Introduction
Chronic myeloid leukemia (CML) is a
myeloproliferative disorder characterized by the BCR-ABL gene
rearrangement (1,2). As a driving force for leukemogenesis
of CML, the activated tyrosine kinase of BCR-ABL stimulates
multiple signaling pathways that confers growth advantage and
counteracts apoptosis (3–5). The most prominent downstream pathways
upregulated by BCR-ABL include the Ras/Raf/MAPK, PI3K/Akt, and
JAK/STAT pathways. Imatinib mesylate, a selective BCR-ABL tyrosine
kinase inhibitor (TKI), has become the standard therapy in CML. It
induces durable cytogenetic remissions in the majority of
chronic-phase patients with CML, but a significant proportion of
the patients experiences drug-resistance, mainly as a consequence
of BCR-ABL point mutations (6).
Among BCR-ABL point mutations, T315I mutant remains a crucial
clinical challenge, because it is resistant to imatinib and second
generation TKIs (7,8). Ponatinib, a third-generation TKI, has
an antileukemia activity against CML with unmutated or mutated
BCR-ABL including T315I. However, its use clinically is limited by
serious side effects such as vascular occlusion, heart failure and
hepatotoxicity (9,10).
As an ATP-dependent molecule chaperon, heat shock
protein 90 (Hsp90) is associated with many different client
oncoproteins such as BCR-ABL, Raf, ErbB and Akt (11). Disruption of Hsp90 function by
specific inhibitors leads to the destabilization and degradation of
its client oncoproteins, thereby inhibiting cell growth and
inducing apoptosis in cancer cells (12). Thus, Hsp90 represents a promising
molecular target for cancer therapy. In this regard, it was
demonstrated that a prototypical Hsp90 inhibitor geldanamycin and
its analogue 17-AAG downregulated BCR-ABL levels and induced
apoptosis of CML cells (13).
Similar results have been obtained with some synthetic Hsp90
inhibitors such as AUY922A and EC141 (14–16)
suggesting that targeting Hsp90 might be a promising therapeutic
approach to treat CML.
BIIB021, the first fully synthetic inhibitor, has a
high binding affinity for Hsp90, and induces degradation of Hsp90
client proteins including HER-2, Akt and Raf-1 (17). It was reported that BIIB021 was
more active than 17-AAG against tumor cells with acquired multidrug
resistance (18). Preclinical data
have also demonstrated the potent anticancer activity in various
solid tumors and hematological malignancies (18–20).
In a phase I clinical trial performed in patients with advanced
solid tumors, BIIB021 safety profile was displayed and a phase II
study documented objective responses in refractory gastrointestinal
stromal tumor patients (21,22).
We recently reported that BIIB021 mediates its antileukemic
activity via inhibiting PI3K/Akt pathway and disrupting p53-MDM2
interaction (23,24). These results indicated the multiple
biological functions of BIIB021. However, little is known about the
effects of BIIB021 on CML cells.
In this study, we investigated the biological
effects of BIIB021 on CML cells. We found that BIIB021 induces
potent cytotoxicity against imatinib-sensitive and -resistant CML
cells as well as leukemic cells with T315I-mutant BCR-ABL. BIIB021
also induces proteasomal degradation of BCR-ABL. Interestingly,
treatment with BIIB021 results in a cytoprotective autophagy, which
might be independent of Beclin-1 but dependent on mTOR-Ulk1
pathway.
Materials and methods
Cell culture and reagents
Human CML cell line K562 was obtained from American
Type Culture Collection (ATCC, Rockville, MD, USA). K562/G, an
imatinib-resistant cell line, was kindly provided by Institute of
Hematology, Chinese Academy of Medical Sciences (Tianjin, China).
Murine leukemic 32D cells with wild-type (wt) BCR-ABL (32Dp210) and
32Dp210-T315I (32D cells carrying T315I mutation) were provided by
Professir L. Qiu (Harbin Institute of Hematology and Oncology,
Harbin, China). All cell lines were cultured in RPMI-1640 medium
(Gibco-RRL, Grand Island, NY, USA) supplemented with 10% fetal
bovine serum (Gibco) at 37ºC in a humidified atmosphere of 5%
CO2. BIIB021, MG-132, 3-methyladenine (3-MA), z-DEVD-fmk
and z-VAD-fmk were purchased from Selleck Chemicals (Houston, TX,
USA). Insulin-like growth factor 1 (IGF-1) was purchased from
Peprotech (Rocky Hill, NJ, USA). Bafilomycin A1 (Baf A1) was
obtained from LC Laboratories (Woburn, MA, USA). Chloroquine (CQ)
and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All
antibodies used in the western blot analysis were purchased from
Cell Signaling Technology (Beverly, MA, USA) with the exception of
p-STAT3 Ser727, p-STAT3 Tyr705, Ulk1 (Abcam, Cambridge, UK) and
Lamin B1 (Proteintech, Chicago, IL, USA).
Cell viability assays
The survival of CML cells was determined by means of
the MTT assay. Cells were cultured at a density of 1×105
cells/ml in a 96-well plate with BIIB021 (50–800 nM) for the
indicated times. Following incubation, 20 μl MTT solutions (5
mg/ml) were added to each well and then the plates were incubated
for an additional 4 h at 37ºC. After supernatant was removed, 200
μl dimethylsulfoxide (DMSO) was added. A spectrophotometry was
applied to detect the absorbance at a wavelength of 570 nm. Each
assay was performed three times.
Annexin V/PI binding assays
Cells were cultured at a density of 1×105
cells/ml in a 6-well plate and treated with different
concentrations of BIIB021. After 48-h incubation at 37ºC, the cells
were washed, resuspended in 500 μl of binding buffer and stained
with 5 μl of Annexin V-FITC and 10 μl of prop-idium iodide (PI)
(Biouniquer, Suzhou, China) for 15 min in the dark. Then cells were
examined by flow cytometry (Accuri C6, BD, Franklin Lakes, NJ,
USA).
Real-time PCR
Total RNA was isolated and quantitative real-time
PCR was performed as previously described (25) using the primers (Sangon Biotech,
Shanghai, China): 5′-TCC GCT GAC CAT CAA YAA GGA-3′ (forward) and
5′-CAC TCA GAC CCT GAG GCT CAA-3′ (reverse) for p210BCR-ABL and
5′-GTC ATC ACC ATT GGC AAT GAG-3′ (forward) and 5′-CGT CAC ACT TCA
TGA TGG AGT T-3′ (reverse) for GAPDH. The amount of p210BCR-ABL was
analyzed by the comparative CT method taking GAPDH as the
control.
Western blot analysis
Following treatment at the indicated time and doses
of BIIB021, cells were collected and lysed at 4ºC in lysis buffer.
Protein concentration of samples was measured by bicinchoninic acid
(BCA) method. The protein samples were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and then electroblotted
onto hybond-polyvinylidene fluoride membranes. The membranes were
blocked in 5% non-fat milk for 2 h and incubated with primary
antibodies at 4ºC overnight. The membranes were washed and
incubated with secondary antibody conjugated with horseradish
peroxidase (1:5,000, Cell Signaling Technology). ECL detecting kit
was applied to visualize results. The primary antibodies used in
this study included actin, caspase-9, caspase-3, poly(ADP ribose)
polymerase (PARP), Bcl-2, Bax, Bak, Bad, Mcl-1, Bcl-XL, c-ABL,
p-BCR Tyr177, JAK2, STAT5, p-STAT5 Tyr694, STAT3, p-STAT3 Ser727,
p-STAT3 Tyr705, EKR1/2, p-EKR1/2 Thr202/Tyr204, Akt, β-catenin,
non-phospho-β-catenin (Ser33/37/Thr41), c-Myc, Lamin B1, LC3I/II,
p62, Beclin-1, mTOR, p-mTOR Ser2448, p70S6K, p-p70S6K Thr389, Ulk1,
p-Ulk1 Ser757, AMPKα, p-AMPKαThr172.
Preparation of cytoplasm and nuclear
fractions
The cytoplasm and nuclear proteins were extracted
using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo
Fisher Scientific, Rockford, IL, USA) according to the
manufacturer's instructions. BIIB021-treated cells were pelleted by
centrifugation and rinsed with PBS. Then, cells were suspended in
ice-cold Cytoplasmic Extraction Reagent I, and the tube was
vortexed for 15 sec. After incubation on ice for 10 min, the tube
was added in Cytoplasmic Extraction Reagent II, vortexed, incubated
and centrifuged at 16,000 g for 5 min. The supernatant was
transferred to a fresh tube and referred to as cytoplasm extract.
The insoluble fraction, containing nuclei, was suspended in
ice-cold nuclear extraction reagent, placed on ice and continued
vortexing for 15 sec every 10 min, for a total of 40 min. After
centrifugation at 16,000 g for 10 min, the supernatant was
transferred to a new tube and kept as nuclear fraction.
Detection of acidic vesicular
organelles
To detect the presence of acidic vesicular
organelles (AVOs), cells were treated with BIIB021 for 48 h. Next,
cells were stained with acridine orange (AO 1 μg/ml, 37ºC for 15
min, Sigma) or monodan-sylcadaverine (MDC, 0.1 mM, 37ºC for 60 min,
Sigma), fixed with 4% paraformaldehyde and examined under a
fluorescence microscope (Olympus, Tokyo, Japan).
Immunofluorescence studies
Cells were treated with or without BIIB021 for 24 h
and then fixed with 4% parafor-maldehyde. After permeablized by
0.3% Triton X-100 and incubated with goat serum, cells were stained
with anti-β-catenin antibody (Abcam, 1:200 dilution) overnight at
4ºC. Then, cells were incubated with a goat anti-rabbit antibody as
secondary antibody (1:500) at 37ºC for 1 h and 1 μM DAPI
(SouthernBiotech, Birmingham, AL, USA) for 10 min. Finally, samples
were examined with a Nikon confocal microscope (Nikon C1-Si,
Japan).
Approximately 105 cells were treated with
or without BIIB021 for 24 h. After washing with PBS, cells were
incubated with MitoTracker (Invitrogen, Carlsbad, CA, USA) at a
concentration of 100 nM (37ºC for 0.5 h) to visualize mitochondria.
Then cells were fixed with 4% paraformaldehyde and following
experiments were performed as described above except the primary
antibody (cytochrome c, Abcam, 1:200 dilution) and secondary
antibody (goat anti-mouse antibody, 1:500).
Staining autophagosomes with mRFP-GFP-LC3
and confocal microscopy
To detect the presence of LC3, leukemia cells were
transfected with adenovirus encoding red and green fluorescent
protein-LC3 (AdmRFP-GFP-LC3) at a concentration of 800 virus
particles/cell. After centrifugation at 500 g for 1 h and
incubation for 3 h at 37ºC, cells were exposed to BIIB021 for 48 h.
Cells were fixed with 4% paraformaldehyde. The fluorescence of
mRFP-GFP-LC3 was viewed under a confocal microscope (Nikon C1-Si,
Japan).
Caspase-3 knockdown by short hairpin
RNA
Three lentiviral vectors containing shRNA against
caspase-3 and a negative control scramble shRNA were purchased from
Hanheng Biotech (Shanghai, China). K562 cells (2×104
cells/well) were seeded in a 96-well plate and transfected with
lentivirus at a concentration of 100 virus particles/cell. After
centrifugation at 800 g for 1 h, cells were cultured at 37ºC. Then
cells were selected in medium containing puromycin (2 μg/ml).
Protein expression of caspase-3 was determined by western blot
analysis. The sequence of shRNA targeting caspase-3 was
5′-GGAAGCGAATCAATGGACTCTGGAA-3′.
Statistical analysis
Experimental results are statistically presented as
the mean ± standard deviation. Data were analyzed by one-way
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
BIIB021 inhibits proliferation and
induces apoptosis in both imatinib-sensitive and -resistant CML
cells
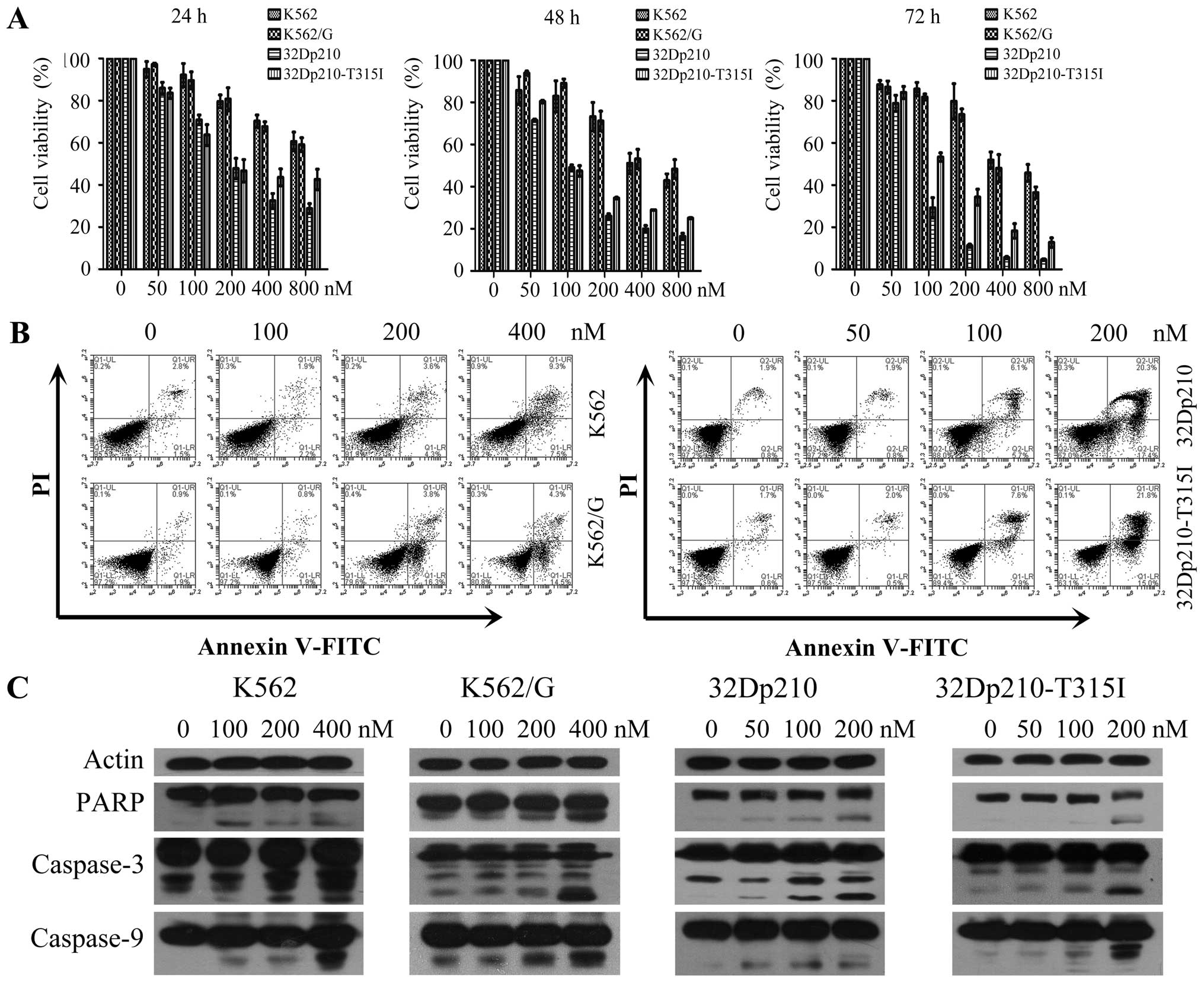
To investigate the effects of BIIB021 on the
cellular proliferation of CML cells, human CML cell lines K562,
K562/G and a pair of murine 32D leukemic cell lines 32Dp210, and
32Dp210-T315I were treated with different concentrations of BIIB021
for 24, 48 or 72 h, respectively. An MTT assay was used to
determine the cell viability. The results showed that BIIB021
effectively inhibited the proliferation of CML cell lines in a
dose- and time-dependent manner. The 50% inhibition
(IC50) of BIIB021 in K562, K562/G, 32Dp210, and
32Dp210-T315I cell lines at 48 h was observed at 513.99, 603.53,
110.08 and 148.07 nM, respectively (Fig. 1A). In order to characterize the
cytotoxicity of BIIB021 against CML cells, we next performed
Annexin V apoptosis assays in K562, K562/G, 32Dp210, and
32Dp210-T316I cells treated with multiple doses of BIIB021 for 48
h. Treatment with the drug resulted in a marked increase in the
number of early (Annexin V-positive/PI-negative) and late apoptotic
(Annexin V-positive/PI-positive) cells at 100 and 200 nM
concentrations (Fig. 1B). We
further tested molecules that control apoptosis. Western blot
analysis was used to confirm our flow cytometric findings by
showing that BIIB021 treatment activated key molecules in the
apoptosis pathway, namely, caspase-9, -3 and PARP (Fig. 1C).
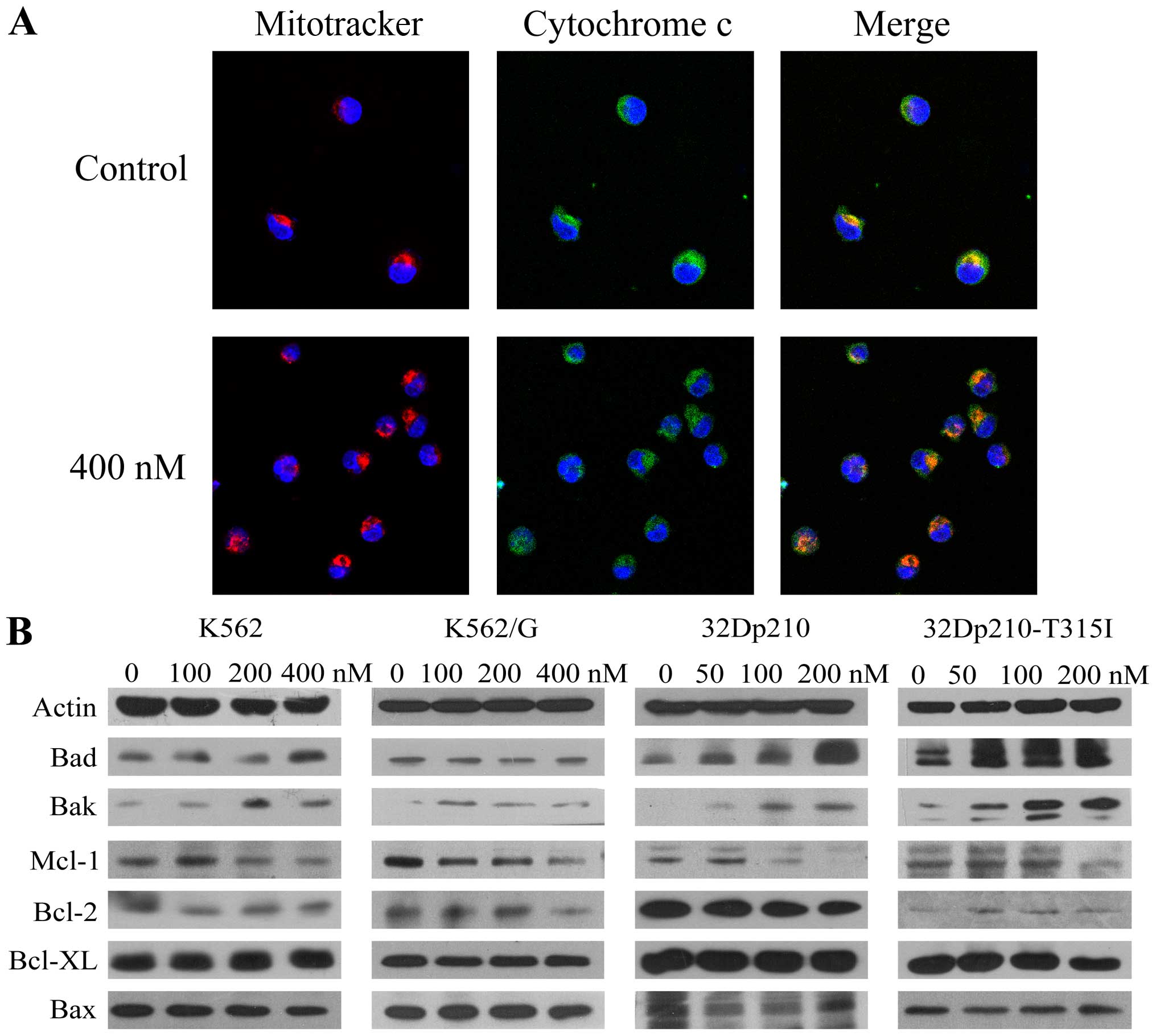
Based on the observation of activation of caspase-9,
we monitored the release of cytochrome c from mitochondria
in K562 cells after treatment with BIIB021 (400 nM) by confocal
microscopy. A significant increase was observed in cytochrome
c released from dsRed Mitotracker-tagged mitochondria
(Fig. 2A). Furthermore, western
blot analysis revealed that BIIB021 treatment resulted in
upregulation of pro-apoptotic protein Bak and Bad, which are
associated with the function of mitochondria, while anti-apoptotic
protein Mcl-1 was downregulated (Fig.
2B). Taken together, these findings suggested that the
mitochondria-dependent intrinsic apoptotic pathway is involved in
BIIB021-induced cell death.
BIIB021 effectively inhibits BCR-ABL and
its downstream molecules
It has been shown that rapid degradation of BCR-ABL
protein could be observed in the leukemic cells treated with Hsp90
inhibitors (16). In this study we
observed a dose-dependent inhibition of BCR-ABL protein when the
CML cell lines, which express wild-type or T315I BCR-ABL, were
treated with BIIB021, and that the depletion of BCR-ABL protein was
accompanied by a concomitant decrease in phosphorylation of BCR-ABL
protein on Tyr177 (Fig. 3A). To
investigate how the agent alters BCR-ABL expression, we examined
mRNA expression levels of BCR-ABL in response to BIIB021 (Fig. 3B). The results showed that the
BCR-ABL mRNA level slightly increased, suggesting BIIB021 acts on
BCR-ABL expression at the post-transcriptional level. Indeed,
BIIB021-mediated downregulation of BCR-ABL was partly reversed by
MG-132, a proteasome inhibitor, whereas CQ, a lysosome inhibitor,
and Baf A1 did not show a similar effect (Fig. 3C and D). These findings suggested
that proteasome pathway is involved in the BIIB021-mediated
downregulation of BCR-ABL. We next evaluated the effects of BIIB021
on downstream pathways, which are stimulated by BCR-ABL (3–5).
Treatment with BIIB021 clearly decreased the phosphorylation of
STAT3, STAT5 and ERK1/2. Additionally, BIIB021 induced
downregulation of total levels of JAK2 and Akt (Fig. 3E). Taken together, we demonstrated
that BIIB021 effectively inhibits the protein expression of BCR-ABL
and its downstream signaling mediators.
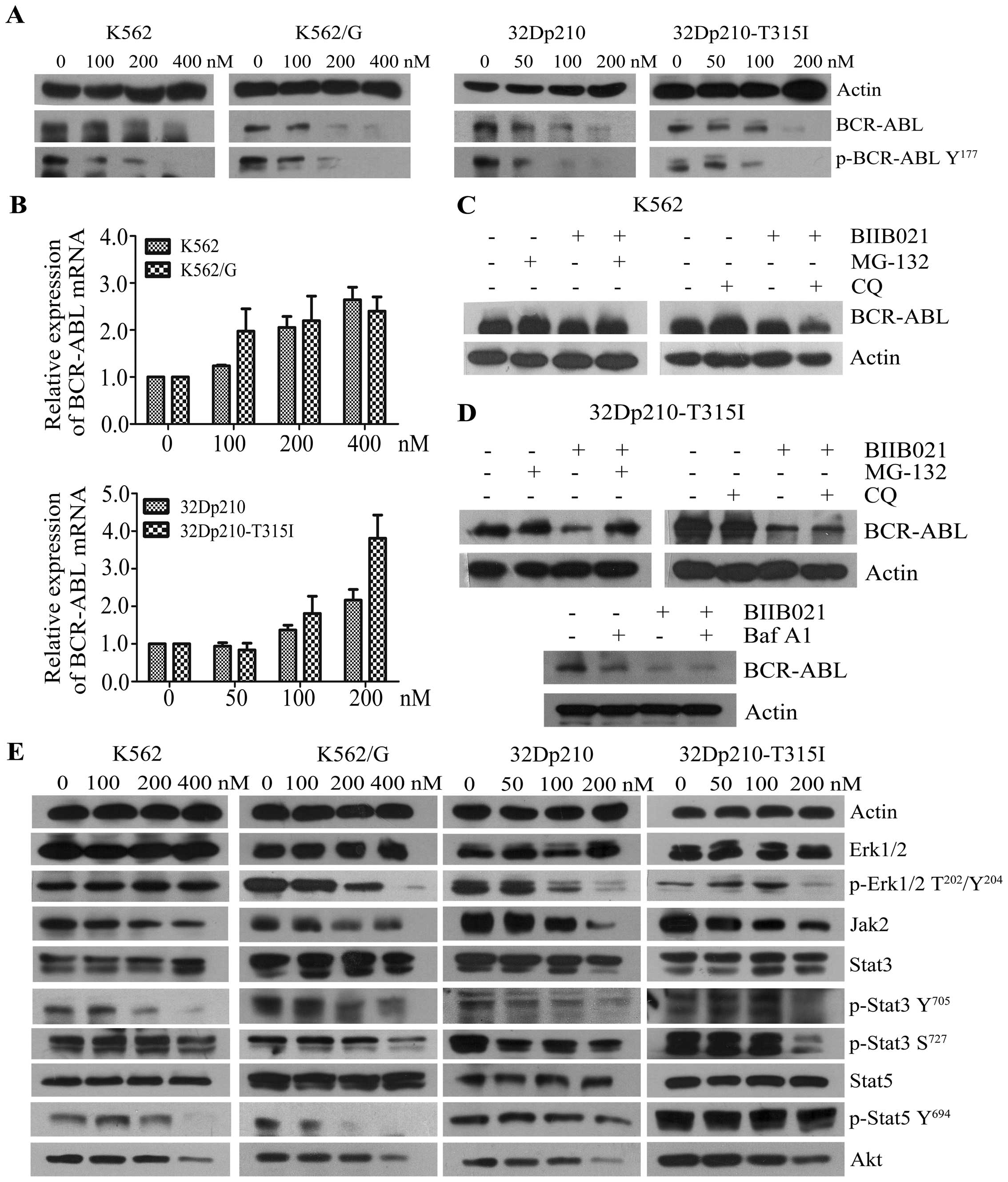 | Figure 3BIIB021 inhibits the expression of
BCR-ABL and its downstream signaling mediators. (A) Four kinds of
leukemic cell lines were treated with BIIB021 at the indicated
doses for 24 h. Whole-cell lysates were extracted to assess the
levels of BCR-ABL and phosphorylated (p)-BCR-ABL (Tyr177). (B)
Leukemic cell lines were treated with BIIB021 at the indicated
concentrations for 24 h. The expression levels of mRNA were
evaluated by real-time PCR. The relative changes in gene expression
are shown compared to the untreated controls. (C) After pretreated
with MG-132 (1.0 μM), or with CQ (25 μM) for 4 h, K562 cells were
treated with BIIB021 for 20 h. Whole-cell lysates were then
prepared and analyzed for expression of BCR-ABL by western
blotting. (D) 32Dp210-T315I cells were pretreated with MG-132, CQ,
or with Baf A1 (25 nM) for 4 h. Cell lysates were subjected to
western blotting to determine expression of BCR-ABL. (E) After
incubation of CML cell lines with the indicated concentrations of
BIIB021, whole-cell lysates were analyzed for p-Erk1/2
(Thr202/Tyr204), total Erk1/2, Jak2, p-Stat3 (Tyr705 and Ser727),
total Stat3, p-Stat5 (Tyr694), total Stat5 and Akt expression by
western blot analysis. |
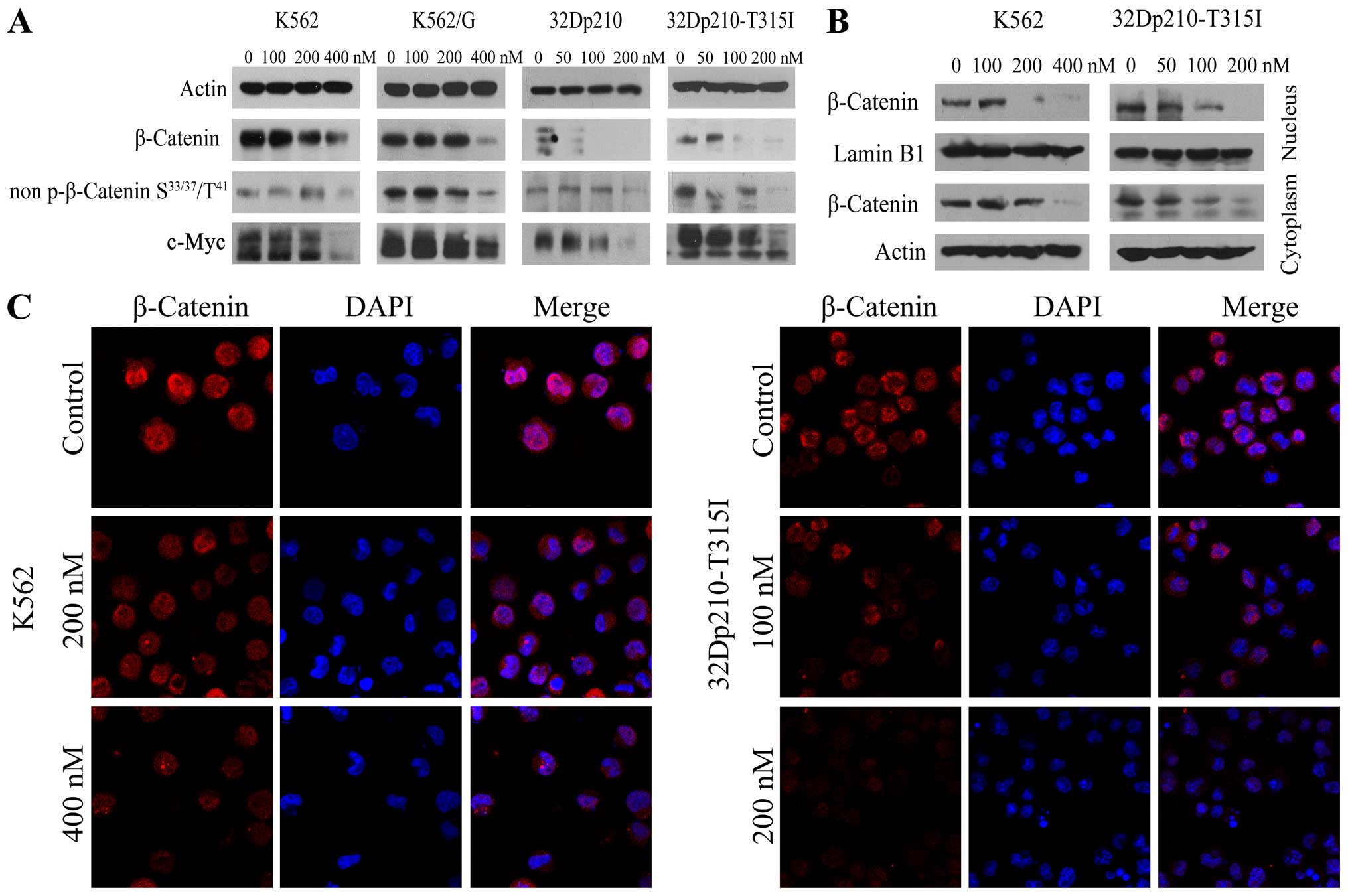
BIIB021 inhibits β-catenin/c-Myc pathway
in BCR-ABL positive cells
Since it has been shown that Wnt/β-catenin signaling
is required for leukemic stem cell maintenance in CML (26), and the tyrosine kinase activity of
BCR-ABL is required to phosphorylate β-catenin (27), experiments were performed to
ascertain effects of BIIB021 on β-catenin signaling. Western blot
analysis revealed that treatment with BIIB021 caused a significant
reduction in levels of β-catenin and non-phospho (Active) β-catenin
(Ser33/37/Thr41), together with a marked downregulation of c-Myc, a
downstream molecule of β-catenin signaling (Fig. 4A). To determine the intracellular
localization of β-catenin protein in untreated and BIIB021-treated
K562 and 32Dp210-T315I cells, we obtained nuclear and cytoplasmic
protein extracts. Compared with untreated control, the levels of
β-catenin were significantly decreased in both nuclear and
cytoplasmic extracts from BIIB021-treated cells (Fig. 4B). This effect was confirmed by
confocal microscopy analysis (Fig.
4C).
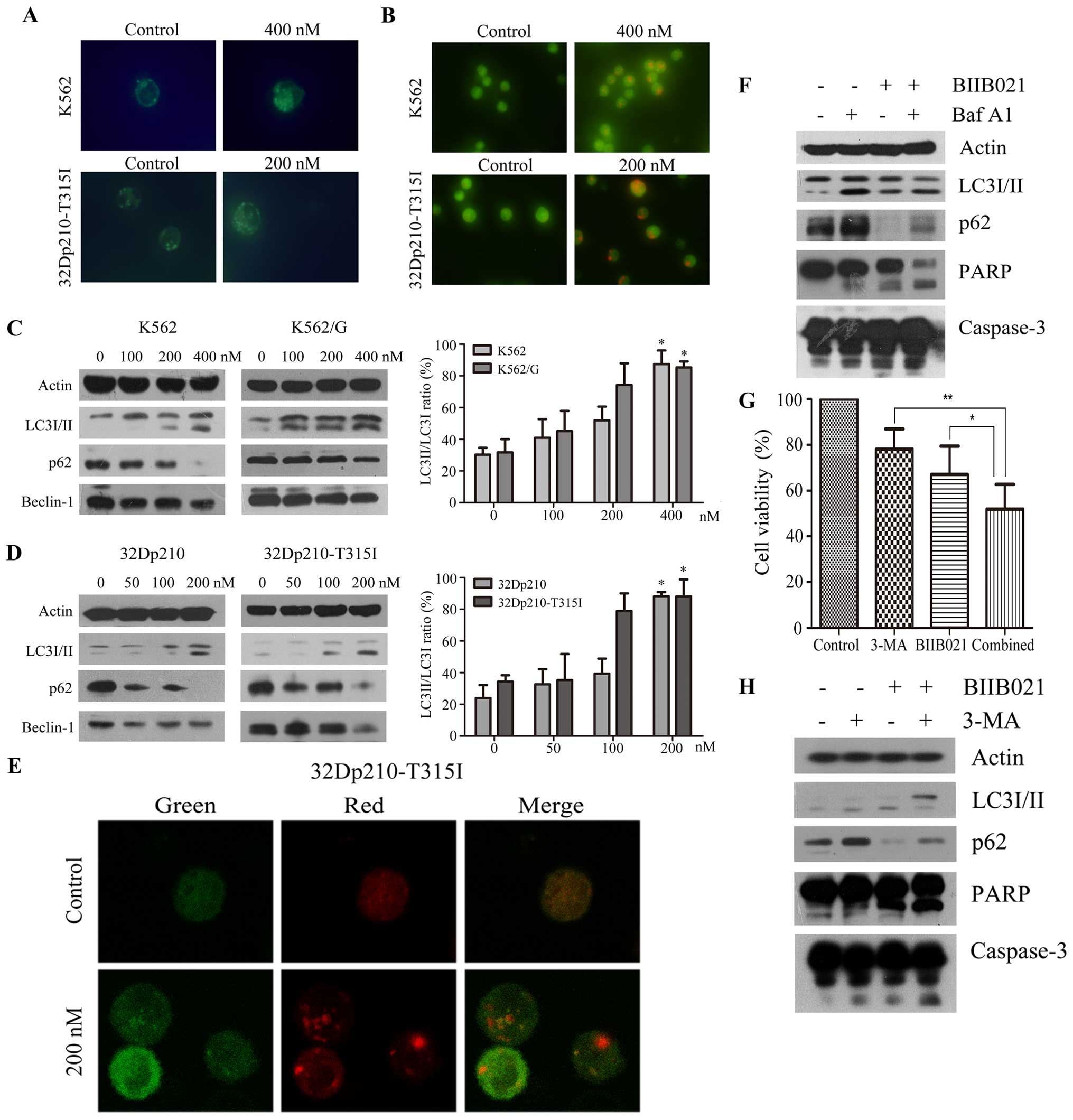
BIIB021 induces Beclin-1-independent
autophagy in CML cells
It was reported that Hsp90 inhibitor geldanamycin
can induce autophagy (28). We
then investigated whether BIIB021 could induce autophagy in K562
and 32Dp210-T315I cells. As shown in Fig. 5A, MDC fluorescence was observed in
control and BIIB021-treated CML cells. However, BIIB021-treated
cells displayed more frequent accumulation of MDC than control
cells. Acridine orange staining of BIIB021-treated CML cells also
showed significantly increased formation of acidic vesicles
(Fig. 5B). Consistent with these
data, western blot analysis showed that increased conversion of
LC3-I to LC3-II, and decreased p62 (SQSTM1) protein levels, in a
dose-dependent manner, were found in the BIIB021-treated cells
(Fig. 5C and D). Unexpectedly, we
found downregulation of Beclin-1, a key protein inducing autophagy,
in the BIIB021-treated cells (Fig. 5C
and D). These experiments collectively demonstrated induction
of autophagy by BIIB021, which may be via a Beclin-1-independent
mechanism.
We next examined alteration of mRFP-GFP-LC3
fluorescent signals by confocal microscopy (29) to analyze autophagic flux. An
increase in number of acidic autophagolysosomes (red fluorescence)
and non-acidic autophagolysosomes (yellow) was observed in
BIIB021-treated 32Dp210-T315I cells (Fig. 5E). To further determine the
autophagic flux into the lysosomal compartment, we analyzed LC3-II
and p62 in cells co-treated with Baf A1, an inhibitor of vacuolar
H+ ATPase that leads to accumulation of autophagic
vacuoles by blocking their fusion with lysosomes (Fig. 5F). The levels of LC3-II and p62
protein determined by immunodetection were increased in the
presence of Baf A1, supporting the notion that BIIB021 causes
activation of autophagy by promoting the synthesis of autophagosome
and increasing autophagic flux. We further investigated whether
BIIB021-induced autophagy acted as a cytoprotective mechanism. For
this purpose, we inhibited autophagy in 32Dp210-T315I cells by
using 2.5 mM 3-MA, a specific inhibitor of autophagic
sequestration, and analyzed the effects on the levels of LC3-II and
p62, as well as BIIB021-induced cell death. As shown in Fig. 5G, inhibition of cellular
proliferation by BIIB021 following 3-MA pretreatment was
significantly higher than that in the absence of 3-MA (P<0.05).
Western blot analysis indicated that 3-MA inhibited the conversion
of LC3-I to LC3-II and reversed the reduction of p62 proteins.
Furthermore, pretreatment of cells with 3-MA strongly increased the
cleavage of caspase-3 and PARP induced by BIIB021 (Fig. 5H).
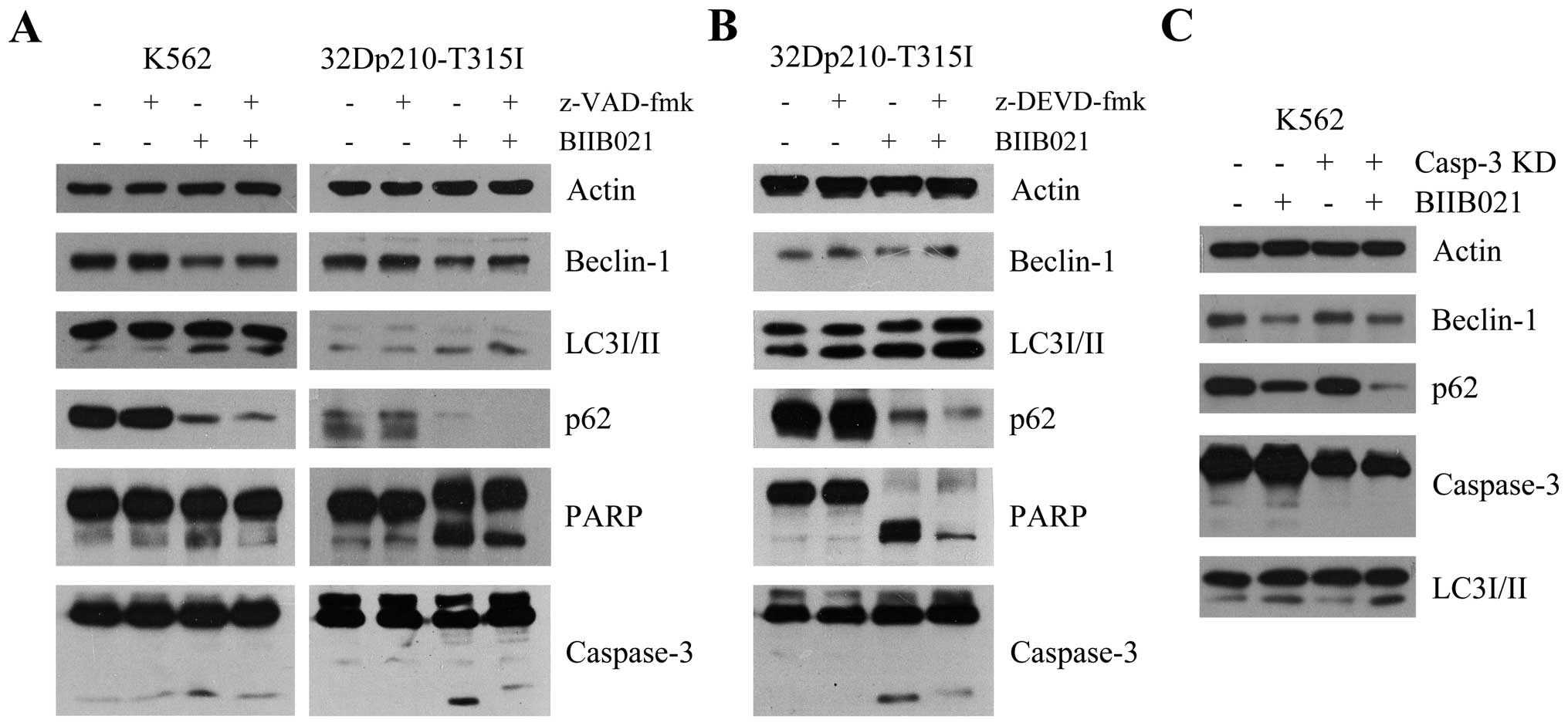
BIIB021-mediated caspase activation
contributes to inhibition of Beclin-1 expression
Recent studies suggested that the activation of
caspase at the onset of apoptosis could mediate cleavage of
Beclin-1 and thereby inhibit Beclin-1-induced autophagy (30,31).
To investigate if BIIB021 causes inhibition of Beclin-1 in CML
cells in association with the activation of caspase, we incubated
K562 and 32Dp210-T315I cells with the broad caspase inhibitor
z-VAD-fmk and BIIB021. As shown in Fig. 6A, pretreatment with z-VAD-fmk
resulted in a partial reversal of inhibition of BIIB021, in
parallel with an enhanced autophagy as evidenced by increased
amount of LC3-II and decreased amount of p62. In contrast,
BIIB021-induced apoptosis was inhibited by z-VAD-fmk. Similar
results were found in 32Dp210-T315I cells pretreated with a
specific caspase-3 inhibitor z-DEVD-fmk (Fig. 6B), and in K562 cells where
caspase-3 was knocked down by shRNA transfection (Fig. 6C).
BIIB021 induces autophagy by affecting
Ulk1 and negatively regulating mTOR
Autophagy depends on the hierarchically ordered
activity of autophagy-related (ATG) proteins which were controlled
by the main autophagy repressor, mTOR that prevents Ulk1 activation
by phosphorylating Ulk1 at Ser757. In contrast, AMPK promotes
autophagy by activating Ulk1 through phosphorylation of Ser317 and
Ser777 (32,33). Accordingly, we determined whether
the mTOR-Ulk1 pathway or AMPK-Ulk1 pathway was involved in
BIIB021-induced autophagy (Fig.
7A). There was no significant decrease in the amount of AMPK
protein and phosphorylation of AMPK Thr172 in the BIIB021-treated
cells. However, western blot analysis revealed that, along with
downregulation of Akt, BIIB021 dose-dependently inhibited the
phosphorylation of mTOR Ser2448 and its downstream p70S6K Thr389 in
K562 and 32Dp210-T315I cells. Importantly, treatment with BIIB021
also resulted in a significant decrease of phosphorylation of Ulk1
Ser757. Since Akt is upstream of mTOR, we then examined whether Akt
activator IGF-1 attenuates BIIB021-induced inhibition of Akt,
phosphorylation of mTOR Ser2448 and Ulk1 Ser757 in CML cells
(Fig. 7B). Pretreatment with IGF-1
markedly restored the levels of Akt protein and phosphorylation of
mTOR Ser2448, p70S6K Thr389 and Ulk1 Ser757. Moreover, IGF-1
pretreatment obviously decreased BIIB021-induced expression of
LC3-II and downregulation of p62, indicating that inhibition of
Akt-mTOR by BIIB021 reduced autophagy via reactivating Ulk1.
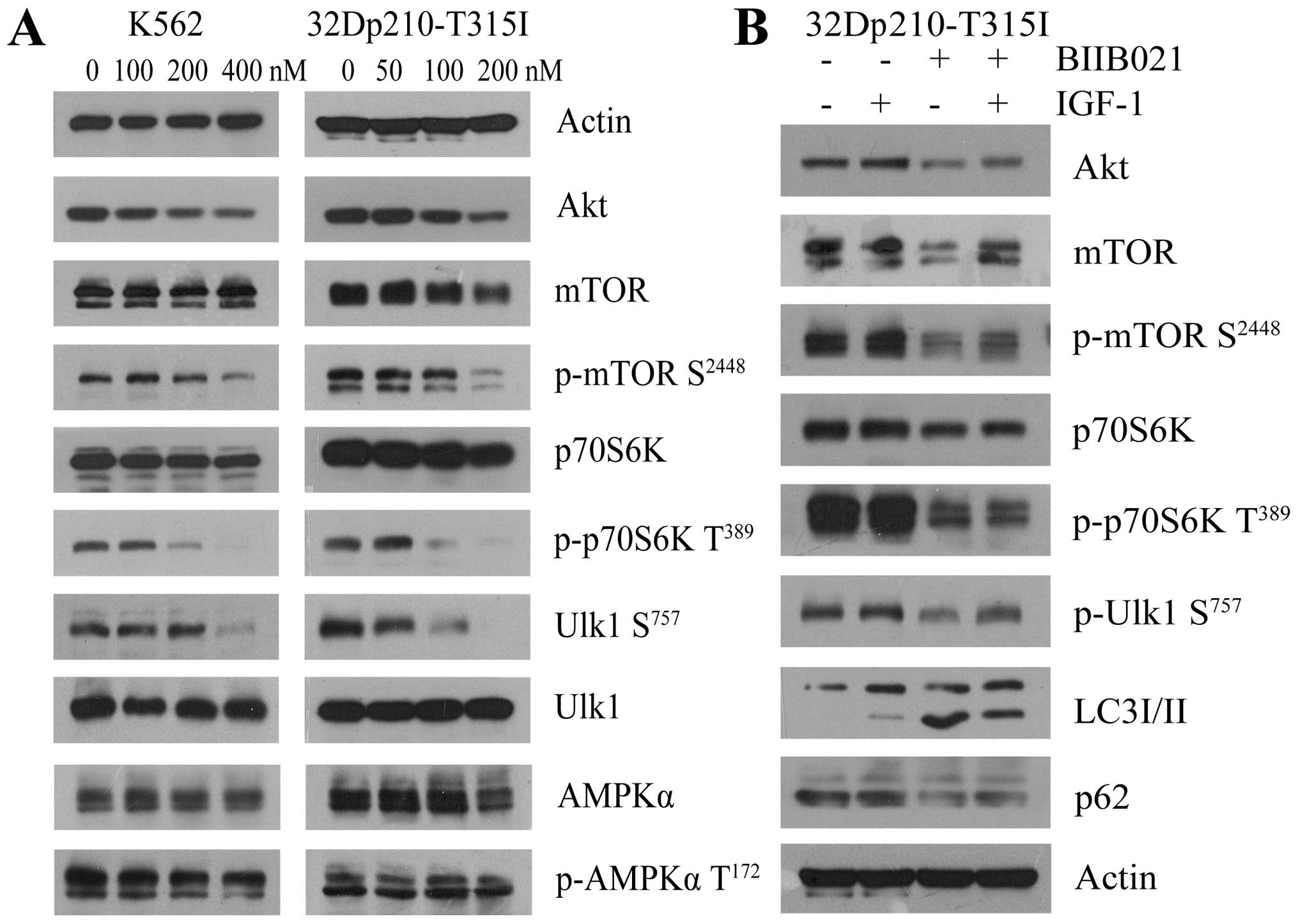 | Figure 7BIIB021 regulates the Akt-mTOR
pathway to initiate autophagy. (A) K562 and 32Dp210-T315I cells
were treated with BIIB021 at the indicated doses for 24 h. Whole
cells were lysed to evaluate the expression levels of Akt, total
mTOR, p-mTOR (Ser2448), total p70S6K, p-p70S6K (Thr389), total
Ulk1, p-Ulk1 (Ser757), AMPKα, and p-AMPKα (Thr172) by western blot
analysis. (B) After pretreated with IGF-1 (200 ng/ml) for 4 h,
32Dp210-T315I cells were exposed to BIIB021 (200 nM) for 24 h.
Then, whole-cell lysates were extracted to analyze the protein
levels of Akt, total mTOR, p-mTOR (Ser2448), total p70S6K, p-p70S6K
(Thr389), p-Ulk1 (Ser757), LC3 conversion, and p62 by western blot
analysis. |
Discussion
Hsp90 has recently been considered as a promising
target for therapeutic intervention in a variety of cancers
(12). The biological activity of
Hsp90 inhibitors towards CML has been demonstrated in vitro
and in murine xenograft models (14–16,34,35).
BIIB021 is the first oral, synthetic Hsp90 inhibitor to enter the
clinic for treatment of solid tumors and lymphoma (20,21).
However, little is known regarding the potential activity of
BIIB021 on CML. In this study, we investigate the potential
antitumor effects of BIIB021 on imatinib-sensitive and -resistant
CML cell lines as well as leukemic cells bearing T315I mutation. We
found that BIIB021 effectively inhibited the proliferation of CML
cells, and clearly indicated that by lowering BCR-ABL, BIIB021
induced release of cytochrome c, which promotes
consequential activation of caspase-9, -3 and PARP, thereby
inducing apoptosis. Additionally, BIIB021 treatment resulted in the
accumulation of proapoptotic proteins Bad and Bak, which are
involved in mitochondrial outer membrane permeabilization, a
critical event responsible for caspase activation in the intrinsic
pathway (36). These results are
consistent with a previous report showing that novobiocin, a new
Hsp90 inhibitor, induces the mitochondrial pathway of apoptosis in
CML cells (15).
Degradation of BCR-ABL oncoproteins by either Hsp90
inhibitors via the proteasome pathway (15,34)
or arsenic and imatinib through the lysosome pathway (37,38)
in CML has been shown to overcome resistance to TKIs. This
represents an alternative treatment strategy that does not rely
solely on kinase domain inhibition. In this study, we show that
treatment with BIIB021 caused a significant reduction in the levels
of both total and phosphorylation of BCR-ABL protein in CML cells
harboring wild-type BCR-ABL or BCR-ABL-T315I mutation, even though
the levels of BCR-ABL mRNA expression did not differ from untreated
control. Furthermore, inhibition of BCR-ABL could be partly
reversed by the proteasome inhibitor MG-132, but not by the
lysosome inhibitor CQ and Baf A1. These data suggest that
proteasome pathway is involved in BIIB021-mediated degradation of
BCR-ABL oncoproteins. On the contrary, imatinib can not eliminate
the primitive BCR-ABL positive stem cells of CML although it has
shown remarkable efficacy in the treatment of CML (39,40).
β-catenin, the central mediator of the Wnt/β-catenin signaling, is
involved in transcriptional regulation and chromatin modification,
and plays an important role in survival/self-renewal of dividing
BCR-ABL positive stem/progenitors (26,27,41).
Deletion of β-catenin has been shown to reduce survival and
self-renewal of CML quiescent stem cells and synergize with
imatinib to abrogate CML stem cells (26,41).
These results demonstrate that BIIB021 dose-dependently decreased
β-catenin expression and concomitantly decreased the levels of its
downstream c-myc. Confocal microscopy analysis showed that BIIB021
was able to completely abolish the nuclear accumulation of
β-catenin.
Treatments with antileukemic agents, including TKI,
have been shown to induce cellular autophagy in CML (37,38,42,43).
In this respect, autophagy exerts cytoprotective (42,43)
or beneficial actions (autophagic cell death) (37). The molecular regulation of
autophagy and potential efficacy of autophagy inhibition in CML
have been reviewed in detail (44). In this study, we found that cells
treated with BIIB021 showed pronounced autophagy evidenced by
increased formation of AVOs and levels of LC3-II in cells. In order
to investigate whether autophagy underlies cell death or protective
response, we blocked autophagy by Baf A1 or 3-MA. Inhibition of
autophagy significantly enhanced cell death in the BIIB021-treated
CML cells, and this cell death depended on caspase activation,
suggesting that a combination of BIIB021 and autophagy inhibitor
could be beneficial for the treatment of CML. We also attempted to
address the key question as to the signals delivered by BIIB021
that induce autophagy. Can et al (45) reported that Beclin-1, a critical
regulator of autophagy initiation, is required for autophagy
induction by imatinib. However, there was downregulation of
Beclin-1 proteins in the cells treated with BIIB021. Recent
evidence indicates that after initiating apoptosis by chemotherapy,
Beclin-1 is cleaved by caspase and the N-terminal fragment of
Beclin-1 can suppress autophagy (30,31).
Our results demonstrated that the addition of the caspase inhibitor
z-VAD-fmk and caspase-3 inhibition by z-DEVD-fmk or by siRNA partly
abrogated BIIB021-induced downregulation of Beclin-1 and enhanced
autophagy. Together, these data suggest that activation of caspase
is associated with downregulation of Beclin-1, and BIIB021 triggers
autophagy through Beclin-1 independent pathway.
It is well known that mTOR complex 1 (mTORC1) also
acts as a negative regulator of autophagy by phophorylating and
inhibiting Ulk1 (32,33). In acute myeloid leukemia cells,
autophagy can be elicited for treatment with dual mTORC1 and mTORC2
inhibitors such as OSI-027, AZD-2014 and AZD8055 (46,47).
We showed for the first time that mTOR-Ulk1 pathway might be
involved in the initiation of autophagy because BIIB021 strongly
inhibited phosphorylation of mTOR Ser2448, a marker for mTORC1
activity (48) which was
accompanied by a decreased level of phosphor-Ulk1 Ser757. This
effect may results in Ulk1 activation and induction of autophagy
(33). Also, BIIB021 inhibited the
level of Akt protein, an upstream molecule of mTOR. Furthermore,
our results show that Akt activator IGF-1 not only rescued
BIIB021-induced inhibition of Akt-mTOR pathway, but also suppressed
the occurrence of autophagy. Collectively, present findings suggest
that suppression of mTORC1 by BIIB021 contributed to the induction
of autophagy.
In conclusion, these results suggest that BIIB021
stimulates a multifaceted effector mechanism, all parts of which
are required for induction of cell death in both imatinib-sensitive
and -resistant CML cells, including leukemic cells harboring
T315I-mutant BCR-ABL. Also, BIIB021 significantly induces
autophagy, which is Beclin-1 independent, but associated with
downregulation of Akt-mTOR pathway and activation of Ulk1.
Inhibition of autophagy enhances the sensitivity of CML cells to
BIIB021. These data suggest the possibility of combining BIIB021
with autophagy inhibitors in a regimen that would optimize the
antileukemic activity against CML.
Acknowledgements
This study was supported by National Natural Science
Foundation of China (nos. 81370645, 81500110 and 81500111),
Doctoral Fund of Ministry of Education of China (no.
20120101110010), Funds of Science Technology Department of Zhejiang
Province (nos. 2012C13021-2 and 2014C33235), and Funds of the
Hangzhou Science and Technology Bureau (no. 20120633B15).
References
|
1
|
Clark SS, Crist WM and Witte ON: Molecular
pathogenesis of Ph-positive leukemias. Annu Rev Med. 40:113–122.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Danisz K and Blasiak J: Role of
anti-apoptotic pathways activated by BCR/ABL in the resistance of
chronic myeloid leukemia cells to tyrosine kinase inhibitors. Acta
Biochim Pol. 60:503–514. 2013.PubMed/NCBI
|
|
3
|
Steelman LS, Pohnert SC, Shelton JG,
Franklin RA, Bertrand FE and McCubrey JA: JAK/STAT, Raf/MEK/ERK,
PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis.
Leukemia. 18:189–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Skorski T, Bellacosa A,
Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, Trotta R,
Wlodarski P, Perrotti D, Chan TO, et al: Transformation of
hematopoietic cells by BCR/ABL requires activation of a
PI-3k/Akt-dependent pathway. EMBO J. 16:6151–6161. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin TS, Mahajan S and Frank DA: STAT
signaling in the pathogenesis and treatment of leukemias. Oncogene.
19:2496–2504. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Deininger M, Buchdunger E and Druker BJ:
The development of imatinib as a therapeutic agent for chronic
myeloid leukemia. Blood. 105:2640–2653. 2005. View Article : Google Scholar
|
|
7
|
Talpaz M, Shah NP, Kantarjian H, Donato N,
Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickardt E,
et al: Dasatinib in imatinib-resistant Philadelphia
chromosome-positive leukemias. N Engl J Med. 354:2531–2541. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kantarjian H, Giles F, Wunderle L, Bhalla
K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W,
et al: Nilotinib in imatinib-resistant CML and Philadelphia
chromosome-positive ALL. N Engl J Med. 354:2542–2551. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hoy SM: Ponatinib: A review of its use in
adults with chronic myeloid leukaemia or Philadelphia
chromosome-positive acute lymphoblastic leukaemia. Drugs.
74:793–806. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cortes JE, Kantarjian H, Shah NP, Bixby D,
Mauro MJ, Flinn I, O'Hare T, Hu S, Narasimhan NI, Rivera VM, et al:
Ponatinib in refractory Philadelphia chromosome-positive leukemias.
N Engl J Med. 367:2075–2088. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bishop SC, Burlison JA and Blagg BS:
Hsp90: A novel target for the disruption of multiple signaling
cascades. Curr Cancer Drug Targets. 7:369–388. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Isaacs JS, Xu W and Neckers L: Heat shock
protein 90 as a molecular target for cancer therapeutics. Cancer
Cell. 3:213–217. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nimmanapalli R, O'Bryan E and Bhalla K:
Geldanamycin and its analogue
17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and
induces apoptosis and differentiation of Bcr-Abl-positive human
leukemic blasts. Cancer Res. 61:1799–1804. 2001.PubMed/NCBI
|
|
14
|
Tao W, Chakraborty SN, Leng X, Ma H and
Arlinghaus RB: HSP90 inhibitor AUY922 induces cell death by
disruption of the Bcr-Abl, Jak2 and HSP90 signaling network complex
in leukemia cells. Genes Cancer. 6:19–29. 2015.PubMed/NCBI
|
|
15
|
Wu LX, Xu JH, Zhang KZ, Lin Q, Huang XW,
Wen CX and Chen YZ: Disruption of the Bcr-Abl/Hsp90 protein
complex: A possible mechanism to inhibit Bcr-Abl-positive human
leukemic blasts by novobiocin. Leukemia. 22:1402–1409. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tong WG, Estrov Z, Wang Y, O'Brien S,
Faderl S, Harris DM, Van Pham Q, Hazan-Halevy I, Liu Z, Koch P, et
al: The synthetic heat shock protein 90 (Hsp90) inhibitor EC141
induces degradation of Bcr-Abl p190 protein and apoptosis of
Ph-positive acute lymphoblastic leukemia cells. Invest New Drugs.
29:1206–1212. 2011. View Article : Google Scholar
|
|
17
|
Lundgren K, Zhang H, Brekken J, Huser N,
Powell RE, Timple N, Busch DJ, Neely L, Sensintaffar JL, Yang YC,
et al: BIIB021, an orally available, fully synthetic small-molecule
inhibitor of the heat shock protein Hsp90. Mol Cancer Ther.
8:921–929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang H, Neely L, Lundgren K, Yang YC,
Lough R, Timple N and Burrows F: BIIB021, a synthetic Hsp90
inhibitor, has broad application against tumors with acquired
multidrug resistance. Int J Cancer. 126:1226–1234. 2010.
|
|
19
|
Chen W, Sin SH, Wen KW, Damania B and
Dittmer DP: Hsp90 inhibitors are efficacious against Kaposi Sarcoma
by enhancing the degradation of the essential viral gene LANA, of
the viral co-receptor EphA2 as well as other client proteins. PLoS
Pathog. 8:e10030482012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Suzuki M, Takeda T, Nakagawa H, Iwata S,
Watanabe T, Siddiquey MN, Goshima F, Murata T, Kawada J, Ito Y, et
al: The heat shock protein 90 inhibitor BIIB021 suppresses the
growth of T and natural killer cell lymphomas. Front Microbiol.
6:2802015.PubMed/NCBI
|
|
21
|
Saif MW, Takimoto C, Mita M, Banerji U,
Lamanna N, Castro J, O'Brien S, Stogard C and Von Hoff D: A phase
1, dose-escalation, pharmacokinetic and pharmacodynamic study of
BIIB021 administered orally in patients with advanced solid tumors.
Clin Cancer Res. 20:445–455. 2014. View Article : Google Scholar
|
|
22
|
Dickson MA, Okuno SH, Keohan ML, Maki RG,
D'Adamo DR, Akhurst TJ, Antonescu CR and Schwartz GK: Phase II
study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal
tumors. Ann Oncol. 24:252–257. 2013. View Article : Google Scholar
|
|
23
|
Lin S, Li J, Zhou W, Qian W, Wang B and
Chen Z: BIIB021, an Hsp90 inhibitor, effectively kills a
myelodysplastic syndrome cell line via the activation of caspases
and inhibition of PI3K/Akt and NF-κB pathway proteins. Exp Ther
Med. 7:1539–1544. 2014.PubMed/NCBI
|
|
24
|
Li M, Zhang X, Zhou WJ, Chen YH, Liu H,
Liu L, Yang CM and Qan WB: Hsp90 inhibitor BIIB021 enhances
triptolide-induced apoptosis of human T-cell acute lymphoblastic
leukemia cells in vitro mainly by disrupting p53-MDM2 balance. Acta
Pharmacol Sin. 34:1545–1553. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qian W, Liu J, Jin J, Ni W and Xu W:
Arsenic trioxide induces not only apoptosis but also autophagic
cell death in leukemia cell lines via up-regulation of Beclin-1.
Leuk Res. 31:329–339. 2007. View Article : Google Scholar
|
|
26
|
Heidel FH, Bullinger L, Feng Z, Wang Z,
Neff TA, Stein L, Kalaitzidis D, Lane SW and Armstrong SA: Genetic
and pharmacologic inhibition of β-catenin targets
imatinib-resistant leukemia stem cells in CML. Cell Stem Cell.
10:412–424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Coluccia AM, Vacca A, Duñach M, Mologni L,
Redaelli S, Bustos VH, Benati D, Pinna LA and Gambacorti-Passerini
C: Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia
through its tyrosine phosphorylation. EMBO J. 26:1456–1466. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mori M, Hitora T, Nakamura O, Yamagami Y,
Horie R, Nishimura H and Yamamoto T: Hsp90 inhibitor induces
autophagy and apoptosis in osteosarcoma cells. Int J Oncol.
46:47–54. 2015.
|
|
29
|
Del Bufalo D, Desideri M, De Luca T, Di
Martile M, Gabellini C, Monica V, Busso S, Eramo A, De Maria R,
Milella M, et al: Histone deacetylase inhibition synergistically
enhances pemetrexed cytotoxicity through induction of apoptosis and
autophagy in non-small cell lung cancer. Mol Cancer. 13:2302014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li H, Wang P, Sun Q, Ding WX, Yin XM,
Sobol RW, Stolz DB, Yu J and Zhang L: Following cytochrome c
release, autophagy is inhibited during chemotherapy-induced
apoptosis by caspase 8-mediated cleavage of Beclin 1. Cancer Res.
71:3625–3634. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wirawan E, Vande Walle L, Kersse K,
Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R,
Verspurten J, Declercq W, et al: Caspase-mediated cleavage of
Beclin-1 inactivates Beclin-1-induced autophagy and enhances
apoptosis by promoting the release of proapoptotic factors from
mitochondria. Cell Death Dis. 1:e182010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar :
|
|
33
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peng C, Brain J, Hu Y, Goodrich A, Kong L,
Grayzel D, Pak R, Read M and Li S: Inhibition of heat shock protein
90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia
and suppresses leukemic stem cells. Blood. 110:678–685. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Barnes DJ, De S, van Hensbergen P,
Moravcsik E and Melo JV: Different target range and cytotoxic
specificity of adaphostin and
17-allylamino-17-demethoxygeldanamycin in imatinib-resistant and
sensitive cell lines. Leukemia. 21:421–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Green DR and Kroemer G: Pharmacological
manipulation of cell death: Clinical applications in sight? J Clin
Invest. 115:2610–2617. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Goussetis DJ, Gounaris E, Wu EJ, Vakana E,
Sharma B, Bogyo M, Altman JK and Platanias LC: Autophagic
degradation of the BCR-ABL oncoprotein and generation of
antileukemic responses by arsenic trioxide. Blood. 120:3555–3562.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Elzinga BM, Nyhan MJ, Crowley LC,
O'Donovan TR, Cahill MR and McKenna SL: Induction of autophagy by
Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates
Bcr-Abl protein. Am J Hematol. 88:455–462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Graham SM, Jørgensen HG, Allan E, Pearson
C, Alcorn MJ, Richmond L and Holyoake TL: Primitive, quiescent,
Philadelphia-positive stem cells from patients with chronic myeloid
leukemia are insensitive to STI571 in vitro. Blood. 99:319–325.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bhatia R, Holtz M, Niu N, Gray R, Snyder
DS, Sawyers CL, Arber DA, Slovak ML and Forman SJ: Persistence of
malignant hematopoietic progenitors in chronic myelogenous leukemia
patients in complete cytogenetic remission following imatinib
mesylate treatment. Blood. 101:4701–4707. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Neviani P, Harb JG, Oaks JJ, Santhanam R,
Walker CJ, Ellis JJ, Ferenchak G, Dorrance AM, Paisie CA, Eiring
AM, et al: PP2A-activating drugs selectively eradicate
TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest.
123:4144–4157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tong Y, Liu YY, You LS and Qian WB:
Perifosine induces protective autophagy and upregulation of ATG5 in
human chronic myelogenous leukemia cells in vitro. Acta Pharmacol
Sin. 33:542–550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kamitsuji Y, Kuroda J, Kimura S, Toyokuni
S, Watanabe K, Ashihara E, Tanaka H, Yui Y, Watanabe M, Matsubara
H, et al: The Bcr-Abl kinase inhibitor INNO-406 induces autophagy
and different modes of cell death execution in Bcr-Abl-positive
leukemias. Cell Death Differ. 15:1712–1722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Helgason GV, Karvela M and Holyoake TL:
Kill one bird with two stones: Potential efficacy of BCR-ABL and
autophagy inhibition in CML. Blood. 118:2035–2043. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Can G, Ekiz HA and Baran Y: Imatinib
induces autophagy through BECLIN-1 and ATG5 genes in chronic
myeloid leukemia cells. Hematology. 16:95–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Willems L, Chapuis N, Puissant A, Maciel
TT, Green AS, Jacque N, Vignon C, Park S, Guichard S, Herault O, et
al: The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor
activity in acute myeloid leukemia. Leukemia. 26:1195–1202. 2012.
View Article : Google Scholar
|
|
47
|
Altman JK, Szilard A, Goussetis DJ,
Sassano A, Colamonici M, Gounaris E, Frankfurt O, Giles FJ, Eklund
EA, Beauchamp EM, et al: Autophagy is a survival mechanism of acute
myelogenous leukemia precursors during dual mTORC2/mTORC1
targeting. Clin Cancer Res. 20:2400–2409. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Park S, Chapuis N, Tamburini J, Bardet V,
Cornillet-Lefebvre P, Willems L, Green A, Mayeux P, Lacombe C and
Bouscary D: Role of the PI3K/AKT and mTOR signaling pathways in
acute myeloid leukemia. Haematologica. 95:819–828. 2010. View Article : Google Scholar :
|