Introduction
Cervical cancer, with an estimated global incidence
of 528,000 cases and approximately 266,000 deaths every year, is
one of the most common types of cancer among women world-wide
(1). It has been well recognized
that persistent infection with high-risk human papillomaviruses
(HR-HPVs), such as HPV16, HPV18 and HPV31, is the most important
risk factor of cervical cancer (2). E6 and E7, two oncoproteins encoded by
HR-HPVs, are believed to be crucial for the development and
progression of cervical cancer. E6 deregulates host genes by
inactivating the tumor suppressor p53, which serves as genome
safeguard via inducing DNA repair, cell cycle arrest and apoptosis
(3). E7 can destabilize another
tumor suppressor RB1, to release the E2F family, leading to
deregulation of cell cycle progression (4).
MicroRNAs (miRNAs) are a class of small non-coding
endogenous RNAs of 21–25 nucleotides in size that modulate gene
expression. Mature miRNAs may inhibit translation by interacting
preferentially with 3′-untranslated region (3′-UTR) of target mRNAs
(5). Recent studies show that
miRNAs mediate important biological activities such as cellular
proliferation, differentiation and apoptosis. Thus, dysregulated
miRNA expression is linked to the development of a number of
diseases, including human cancers (5,6).
miRNAs can act as oncogenes or tumor suppressors by regulating
different pathways (7–9). Accumulating evidence indicates that
the expression of cellular miRNA are deregulated in response to
virus infection (10,11). However, the function of miRNAs
involved in virus-mediated cervical carcinogenesis remains largely
undefined.
Peroxisome proliferator-activated receptor gamma
(PPARγ) belongs to the nuclear receptor superfamily (12). It functions as a transcription
factor or regulator of gene expression. Thus, PPARγ has been
implicated to modulate various biological process and play a
significant role in several diseases, such as obesity, diabetes,
atherosclerosis and a variety of cancers (13). Due to the ability of promoting
apoptosis and differentiation as well as inhibiting proliferation
and growth, PPARγ is suggested to function as a tumor suppressor
(14–17). Cervical cancer tissues, in
particular, were found to express lower levels of PPARγ than normal
cervical tissues (18). Therefore,
PPARγ is considered to exert antitumor roles in cervical cancer.
However, the detailed function of PPARγ in cervical cancer has not
been well elucidated.
A previous report indicated that the activation of
PPARγ inhibits growth of breast cancer cells by repressing NHE1
expression (19).
Na+/H+ exchanger isoform 1 (NHE1) is a pH
regulator that mediate the electroneutral exchange of extracellular
Na+ for intracellular H+ across the cell
membrane (20). NHE1 activation
results in cytosolic alkalinization, which is commonly regarded as
an early phenotype to most carcinoma cells (20,21).
It has been shown that HPV16 E7 stimulates NHE1 activity to
alkalinize pHi in NIH3T3 cells (21). Although both HPV16 E7 and PPARγ
have been implicated to be associated with NHE1 activity, the
correlation between HPV16 E7, PPARγ and NHE1 has not been confirmed
and the underlying mechanism remains unknown.
In the present study, we found that miR-27b was
upregulated by HPV16 E7 to suppress the expression of PPARγ and
increase the level of NHE1. Furthermore, we observed that miR-27b
enhanced the proliferation and invasion of cervical cancer cells.
Consequently, the HPV16 E7-miR-27b-PPARγ-NHE1 pathway is
established and its role in HPV-related carcinogenesis in cervical
cancer cells is shown.
Materials and methods
Tissue samples and cell lines
Clinical samples were obtained from six
HPV16-positive cervical cancer patients treated at the Guangzhou
Nanfang Hospital in China. All of the samples were collected with
informed consent of patients and all of the experiments were
approved by the Internal Review and Ethics Boards of Nanfang
Hospital.
HPV16-positive human cervical carcinoma cell lines
CaSki and SiHa, and HPV-negative cervical cancer cell line C33A,
were purchased from the Cell Bank of the Chinese Academy of
Sciences (Shanghai, China). CaSki cells were cultured in RPMI-1640
medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco) at 37°C and 5% CO2. SiHa and
C33A cells were grown in Dulbecco's modified Eagle's medium (DMEM;
Gibco) with 10% FBS at 37°C and 5% CO2.
Transfection
All siRNAs, hsa-miR-27b mimics and hsa-miR-27b
inhibitors were purchased from Shanghai GenePharma Co., Ltd.
(Shanghai, China). The full-length of HPV16 E6 and E7 cDNA was
subcloned into the pEGFP vector (Invitrogen, Grand Island, NY,
USA). The target sequences of siRNAs were as follows: siRNA-198
(22) targeting HPV16 E6 and E7:
5′-GCA CAC ACG UAG ACA UUC G-3′; PPARγ: 5′-GAG GGC GAT CTT GAC AGG
AAA-3′. Cells were seeded into 6-well plates and grown to 50–60%
confluence. Then cells were transfected with the respective siRNAs,
plasmids, mimics or inhibitors using Lipofectamine 2000
(Invitrogen) according to the manufacturer's instructions. The RNA
level was assessed by real-time PCR at 48 h after transfection and
protein level was assayed by western blot analysis at 72 h after
transfection.
RNA isolation and real-time PCR
Total RNA was extracted by TRIzol (Takara, Shiga,
Japan) according to the manufacturer's instructions and then
reverse transcribed using PrimeScript™ RT reagent kit (Takara) to
generate cDNA. cDNA was amplified using SYBR® Premix Ex
Taq™ (Takara). For real-time PCR analysis of HPV16 E6, HPV16 E7,
PPARγ, NHE1 mRNAs, the following primers were used: PPARγ: forward,
5′-CTC TCC GTA ATG GAA GAC CAC T-3′ and reverse, 5′-TCT GCA ACC ACT
GGA TCT GTT C-3′; NHE1: forward, 5′-GCC TTC TCT CTG GGC TAC CT-3′
and reverse, 5′-CTT GTC CTT CCA GTG GTG GT-3′; β-actin: forward,
5′-TGG CAC CCA GCA CAA TGA A-3′ reverse, 5′-CTA AGT CAT AGT CCG CCT
AGA AGC A-3′. β-actin was used as a loading control. For real-time
PCR analysis of hsa-miR-27b, cDNA was prepared from 2000 ng total
RNA using All-in-One™ miRNA First-Strand cDNA Synthesis kit
(GeneCopoeia, Guangzhou China) and was amplified using All-in-One™
miRNA qPCR kit (GeneCopoeia). U6 was used as miRNA reference gene.
Primers for hsa-miR-27b were purchased as kits from GeneCopoeia.
Each reaction was done in triplicate and 2−ΔΔCT method
was used to calculate fold changes (23).
Western blot analysis
A total of 50 μg of total cellular protein was
extracted by Lysis buffer (Beyotime Institute of Biotechnology,
Haimen, China), separated on 12% sodium dodecyl
sulfate-polyacrylamide gels, transferred to PVDF membranes
(Millipore, Billerica, MA, USA). The membranes were incubated
overnight at 4°C with anti-PPARγ (1:1,000; Proteintech Group Inc.,
Chicago, IL, USA), anti-NHE1 (1:1,000; Proteintech Group Inc.) and
anti-β-actin (1:1,000; Cell Signaling Technology, Danvers, MA, USA)
and subsequently incubated with secondary anti-rabbit goat
peroxidase antibody (1:10,000; Bioworld, Visalia, CA, USA). Protein
was detected using ECL reagents (Thermo Fisher Scientific, Waltham,
MA, USA). β-actin was used to demonstrate equal loading.
Cell proliferation assay
In vitro proliferation activities were
measured by Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto,
Japan). Cells were seeded in 96-well plates at a density of 5,000
cells/well with 100 μl complete culture medium and allowed to
adhere overnight prior to transfection as described above. At 24,
48, 72 and 96 h after transfection, the media was removed and cells
were treated with 10% CCK-8 in basic media for 1–4 h at 37°C. The
absorbance of all wells was recorded at 450 nm. All samples were
run in triplicate independently.
Transwell invasion assay
Cell invasion assay was conducted in 24-well plates
using a Transwell invasion system (Corning, Corning, NY, USA)
following the manufacturer's protocols. Approximately 200,000 cells
in serum-free media were added into the top chamber, and bottom
chamber was filled with media containing 30% FBS.
Statistical analysis
All statistical analyses were undertaken by one-way
ANOVA or Student's t-test using Graphpad Prism 6.01. (GraphPad
Software, San Diego, CA, USA). P<0.05 was considered
statistically significant.
Results
HPV16 E7 promotes miR-27b expression in
cervical cancer cell lines
In order to understand the way miRNAs are involved
in HPV16 oncogene-induced carcinoma progression, we knocked down
the expression of E6 and E7 using a reported siRNA-198 (22) in the HPV16-positive human cervical
cancer cell line, CaSki, and then miRNA microarray analysis was
used to search miRNAs regulated by HPV16 E6 and E7. The results
showed that knockdown of HPV16 E6 and E7 led to significant
downregulation of miR-27b in CaSki cells (data not shown), which
was validated by real-time PCR (Fig.
1A). Similar results were also observed in the HPV16-positive
cervical cancer cell line SiHa (Fig.
1A). We then transfected CaSki and SiHa cells with E6 or E7
plasmids to determine which viral oncogene regulated the expression
of miR-27b. As shown in Fig. 1B,
an increase of miR-27b was only observed in cells transfected with
HPV16 E7 plasmids, not E6 plasmids, indicating that it was HPV16 E7
that induced miR-27b expression in cervical cancer cells.
Furthermore, the basal levels of miR-27b in CaSki and SiHa cells
were also higher than HPV-negative C33A cells (Fig. 1C), which was consistent with a
recent report (24).
miR-27b represses PPAR,γ expression in
cervical cancer cells
Given the fact that PPARγ was one of the direct
targets of miR-27b in adipocytes and macrophages (25,26),
we examined whether miR-27b also regulated endogenous PPARγ levels
in cervical cancer by transfecting miR-27b mimics or inhibitors
into CaSki cells. As shown in Fig.
2, the overexpression of miR-27b reduced both mRNA and protein
levels of PPARγ. The opposite result was observed after inhibiting
miR-27b expression. Together, these results suggest that PPARγ is
also targeted by miR-27b in cervical cancer cells.
PPARγ inhibits tumor progression in
cervical cancer cells
PPARγ has been shown in breast cancer cells to
directly downregulate NHE1, which is a well characterized oncogenic
pH regulator (19). To examine
whether PPARγ also affected the expression of NHE1 in cervical
cancer cells, CaSki cells were transfected with PPARγ siRNA,
followed by the measurement of NHE1 mRNA and protein levels using
RT-PCR and western blot analysis. The results indicated that the
inhibition of PPARγ was able to increase both mRNA and protein
expression level of NHE1 in cervical cancer cells (Fig. 3A). PPARγ has been reported to be
involved in tumor cell proliferation and invasion (13). To investigate the effects of PPARγ
on proliferation of cervical cancer cells, CaSki cells were
transfected with siRNA targeting PPARγ and the changes in cell
proliferation were analyzed using the CCK-8 assay. The results
showed that inhibition of PPARγ promoted proliferation of CaSki
cells (Fig. 3B). We then
determined the roles of PPARγ in invasion ability of cervical
cancer cells. SiHa cells were transfected with PPARγ siRNA,
followed by Transwell assay. As shown in Fig. 3C, knockdown of PPARγ resulted in
the induction of invasion.
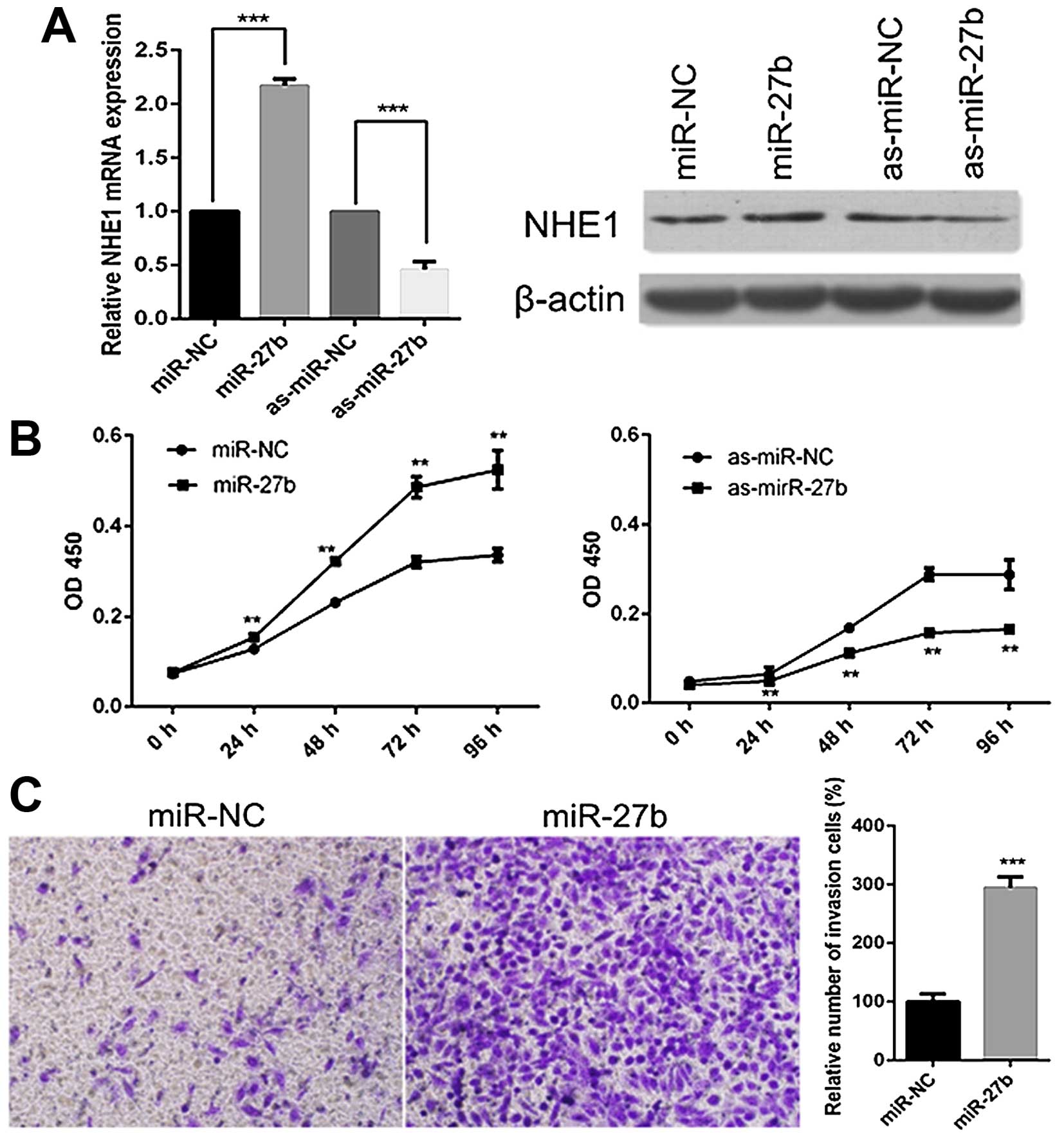
miR-27b has a tumor-promoting role in
cervical cancer cells
Since PPARγ, the target of miR-27b, has been
indicated as a tumor suppressor in cervical cancer in the present
study, we then investigated the roles of miR-27b in cervical cancer
cells by transfecting miR-27b mimics or inhibitors into CaSki or
SiHa cells. In accordance with the results of PPARγ inhibition, the
miR-27b overexpression significantly increased the levels of NHE1,
while miR-27b repression led to a reduction of NHE1 both at mRNA
and protein levels (Fig. 4A).
Next, we assessed the effects of miR-27b on the proliferation of
CaSki cells. CCK-8 assay revealed that overexpression of miR-27b
enhanced the proliferation, whereas miR-27b inhibition reduced the
proliferation of CaSki cells (Fig.
4B). Furthermore, we tested the ability of miR-27b in invasion
of SiHa cells. As seen in Fig. 4C,
the treatment with miR-27b mimics significantly increased the
number of SiHa cells that passed through the Transwell chamber.
These results suggest the tumor-promoting role of miR-27b in
cervical cancer.
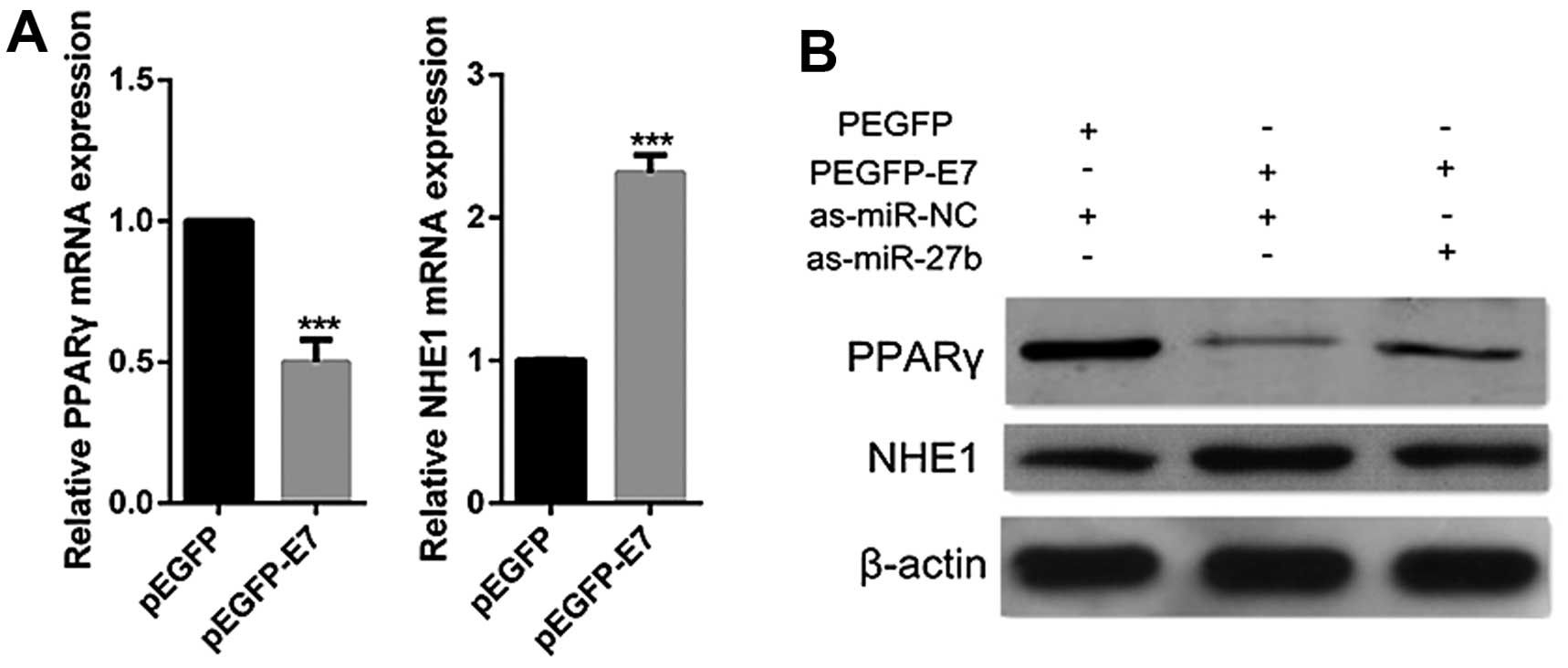
HPV16 E7 inhibits PPARγ expression and
promotes NHE1 expression via promotion of miR-27b in cervical
cancer cells
HPV16 E7 acts as an oncogenic promoter and is
considered to be crucial for human cervical tumorigenic process
(27). HPV16 E7 upregulates
miR-27b which suppresses the expression of PPARγ, it is therefore
hypothesized that HPV16 E7 could inhibit PPARγ expression. To
confirm this hypothesis, CaSki cells were transfected with E7
plasmids. Decrease of PPARγ expression was observed in HPV16 E7
overexpressed cells by real-time PCR and western blot analyses
(Fig. 5). Besides, NHE1 was
reported previously to be upregulated by HPV16 E7 (21,28).
In the present study, we also observed that the overexpression of
HPV16 E7 led to increase of NHE1 (Fig.
5). However, when cells were co-transfected with HPV16 E7
plasmids and miR-27b inhibitors, the inhibition of miR-27b
dramatically abolished the ability of HPV16 E7 to suppress PPARγ or
induce NHE1 expression (Fig. 5B).
Taken together, these data indicate that HPV16 E7 modulates the
expression of PPARγ and NHE1 through miR-27b regulation.
Increase in expression of miR-27b and
NHE1, and decrease in expression of PPARγ are observed in
HPV16-positive cervical cancer tumor samples
We then measured the expression of miR-27b, PPARγ
and NHE1 by real-time PCR using six pairs of HPV16-positive
cervical cancer tissue samples. The results showed that the levels
of miR-27b and NHE1 were significantly higher in cervical tumor
tissues than in adjacent normal cervical tissues, while PPARγ
expression was decreased in cervical tumor tissues (Fig. 6). Again, these clinical data
provide strong support for our hypothesis that the reduced
expression of PPARγ and enhanced expression of miR-27b and NHE1 are
likely associated with the positive status of HPV16.
Discussion
Cervical cancer is one of the most highly malignant
and lethal types of cancer in women worldwide. HR-HPV infection has
been widely recognized as a leading cause of cervical cancer
(11). Two viral oncogenes, HR-HPV
E6 and E7, have been considered to play a critical role in
HPV-associated cervical cancer pathogenesis. They deregulate
multiple genes that are essential for host cell biological
processes (27). MicroRNAs
represent a class of non-coding RNAs that regulate gene expression
at the post-transcriptional level. Substantial numbers of cellular
miRNAs exhibited altered expression due to HPV infection (11). Thus, it is conceivable that
aberrant expression of miRNAs is associated with HPV E6 and E7.
Recent reports have suggested that HPV E7 deregulates the
expression of miRNAs such as miR-15a/miR-16-1, miR-203 and miR-15b
(11,29,30).
Similarly, in the present study, we found that miR-27b was
upregulated by HPV16 E7. The miR-27 family is shown to be induced
upon inflammation in macrophages and could inhibit adipocyte
differentiation (25,26,31).
However, the roles of miR-27b in tumor development are
controversial. It has been suggested that miR-27b acts as a tumor
suppressor. A recent report has shown that miR-27b inhibits tumor
progression and angiogenesis in colorectal cancer by targeting
VEGFC (32). It is also shown that
miR-27b targets LIMK1 to inhibit growth and invasion of NSCLC cells
(33). However, several lines of
evidence have suggested that miR-27b functions as an oncogene. One
report showed that miR-27b could promote the proliferation and
invasion of breast cancer cells by inhibiting the expression of
ST-14 (34). It has been also
found that the inhibition of miR-27b promotes apoptosis and
negatively regulates the growth and invasion of glioma cells
(35). Nevertheless, the
expression condition and detailed roles of miR-27b in cervical
cancer are poorly understood, except that miR-27b has been
previously reported to be at much higher levels in HPV16-positive
cervical carcinoma cells than in HPV-negative cervical carcinoma
cells (24). According to the
results of the present study, it is suggested that higher basal
level of miR-27b in HPV16-positive cells is contributed to HPV16
E7. Subsequently, we found that miR-27b expression levels were
higher in cervical cancer tissues than adjacent normal tissues and
miR-27b enhanced proliferation and invasion of cervical cancer cell
lines, indicating that miR-27b serves as an oncogene in cervical
cancer.
It has been previously reported that miR-27b targets
PPARγ directly in adipocytes and macrophages (25,26).
Similar results were found in the present study that miR-27b
suppressed PPARγ at mRNA and protein levels in CaSki cells. PPARγ
is a member of nuclear receptor superfamily and is suggested to be
involved in cancer progression (13). PPARγ probably acts as a tumor
suppressor in various types of cancer (13). The activation of PPARγ interferes
with proliferation and invasion in glioblastoma, as well as reduces
growth and expansion of brain tumor stem cells (36,37).
It is also suggested that PPARγ activators induce differentiation
and apoptosis in non-small-cell lung cancer cells (38). In gastric cancer cell lines PPARγ
promoted apoptosis and induced G1 cell cycle arrest (39,40).
Anticancer effects of PPARγ ligands have also been reported in
pancreatic cancer cells as PPARγ activation represses cell growth
and attenuates the capacity of migration and invasion (41,42).
Regarding the roles of PPARγ in cervical cancer, one report shows
that PPARγ is less abundant in cervical carcinoma tissues than in
normal cervical tissues (18),
which is confirmed in the present study. Another study showed that
treatment with PPARγ agonist in vitro induced apoptosis and
suppressed proliferation of cervical cancer cells (43). In this study, we found that the
inhibition of PPARγ by siRNA promoted proliferation and invasion of
cervical cancer cells, implicating the antitumor roles of PPARγ in
cervical cancer. In addition, we observed that miR-27b, which was
positively regulated by HPV16 E7, inhibited the expression of
PPARγ. Furthermore, the overexpression of HPV16 E7 suppressed the
expression of PPARγ depending on the existence of miR-27b. These
findings suggest a link between HPV16 E7 and PPARγ whereby HPV16 E7
is able to repress the expression of PPARγ through the promotion of
miR-27b.
NHE1 is an integral membrane transport protein
involved in regulating cytoplasmic pH. The activation of NHE1 could
result in intracellular alkalinization and extracellular
acidification. This dyregulation in pH by NHE1 takes place very
early in cancer progression and subsequently drives behavior such
as enhanced proliferation and growth, dyregulation of cell cycle
progression, resistance to apoptosis, facilitation of migration and
invasion, which is essential for the development and maintenance of
transformed phenotype (20).
Therefore, NHE1 is well characterized to be an oncogene. In the
present study, we found that the levels of NHE1 were significantly
higher in cervical cancer tissues than those in normal adjacent
tissues. It has been reported that NHE1 enhanced proliferation,
migration and invasion in cervical cancer cell lines (44–46),
suggesting the carcinogenic role of NHE1 in cervical cancer. A
recent study showed that NHE1 is directly targeted by PPARγ in
breast cancer cells (19).
Similarly, in this study, the repression of PPARγ resulted in
significant increase of NHE1, indicating that PPARγ also inhibited
NHE1 in cervical cancer cells. The findings also illustrated
ectopic expression of miR-27b induced NHE1 expression, while
suppression of miR-27b led to the downregulation of NHE1. As for
the relationship between E7 and NHE1, our results showed that HPV16
E7 could upregulate NHE1. Actually, the induction of NHE1
expression by HPV16 E7 was previously reported (21,28).
HPV16 E7 may activate NHE1 through a PKA-RhoA-induced inhibition of
p38α (28). Nevertheless, the
present study revealed another mechanism underlying HPV16
E7-mediated upregulation of NHE1 whereby HPV16 E7 upregulates NHE1
via inducing the expression of miR-27b.
Numerous studies have focused on exploiting the
clinical potential of PPAR or NHE1 in anti-cancer treatment
(13,47). The intelligent use of optimized
compounds derived from PPAR agonist and the development of NHE1
inhibitors could be avenues for anti-cancer drug design. In the
present study, we observed the levels of miR-27b were consistently
correlated with those of NHE1 but inversely correlated with PPARγ
in cervical cancer tissues. Besides, miR-27b suppresses the
expression of PPARγ and upregulates the levels of NHE1, suggesting
that miR-27b may be a promising therapeutic target for treatment of
cervical cancer.
In a conclusion, our findings demonstrate that HPV16
E7 upregulates miR-27b, which in turn decreases the expression of
PPARγ to enhance cervical cancer progression. We believe that an
understanding of the roles of E7-miR-27b-PPARγ-NHE1 pathway holds
much promise for the development of future treatments for cervical
cancer.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81371901) and the Doctoral
Program of Ministry of Education (no. 20134433110010).
References
|
1
|
Comprehensive Cervical Cancer Control. A
Guide to Essential Practice. World Health Organization; 2nd
edition. Geneva: 2014
|
|
2
|
Walboomers JM, Jacobs MV, Manos MM, Bosch
FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ and Muñoz N:
Human papillomavirus is a necessary cause of invasive cervical
cancer worldwide. J Pathol. 189:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tommasino M, Accardi R, Caldeira S, Dong
W, Malanchi I, Smet A and Zehbe I: The role of TP53 in cervical
carcinogenesis. Hum Mutat. 21:307–312. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hwang SG, Lee D, Kim J, Seo T and Choe J:
Human papillomavirus type 16 E7 binds to E2F1 and activates
E2F1-driven transcription in a retinoblastoma protein-independent
manner. J Biol Chem. 277:2923–2930. 2002. View Article : Google Scholar
|
|
5
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Di Leva G and Croce CM: Roles of small
RNAs in tumor formation. Trends Mol Med. 16:257–267. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar
|
|
10
|
Lin Z and Flemington EK: miRNAs in the
pathogenesis of oncogenic human viruses. Cancer Lett. 305:186–199.
2011. View Article : Google Scholar :
|
|
11
|
Zheng ZM and Wang X: Regulation of
cellular miRNA expression by human papillomaviruses. Biochim
Biophys Acta. 1809:668–677. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mangelsdorf DJ, Thummel C, Beato M,
Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M,
Chambon P, et al: The nuclear receptor superfamily: The second
decade. Cell. 83:835–839. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Youssef J and Badr M: Peroxisome
proliferator-activated receptors and cancer: Challenges and
opportunities. Br J Pharmacol. 164:68–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim KY, Kim SS and Cheon HG: Differential
anti-proliferative actions of peroxisome proliferator-activated
receptor-gamma agonists in MCF-7 breast cancer cells. Biochem
Pharmacol. 72:530–540. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sarraf P, Mueller E, Jones D, King FJ,
DeAngelo DJ, Partridge JB, Holden SA, Chen LB, Singer S, Fletcher
C, et al: Differentiation and reversal of malignant changes in
colon cancer through PPARgamma. Nat Med. 4:1046–1052. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tontonoz P, Singer S, Forman BM, Sarraf P,
Fletcher JA, Fletcher CD, Brun RP, Mueller E, Altiok S, Oppenheim
H, et al: Terminal differentiation of human liposarcoma cells
induced by ligands for peroxisome proliferator-activated receptor
gamma and the retinoid X receptor. Proc Natl Acad Sci USA.
94:237–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cellai I, Benvenuti S, Luciani P, Galli A,
Ceni E, Simi L, Baglioni S, Muratori M, Ottanelli B, Serio M, et
al: Antineoplastic effects of rosiglitazone and PPARgamma
transactivation in neuroblastoma cells. Br J Cancer. 95:879–888.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jung TI, Baek WK, Suh SI, Jang BC, Song
DK, Bae JH, Kwon KY, Bae JH, Cha SD, Bae I, et al: Down-regulation
of peroxisome proliferator-activated receptor gamma in human
cervical carcinoma. Gynecol Oncol. 97:365–373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kumar AP, Quake AL, Chang MK, Zhou T, Lim
KS, Singh R, Hewitt RE, Salto-Tellez M, Pervaiz S and Clément MV:
Repression of NHE1 expression by PPARgamma activation is a
potential new approach for specific inhibition of the growth of
tumor cells in vitro and in vivo. Cancer Res. 69:8636–8644. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reshkin SJ, Cardone RA and Harguindey S:
Na+-H+ exchanger, pH regulation and cancer.
Recent Pat Anticancer Drug Discov. 8:85–99. 2013. View Article : Google Scholar
|
|
21
|
Reshkin SJ, Bellizzi A, Caldeira S,
Albarani V, Malanchi I, Poignee M, Alunni-Fabbroni M, Casavola V
and Tommasino M: Na+/H+ exchanger-dependent
intracellular alkalinization is an early event in malignant
transformation and plays an essential role in the development of
subsequent transformation-associated phenotypes. FASEB J.
14:2185–2197. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang S, Tao M, McCoy JP Jr and Zheng ZM:
The E7 oncoprotein is translated from spliced E6*I transcripts in
high-risk human papillomavirus type 16- or type 18-positive
cervical cancer cell lines via translation reinitiation. J Virol.
80:4249–4263. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Martinez I, Gardiner AS, Board KF, Monzon
FA, Edwards RP and Khan SA: Human papillomavirus type 16 reduces
the expression of microRNA-218 in cervical carcinoma cells.
Oncogene. 27:2575–2582. 2008. View Article : Google Scholar :
|
|
25
|
Karbiener M, Fischer C, Nowitsch S,
Opriessnig P, Papak C, Ailhaud G, Dani C, Amri EZ and Scheideler M:
microRNA miR-27b impairs human adipocyte differentiation and
targets PPARgamma. Biochem Biophys Res Commun. 390:247–251. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jennewein C, von Knethen A, Schmid T and
Brüne B: MicroRNA-27b contributes to lipopolysaccharide-mediated
peroxisome proliferator-activated receptor gamma (PPARgamma) mRNA
destabilization. J Biol Chem. 285:11846–11853. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ghittoni R, Accardi R, Hasan U, Gheit T,
Sylla B and Tommasino M: The biological properties of E6 and E7
oncoproteins from human papillomaviruses. Virus Genes. 40:1–13.
2010. View Article : Google Scholar
|
|
28
|
Cardone RA, Busco G, Greco MR, Bellizzi A,
Accardi R, Cafarelli A, Monterisi S, Carratù P, Casavola V,
Paradiso A, et al: HPV16 E7-dependent transformation activates NHE1
through a PKA-RhoA-induced inhibition of p38alpha. PLoS One.
3:e35292008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Myklebust MP, Bruland O, Fluge Ø,
Skarstein A, Balteskard L and Dahl O: MicroRNA-15b is induced with
E2F-controlled genes in HPV-related cancer. Br J Cancer.
105:1719–1725. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Melar-New M and Laimins LA: Human
papillomaviruses modulate expression of microRNA 203 upon
epithelial differentiation to control levels of p63 proteins. J
Virol. 84:5212–5221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim SY, Kim AY, Lee HW, Son YH, Lee GY,
Lee JW, Lee YS and Kim JB: miR-27a is a negative regulator of
adipocyte differentiation via suppressing PPARgamma expression.
Biochem Biophys Res Commun. 392:323–328. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ye J, Wu X, Wu D, Wu P, Ni C, Zhang Z,
Chen Z, Qiu F, Xu J and Huang J: miRNA-27b targets vascular
endothelial growth factor C to inhibit tumor progression and
angiogenesis in colorectal cancer. PLoS One. 8:e606872013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wan L, Zhang L, Fan K and Wang J: MiR-27b
targets LIMK1 to inhibit growth and invasion of NSCLC cells. Mol
Cell Biochem. 390:85–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Y, Rathinam R, Walch A and Alahari
SK: ST14 (suppression of tumorigenicity 14) gene is a target for
miR-27b, and the inhibitory effect of ST14 on cell growth is
independent of miR-27b regulation. J Biol Chem. 284:23094–23106.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen L, Li H, Han L, Zhang K, Wang G, Wang
Y, Liu Y, Zheng Y, Jiang T, Pu P, et al: Expression and function of
miR-27b in human glioma. Oncol Rep. 26:1617–1621. 2011.PubMed/NCBI
|
|
36
|
Grommes C, Landreth GE, Sastre M, Beck M,
Feinstein DL, Jacobs AH, Schlegel U and Heneka MT: Inhibition of in
vivo glioma growth and invasion by peroxisome
proliferator-activated receptor gamma agonist treatment. Mol
Pharmacol. 70:1524–1533. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chearwae W and Bright JJ: PPARgamma
agonists inhibit growth and expansion of CD133+ brain
tumour stem cells. Br J Cancer. 99:2044–2053. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chang TH and Szabo E: Induction of
differentiation and apoptosis by ligands of peroxisome
proliferator-activated receptor gamma in non-small cell lung
cancer. Cancer Res. 60:1129–1138. 2000.PubMed/NCBI
|
|
39
|
Sato H, Ishihara S, Kawashima K, Moriyama
N, Suetsugu H, Kazumori H, Okuyama T, Rumi MA, Fukuda R, Nagasue N,
et al: Expression of peroxisome proliferator-activated receptor
(PPAR) gamma in gastric cancer and inhibitory effects of PPARgamma
agonists. Br J Cancer. 83:1394–1400. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen YX, Zhong XY, Qin YF, Bing W and He
LZ: 15d-PGJ2 inhibits cell growth and induces apoptosis of MCG-803
human gastric cancer cell line. World J Gastroenterol. 9:2149–2153.
2003.PubMed/NCBI
|
|
41
|
Motomura W, Okumura T, Takahashi N, Obara
T and Kohgo Y: Activation of peroxisome proliferator-activated
receptor gamma by troglitazone inhibits cell growth through the
increase of p27KiP1 in human. Pancreatic carcinoma cells. Cancer
Res. 60:5558–5564. 2000.PubMed/NCBI
|
|
42
|
Motomura W, Nagamine M, Tanno S, Sawamukai
M, Takahashi N, Kohgo Y and Okumura T: Inhibition of cell invasion
and morphological change by troglitazone in human pancreatic cancer
cells. J Gastroenterol. 39:461–468. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen HM, Zhang DG, Wu JX, Pei DS and Zheng
JN: Ubiquitination of p53 is involved in troglitazone induced
apoptosis in cervical cancer cells. Asian Pac J Cancer Prev.
15:2313–2318. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li QH, Wang LH, Lin YN, Chang GQ, Li HW,
Jin WN, Hu RH and Pang TX: Nuclear accumulation of calcineurin B
homologous protein 2 (CHP2) results in enhanced proliferation of
tumor cells. Genes Cells. 16:416–426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chiang Y, Chou CY, Hsu KF, Huang YF and
Shen MR: EGF upregulates Na+/H+ exchanger
NHE1 by post-translational regulation that is important for
cervical cancer cell invasiveness. J Cell Physiol. 214:810–819.
2008. View Article : Google Scholar
|
|
46
|
Lin Y, Wang J, Jin W, Wang L, Li H, Ma L,
Li Q and Pang T: NHE1 mediates migration and invasion of HeLa cells
via regulating the expression and localization of MT1-MMP. Cell
Biochem Funct. 30:41–46. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Loo SY, Chang MK, Chua CS, Kumar AP,
Pervaiz S and Clement MV: NHE-1: A promising target for novel
anti-cancer therapeutics. Curr Pharm Des. 18:1372–1382. 2012.
View Article : Google Scholar : PubMed/NCBI
|