1 might play
critical roles in p53-dependent vitamin C-mediated cell cycle
arrest and apoptosis.
p34SEI-1 is involved in
vitamin C-induced cell death
To determine whether p34SEI-1 is involved
in the p53-dependent vitamin C cytotoxicity, we examined whether
the altered expression of p34SEI-1 modulates the vitamin
C-mediated cell death in a p53-dependent manner. In
HCT116+/+ cells, p34SEI-1 overexpression
alleviated cell death after vitamin C treatment compared with
vector only transfected control cells (Fig. 2A). In contrast, p34SEI-1
suppression in HCT116−/− cells increased cell death
compared with the control cells (Fig.
2A). These observations indicate that the p34SEI-1
negatively affects vitamin C-induced cell death. The next question
was how vitamin C downregulates p34SEI-1 expression in a
p53-dependent manner. Our result showed that p34SEI-1
mRNA level was not changed upon vitamin C treatment in either
HCT116+/+ or HCT116−/− cells (Fig. 2B). However, p34SEI-1
protein level was greatly decreased at 8 h after vitamin C
treatment but it was not changed by addition of MG132 (Fig. 2C). The data suggest that vitamin C
downregulated p34SEI-1 at the protein level
proteasome-dependently. To identify the E3 ligase responsible for
the p34SEI-1 ubiquitination/degradation in the vitamin C
treated cells, we initially focused on SIAH1 E3 ubiquitin ligase
because SIAH1 expression is strongly dependent on p53 (24,32,34).
Our data showed that SIAH1 protein level was significantly
increased in HCT116+/+ cells, but not in
HCT116−/− cells upon treatment of vitamin C, in which
inverse relationship was found between SIAH1 and
p34SEI-1 expression levels (Fig. 2D). This result implies that the
SIAH1 might be responsible for the p34SEI-1 degradation
in p53 expressing cells in response to vitamin C treatment. Our
collective data support the view that p34SEI-1 is
involved in vitamin C-induced cell death in p53 wild-type
cells.
Vitamin C induces polyubiquitination and
degradation of p34SEI-1 in a p53-dependent manner
In order to reconfirm the essential function of p53
in the p34SEI-1 downregulation via the SIAH1 E3 ligase,
HCT116−/− and HCT116+/+ cells were
transfected with a p53 overexpressing vector (pcDNA3.1/p53) or a
p53 silencing vector (pLKO.1/p53-shRNA), respectively, and then the
SIAH1 and p34SEI-1 expression levels were checked after
the vitamin C treatment. Upon the reintroduction of the wild-type
p53 into the p53-deficient HCT116−/−, vitamin C was able
to downregulate intracellular p34SEI-1 such as the case
of HCT116+/+cells (Fig.
3A). On the contrary, the p53 silenced HCT116+/+
cells exhibited the similar result to that of HCT116−/−
cells (Fig. 3A). This result
suggests that p53 is required for SIAH1 upregulation and
p34SEI-1 downregulation in response to the vitamin C. To
further elucidate whether the proteasome-dependent degradation of
p34SEI-1 is dependent on p53 under vitamin C treated
condition, immunoprecipitation was employed to examine whether
vitamin C can induce 34SEI-1 polyubiquitination
depending on p53. As shown in Fig.
3B, the vitamin C treatment significantly induced
polyubiquitination of the endogenous p34SEI-1 in the
HCT116+/+ and p53 overexpressing HCT116−/−
cells compared with the control cells. However, this effect was not
detected in the HCT116−/− and p53 silenced
HCT116+/+ cells (Fig.
3B). Our immunoprecipitation data also revealed that SIAH1
directly interacts with p34SEI-1 (Fig. 3B). Collectively, these results
suggest that p53 induce the direct interaction of SIAH1 with
p34SEI-1 and subsequent p34SEI-1
ubiquitination/degradation under the conditions of a vitamin C
treatment. In addition, we performed similar experiment in a
wild-type p53 expressing MCF7 breast cancer cell line.
p34SEI-1 was also polyubiquitinated and degraded by the
vitamin C in a wild-type p53 expressing MCF7 cells, but not in the
p53 silenced MCF7 cells (data not shown). Altogether, our results
strongly suggest that p53 induces the p34SEI-1
polyubiquitination and subsequent degradation under the conditions
of vitamin C treatment.
SIAH1 is responsible for
polyubiquitination/degradation of p34SEI-1
In order to confirm the SIAH1 requirement for the
p53-dependent p34SEI-1 polyubiquitination/degradation,
the effect of SIAH1 on the expression and polyubiquitination of
p34SEI-1 was analyzed in the SIAH1 suppressed
HCT116+/+ cells after the vitamin C treatment. The
vitamin C treatment did not induce the downregulation and
polyubiquitination of p34SEI-1 in SIAH1 deficient
HCT116+/+ cells even in the presence of the wild-type
p53 (Fig. 4A and B). This result
strongly suggests that SIAH1 is critically required for
p53-mediated p34SEI-1 polyubiquitination/degradation. In
an extended study, p34SEI-1 protein level was decreased
in wild-type SIAH1 expressing HCT116+/+ cells, but not
in ligase deficient mutant-type SIAH1C44S expressing
cells (Fig. 4C). Our data also
show that p34SEI-1 polyubiquitination was more increased
in wild-type SIAH1 expressing cells compared to control and
SIAH1C44S expressing HCT116+/+ cells
(Fig. 4D). Taken together, these
results strongly suggest that vitamin C-mediated
p34SEI-1 polyubiquitination/degradation is achieved in a
SIAH1-dependent manner.
Discussion
We previously reported that p53 makes cancer cells
more sensitive to vitamin C treatment and therefore increases its
cytotoxicity (13). According to
our data, vitamin C induces the MDM2 polyubiquitination/degradation
and consequently stabilizes p53 (13). It is in turn considered to modify
the expression levels of different target proteins that are
involved in many other signaling pathways to render cancer cells
more susceptible to vitamin C. In our present study, our collective
data support the view that p53 induces SIAH1-mediated
polyubiquitination/degradation of the p34SEI-1 oncogenic
protein upon vitamin C treatment. However, there are controversial
studies on the dependence of SIAH1 expression on p53. For example,
tumor suppressor HIPK2 stability is regulated by SIAH1-mediated
polyubiquitination/degradation under stress conditions, in which
SIAH1 expression level is increased by p53 (34). On the contrary, Frew et al
proposed that p53 overexpression has no effect on the expression of
SIAH1 genes in a p53-null mouse erythroleukemic cell line (47). We considered that different stimuli
or different cell types may evoke these discrepant results.
It has been reported that oxidative stress inducing
compounds, sodium nitroprusside or cucurbitacin B can activate p53,
which inhibits the cell cycle at the G2 phase (48–50).
High dose of vitamin C also causes oxidative stress to cancer cells
and functions as anticancer therapeutic agent (7,11).
Our current data showed that pharmacological concentration of
vitamin C induces cell cycle arrest at the G2 phase as well as
apoptosis in p53-dependent manner. p34SEI-1 is
well-known as a positive regulator of the cell cycle (15) and SIAH1 expression is also closely
related to G2 arrest (30). These
data imply that vitamin C inducible G2 arrest might be at least
partly influenced by SIAH1-mediated p34SEI-1 degradation
in the presence of p53.
In conclusion, our previous and current data suggest
that wild-type p53 is the prerequisite factor for stronger
anticancer effects of vitamin C, in which vitamin C cytotoxicity
appears to be achieved at least partly through the downregulation
of the p34SEI-1 in a SIAH1-dependent manner. Therefore,
p34SEI-1 can be developed as a new target protein for an
efficient therapeutic agent against various cancers.
Acknowledgements
This study was supported by the grant from Sookmyung
Women’s University (2012).
Abbreviations:
|
p34SEI-1
|
34 kDa protein encoding SEI-1
(Selected with Ink4a-1 as bait) gene
|
|
SIAH1
|
seven in absentia homolog 1
|
References
|
1
|
Du J, Cullen JJ and Buettner GR: Ascorbic
acid: chemistry, biology and the treatment of cancer. Biochim
Biophys Acta. 1826:443–457. 2012.PubMed/NCBI
|
|
2
|
Head KA: Ascorbic acid in the prevention
and treatment of cancer. Altern Med Rev. 3:174–186. 1998.PubMed/NCBI
|
|
3
|
Cameron E and Pauling L: Supplemental
ascorbate in the supportive treatment of cancer: prolongation of
survival times in terminal human cancer. Proc Natl Acad Sci USA.
73:3685–3689. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Q, Espey MG, Sun AY, et al:
Pharmacologic doses of ascorbate act as a prooxidant and decrease
growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci
USA. 105:11105–11109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Laurenzi V, Melino G, Savini I,
Annicchiarico-Petruzzelli M, Finazzi-Agro A and Avigliano L: Cell
death by oxidative stress and ascorbic acid regeneration in human
neuroectodermal cell lines. Eur J Cancer. 31A:463–466. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Verrax J and Calderon PB: Pharmacologic
concentrations of ascorbate are achieved by parenteral
administration and exhibit antitumoral effects. Free Radic Biol
Med. 47:32–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Q, Espey MG, Krishna MC, et al:
Pharmacologic ascorbic acid concentrations selectively kill cancer
cells: action as a pro-drug to deliver hydrogen peroxide to
tissues. Proc Natl Acad Sci USA. 102:13604–13609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoffer LJ, Levine M, Assouline S, et al:
Phase I clinical trial of i.v ascorbic acid in advanced malignancy.
Ann Oncol. 19:1969–1974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Park S, Ahn ES, Lee S, et al: Proteomic
analysis reveals upregulation of RKIP in S-180 implanted BALB/C
mouse after treatment with ascorbic acid. J Cell Biochem.
106:1136–1145. 2009. View Article : Google Scholar
|
|
10
|
Belin S, Kaya F, Duisit G, Giacometti S,
Ciccolini J and Fontes M: Antiproliferative effect of ascorbic acid
is associated with the inhibition of genes necessary to cell cycle
progression. PLoS One. 4:e44092009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen Q, Espey MG, Sun AY, et al: Ascorbate
in pharmacologic concentrations selectively generates ascorbate
radical and hydrogen peroxide in extracellular fluid in vivo. Proc
Natl Acad Sci USA. 104:8749–8754. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong SW, Jin DH, Hahm ES, et al: Ascorbate
(vitamin C) induces cell death through the apoptosis-inducing
factor in human breast cancer cells. Oncol Rep. 18:811–815.
2007.PubMed/NCBI
|
|
13
|
Kim J, Lee SD, Chang B, et al: Enhanced
antitumor activity of vitamin C via p53 in cancer cells. Free Radic
Biol Med. 53:1607–1615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hsu SI, Yang CM, Sim KG, Hentschel DM,
O’Leary E and Bonventre JV: TRIP-Br: a novel family of PHD zinc
finger- and bromodomain-interacting proteins that regulate the
transcriptional activity of E2F-1/DP-1. EMBO J. 20:2273–2285. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Muscarella P, Joo SH, et al:
Dissection of CDK4-binding and transactivation activities of
p34(SEI-1) and comparison between functions of p34(SEI-1) and
p16(INK4A). Biochemistry. 44:13246–13256. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ried T, Petersen I, Holtgreve-Grez H, et
al: Mapping of multiple DNA gains and losses in primary small cell
lung carcinomas by comparative genomic hybridization. Cancer Res.
54:1801–1806. 1994.PubMed/NCBI
|
|
17
|
Tang TC, Sham JS, Xie D, et al:
Identification of a candidate oncogene SEI-1 within a minimal
amplified region at 19q13.1 in ovarian cancer cell lines. Cancer
Res. 62:7157–7161. 2002.PubMed/NCBI
|
|
18
|
Hong SW, Shin JS, Lee YM, et al: p34
(SEI-1) inhibits ROS-induced cell death through suppression of
ASK1. Cancer Biol Ther. 12:421–426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hong SW, Kim CJ, Park WS, et al:
p34SEI-1 inhibits apoptosis through the stabilization of
the X-linked inhibitor of apoptosis protein: p34SEI-1 as
a novel target for anti-breast cancer strategies. Cancer Res.
69:741–746. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roos-Mattjus P and Sistonen L: The
ubiquitin-proteasome pathway. Ann Med. 36:285–295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ciechanover A and Schwartz AL: The
ubiquitin system: pathogenesis of human diseases and drug
targeting. Biochim Biophys Acta. 1695:3–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Komander D and Rape M: The ubiquitin code.
Annu Rev Biochem. 81:203–229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Berkers CR and Ovaa H: Drug discovery and
assay development in the ubiquitin-proteasome system. Biochem Soc
Trans. 38:14–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Matsuzawa S, Takayama S, Froesch BA,
Zapata JM and Reed JC: p53-inducible human homologue of Drosophila
seven in absentia (Siah) inhibits cell growth: suppression by
BAG-1. EMBO J. 17:2736–2747. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roperch JP, Lethrone F, Prieur S, et al:
SIAH-1 promotes apoptosis and tumor suppression through a network
involving the regulation of protein folding, unfolding, and
trafficking: identification of common effectors with p53 and
p21(Waf1). Proc Natl Acad Sci USA. 96:8070–8073. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qi J, Kim H, Scortegagna M and Ronai ZA:
Regulators and effectors of Siah ubiquitin ligases. Cell Biochem
Biophys. 67:15–24. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nemani M, Linares-Cruz G,
Bruzzoni-Giovanelli H, et al: Activation of the human homologue of
the Drosophila sina gene in apoptosis and tumor suppression. Proc
Natl Acad Sci USA. 93:9039–9042. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Telerman A and Amson R: The molecular
programme of tumour reversion: the steps beyond malignant
transformation. Nat Rev Cancer. 9:206–216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu H, Lin Y, Shi Y, et al: SIAH-1
interacts with mammalian polyhomeotic homologues HPH2 and affects
its stability via the ubiquitin-proteasome pathway. Biochem Biophys
Res Commun. 397:391–396. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoshibayashi H, Okabe H, Satoh S, et al:
SIAH1 causes growth arrest and apoptosis in hepatoma cells through
beta-catenin degradation-dependent and -independent mechanisms.
Oncol Rep. 17:549–556. 2007.PubMed/NCBI
|
|
31
|
Kim SY, Choi DW, Kim EA and Choi CY:
Stabilization of HIPK2 by escape from proteasomal degradation
mediated by the E3 ubiquitin ligase Siah1. Cancer Lett.
279:177–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matsuzawa SI and Reed JC: Siah-1, SIP, and
Ebi collaborate in a novel pathway for beta-catenin degradation
linked to p53 responses. Mol Cell. 7:915–926. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matsuo K, Satoh S, Okabe H, et al: SIAH1
inactivation correlates with tumor progression in hepatocellular
carcinomas. Genes Chromosomes Cancer. 36:283–291. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Winter M, Sombroek D, Dauth I, et al:
Control of HIPK2 stability by ubiquitin ligase Siah-1 and
checkpoint kinases ATM and ATR. Nat Cell Biol. 10:812–824. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Herst PM, Broadley KW, Harper JL and
McConnell MJ: Pharmacological concentrations of ascorbate
radiosensitize glioblastoma multiforme primary cells by increasing
oxidative DNA damage and inhibiting G2/M arrest. Free Radic Biol
Med. 52:1486–1493. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang JS, Cho D, Kim YI, et al: Sodium
ascorbate (vitamin C) induces apoptosis in melanoma cells via the
down-regulation of transferrin receptor dependent iron uptake. J
Cell Physiol. 204:192–197. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thomas CG, Vezyraki PE, Kalfakakou VP and
Evangelou AM: Vitamin C transiently arrests cancer cell cycle
progression in S phase and G2/M boundary by modulating the kinetics
of activation and the subcellular localization of Cdc25C
phosphatase. J Cell Physiol. 205:310–318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Khanna KK, Keating KE, Kozlov S, et al:
ATM associates with and phosphorylates p53: mapping the region of
interaction. Nat Genet. 20:398–400. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oda K, Arakawa H, Tanaka T, et al:
p53AIP1, a potential mediator of p53-dependent apoptosis, and its
regulation by Ser-46-phosphorylated p53. Cell. 102:849–862. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Perfettini JL, Castedo M, Nardacci R, et
al: Essential role of p53 phosphorylation by p38 MAPK in apoptosis
induction by the HIV-1 envelope. J Exp Med. 201:279–289. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim JE, Jin DH, Lee SD, et al: Vitamin C
inhibits p53-induced replicative senescence through suppression of
ROS production and p38 MAPK activity. Int J Mol Med. 22:651–655.
2008.PubMed/NCBI
|
|
42
|
Cheung HH, LaCasse EC and Korneluk RG:
X-linked inhibitor of apoptosis antagonism: strategies in cancer
treatment. Clin Cancer Res. 12:3238–3242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Schimmer AD, Dalili S, Batey RA and Riedl
SJ: Targeting XIAP for the treatment of malignancy. Cell Death
Differ. 13:179–188. 2006. View Article : Google Scholar
|
|
44
|
Fraser M, Leung BM, Yan X, Dan HC, Cheng
JQ and Tsang BK: p53 is a determinant of X-linked inhibitor of
apoptosis protein/Akt-mediated chemoresistance in human ovarian
cancer cells. Cancer Res. 63:7081–7088. 2003.PubMed/NCBI
|
|
45
|
Srinivasula SM, Hegde R, Saleh A, et al: A
conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO
regulates caspase activity and apoptosis. Nature. 410:112–116.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Datta R, Oki E, Endo K, Biedermann V, Ren
J and Kufe D: XIAP regulates DNA damage-induced apoptosis
downstream of caspase-9 cleavage. J Biol Chem. 275:31733–31738.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Frew IJ, Dickins RA, Cuddihy AR, et al:
Normal p53 function in primary cells deficient for Siah genes. Mol
Cell Biol. 22:8155–8164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cardaci S, Filomeni G, Rotilio G and
Ciriolo MR: Reactive oxygen species mediate p53 activation and
apoptosis induced by sodium nitroprusside in SH-SY5Y cells. Mol
Pharmacol. 74:1234–1245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Guo J, Wu G, Bao J, Hao W, Lu J and Chen
X: Cucurbitacin B induced ATM-mediated DNA damage causes G2/M cell
cycle arrest in a ROS-dependent manner. PLoS One. 9:e881402014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang J, Wu LJ, Tashino S, Onodera S and
Ikejima T: Protein tyrosine kinase pathway-derived ROS/NO
productions contribute to G2/M cell cycle arrest in
evodiamine-treated human cervix carcinoma HeLa cells. Free Radic
Res. 44:792–802. 2010. View Article : Google Scholar : PubMed/NCBI
|