Introduction
Mitochondria are highly dynamic organelles that
continuously elongate and divide to form a network throughout the
cell. The shape, location and function of mitochondria are defined
by an equilibrium between opposing fusion and fission events
(1). Mitochondrial dynamics are
crucial to homeostasis and cellular energy production (2,3).
Mitochondrial fusion and fission are precisely controlled by
various mitochondria-shaping proteins (4–6). In
mammalian cells, three large GTPases, mitofusin 1 (Mfn1), mitofusin
2 (Mfn2) and optic atrophy 1 (Opa1), are essential for
mitochondrial fusion. Mfns are integrated into the outer
mitochondrial membrane (OMM) and form homo- and hetero-oligomers,
which promote the tethering and fusion of OMMs from two different
mitochondria (1). Opa1 localizes
in complexes at the inner mitochondrial membrane (IMM) and drives
fusion on the IMM (7). A GTPase
cytosolic dynamin-related protein 1 (Drp1) mediates mitochondrial
fission in mammalian cells. To constrict and cut mitochondria
during mitochondrial fission, Drp1, which is located in the
cytosol, needs to be activated and assembled onto mitochondria
(8). Fisson 1 (Fis1), which is
located on the OMM, has been suggested to be a Drp1 receptor and
required for mitochondrial fission (1,9).
However, its action mechanism remains highly controversial
(10).
Various stimuli such as anticancer agents, hypoxia
or radiation can induce mitochondrial dysfunction. Damaged
mitochondria may be repaired or removed. Mitochondrial fission is a
crucial mechanism for removing dysfunctional mitochondria via
mitophagy (11). Crosstalk between
apoptosis, mitophagy and mitochondrial dynamics seems to be
critical to the overall fate of cells, i.e. death or survival
(12). Previous studies reported
that apoptosis-inducing agents generally induce mitochondrial
fragmentation (13–16).
Previous studies have reported that a Gamitrinib
variant containing triphenylphosphonium (G-TPP) readily accumulated
in the mitochondria of normal or tumor cells and inhibited the
tumor necrosis factor receptor-associated protein 1 (TRAP1) and
mitochondrial heat shock protein 90 (Hsp90) inside the
mitochondria. G-TPP reduced the IMM potential and caused the
discharge of apoptogenic proteins into the cytosol by activating
cyclophilin D-dependent mitochondrial permeability in various
cancer cells, which resulted in apoptosis (17,18).
Additionally, G-TPP binds to mitochondrial Hsp90, which causes
apoptosis by activating cyclophilin D-dependent mitochondrial
permeability transition in tumor cells (18,19).
We observed that G-TPP induces cell death and causes
Drp1-mediated mitochondrial elongation in Hep3B cells by increasing
the reactive oxygen species (ROS) level.
Materials and methods
Cell culture
Hep3B cells obtained from the American Type Culture
Collection were maintained in Dulbecco's modified Eagle's medium
(DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and
1% (v/v) penicillin-streptomycin (PS) at 37°C in a 5%
CO2 humid atmosphere. After 48 h of culture, the medium
was removed from the Hep3B cells, which were then washed with PBS
and incubated in the same fresh medium.
The establishment of
parkin-YFP-overexpressing Hep3B cells
The parkin-YFP plasmid was provided by Dr J. Chung
(Seoul National University, Seoul, Korea). To establish cell lines
that stably expressed parkin-YFP, Hep3B cells were seeded into
6-well plates (2×105 cells/well) for 24 h prior to
transfection. The cells were transfected with 2 μg of parkin-YFP
plasmid using Lipofectamine 2000 according to the manufacturer's
instructions. Stably transfected Hep3B/parkin-YFP cells were
selected by incubating the cells in medium containing 500 μg/ml of
neomycin sulfate (G418) for 2 weeks. The over-expression of
parkin-YFP was confirmed by observing cells under Zeiss LSM 700
laser-scanning confocal microscope (Goettingen, Germany).
Transfection of silencing RNA
(siRNA)
Hep3B cells growing into 6-well plates
(0.8×105 cells/well) were transfected with siRNA. siRNAs
against the human Mfn1, Opa1, Drp1 transcripts were purchased from
Dharmacon (ON-TARGETplus SMARTpool siRNA) and used at a
concentration of 10 nM. As a negative control, the same nucleotide
was scrambled to generate a non-targeting siRNA. The siRNAs were
transfected into Hep3B cells using Lipofectamine®
RNAiMAX per the manufacturer's instructions.
Reagents
The reagents were obtained from commercial sources:
DMEM and FBS were obtained from Gibco-BRL (Gaithersburg, MD, USA);
rabbit polyclonal antibodies to human Mfn1 (sc-50330), Tom20
(sc-11415) and cyclin B1 (sc-752), and mouse monoclonal antibody to
human CDK1 (sc-54) were obtained from Santa Cruz Biotechnology
(Santa Cruz, CA, USA); mouse monoclonal antibodies to human Drp1
(61112) and Opa1 (612606) were obtained from BD Transduction
Laboratories (Lexington, KY, USA); rabbit polyclonal antibodies
against human caspase-3 (#9662), caspase-7 (#9492) and p-Drp1
(Ser616) (#3455) as well as HRP-conjugated goat anti-rabbit and
horse anti-mouse IgG antibodies were obtained from Cell Signaling
Technologies (Danvers, MA, USA), as was also the RIPA buffer. Texas
Red-conjugated goat anti-rabbit IgG antibody was obtained from
Vector (Burlingame, CA, USA); mouse monoclonal antibodies against
human β-actin (A5441) and α-tubulin (T5168) as well as Hoechst
33342, protein-A agarose, N-acetyl-L-cysteine (NAC),
propidium iodide (PI), carbonyl cyanide m-chlorophenylhydrazone
(CCCP) and the Annexin V-FITC apoptosis detection kit were obtained
from Sigma-Aldrich (Irvine, CA, USA). G418 was purchased from
Duchefa (Haarlem, The Netherlands).
5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl benzimidazole
carbocyanine iodide (JC-1) and MitoSOX™ Red reagent were purchased
from Molecular Probes (Eugene, OR, USA). Lipofectamine®
RNAiMAX reagent and Lipofectamine 2000 were obtained from
Invitrogen (Carlsbad, CA, USA). The SuperSignal West Pico enhanced
chemiluminescence western blotting detection reagent was purchased
from Pierce (Rockford, IL, USA) and RNase A was purchased from
Biosesang (Biosesang, Inc., Korea).
Chemical synthesis of G-TPP
G-TTP was synthesized by LegoChem Biosciences Inc.
(Daejeon, Korea) as previously described (18).
Cell viability assay
The cell viability was assessed using the Vi-Cell
cell counter (Beckman Coulter, Miami, FL, USA) to perform an
automated trypan blue exclusion assay.
Western blot analysis
The cells were washed twice with ice-cold PBS,
resuspended in RIPA buffer and incubated at 4°C for 30 min. The
lysates were centrifuged at 14,000 rpm for 20 min at 4°C. The
protein concentrations of the cell lysates were determined with the
Bradford protein assay reagent (Bio-Rad), and 30 μg of protein was
loaded onto 7.5–15% SDS/PAGE gels. The proteins were transferred to
nitrocellu-lose membranes (Amersham Pharmacia Biotech, Piscataway,
NJ, USA). The bands were visualized by incubation with 1:1,000
dilutions of primary antibody overnight at 4°C, followed by
incubation with 1:2,000 dilutions of secondary antibody at room
temperature for 1 h. The antibody signal was developed using the
SuperSignal West Pico-enhanced chemiluminescence substrate and
detected with a LAS-4000PLUS (Fuji Photo Film, Tokyo, Japan).
Subcellular fractionation
The cells (3×107 cells) were washed in
ice-cold Tris-based Mg2+/Ca2+-free buffer
(135 mM NaCl, 5 mM KCl and 25 mM Tris-HCl pH 7.4). The
mitochondrial and cytosolic fractions were isolated using the
Mitochondrial Fractionation kit (Active Motif, Carlsbad, CA, USA).
The cells were resuspended in cytosolic buffer and incubated on ice
for 15 min. The cells were then homogenized on ice with a
homogenizer operated at 60 strokes. The lysates were centrifuged at
3,000 rpm and 4°C for 15 min. The supernatant contained the
cytosol, including the mitochondria. The supernatant was
transferred to a microcentrifuge tube and centrifuged at 13,000 rpm
and 4°C for 30 min to pellet the mitochondria. The mitochondrial
pellets were lysed with mitochondrial buffer on ice for 15 min and
then centrifuged at 13,000 rpm for 30 min at 4°C.
Co-immunoprecipitation (Co-IP)
After being incubated with antibodies, the cell
extracts were precipitated with protein A-Sepharose beads for 3 h
and washed 3 times with an extraction buffer prior to boiling them
in the SDS sample buffer. The immunoprecipitated proteins were
separated using SDS-PAGE, and a western blot analysis was performed
as described above.
Observation and quantification of
mitochondrial morphology using confocal microscopy
The cells were cultured on coverslips and fixed with
4% paraformaldehyde for 1 h. The cells were then permeabilized with
0.2% Triton X-100 for 15 min and incubated with Tom20 antibody for
1 h at room temperature (RT). They were washed 3 times with PBS for
5 min each, incubated with a Texas Red-conjugated secondary
antibody for 1 h at room temperature and counterstained with
Hoechst 33342. A Zeiss LSM 700 laser-scanning confocal microscope
at a magnification of ×40 (0.55 numerical aperture) was used to
obtain and analyze the fluorescent images. The cells were divided
into the following 3 groups based on their mitochondrial
morphology: fragmented, cells that primarily contained mitochondria
shorter than 2 μm; tubular and donuts, cells that primarily
contained mitochondria between ~2 and 5 μm long; and elongated,
cells that primarily contained mitochondria >5 μm. Three
independent experiments were conducted, and 100 cells were scored
per experiment.
Quantification of DNA hypoploidy and cell
cycle phase analysis by flow cytometry
Ice-cold 95% ethanol containing 0.5% Tween-20 was
added to the cell suspension to a final concentration of 70%
ethanol. The fixed cells were pelleted and washed in 1% BSA-PBS
solution. They were re-suspended in 1 ml of PBS containing 11
Kunitz U/ml RNase A, incubated at 37°C for 1 h, washed once with
BSA-PBS, re-suspended in PI solution (10 μg/ml), and incubated in
the dark at 4°C for 30 min. The cells were then washed with PBS,
and the DNA content was measured on an Epics XL (Beckman Coulter).
The data were analyzed using the MultiCycle software, which allowed
the simultaneous estimation of cell cycle parameters and
apoptosis.
Mitochondrial membrane potential (MMP)
assay
To measure the MMP, the cells were trypsinized,
collected, stained with JC-1 and subjected to flow cytometry using
an Epics XL flow cytometer (Beckman Coulter). The data were
acquired and analyzed using the EXPO32 ADC XL 4 color software
program.
Flow cytometric analysis of Annexin
V-FITC
The cells were trypsinized, collected and stained
with the Annexin V-FITC apoptosis detection kit according to the
manufacturer's instructions. After centrifugation, cells were
resuspended in PBS and analyzed with Epics XL.
Measurement of ROS levels
The cells were trypsinized, collected and stained
with 5 μM MitoSox for 30 min at 37°C. After centrifugation, the
cells were resuspended in PBS and analyzed with Epics XL.
Statistical analysis
At least three independent experiments were carried
out in vitro. The results are expressed as the means ± SD
from three experiments. The significance of differences was
determined using the paired Kruskal-Wallis non-parametric test. A
p-value <0.05 was considered significant.
Results
G-TPP induces cell death and
mitochondrial elongation in Hep3B cells
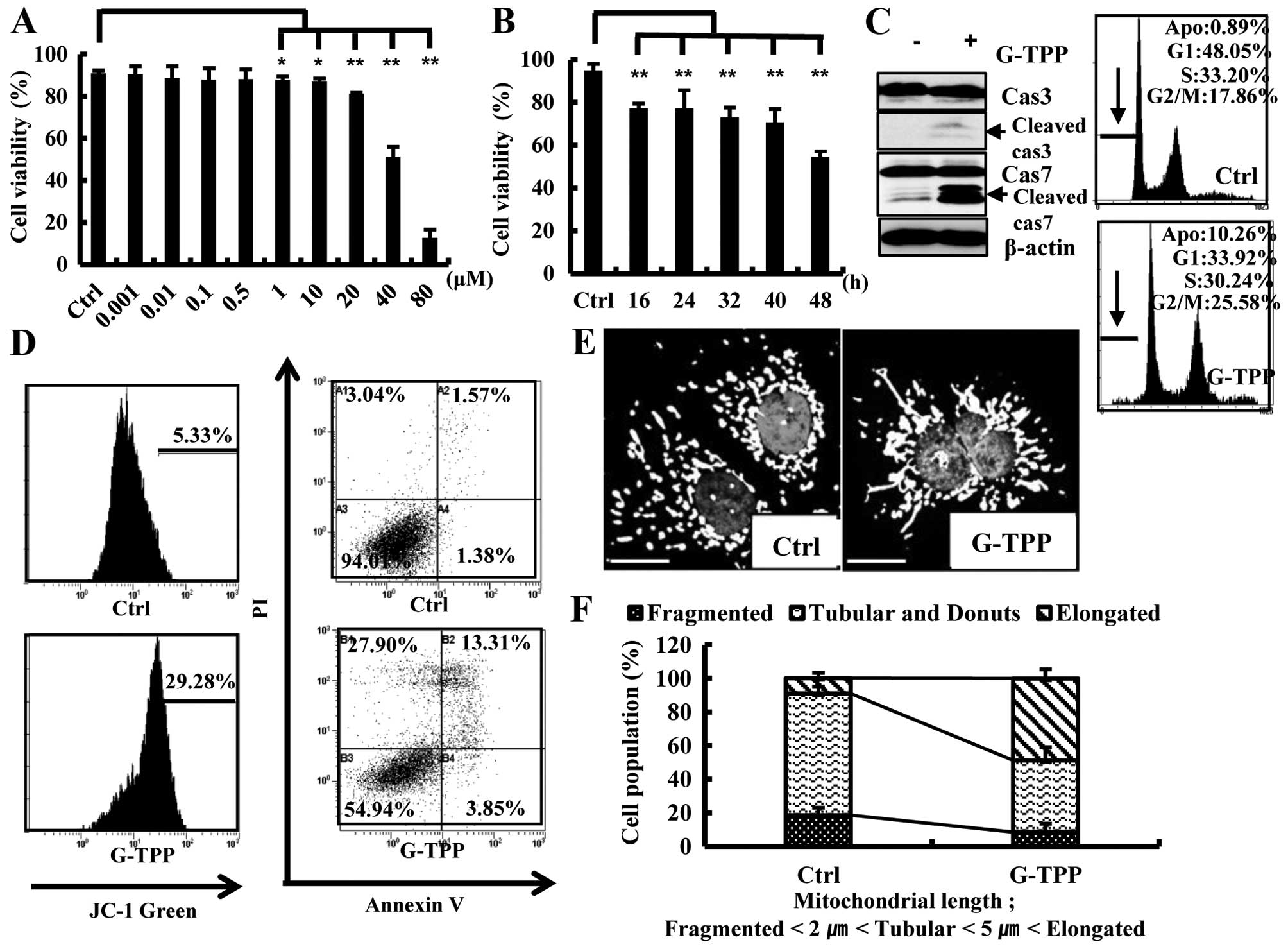
Treating Hep3B cells with 1–80 μM G-TPP for 48 h
significantly reduced their viability in a dose-dependent manner
(Fig. 1A). Because the viability
of Hep3B cells treated with 40 μM G-TTP for 48 h was ~50%, this
concentration was used for further studies. G-TPP treatment reduced
the viability of Hep3B cells in a time-dependent manner (Fig. 1B). To examine whether the reduced
viability of G-TTP-treated Hep3B cells was due to apoptotic cell
death, we performed various apoptosis assays. The caspase cleavage
and mitochondrial membrane potential assays indicated that G-TPP at
least partly induced Hep3B cell death via apoptosis (Fig. 1C and D). Notably, G-TPP treatment
significantly increased the population of Hep3B cells that
contained elongated mitochondria (Fig.
1E and F).
Drp1 is involved in G-TPP-induced
mitochondrial elongation
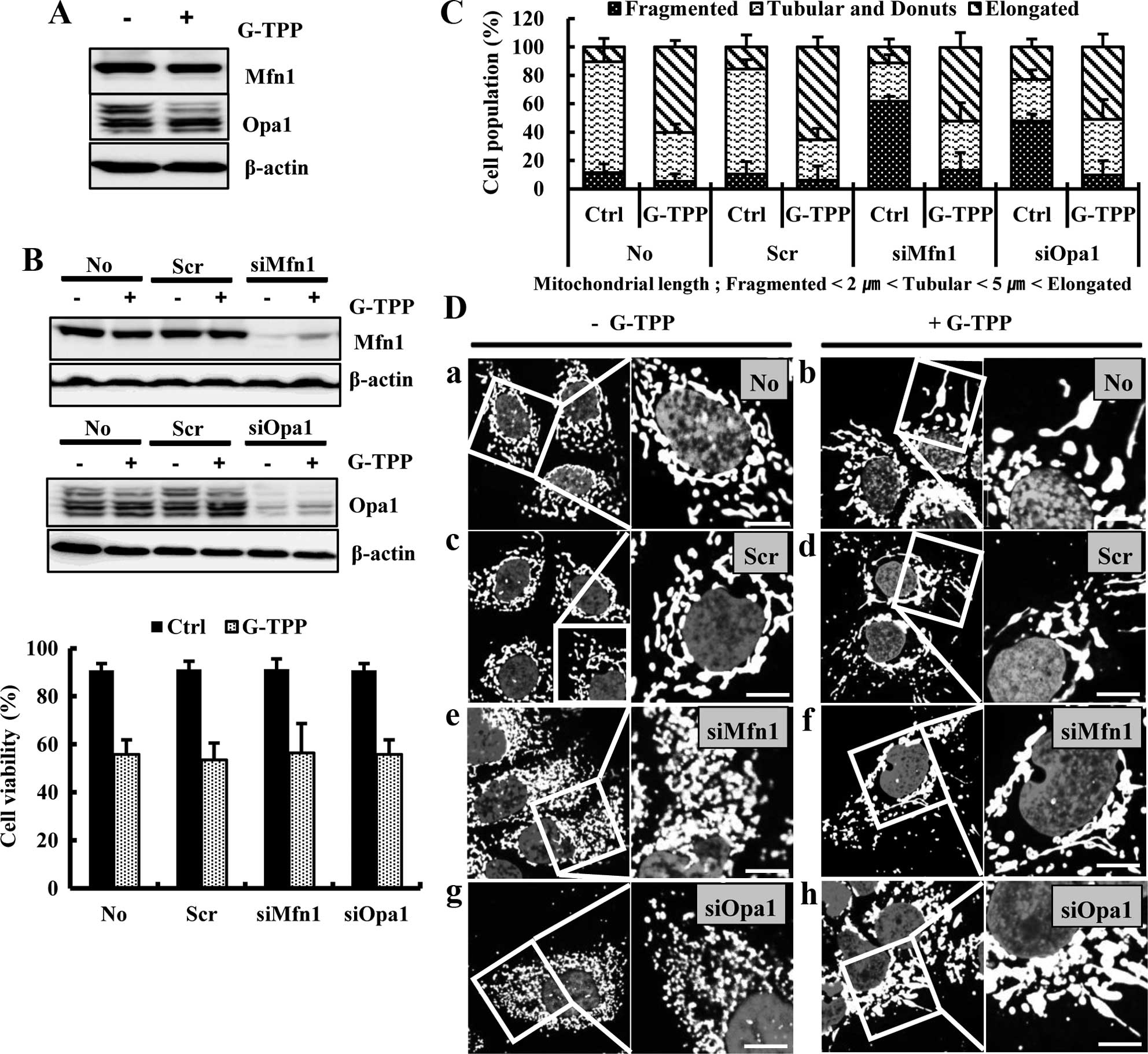
We examined whether the G-TPP-induced mitochondrial
elongation was mediated by alterations in the mitochondrial
fusion-regulating proteins Mfn1 and Opa1. The western blot assay
showed that G-TPP did not increase the expression levels of Mfn1
and Opa1 (Fig. 2A). We then
examined the effect of Mfn1 or Opa1 depletion on G-TPP-induced cell
death. A viability assay showed that neither siMfn1 nor siOpa1
significantly alter G-TPP-induced cell death (Fig. 2B). As predicted, both siMfn1 and
siOpa1 significantly increased the population of G-TPP-untreated
Hep3B cells that contained fragmented mitochondria. However,
treatment with G-TPP significantly increased the populations of
both Mfn1- and Opa1-depleted Hep3B cells that contained elongated
mitochondria (Fig. 2C and D).
These data indicate that G-TPP-induced mitochondrial elongation is
not mediated by an increase in the mitochondrial fusion proteins
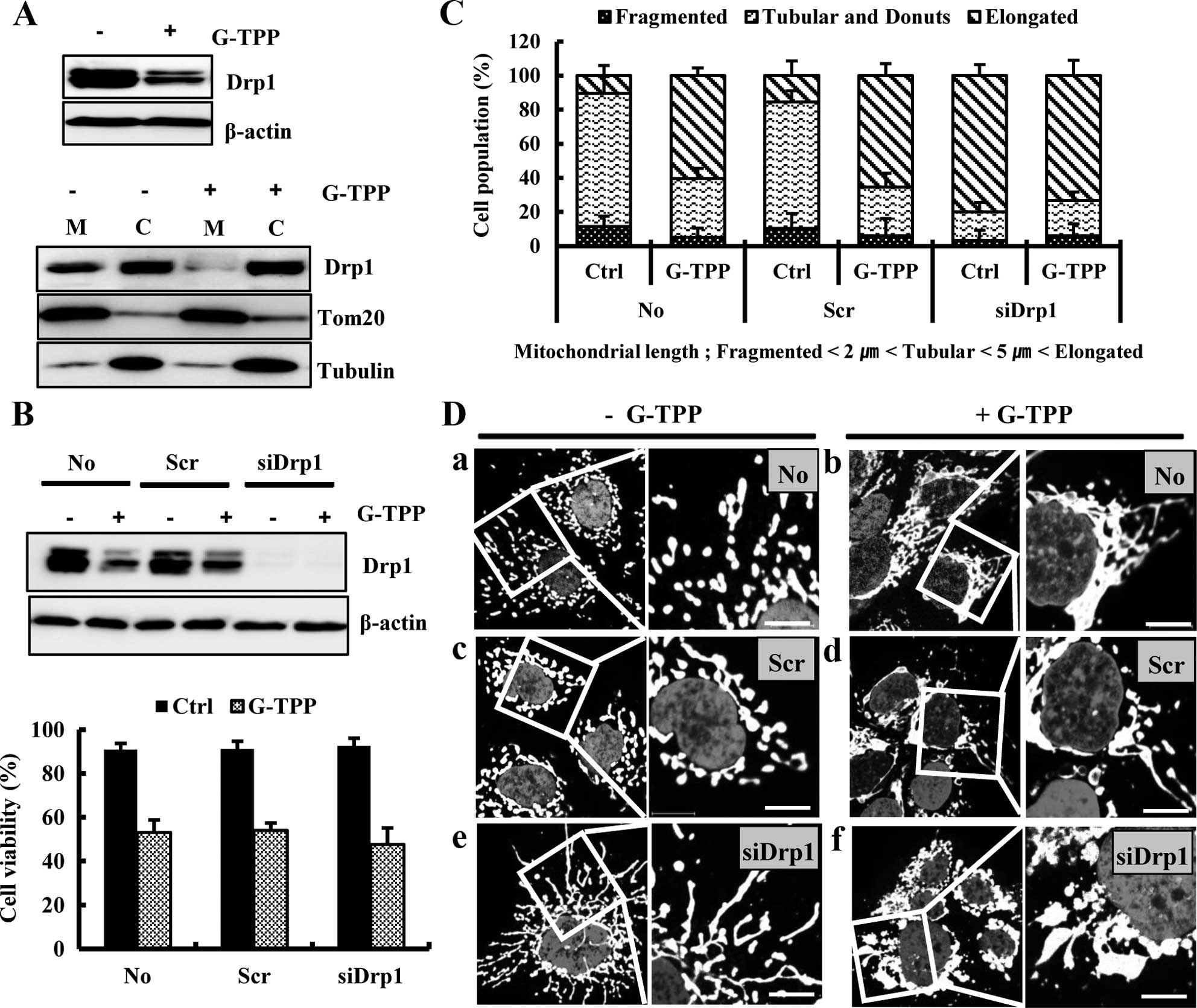
Mfn1 and Opa1. We next examined whether the G-TPP-induced
mitochondrial elongation was mediated by alterations in the
fission-regulating protein Drp1. The western blot assay showed that
treatment with G-TPP markedly decreased the Drp1 level in Hep3B
cells, particularly in the mitochondria (Fig. 3A). We next examined the effect of
Drp1 depletion on G-TPP-induced cell death. siDrp1 did not
significantly alter the viability of Hep3B cells treated with G-TPP
(Fig. 3B). However, siDrp1
significantly increased the populations of both G-TPP-treated and
untreated Hep3B cells that contained elongated mitochondria
compared with scrambled siRNA (Fig.
3C). Confocal microscopy demonstrated that mitochondria are
more aggregated in Hep3B cells treated with G-TPP plus siDrp1 than
cells treated with G-TPP plus scrambled siRNA (Fig. 3D). These data indicated that
G-TPP-induced mitochondrial elongation was caused by Drp1
reduction.
G-TPP suppresses the translocation of
Drp1 into mitochondria in parkin-overexpressing Hep3B cells
We investigated whether G-TPP suppressed the
translocation of Drp1 into mitochondria in parkin-overexpressing
Hep3B cells. We observed that parkin translocated into the
mitochondria of parkin-overexpressing Hep3B cells, irrespective of
G-TPP treatment. Additionally, parkin translocated into the
fragmented mitochondria of parkin-overexpressing Hep3B cells
treated with the representative mitophagy inducer CCCP (Fig. 4A). A western blot assay
demonstrated that G-TPP decreased the expression level of Drp1 not
only in Hep3B cells but also in parkin-overexpressing Hep3B cells.
G-TPP also inhibited the translocation of Drp1 into mitochondria in
Hep3B cells. Importantly, CCCP increased the expression level of
Drp1 and augmented the mitochondrial translocation of Drp1 in the
parkin-overexpressing Hep3B cells. G-TPP inhibited the
translocation of Drp1 into mitochondria in the
parkin-overexpressing Hep3B cells (Fig. 4B). The population of cells that
contained elongated or fragmented mitochondria did not differ
between control Hep3B and parkin-overexpressing Hep3B cells treated
with G-TPP (Fig. 4C). Furthermore,
the overexpression of parkin did not affect the level of
G-TPP-induced cell death (Fig.
4D). These data indicate that G-TPP inhibited the mitochondrial
translocation of Drp1 even in the parkin-overexpressing Hep3B
cells, which may impair parkin-mediated mitophagy.
G-TPP reduces the interaction of CDK1
with cyclin B1 and the activating phosphorylation of Drp1
(Ser616)
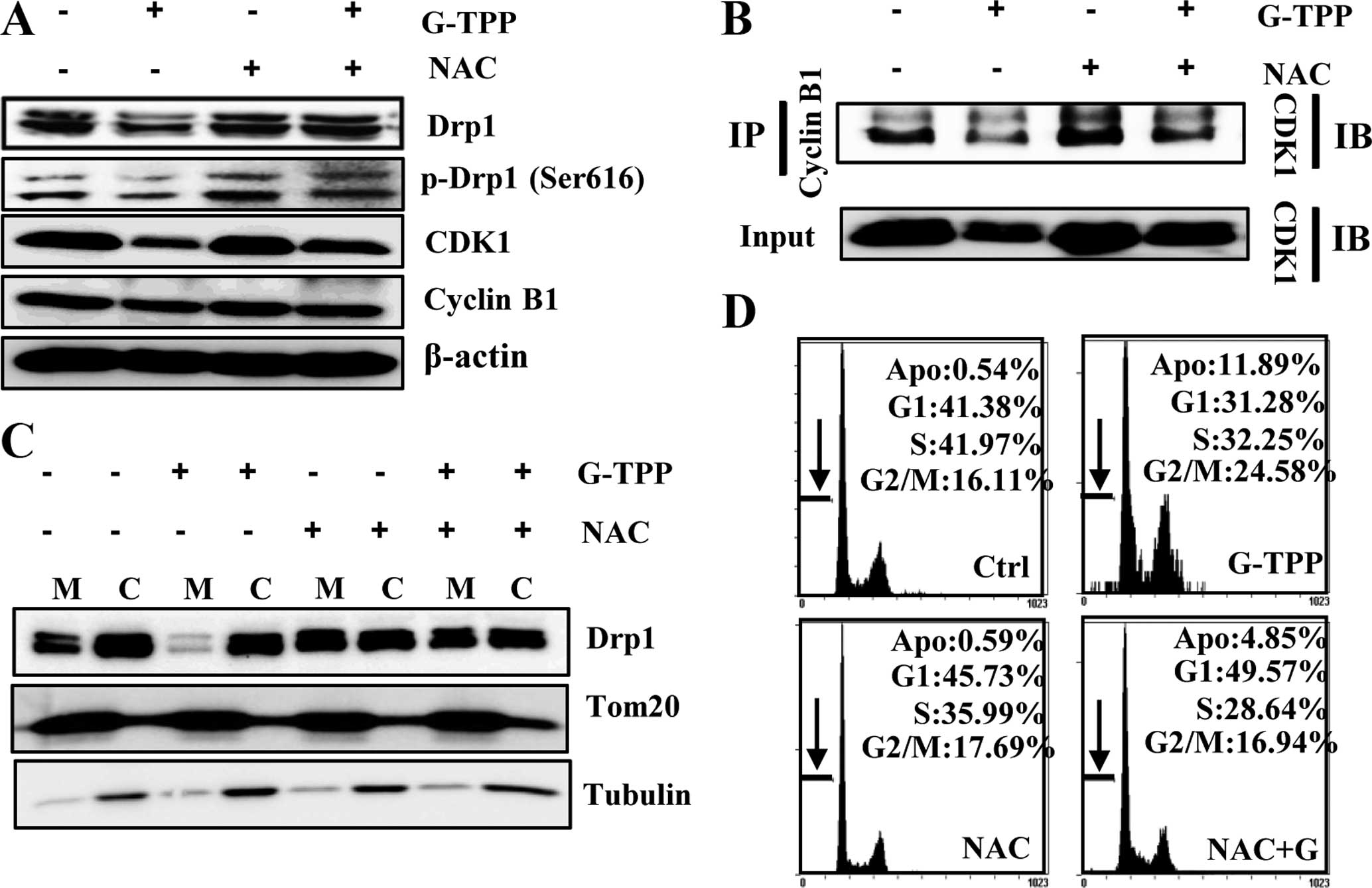
Flow cytometry revealed an increase in the
percentage of G2-M phase cells and a concomitant decrease in the
percentage of the G1 phase cells (Fig.
1C), which indicated that G-TPP induced G2-M phase cell cycle
arrest in Hep3B cells. Therefore, we examined the association
between mitochondrial elongation and cell cycle progression in
G-TPP-treated Hep3B cells. We examined the level of CDK1 and cyclin
B1 with a western blotting, which showed that G-TPP markedly
decreased the expression level of CDK1. G-TPP also markedly reduced
the activating phosphorylation of Drp1 (Ser616) (Fig. 5A). Because the CDK1-cyclin B1
complex is known to be associated with the activation of Drp1 via
the phosphorylation of Drp1 at Ser616 (pDrp1-Ser616) (20), we examined the effect of G-TPP on
the formation of the CDK1-cyclin B1 complex. The
co-immunoprecipitation results indicated that G-TPP reduced the
level of CDK1-cyclin B1 complex formation (Fig. 5B). These data indicated that G-TPP
reduced the interaction between CDK1 and cyclin B1 and thereby
inhibited its Drp1 activation and mitochondrial localization, which
induced mitochondrial elongation.
ROS mediates G-TPP-induced cell death and
mitochondrial elongation
Next, we examined the involvement of ROS in
G-TPP-induced cell death and mitochondrial elongation in Hep3B
cells. We observed that G-TPP increased the ROS level in Hep3B
cells, and the ROS scavenger NAC remarkably inhibited the level of
ROS in the G-TPP-treated Hep3B cells (Fig. 6A). Importantly, NAC significantly
suppressed G-TPP-induced cell death (Fig. 6B). Various apoptosis assays showed
that NAC suppressed G-TPP-induced apoptosis (Fig. 6C and D). NAC also significantly
decreased the population of Hep3B cells treated with G-TPP cells
that contained elongated mitochondria (Fig. 6E and F). These data indicated that
ROS mediates mitochondrial elongation and cell death in
G-TPP-treated Hep3B cells. To this end, we examined whether G-TPP
reduced the interaction of CDK1 with cyclin B1 and the
phosphorylation of Drp1 (pDrp1-Ser616) via ROS. Noticeably, NAC
recovered the expression levels of Drp1, CDK1 and p-Drp1 (Ser616)
(Fig. 7A); suppressed the
G-TPP-induced dissociation of CDK1 from cyclin B1 in Hep3B cells
(Fig. 7B); and recovered the
recruitment of Drp1 to mitochondria in G-TPP-treated Hep3B cells
(Fig. 7C). Flow cytometry revealed
that NAC suppressed G-TPP-induced G2-M arrest (Fig. 7D). These results indicate that ROS
played a pivotal role in mitochondrial elongation, which is
mediated by the reduced association of CDK1 with cyclin B1 and
decreased Drp1 phosphorylation at Ser616 in G-TPP-treated Hep3B
cells.
Discussion
Previous studies reported that the targeted
inhibition of mitochondrial Hsp90 using G-TPP induces cell death
via ER- and calcium-mediated stress in various cancer cells
(21,22).Because apoptosis-inducing agents
generally induce mitochondrial fragmentation (13–16),
we first predicted that G-TPP would induce mitochondrial
fragmentation in Hep3B cells. However, we observed that G-TPP
induces mitochondrial elongation in Hep3B cells. Mitochondrial
fragmentation by apoptotic stimuli depends on the regulation of the
mitochondrial fusion-fission balance, which is primarily mediated
by the mitochondrial fission machinery (15,23).
However, some studies showed that apoptosis-inducing agents could
result in mitochondrial elongation. A previous study reported that
HDAC inhibitors, which induce apoptosis, caused mitochondrial
elongation in various cells in addition to inducing apoptosis
(24). Our data suggest that G-TPP
induces mitochondrial elongation by reducing and inactivating the
mitochondrial fission-regulating protein Drp1 and increasing the
ROS level.
Because cellular homeostasis is tightly linked to
mitochondrial function, the cell must eliminate mitochondria
damaged by various stimuli, such as anticancer agents, oxidative
stress and starvation. Dysfunctional mitochondria are eliminated
via ‘mitophagy’, a process by which cells selectively remove
depolarized mitochondria (25).
Parkin, an E3 ligase that was originally discovered as mutated in
monogenic forms of Parkinson's disease, has been shown to
selectively recognize and eliminate damaged mitochondria (26).
Mitophagy and mitochondrial dynamics are closely
correlated (11,27–30).
Several previous studies demonstrated that mitochondrial fission is
linked to the function of parkin, and parkin recruitment to
mitochondria may be a consequence of depolarization-induced
fragmentation (31,32). Whereas staurosporine induced
mitochondrial fragmentation and mitophagy in HeLa cells, Drp1
overexpression impaired mitophagy and mitochondrial fission,
indicating that mitochondrial fission is required for mitophagy
(29). However, a previous study
showed that excessive mitochondrial fragmentation alone is
insufficient to recruit parkin (26). Mitophagy can be induced independent
of parkin (33). While most recent
studies have focused on parkin/PINK1-dependent mitophagy, some have
examined the parkin/PINK1-independent mechanisms of mitophagy
(34). The present study suggests
that G-TPP induces mitochondrial elongation in Hep3B cells by
inhibiting the mitochondrial translocation of Drp1, even in
parkin-overexpressing cells, which may impair mitophagy. We assume
that the induction of apoptosis in G-TPP-treated Hep3B cells is at
least partly due to the inefficient removal of dysfunctional
mitochondria.
The equal distribution of mitochondria between
daughter cells during mitosis is important. Various studies
reported that mitochondrial dynamics are integrated with cell cycle
progression (35–37). Mitochondrial hyperfusion is known
to promote a defect in cell cycle progression characterized by an
inability of cells to exit the G2 phase (37). In eukaryotic cells, several cyclin
family members regulate the progression from the G2 to the M phase.
Cyclin B1, together with CDK1, promotes the G2/M transition
(38). In particular, loss of Drp1
induced G2 phase arrest (37). The
activation of CDK1 and cyclin B1 is a key factor for the G2/M phase
transition (36). CDK1-cyclin B1
phosphorylates Drp1 at Ser616 and activates the mitochondrial
fission machinery (39–41). This study suggests that G-TPP
induces G2 phase arrest via the dissociation of the CDK1-cyclin B1
complex and inhibits Drp1 translocation to the mitochondria.
ROS regulate a wide range of biological processes,
including oxygen sensing, immune responses, cell proliferation and
differentiation (42,43). The direct or indirect accumulation
of ROS lead to apoptosis (44).
ROS are primarily produced in mitochondria (45) and mediate mitochondrial dynamics.
Numerous studies demonstrated that increased ROS levels mediate
mitochondrial fission (46,47).
ROS also mediate mitochondrial elongation. A previous study showed
that oxidative stress converted elongated tubules into large
spheres in fibroblasts (48).
Another study reported that the mitochondria of hypoxia-induced
chemotherapy-resistant cells undergo a HIF-1-dependent and
mitofusin-1-mediated changes in morphology from a tubular network
to an enlarged phenotype (49).
Moreover, ROS mediate the formation of elongated mitochondria
during cellular senescence (50).
Our study showed that G-TPP induced mitochondrial elongation in
Hep3B cells by increasing the ROS level.
In conclusion, our results indicated that G-TPP
induces cell death and causes Drp1-inhibited mitochondrial
elongation in Hep3B cells by increasing the ROS level.
Acknowledgements
This study was supported by a National Research
Foundation of Korea (NRF) grant funded by the Korean government
(MSIP) (no. 2015 008728).
References
|
1
|
Campello S, Strappazzon F and Cecconi F:
Mitochondrial dismissal in mammals, from protein degradation to
mitophagy. Biochim Biophys Acta. 1837:451–460. 2014. View Article : Google Scholar
|
|
2
|
Picard M, Shirihai OS, Gentil BJ and
Burelle Y: Mitochondrial morphology transitions and functions:
Implications for retrograde signaling? Am J Physiol Regul Integr
Comp Physiol. 304:R393–R406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Harbauer AB, Zahedi RP, Sickmann A,
Pfanner N and Meisinger C: The protein import machinery of
mitochondria - a regulatory hub in metabolism, stress, and disease.
Cell Metab. 19:357–372. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karbowski M and Youle RJ: Dynamics of
mitochondrial morphology in healthy cells and during apoptosis.
Cell Death Differ. 10:870–880. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cerveny KL, Tamura Y, Zhang Z, Jensen RE
and Sesaki H: Regulation of mitochondrial fusion and division.
Trends Cell Biol. 17:563–569. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Benard G and Karbowski M: Mitochondrial
fusion and division: Regulation and role in cell viability. Semin
Cell Dev Biol. 20:365–374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ishihara N, Fujita Y, Oka T and Mihara K:
Regulation of mitochondrial morphology through proteolytic cleavage
of OPA1. EMBO J. 25:2966–2977. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smirnova E, Griparic L, Shurland DL and
van der Bliek AM: Dynamin-related protein Drp1 is required for
mitochondrial division in mammalian cells. Mol Biol Cell.
12:2245–2256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Landes T and Martinou JC: Mitochondrial
outer membrane permeabilization during apoptosis: The role of
mitochondrial fission. Biochim Biophys Acta. 1813:540–545. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Otera H, Wang C, Cleland MM, Setoguchi K,
Yokota S, Youle RJ and Mihara K: Mff is an essential factor for
mitochondrial recruitment of Drp1 during mitochondrial fission in
mammalian cells. J Cell Biol. 191:1141–1158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Twig G, Elorza A, Molina AJ, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang IH, Chen HY, Wang YH, Chang KW, Chen
YC and Chang CR: Resveratrol modulates mitochondria dynamics in
replicative senescent yeast cells. PLoS One. 9:e1043452014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pinton P, Ferrari D, Rapizzi E, Di
Virgilio F, Pozzan T and Rizzuto R: The Ca2+
concentration of the endoplasmic reticulum is a key determinant of
ceramide-induced apoptosis: Significance for the molecular
mechanism of Bcl-2 action. EMBO J. 20:2690–2701. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Breckenridge DG, Stojanovic M, Marcellus
RC and Shore GC: Caspase cleavage product of BAP31 induces
mitochondrial fission through endoplasmic reticulum calcium
signals, enhancing cytochrome c release to the cytosol. J Cell
Biol. 160:1115–1127. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bai X, Yan Y, Canfield S, Muravyeva MY,
Kikuchi C, Zaja I, Corbett JA and Bosnjak ZJ: Ketamine enhances
human neural stem cell proliferation and induces neuronal apoptosis
via reactive oxygen species-mediated mitochondrial pathway. Anesth
Analg. 116:869–880. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Atay C, Ugurlu S and Ozören N: Shock the
heat shock network. J Clin Invest. 119:445–448. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kang BH, Plescia J, Song HY, Meli M,
Colombo G, Beebe K, Scroggins B, Neckers L and Altieri DC:
Combinatorial drug design targeting multiple cancer signaling
networks controlled by mitochondrial Hsp90. J Clin Invest.
119:454–464. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mayer MP, Prodromou C and Frydman J: The
Hsp90 mosaic: A picture emerges. Nat Struct Mol Biol. 16:2–6. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamano K and Youle RJ: Coupling
mitochondrial and cell division. Nat Cell Biol. 13:1026–1027. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park HK, Lee JE, Lim J and Kang BH:
Mitochondrial Hsp90s suppress calcium-mediated stress signals
propagating from mitochondria to the ER in cancer cells. Mol
Cancer. 13:1482014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Siegelin MD, Dohi T, Raskett CM, Orlowski
GM, Powers CM, Gilbert CA, Ross AH, Plescia J and Altieri DC:
Exploiting the mitochondrial unfolded protein response for cancer
therapy in mice and human cells. J Clin Invest. 121:1349–1360.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Frank S, Gaume B, Bergmann-Leitner ES,
Leitner WW, Robert EG, Catez F, Smith CL and Youle RJ: The role of
dynamin-related protein 1, a mediator of mitochondrial fission, in
apoptosis. Dev Cell. 1:515–525. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee JS, Yoon YG, Yoo SH, Jeong NY, Jeong
SH, Lee SY, Jung DI, Jeong SY and Yoo YH: Histone deacetylase
inhibitors induce mitochondrial elongation. J Cell Physiol.
227:2856–2869. 2012. View Article : Google Scholar
|
|
25
|
Abeliovich H: Mitophagy: The life-or-death
dichotomy includes yeast. Autophagy. 3:275–277. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Narendra D, Tanaka A, Suen DF and Youle
RJ: Parkin is recruited selectively to impaired mitochondria and
promotes their autophagy. J Cell Biol. 183:795–803. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Twig G and Shirihai OS: The interplay
between mitochondrial dynamics and mitophagy. Antioxid Redox
Signal. 14:1939–1951. 2011. View Article : Google Scholar :
|
|
28
|
Ni HM, Williams JA and Ding WX:
Mitochondrial dynamics and mitochondrial quality control. Redox
Biol. 4:6–13. 2015. View Article : Google Scholar :
|
|
29
|
Arnoult D, Rismanchi N, Grodet A, Roberts
RG, Seeburg DP, Estaquier J, Sheng M and Blackstone C:
Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated
mitochondrial fission and mitoptosis during programmed cell death.
Curr Biol. 15:2112–2118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bernhardt D, Müller M, Reichert AS and
Osiewacz HD: Simultaneous impairment of mitochondrial fission and
fusion reduces mitophagy and shortens replicative lifespan. Sci
Rep. 5:78852015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deng H, Dodson MW, Huang H and Guo M: The
Parkinson's disease genes pink1 and parkin promote mitochondrial
fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci
USA. 105:14503–14508. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Poole AC, Thomas RE, Andrews LA, McBride
HM, Whitworth AJ and Pallanck LJ: The PINK1/Parkin pathway
regulates mitochondrial morphology. Proc Natl Acad Sci USA.
105:1638–1643. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kageyama Y, Hoshijima M, Seo K, Bedja D,
Sysa-Shah P, Andrabi SA, Chen W, Höke A, Dawson VL, Dawson TM, et
al: Parkin-independent mitophagy requires Drp1 and maintains the
integrity of mammalian heart and brain. EMBO J. 33:2798–2813. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hirota Y, Kang D and Kanki T: The
physiological role of mitophagy: New insights into phosphorylation
events. Int J Cell Biol. 2012:3549142012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mitra K, Wunder C, Roysam B, Lin G and
Lippincott-Schwartz J: A hyperfused mitochondrial state achieved at
G1-S regulates cyclin E buildup and entry into S phase. Proc Natl
Acad Sci USA. 106:11960–11965. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Qian W, Choi S, Gibson GA, Watkins SC,
Bakkenist CJ and Van Houten B: Mitochondrial hyperfusion induced by
loss of the fission protein Drp1 causes ATM-dependent G2/M arrest
and aneuploidy through DNA replication stress. J Cell Sci.
125:5745–5757. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Westrate LM, Sayfie AD, Burgenske DM and
MacKeigan JP: Persistent mitochondrial hyperfusion promotes G2/M
accumulation and caspase-dependent cell death. PLoS One.
9:e919112014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang WW, Ko SW, Tsai HY, Chung JG, Chiang
JH, Chen KT, Chen YC, Chen HY, Chen YF and Yang JS: Cantharidin
induces G2/M phase arrest and apoptosis in human colorectal cancer
colo 205 cells through inhibition of CDK1 activity and
caspase-dependent signaling pathways. Int J Oncol. 38:1067–1073.
2011.PubMed/NCBI
|
|
39
|
Cribbs JT and Strack S: Reversible
phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and
calcineurin regulates mitochondrial fission and cell death. EMBO
Rep. 8:939–944. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang CR and Blackstone C: Cyclic
AMP-dependent protein kinase phosphorylation of Drp1 regulates its
GTPase activity and mitochondrial morphology. J Biol Chem.
282:21583–21587. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Taguchi N, Ishihara N, Jofuku A, Oka T and
Mihara K: Mitotic phosphorylation of dynamin-related GTPase Drp1
participates in mitochondrial fission. J Biol Chem.
282:11521–11529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang Y, Bazhin AV, Werner J and
Karakhanova S: Reactive oxygen species in the immune system. Int
Rev Immunol. 32:249–270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Matsuzawa A and Ichijo H: Redox control of
cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase
pathway in stress signaling. Biochim Biophys Acta. 1780:1325–1336.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Y, Du Y, Le W, Wang K, Kieffer N and
Zhang J: Redox control of the survival of healthy and diseased
cells. Antioxid Redox Signal. 15:2867–2908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chandel NS: Mitochondria as signaling
organelles. BMC Biol. 12:342014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu T, Robotham JL and Yoon Y: Increased
production of reactive oxygen species in hyperglycemic conditions
requires dynamic change of mitochondrial morphology. Proc Natl Acad
Sci USA. 103:2653–2658. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu T, Sheu SS, Robotham JL and Yoon Y:
Mitochondrial fission mediates high glucose-induced cell death
through elevated production of reactive oxygen species. Cardiovasc
Res. 79:341–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kageyama Y, Zhang Z, Roda R, Fukaya M,
Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M and
Sesaki H: Mitochondrial division ensures the survival of
postmitotic neurons by suppressing oxidative damage. J Cell Biol.
197:535–551. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chiche J, Rouleau M, Gounon P,
Brahimi-Horn MC, Pouysségur J and Mazure NM: Hypoxic enlarged
mitochondria protect cancer cells from apoptotic stimuli. J Cell
Physiol. 222:648–657. 2010.
|
|
50
|
Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung
HY, Rojo M, Malka F, Jou MJ, Martinou JC and Yoon G: Formation of
elongated giant mitochondria in DFO-induced cellular senescence:
Involvement of enhanced fusion process through modulation of Fis1.
J Cell Physiol. 209:468–480. 2006. View Article : Google Scholar : PubMed/NCBI
|