Introduction
Gastric cancer (GC) is the fourth most common
malignancy in the world (1). To
date, systemic chemotherapy in addition to surgery has improved the
survival in some patients with advanced GC, but treatments for
patients with far advanced or potentially aggressive GC often
result in poor survival owing to metastases to distant organs
(2,3). At present, the underlying mechanisms
that accelerate invasion or metastasis in GC are not well
documented.
The hypoxic environment is substantial in solid
tumors, where it accelerates their malignant behavior (4–8).
Hypoxia inducible factor (HIF)-1α is a transcription factor that is
induced under hypoxic conditions and monitors the cellular response
to oxygen levels in solid tumors (9,10).
Under normoxia, prolyl hydroxylases (PHDs) hydroxylate proline
residues of HIF-1α in a reaction that uses O2 as a
substrate, and the modified HIF-1α is subsequently degraded by the
ubiquitin proteasome system. Under hypoxia, HIF-1α protein is
stabilized owing to the absence of O2 substrate and
instead forms a heterodimer with the HIF-1β subunit (11–14).
The HIF-1 complex then activates the transcription of hundreds of
target genes and leads to adaptation to the hypoxic environment
(11,12). HIF-1α has been reported to
upregulate target genes related to energy metabolism, angiogenesis,
cell proliferation or survival, invasion or metastasis, and drug
resistance in human cancer cells (13–15).
In GC, previous studies have also reported that HIF-1α activation
following extended hypoxia strongly correlates with an aggressive
tumor phenotype and poor prognosis of patients (16–19).
Therefore HIF-1α is recognized as a central regulator of GC
pathogenesis (14,20).
Reactive oxygen species (ROS) such as superoxide are
generated mainly through the electron transport chain (ETC)
(21,22). Hypoxia is known to stimulate the
ROS production from mitochondrial complex III in the ETC (23). Lower levels of mitochondrial ROS
(mtROS) play important roles as signaling molecules to adapt to
stress and are required for normal cell homeostasis, while
excessive quantities of mtROS directly damage proteins, lipids and
nucleic acids, and lead to cell death (21). Previous reports have shown that
HIF-1α is also stabilized by mtROS, whereby mtROS inactivate PHDs
by decreasing intracellular Fe2+ levels through
oxidization to Fe3+ (24–27).
Macroautophagy (general autophagy) is a dynamic
process through which cytosol and organelles are sequestered into a
double-membrane vesicle called an autophagosome and delivered to
the lysosome for breakdown and recycling during challenging
conditions, such as nutrient depletion or hypoxia. The core process
of general autophagy is mediated by autophagy-related (ATG) genes.
The conversion of LC3-I to LC3-II and degradation of SQSTM1/p62
(referred as p62) are known to indicate the autophagy process
(28–30). Mitochondrial autophagy, designated
as mitophagy, is a selective form of autophagy in which
mitochondria are degraded in autolysosomes (31,32).
Mitophagy represents a critical adaptive mechanism to maintain
oxygen homeostasis and to eliminate old and/or damaged
mitochondrion in response to certain stresses including hypoxia
(33,34). In addition to the core factors in
the autophagy, previous studies have revealed that specific
regulators such as BNIP3, NIX, Parkin, Pink1 and FUNDC1 are
required for promoting mitophagy (31,34–36).
Recently, emerging evidence has revealed that deregulation in key
regulators of mitophagy is found in several cancers and suggested a
possible implication in tumorigenesis (34). For instance, Parkin located at
human chromosome 6q25-q26 is frequently deleted in bladder, lung,
breast and ovarian cancers. These reports highlight the roles of
mitophagy-related factors as potential tumor suppressors (37,38).
However, the mechanisms underlying deregulated mitophagy in
tumorigenesis or cancer progression have not been elucidated.
In the present study, we first compared HIF-1α
expression level with mtROS production and cancer aggressiveness in
three GC cell lines under hypoxic conditions. We evaluated whether
the integrity of mitophagy affects mtROS production and HIF-1α
expression in these cells. This study presents a novel mechanism of
cancer aggressiveness through activated mtROS/HIF-1α interaction,
which originated from impaired mitophagy, in GC cells under hypoxic
conditions.
Materials and methods
Cell culture and exposure to hypoxia
Three human GC cell lines (44As3, 58As9 and MKN45)
were used in this study. The 44As3 and 58As9 cell lines were kindly
provided by Dr K. Yanagihara (National Cancer Center Hospital East,
Chiba, Japan); these two cell lines are scirrhous GC cell lines and
have strong potential for inducing the formation of peritoneal
metastasis in the orthotopic mouse model (39,40).
MKN45 cells were purchased from Cell Bank, RIKEN BioResource Center
(Tsukuba, Japan), and these cells are non-scirrhous GC cells. All
cells were cultured in RPMI-1640 medium (Sigma-Aldrich, St. Louis,
MO, USA) supplemented with 10% heat-inactivated FBS and 100 μg/ml
kanamycin (Meiji, Tokyo, Japan), and maintained under either
normoxic conditions (20% O2 and 5% CO2 in
air) or hypoxic conditions (1% O2, 5% CO2 and
94% N2) in a hypoxic chamber (Astec, Fukuoka,
Japan).
Reagents
DMOG (Sigma-Aldrich) was used as the inhibitor of
PHDs, NAC (Wako, Osaka, Japan) was used as the ROS scavenger and CQ
(Sigma-Aldrich) as the autophagy inhibitor. The reagents were
dissolved in deionized water and used at the indicated
concentration. The working concentrations were determined as DMOG
at 1 mM for 4 h, NAC at 20 mM for 4 h and CQ at 5 μM.
RNA interference for HIF-1α
The siRNA for the human HIF-1α gene
(SASI_Hs02_00332063) and the non-silencing siRNA control
(Mission® siRNA Universal Negative Control) were
purchased from Sigma-Aldrich. The siRNAs were transiently
transfected into 44As3, 58As9 and MKN45 cells using a
MicroPorator-mini (MP-100) (Digital Bio Technology, Seoul, Korea)
in combination with the Neon™ 100-μl kit (Thermo Scientific,
Waltham, MA, USA) according to the manufacturer's instructions. The
transfectants were cultured in complete medium and harvested on
days 1–2 after transfection. The transfected cells were cultured in
complete medium and harvested on days 1–3 for a westren blot
analysis.
Western blot analysis
Whole cell lysates from cultured cells were prepared
using a lysis buffer composed of 150 mM NaCl, 50 mM Tris-HCl (pH
7.6), 0.5% Triton X-100 containing a protease inhibitor cocktail
mix (Roche Diagnostics GmbH, Mannheim, Germany) and 1 mM PMSF
(Sigma-Aldrich). Protein samples (15 μg) were electrophoresed with
NuPAGE 4–12% or 12% Bis-Tris gel (Invitrogen, Waltham, MA, USA) and
were transferred onto polyvinylidene fluoride membranes using the
Trans-Blot®Turbo™ Transfer System (Bio-Rad, Hercules,
CA, USA). After blocking with 5% skim milk for 1 h, the membranes
were incubated with primary antibodies overnight at 4°C. Following
incubation with the corresponding secondary antibodies, the signals
were developed using the Amersham™ ECL Prime Western Blotting
Detection Reagent (GE Healthcare, Buckinghamshire, UK).
Antibodies
The primary antibodies used in this study were mouse
monoclonal anti-HIF-1α antibody (1:1,000, Becton-Dickinson
Biosciences, NJ, USA), rabbit polyclonal anti-SQSTM1/p62 antibody
(1:1,000, Cell Signaling, Danvers, MA, USA), rabbit monoclonal
anti-LC3-I/II antibody (1:1,000, Cell Signaling), and mouse
monoclonal anti-β-actin antibody (1:10,000, Sigma-Aldrich). Goat
anti-rabbit IgG-HRP and goat anti-mouse IgG-HRP (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) were used as secondary
antibodies.
Real-time quantitative
reverse-transcription polymerase chain reaction (RT-qPCR)
Total RNA was extracted from each cell line using an
Isogen II (Nippon Gene, Osaka, Japan) according to the
manufacturer's instructions. For each cell line, 1 μg of RNA was
converted into cDNA using a ReverTra Ace (Toyobo, Osaka, Japan);
reverse transcription reaction kit. The cDNA was used as a template
for the PCR. RT-qPCR was performed by means of the LightCycler™
instrument system using the Light-Cycler-FastStart DNA Master™ SYBR
Green I kit (Roche Diagnostics GmbH). The primer sequences designed
according to cDNA sequences by Ensemble and NCBI Primer-BLAST
system were as follows: HIF-1α sense 5′-CCTATGACCTGCTTGGTGCT-3′ and
antisense 5′-TAT CCAGGCTGTGTCGACTG-3′ (175-bp product); and β-actin
sense 5′-ACTCTTCCAGCCTTCCTTCC-3′ and antisense
5′-GACAGCACTGTCTTGGCGTA-3′ (120-bp product). After performing a
denaturation step at 95°C for 3 min, PCR amplification was
conducted with 50 cycles of 15 sec of denaturation at 95°C, 5 sec
of annealing at 60°C and 10 sec of extension at 72°C. A melting
curve analysis was used to control for the specificity of the
amplification products. The quantitative values were normalized by
the β-actin expression level, which was used as an internal
control. All experiments were performed in triplicate and the mean
values were calculated. The experiments were independently repeated
three times.
Cell survival assay
Cell survival ability was assessed with MTS assay
using the CellTiter 96®AQueous One Solution Cell
Proliferation Assay kit (Promega Corp., Madison, WI, USA). In
brief, 1.5×103 cells per well were seeded in triplicate
in 96-well plates and incubated under hypoxic conditions. Numbers
of viable cells were assessed by measuring the optical density at
490 nm using a plate reader (iMark Microplate Reader, Bio-Rad) in
triplicate at 0, 12, 24 and 48 h under hypoxia. The mean values
were estimated and the relative proliferation values are presented
as the ratio to 0 h. The experiments were independently repeated
three times.
Cell invasion assay
In vitro invasion assays were performed using
the Falcon®Permeable Support for 24-well plates with
transparent PET membrane with 8.0-μm pores (Corning, NY, USA).
Briefly, invasion chambers were coated with 50 μl (1 mg/ml) of
Corning® Matrigel® Basement Membrane Matrix
Phenol Red-Free (Corning) in cold RPMI-1640 medium and dried
overnight. Suspensions of 1×105 cells in 200 μl of
complete RPMI-1640 medium were placed in the upper compartments of
the chamber, while the lower compartments were filled with 800 μl
of conditioned medium obtained from MRC5 fibroblasts. The culture
units were incubated for 24 h at 37°C under normoxic (20%
O2) and hypoxic (1% O2) condition. Thereafter
the non-invasive cells on upper surface of the filters were then
gently removed using a cotton swab moistened with medium. Viable
invaded cells that had infiltrated onto the lower surface of the
filter were fixed with 70% ethanol and the nuclei were stained with
Mayer's hematoxylin (Muto Pure Chemicals, Tokyo, Japan). Number of
the invaded cells was counted under a light microscope. The
experiments were performed in triplicate and independently repeated
at least three times.
Quantitative analysis of intracellular
ROS
Intracellular ROS levels were estimated using the
Total ROS Detection kit (Enzo Life Sciences Inc., Farmingdale, NY,
USA) according to the manufacturer's instructions. In brief, cells
at 50–60% confluence were incubated for 0–48 h under normoxic and
hypoxic conditions. Cells were harvested using 0.05% trypsin-EDTA
(Wako) and stained with ROS detection dye for 30 min at 37°C in the
dark. ROS were measured on a FACSCalibur (Becton-Dickinson, San
Jose, CA, USA), and the intensity levels were analyzed by Cell
Quest softwear program. The experiments were performed in
triplicate and independently repeated at least three times. The
mean fluorescence of ROS production was determined automatically
and presented as the GEO mean.
Immunofluorescence
Cells were immunolabeled with the antibodies and
reagents according to the manufacturer's instructions. In brief,
the medium was removed and cells were washed at least twice. For
staining of mitochondrion and lysosomes, Mitotracker®
Green FM (Thermo Scientific), mitochondrion selective probes, and
Lysotracker® Red DND-99 (Thermo Scientific), lysosome
selective probes, were added to living cells at the final
concentrations of 0.1 and 0.025 μM, respectively. Mitophagy was
assessed by the colocalization of Mitotracker and Lysotracker, as
described elsewhere (42).
To evaluate mtROS, MitoSOX® Red (Thermo
Scientific), a mitochondrial superoxide indicator, was added to
living cells at 5 μM. To monitor autophagy, 5 μl of Premo™
Autophagy Sensor LC3B-RFP (Thermo Scientific) was added to
1×104 cells in media and incubated for 24 h. LC3B
(G120A)-RFP was used for the positive control according to the
manufacturer's instructions. Cell nuclei were counterstained by 1
μM Hoechest 33342 (Thermo Scientific). MitoSOX-Red or LC3B-RFP
positive cells were visualized under an EVOS®
FLoid® Cell Imaging Station (Thermo Scientific) and the
numbers of positive cell were counted.
Statistical analysis
Statistical analysis was performed using the
computer software program SPSS 22 for Windows (SPSS Inc, Chicago,
IL, USA). Comparisons between two groups were made using Student's
t-test and the Mann-Whitney U test. P-values of <0.05 were
considered to be statistically significant.
Results
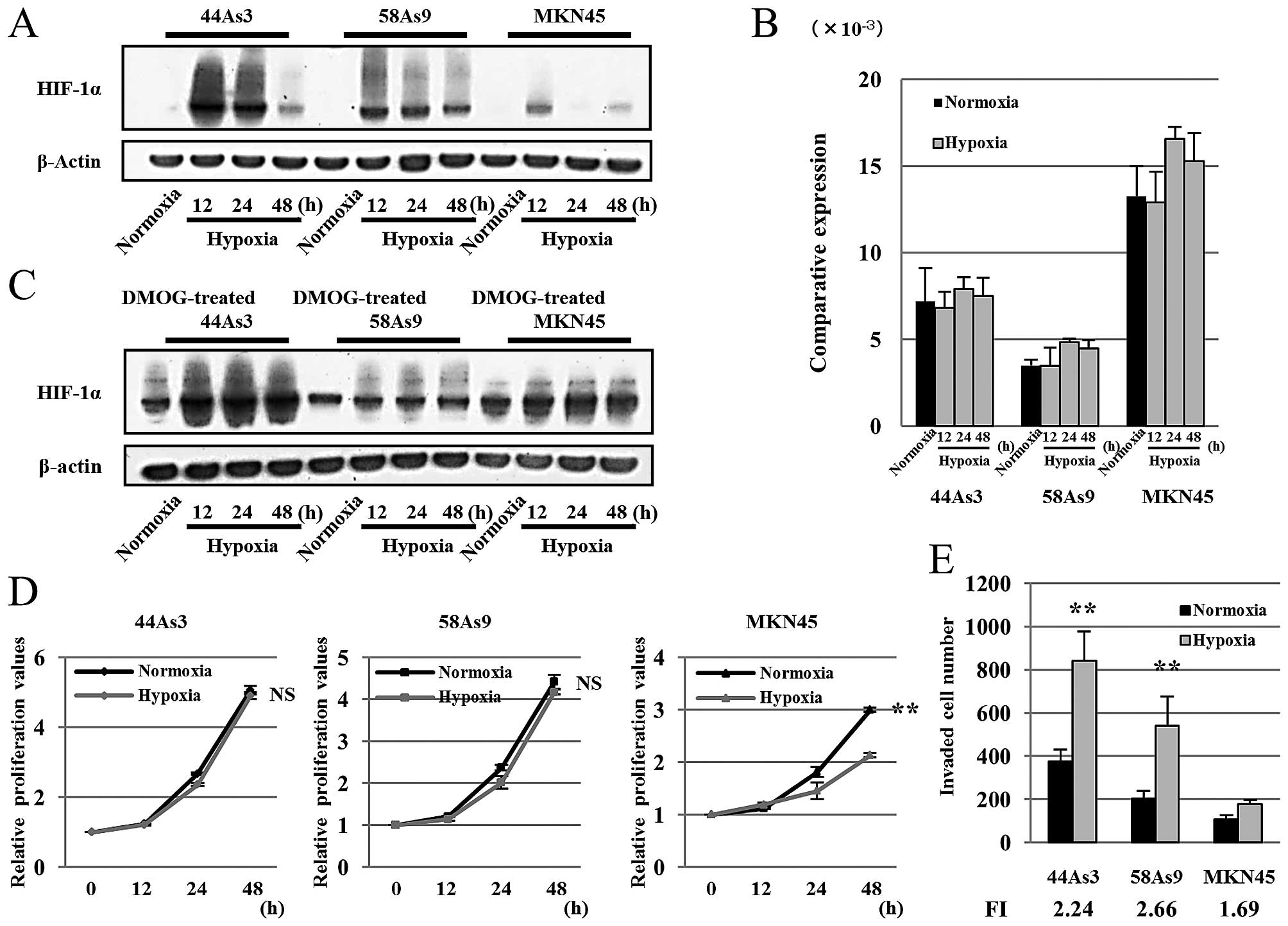
HIF-1α protein levels correlate with cell
survival and invasion activities in GC cell lines under
hypoxia
We first examined the relationship between HIF-1α
expression levels with cell survival and invasion activities among
three GC cell lines, 44As3, 58As9 and MKN45, under hypoxia. HIF-1α
was induced in all cell lines at 12–48 h under hypoxic conditions.
However, the HIF-1α protein levels varied among the cell lines
(Fig. 1A). Expression of HIF-1α
protein, which peaked at 12–24 h of hypoxia, was more strongly
induced in 44As3 and 58As9 cells in comparison to MKN45 cells.
Real-time quantitative reverse-transcription polymerase chain
reaction (RT-qPCR) showed that HIF-1α mRNA was expressed in all
three cell lines under normoxia as well as hypoxia conditions
(Fig. 1B). The strongest mRNA
expression was observed in MKN45 cells among the cell lines.
HIF-1α protein expression was further analyzed in
three GC cell lines with treatment of the PHD inhibitor
dimethyloxallylglycine (DMOG). HIF-1α protein expression was
increased by DMOG treatment in all three GC cell lines under both
normoxia and hypoxia (Fig. 1C).
Next, cell survival was analyzed by MTS assay. Cell viability of
44As3 and 58As9 cells was not affected by hypoxic stimulation. On
the other hand, cell survival of MKN45 cells was suppressed for
24–48 h under hypoxia in comparison to normoxia, and the survival
rate at 48 h was significantly lower under hypoxia than normoxia
(Fig. 1D). We also analyzed cell
invasion activities in the three GC cell lines after 24 h of
normoxia and hypoxia. Hypoxia-induced invasion was evaluated as
fold induction (FI), which was estimated as the ratio of invaded
cell numbers under hypoxia compared with those of normoxia. Results
showed that 44As3 and 58As9 cells exhibited higher FI than MKN45
cells, both overall and under hypoxic condition (Fig. 1E).
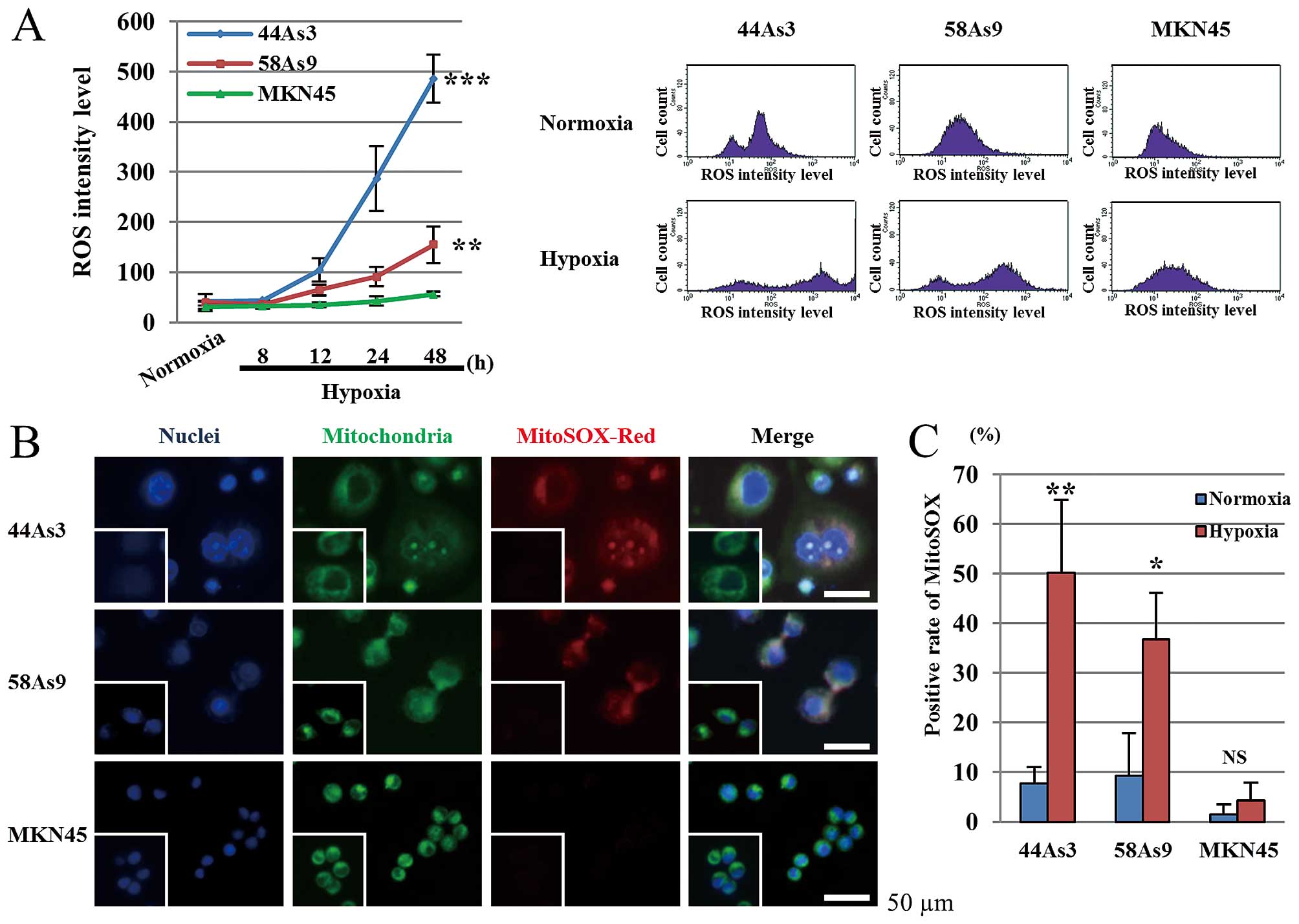
Mitochondrial ROS specifically
accumulates in GC cell lines with aggressive characteristics
We next evaluated the level of intracellular ROS in
three GC cell lines under hypoxia. As shown in Fig. 2A, ROS production was increased in
44As3 and 58As9 cells, but not in MKN45 cells under hypoxia in a
time-dependent manner. ROS levels at 48-h hypoxia were
significantly higher in 44As3 and 58As9 cells compared with MKN45
cells. Mitochondrial ROS (mtROS) production in the three cell lines
was next investigated using MitoSOX-Red staining, a specific
fluorescent marker (Fig. 2B).
Under normoxia conditions, we did not detect any mtROS determined
by MitoSOX-Red staining in all three cell lines. However,
MitoSOX-positive cells were observed in 44As3 and 58As9 cells, but
not in MKN45 cells, after 48-h hypoxia. Merged staining with the
Mitotracker mitochondria marker confirmed ROS production in the
mitochondria of hypoxic 44As3 and 58As9 cells (Fig. 2B). The rate of positive
MitoSOX-stained cells at 48 h was significantly higher under
hypoxia than normoxia conditions in these two cell lines. A lower
frequency of MitoSOX staining was observed in MKN45 cells under
both normoxia and hypoxia (Fig.
2C).
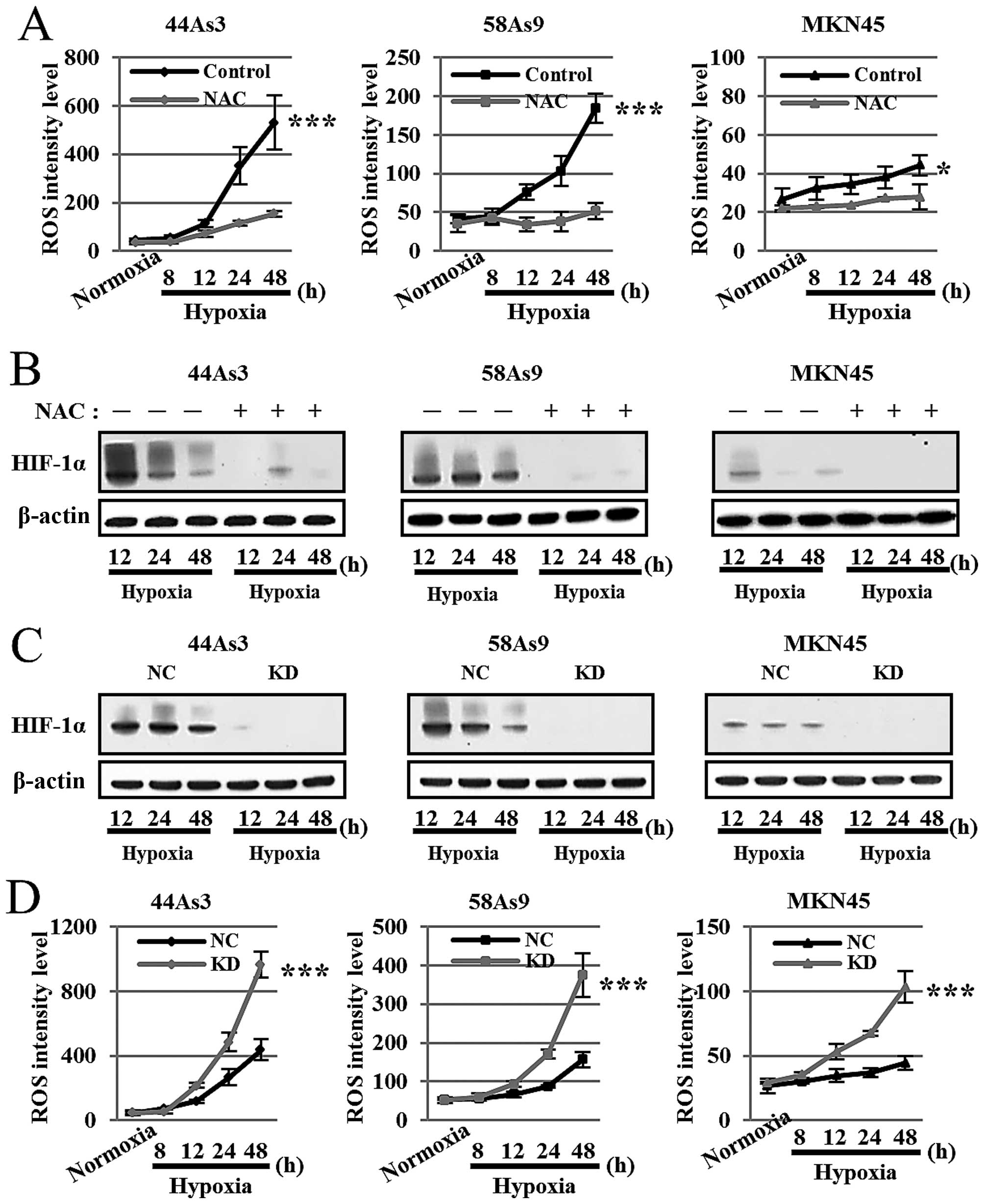
Assessment of the interplay between ROS
and HIF-1α in three GC cell lines under hypoxia
To investigate the ROS/HIF-1α interplay under
hypoxic conditions, we first analyzed HIF-1α expression with or
without the treatment of the antioxidant reagent NAC. As shown in
Fig. 3A, NAC treatment strongly
decreased ROS production in all three cell lines under hypoxia for
12–48 h. There were significant differences in ROS levels at 48-h
hypoxia between NAC treatment and no treatment in all three GC
cells. Furthermore, NAC treatment strongly decreased HIF-1α
expression in all cell lines under hypoxia for 12–48 h (Fig. 3B).
We next evaluated ROS production in the three GC
cell lines knocked down for HIF-1α by siRNA (KD) compared with
negative control siRNA (NC). Western blot analysis confirmed that
HIF-1α expression was strongly knocked down in all three cells
lines transfected with HIF-1α siRNA under hypoxia (Fig. 3C). ROS production was more strongly
elevated in a time-dependent manner in each cell line knocked down
for HIF-1α compared with the corresponding negative control cells
at 12–48-h hypoxia (Fig. 3D).
There were significant differences in ROS levels at 48-h hypoxia
between the HIF-1α knockdown cell lines and negative controls.
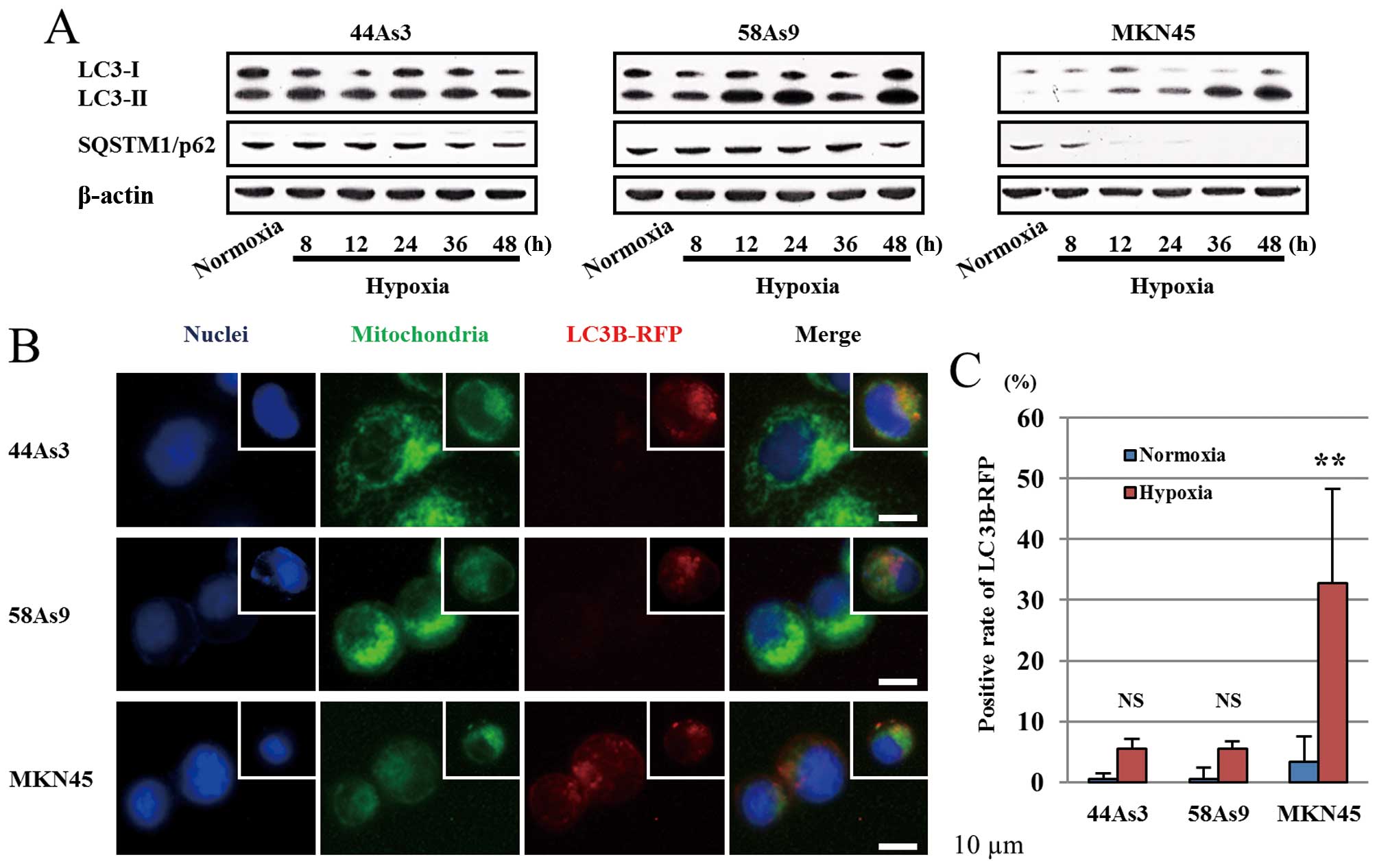
Assessment of general autophagy in three
GC cell lines
We next evaluated autophagy in three GC cell lines
under hypoxic conditions. The conversion of LC3-I to LC3-II as well
as the degradation of p62 was investigated by western blot analysis
(Fig. 4A). From 8 to 48 h under
hypoxia, the expression patterns of LC3-I/II and p62 were not
altered in 44As3 and 58As9 cells. On the other hand, in MKN45
cells, the LC3-II isoform was increased under hypoxia in a
time-dependent manner, resulting in a decreased LC3-I/II ratio.
Moreover, p62 expression disappeared in these cells from 12 h of
hypoxia (Fig. 4A). Autophagy was
also analyzed by fluorescence microscopy of LC3B-RFP (Fig. 4B). LC3B-RFP was not detected in any
of the three cell lines under normoxia (data not shown). Under 48 h
of hypoxia, LC3B-RFP positive cells were observed in MKN45 cells
but not in 44As3 and 58As9 cells (Fig.
4B). Furthermore, we observed co-localization of Mitotracker
and LC3B-RFP in hypoxic MKN45 cells. The positive rate of LC3B-RFP
was significantly higher under hypoxia conditions than in normoxia
in MKN45 cells, while a lower rate was observed in 44As3 and 58As9
cells under both conditions (Fig.
4C).
Assessment of mitophagy in three GC cell
lines
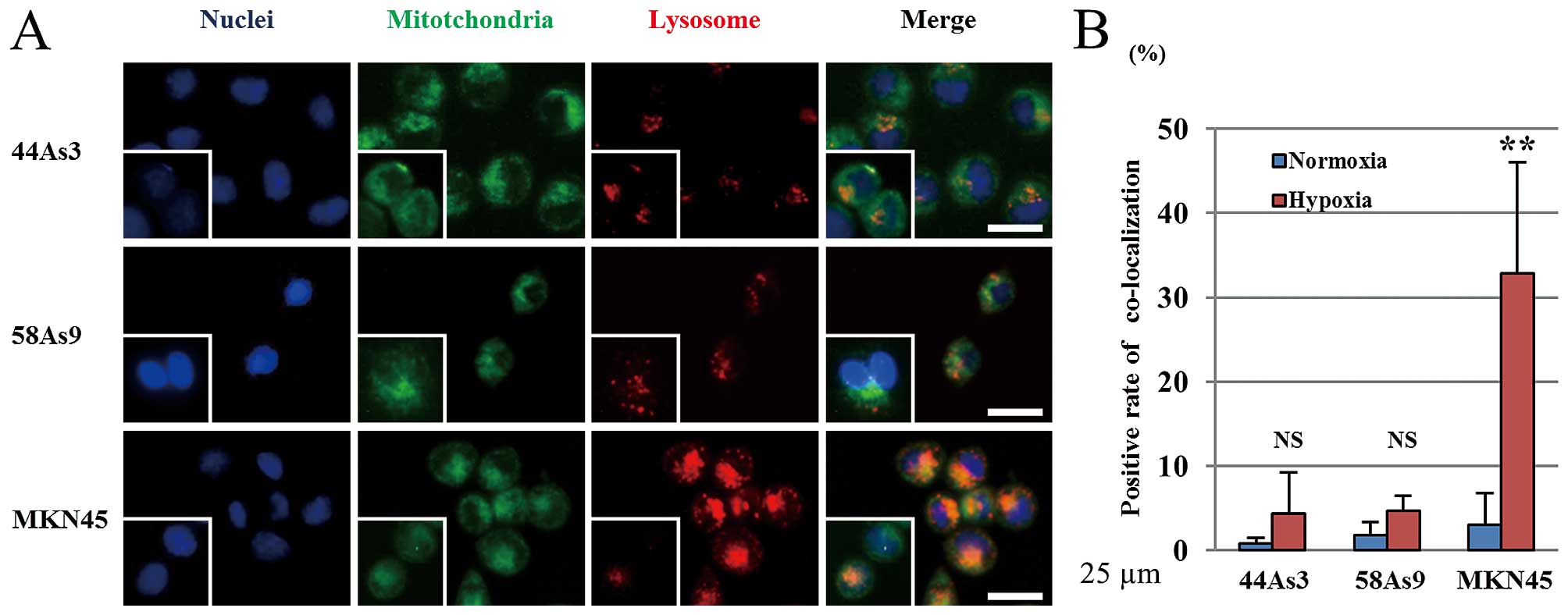
To evaluate the hypoxic induction of mitophagy, we
analyzed the co-localization of the Mitotracker mitochondrial
marker and Lysotracker lysosome marker in the three GC cell lines
(Fig. 5A). Mitotracker-positive
cells were observed in all three cell lines under both normoxia and
hypoxia conditions. In contrast, lysosomes were more strongly
stained by Lysotracker in MKN45 cells under hypoxia than normoxia.
The intensity of lysosomal staining did not differ between the two
conditions in 44As3 and 58As9 cells. The merged image shows the
co-localization of both markers in MKN45 cells (Fig. 5A). The positive rate of
co-localized markers was significantly higher under hypoxia than
normoxia in MKN45 cells, whereas there was no significant
difference between the two conditions in 44As3 and 58As9 cells
(Fig. 5B).
Inhibition of mitophagy increased
malignant characteristics in MKN45 cells under hypoxia
We finally addressed whether inhibition of mitophagy
by the autophagy inhibitor chloroquine diphosphate (CQ) affects ROS
production, HIF-1α expression, cell survival and invasion in
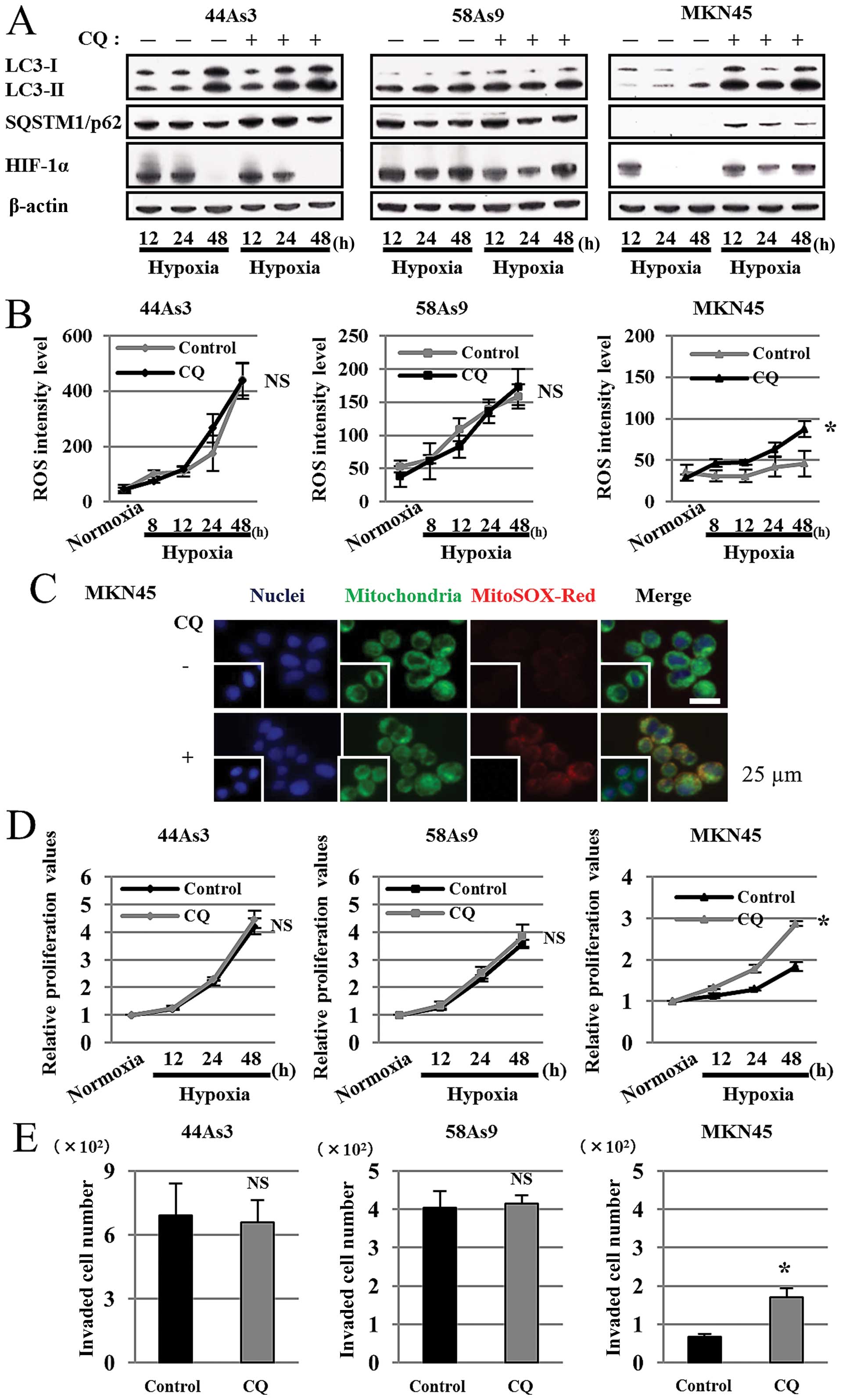
hypoxic GC cell lines (Fig. 6).
Western blot analysis showed that the expression patterns of
LC3-I/II and p62 did not differ between control and CQ treatment in
44As3 and 58As9 cells from 12–48 h of hypoxia. On the contrary,
while we observed a decreased LC3-I/II ratio and p62 degradation in
hypoxic control MKN45 cells, these findings were not observed in
hypoxic MKN45 cells treated with CQ (Fig. 6A). We also evaluated HIF-1α
expression in the three cell lines under hypoxia with or without CQ
treatment. HIF-1α expression was not affected in 44As3 and 58As9
cells with CQ treatment; however, the expression was restored in
MKN45 cells with CQ treatment under 24–48 h of hypoxia (Fig. 6A). FACS analysis showed that ROS
levels did not differ between control and CQ-treated hypoxic 44As3
and 58As9 cells. However, ROS level was elevated in MKN45 cells
with CQ treatment for 8–48 h under hypoxia in comparison to control
cells. There was a significant difference in ROS levels at 48-h
hypoxia between the treatments (Fig.
6B). Immunofluorescence using both Mitotracker and MitoSOX-Red
confirmed ROS production in mitochondria of CQ-treated (+) but not
the untreated (−) MKN45 cells under hypoxia (Fig. 6C). CQ treatment did not affect cell
proliferation of 44As3 or 58As9 cells; however, CQ treatment
increased cell survival in MKN45 cells for 12–48 h under hypoxia.
There was a significant difference at 48-h hypoxia between the
treatments (Fig. 6D). CQ treatment
did not affect cell invasion of 44As3 and 58As9 cells. However,
cell invasion was significantly higher in MKN45 cells with CQ
treatment than those with control treatment at 24 h of hypoxia
(Fig. 6E).
Discussion
We previously analyzed HIF-1α expression in an
immunohistochemical study of 91 GC tissues and found positive
nuclear staining of HIF-1α in 87 of 91 (95.6%) cases, suggesting
that hypoxic environments frequently exist in GC tissues. We also
further revealed that HIF-1α expression level is important to
determine aggressive tumor characteristics such as vessel invasion,
chemoresistance and poor prognosis of patients (19). However, the precise correlation
between HIF-1α expression level and cancer aggressiveness using
human cancer cell lines has not been elucidated. Therefore, in the
present study, we investigated the correlation between HIF-1α
expression level and cancer aggressiveness using three GC cell
lines under hypoxia.
We found that higher levels of HIF-1α expression
were induced by hypoxia in 44As3 and 58As9 cells compared with
MKN45 cells. In vitro cell survival and invasion analysis
also demonstrated that these abilities under hypoxia were stronger
in the 44As3 and 58As9 cells in comparison to MKN45 cells. These
results suggested a positive correlation between HIF-1α expression
level and hypoxia-induced aggressiveness among three GC cell lines.
Notably, RT-qPCR analysis indicated that HIF-1α mRNA levels did not
reflect HIF-1α protein levels in the GC cell lines. However,
treatment with the PHDs inhibitor DMOG strongly restored HIF-1α
protein expression in three GC cell lines under both normoxia and
hypoxia. The results suggest that HIF-1α expression levels in the
GC cell lines under these conditions depend on the PHDs activity,
which determines the HIF-1α protein stability. Previous studies
reported that intracellular ROS increased HIF-1α stability via PHDs
inhibition (24–27) therefore we next compared mtROS
levels among the three GC cell lines under hypoxia conditions. FACS
analysis showed time-dependent accumulation of intracellular ROS in
44As3 and 58As9 cells, but not in MKN45 cells under hypoxia.
Moreover, immunofluorescence confirmed the hypoxia-dependent
accumulation of mtROS in 44As3 and 58As9 cells. These results
indicated that the PHD activity in 44As3 and 58As9 cells might be
more strongly inhibited owing to the increased mtROS than that in
MKN45 cells. Reduced degradation of HIF-1α by the ubiquitin
proteasome system might subsequently occur in 44As3 and 58As9 cells
compared with MKN45 cells under hypoxic conditions. In contrast,
NAC treatment strongly decreased HIF-1α expression in the three GC
cell lines under hypoxia. These results indicated that
re-activation of PHDs might be conversely promoted by ROS
scavenging and increased HIF-1α degradation in the three GC cell
lines under hypoxia. Taken together, ROS-dependent inactivation of
PHDs may determine HIF-1α stabilization under hypoxic conditions.
On the contrary, HIF-1α knockdown increased ROS production in the
three GC cell lines under hypoxic conditions. Previous studies
reported that HIF-1α controls ROS production under hypoxia through
conversion of energy metabolism from oxidative phosphorylation to
glycolysis, which is referred to as the Warburg effect. This
metabolic change enables cancer cells to decrease O2
consumption in oxidative phosphorylation, leading to reduced ROS
production. Furthermore, HIF-1α is known to play a central role in
the Warburg effect by upregulating target genes such as GLUT1,
glycolytic enzymes, PDK1 and LDHA (11–14).
We recently revealed that stable HIF-1α knockdown attenuated the
hypoxic induction of Warburg effect-related genes, and induced
apoptosis owing to excessive ROS production in 58As9 cells
(41). In the present study, we
also noted that transient HIF-1α knockdown decreased cell survival
under hypoxia in the three GC cell lines (data not shown).
Therefore, HIF-1α activation under hypoxia may form a feedback loop
for controlling ROS generation and cell survival in GC cells. Based
on the above findings, the mtROS/HIF-1α interplay may be activated
in 44As3 and 58As9 cells, because these two cell lines possess both
high mtROS production and HIF-1α overexpression. As a result, 44As3
and 58As9 cells with HIF-1α overexpression may accelerate cancer
aggressiveness by more strongly upregulating expression of genes
related to cell survival and invasion under hypoxia, in comparison
to MKN45 with the lower activity of the mtROS/HIF-1α interplay.
To address further mechanisms underlying the
activation of the mtROS/HIF-1α interplay, we focused on mitophagy,
which is essential in mitochondrial quality control, and
hypothesized that the mitophagy process might be impaired, whereby
the mitochondria damaged by mtROS could not be eliminated in
hypoxic 44As3 and 58As9 cells. We first analyzed the expressions of
LC3-I/II and p62, which are recognized as general autophagy markers
(28). Western blot analysis
revealed that the LC3-I/II ratio as well as p62 expression was not
altered in 44As3 and 58As9 cells under hypoxic conditions. In sharp
contrast, a decreased LC3-I/II ratio and loss of p62 were observed
in hypoxic MKN45 cells in a time-dependent manner. These results
indicated that autophagy was induced in MKN45 cells but not in
44As3 or 58As9 cells under hypoxia. Fluorescence microscopy
demonstrated that the staining of LC3B-RFP was strongly increased
by hypoxia in MKN45 cells, but not in the other two cell lines. The
co-localization of LC3B-RFP and Mitotracker suggested
hypoxia-induced mitophagy in MKN45 cells. We further investigated
the lysosomal digestion targeting the mitochondria by assessing
autolysosome formation. The results showed that the co-localization
of Mitotracker and Lysotracker was observed only in hypoxic MKN45
cells, and confirmed that the entire process of mitophagy is intact
in MKN45 cells under hypoxia. The mtROS production by hypoxic
stimulation might be controlled at a low level through the normal
mitophagy process in MKN45 cells. In contrast, mtROS might be
accumulated under hypoxia in 44As3 and 58As9 cells owing to
impaired mitophagy. The increased mtROS might subsequently inhibit
the activity of PHDs, followed by enhanced HIF-1α stability. As a
result, the induced HIF-1α might facilitate the aggressive
characteristics in these two cell lines under hypoxia. To confirm
the mtROS/HIF-1α-induced cancer aggressiveness following impaired
mitophagy, we finally investigated whether the autophagy inhibitor
CQ disturbed the mitophagy process in hypoxic MKN45 cells and
increased cancer aggressiveness. The results showed that the normal
autophagy process was inhibited in hypoxic MKN45 cells with CQ
treatment. Moreover, HIF-1α expression as well as mtROS production
was elevated under hypoxic conditions in the CQ-treated cells. CQ
treatment also increased the cell survival and invasion ability in
MKN45 cells under hypoxia. These results indicated that the
artificial impairment of mitophagy might also increase cancer
aggressiveness via activating the mtROS/HIF-1α interplay in MKN45
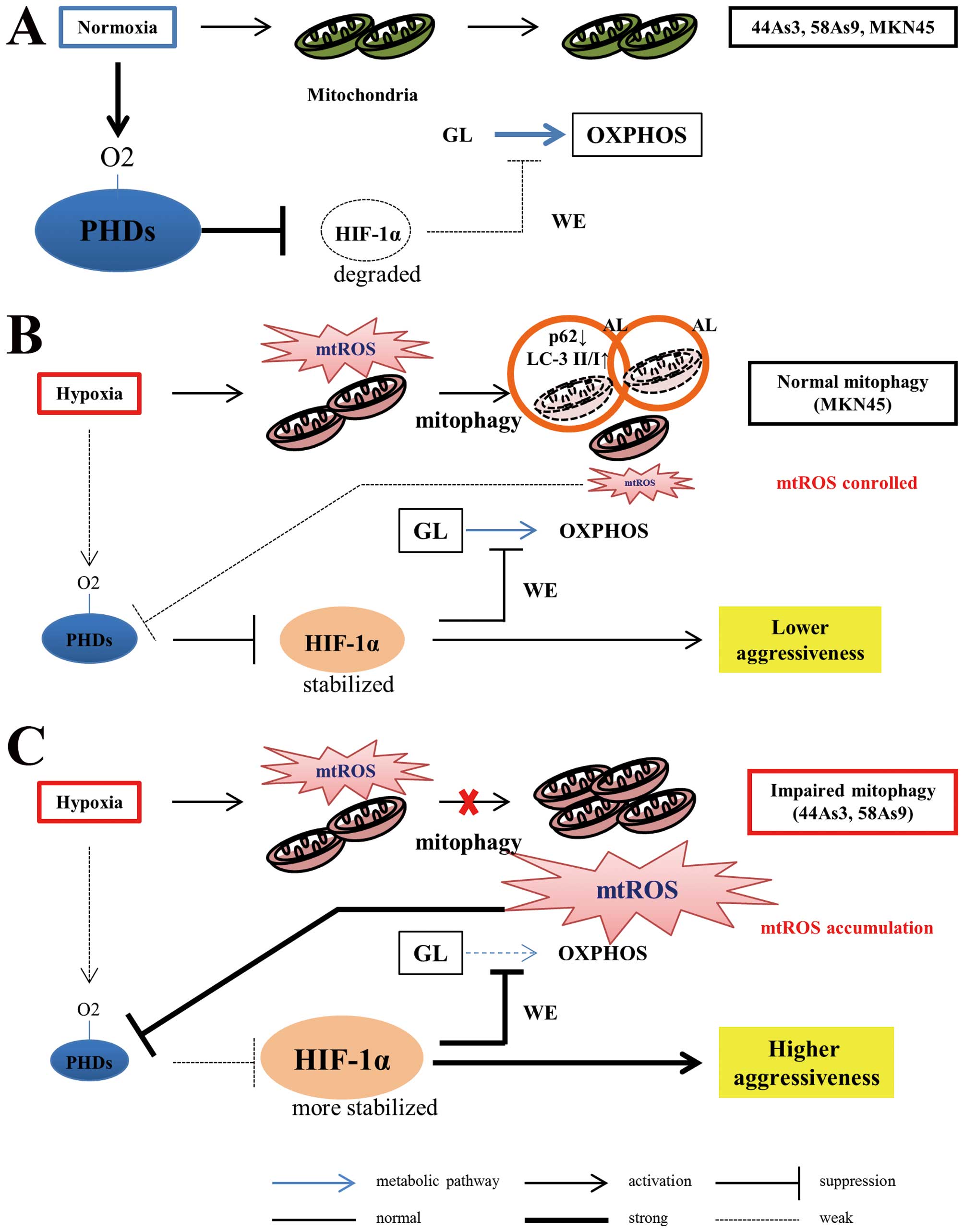
cells. Fig. 7 summarizes a novel
mechanism of hypoxia-induced cancer aggressiveness that originates
from the impairment of mitophagy in GC cells.
In conclusion, the present study demonstrated for
the first time that the integrity of mitophagy determined the
cancer aggressiveness of GC cells under hypoxia via the activation
of the mtROS/HIF-1α interplay. Isolation of abnormality of
expression of genes encoding regular autophagy factors or mitophagy
specific factors might be important for more precise understanding
of this novel theory.
Acknowledgements
This study was supported by JSPS KAKENHI
Grants-in-Aid for Scientific Research on Scientific Research
(Research Project no. 15K10103) (to J.N.).
Abbreviations:
|
GC
|
gastric cancer
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
PHDs
|
prolyl hydroxylases
|
|
ROS
|
reactive oxygen species
|
|
ETC
|
electron transport chain
|
|
mtROS
|
mitochondrial ROS
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
SQSTM1
|
sequestosome 1
|
|
BNIP3
|
BCL2/adenovirus E1 B19 kDa
protein-interacting protein 3
|
|
NIX
|
Nip3-like protein X
|
|
FUNDC
|
FUN14 domain containing 1
|
|
DMOG
|
dimethyloxallylglycine
|
|
NAC
|
N-acetyl-L-cysteine
|
|
CQ
|
chloroquine diphosphate
|
|
siRNA
|
small interfering RNA
|
|
RFP
|
red fluorescent protein
|
|
RT-qPCR
|
real-time quantitative reverse
transcription polymerase chain reaction
|
|
MTS
|
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
|
|
GLUT1
|
glucose transporter 1
|
|
PDK1
|
pyruvate dehydrogenase kinase 1
|
|
LDHA
|
lactate dehydrogenase A
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shiraishi N, Inomata M, Osawa N, Yasuda K,
Adachi Y and Kitano S: Early and late recurrence after gastrectomy
for gastric carcinoma. Univariate and multivariate analyses.
Cancer. 89:255–261. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Macdonald JS, Smalley SR, Benedetti J,
Hundahl SA, Estes NC, Stemmermann GN, Haller DG, Ajani JA,
Gunderson LL, Jessup JM, et al: Chemoradiotherapy after surgery
compared with surgery alone for adenocarcinoma of the stomach or
gastro-esophageal junction. N Engl J Med. 345:725–730. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iyer NV, Kotch LE, Agani F, Leung SW,
Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY,
et al: Cellular and developmental control of O2
homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev.
12:149–162. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vaupel P, Mayer A and Höckel M: Tumor
hypoxia and malignant progression. Methods Enzymol. 381:335–354.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu X and Kang Y: Hypoxia and
hypoxia-inducible factors: Master regulators of metastasis. Clin
Cancer Res. 16:5928–5935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Höckel M and Vaupel P: Tumor hypoxia:
Definitions and current clinical, biologic, and molecular aspects.
J Natl Cancer Inst. 93:266–276. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang BH, Semenza GL, Bauer C and Marti
HH: Hypoxia-inducible factor 1 levels vary exponentially over a
physiologically relevant range of O2 tension. Am J
Physiol. 271:C1172–C1180. 1996.PubMed/NCBI
|
|
10
|
Semenza GL: Defining the role of
hypoxia-inducible factor 1 in cancer biology and therapeutics.
Oncogene. 29:625–634. 2010. View Article : Google Scholar :
|
|
11
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Semenza GL: HIF-1 and tumor progression:
Pathophysiology and therapeutics. Trends Mol Med. 8(Suppl):
S62–S67. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Semenza GL: HIF-1: Upstream and downstream
of cancer metabolism. Curr Opin Genet Dev. 20:51–56. 2010.
View Article : Google Scholar :
|
|
14
|
Kitajima Y and Miyazaki K: The critical
impact of HIF-1α on gastric cancer biology. Cancers (Basel).
5:15–26. 2013. View Article : Google Scholar
|
|
15
|
Ide T, Kitajima Y, Miyoshi A, Ohtsuka T,
Mitsuno M, Ohtaka K, Koga Y and Miyazaki K: Tumor-stromal cell
interaction under hypoxia increases the invasiveness of pancreatic
cancer cells through the hepatocyte growth factor/c-Met pathway.
Int J Cancer. 119:2750–2759. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sumiyoshi Y, Kakeji Y, Egashira A,
Mizokami K, Orita H and Maehara Y: Overexpression of
hypoxia-inducible factor 1alpha and p53 is a marker for an
unfavorable prognosis in gastric cancer. Clin Cancer Res.
12:5112–5117. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nam SY, Ko YS, Jung J, Yoon J, Kim YH,
Choi YJ, Park JW, Chang MS, Kim WH and Lee BL: A hypoxia-dependent
upregulation of hypoxia-inducible factor-1 by nuclear factor-κB
promotes gastric tumour growth and angiogenesis. Br J Cancer.
104:166–174. 2011. View Article : Google Scholar :
|
|
18
|
Stoeltzing O, McCarty MF, Wey JS, Fan F,
Liu W, Belcheva A, Bucana CD, Semenza GL and Ellis LM: Role of
hypoxia-inducible factor 1alpha in gastric cancer cell growth,
angiogenesis, and vessel maturation. J Natl Cancer Inst.
96:946–956. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakamura J, Kitajima Y, Kai K, Hashiguchi
K, Hiraki M, Noshiro H and Miyazaki K: HIF-1alpha is an unfavorable
determinant of relapse in gastric cancer patients who underwent
curative surgery followed by adjuvant 5-FU chemotherapy. Int J
Cancer. 127:1158–1171. 2010. View Article : Google Scholar
|
|
20
|
Rohwer N and Cramer T: HIFs as central
regulators of gastric cancer pathogenesis. Cancer Biol Ther.
10:383–385. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sena LA and Chandel NS: Physiological
roles of mitochondrial reactive oxygen species. Mol Cell.
48:158–167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zepeda AB, Pessoa A Jr, Castillo RL,
Figueroa CA, Pulgar VM and Farías JG: Cellular and molecular
mechanisms in the hypoxic tissue: Role of HIF-1 and ROS. Cell
Biochem Funct. 31:451–459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chandel NS, McClintock DS, Feliciano CE,
Wood TM, Melendez JA, Rodriguez AM and Schumacker PT: Reactive
oxygen species generated at mitochondrial complex III stabilize
hypoxia-inducible factor-1alpha during hypoxia: A mechanism of
O2 sensing. J Biol Chem. 275:25130–25138. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gerald D, Berra E, Frapart YM, Chan DA,
Giaccia AJ, Mansuy D, Pouysségur J, Yaniv M and Mechta-Grigoriou F:
JunD reduces tumor angiogenesis by protecting cells from oxidative
stress. Cell. 118:781–794. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao T, Zhu Y, Morinibu A, Kobayashi M,
Shinomiya K, Itasaka S, Yoshimura M, Guo G, Hiraoka M and Harada H:
HIF-1-mediated metabolic reprogramming reduces ROS levels and
facilitates the metastatic colonization of cancers in lungs. Sci
Rep. 4:37932014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaelin WG Jr: ROS: Really involved in
oxygen sensing. Cell Metab. 1:357–358. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Semenza GL: Hypoxia-inducible factor 1:
Regulator of mitochondrial metabolism and mediator of ischemic
preconditioning. Biochim Biophys Acta. 1813:1263–1268. 2011.
View Article : Google Scholar
|
|
28
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rogov V, Dötsch V, Johansen T and Kirkin
V: Interactions between autophagy receptors and ubiquitin-like
proteins form the molecular basis for selective autophagy. Mol
Cell. 53:167–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lemasters JJ: Selective mitochondrial
autophagy, or mitophagy, as a targeted defense against oxidative
stress, mitochondrial dysfunction, and aging. Rejuvenation Res.
8:3–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim I, Rodriguez-Enriquez S and Lemasters
JJ: Selective degradation of mitochondria by mitophagy. Arch
Biochem Biophys. 462:245–253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chourasia AH, Boland ML and Macleod KF:
Mitophagy and cancer. Cancer Metab. 3:42015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu L, Feng D, Chen G, Chen M, Zheng Q,
Song P, Ma Q, Zhu C, Wang R, Qi W, et al: Mitochondrial
outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in
mammalian cells. Nat Cell Biol. 14:177–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang H, Bosch-Marce M, Shimoda LA, Tan
YS, Baek JH, Wesley JB, Gonzalez FJ and Semenza GL: Mitochondrial
autophagy is an HIF-1-dependent adaptive metabolic response to
hypoxia. J Biol Chem. 283:10892–10903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cesari R, Martin ES, Calin GA, Pentimalli
F, Bichi R, McAdams H, Trapasso F, Drusco A, Shimizu M, Masciullo
V, et al: Parkin, a gene implicated in autosomal recessive juvenile
parkinsonism, is a candidate tumor suppressor gene on chromosome
6q25-q27. Proc Natl Acad Sci USA. 100:5956–5961. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gong Y, Zack TI, Morris LG, Lin K,
Hukkelhoven E, Raheja R, Tan IL, Turcan S, Veeriah S, Meng S, et
al: Pan-cancer genetic analysis identifies PARK2 as a master
regulator of G1/S cyclins. Nat Genet. 46:588–594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yanagihara K, Takigahira M, Tanaka H,
Komatsu T, Fukumoto H, Koizumi F, Nishio K, Ochiya T, Ino Y and
Hirohashi S: Development and biological analysis of peritoneal
metastasis mouse models for human scirrhous stomach cancer. Cancer
Sci. 96:323–332. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Miyake S, Kitajima Y, Nakamura J, Kai K,
Yanagihara K, Tanaka T, Hiraki M, Miyazaki K and Noshiro H: HIF-1α
is a crucial factor in the development of peritoneal dissemination
via natural metastatic routes in scirrhous gastric cancer. Int J
Oncol. 43:1431–1440. 2013.PubMed/NCBI
|
|
41
|
Tanaka T, Kitajima Y, Miyake S, Yanagihara
K, Hara H, Nishijima-Matsunobu A, Baba K, Shida M, Wakiyama K,
Nakamura J, et al: The apoptotic effect of HIF-1α inhibition
combined with glucose plus insulin treatment on gastric cancer
under hypoxic conditions. PLoS One. 10:e01372572015. View Article : Google Scholar
|
|
42
|
Dolman NJ, Chambers KM, Mandavilli B,
Batchelor RH and Janes MS: Tools and techniques to measure
mitophagy using fluorescence microscopy. Autophagy. 9:1653–1662.
2013. View Article : Google Scholar : PubMed/NCBI
|