Introduction
Urinary bladder cancer is the second most common
malignant tumor of the genitourinary tract and ranks fourth among
male cancers (1). Approximately
70% of initially diagnosed tumors are superficial and can be
treated by transurethral resection, while the remaining 30% become
muscle invasive and are associated with a high risk of metastatic
disease (2,3). Systemic chemotherapy is a treatment
option for patients with locally advanced or metastatic disease.
Despite huge efforts to tackle the disease in the past two decades,
current treatments confer only a modest survival benefit upon
bladder cancer patients, and long-term survival of patients
suffering from metastatic disease does not exceed 20% (4); therefore, there is an urgent need for
innovative ideas that deviate from conventional approaches.
The search for novel chemical agents against cancer
has long been the mainstay of cancer research. During recent years,
it has been shown that epigenetic aberrations are involved in
tumorigenesis. Particularly, an imbalance in the equilibrium
between histone acetylation and histone deacetylation has been
proposed as a driving force, causing normal cells to become
malignant. Therefore, modulating acetylation may be an innovative
strategy to treat malignant disease. Acetylation is catalyzed by a
specific enzyme family, histone acetyltransferases (HATs), and
correlates with nucleosome remodeling and transcriptional
activation, whereas deacetylation of histone tails is catalyzed by
histone deacetylases (HDACs) and induces transcriptional repression
through chromatin condensation (5).
Altered expression of different HDACs has been
reported in various human cancers (6–12).
Systemic analysis of the expression levels of HDACs in cultured
cancer cell lines, as well as primary cultures of human cancer
cells and various human tumor biopsy samples, frequently identifies
higher levels of expression than in corresponding normal tissue.
For instance, recent evidence shows that both clinical samples from
patients with urinary bladder cancer and tumor tissues from a mouse
model have demonstrated a significantly increased HDAC expression
compared with surrounding healthy tissue (13). Thus, HDAC inhibition might be an
effective option to treat bladder cancer.
Thus far, 18 HDACs have been identified in mammals
that are classified into four classes based on their homology to
yeast proteins (7,14). Class I HDAC enzymes (HDACs 1, 2, 3
and 8) are widely expressed (12,15),
class II HDAC enzymes (HDACs 4, 5, 6, 7, 9 and 10) have
tissue-specific distribution and are involved in organ development
and function (12), and other
classes are less specific in terms of tissue distribution and
function. HDAC inhibitors (HDACIs) prevent HDACs from removing
acetyl groups, leading to increased acetylation and allowing DNA to
remain transcriptionally active (5). There are many known natural and
synthetic HDACIs, which can be subdivided into five structural
classes, including hydroxamates, cyclic peptides, aliphatic acids,
benzamines and electrophilic ketones. The hydroxamate compound
trichostatin A (TSA) is a potent nanomolar inhibitor of most class
I and class II HDAC enzymes (12,16).
Romidepsin (FK228) is the only cyclic peptide HDACI in clinical
development (17,18), and it potently inhibits class I
HDACs (12,18). Class I HDAC enzymes are
overexpressed in many malignancies, and this overexpression is
often associated with poor prognosis (12). A number of structurally different
HDACIs are in clinical trials for a wide variety of hematologic and
solid neoplasms, including cancer of the lung, breast, pancreas,
and kidney, melanoma, glioblastoma, leukemias, lymphomas and
multiple myeloma (5). Among them,
romidepsin and vorinostat (SAHA) have been approved by the Food and
Drug Administration for the treatment of cutaneous T-cell lymphoma
(CTCL). However, the effect and the mechanism of action of HDACIs
as chemotherapeutic regimens for bladder cancer remain to be
determined.
Herein, we show that the treatment with HDACIs
(romidepsin, TSA and SAHA) inhibited cell growth and proliferation
in a dose-dependent fashion in the urinary bladder cancer cell line
5637. We further analyzed the protein expression patterns in
response to romidepsin and TSA in this cell model system using
quantitative proteomic studies and found that the effect of these
two HDACIs on growth inhibition and cell death is mediated through
modulating the expression of proteins involved in cell cycle
progression, apoptosis, autophagy, reactive oxygen species (ROS)
generation and DNA damage repair in 5637 bladder cancer cells.
Materials and methods
Chemicals and reagents
The minimum essential medium (MEM), fetal bovine
serum (FBS), penicillin-streptomycin (100X), and 0.25% trypsin-EDTA
solution (1X) were obtained from Invitrogen Corp., Life
Technologies (Carlsbad, CA, USA). Trichostatin A (TSA) (>98%
purity) was from Selleckchem (Houston, TX, USA). Romidepsin (FK228)
(>98% purity) was purchased from Apexbio Technology LLC
(Houston, TX, USA). Vorinostat (SAHA) and dimethyl sulfoxide (DMSO)
were from Sigma-Aldrich (St. Louis, MO, USA). Romidepsin, TSA or
SAHA were dissolved in DMSO separately and stored at −20°C. The
CellTiter 96 AQueous One Solution Cell Proliferation Assay was from
Promega Corp. (Madison, WI, USA).
Cell culture and cell viability
assay
The human bladder cancer cell line 5637 was
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). The cell line was grown in MEM, supplemented
with 10% FBS, 50 IU/ml penicillin, and 50 μg/ml streptomycin, at
37°C in a humidified atmosphere with 5% CO2.
The antiproliferative effects of romidepsin, TSA and
SAHA were assessed using an MTS
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)-based
assay (Promega) as previously described (19). In brief, 5637 bladder carcinoma
cells (5×103 cells/well) were evenly distributed in
96-well plates, grown overnight, and then treated with various
concentrations of romidepsin, TSA or SAHA at the indicated
concentrations (0, 0.1 1, 10 and 100 nM, 1, 10 and 100 μM) for 24
or 72 h. At the end of incubation, 20 μl of CellTiter 96 AQueous
One Solution reagent was added to each well of the assay plates
containing the treated and untreated cells in 200 μl of culture
medium, and the plates were incubated at 37°C and 5% CO2
for 2 h. The optical density at 490 nm was determined using a
96-well iMark™ Microplate reader (Bio-Rad Laboratories, Hercules,
CA, USA). Proliferation rates were calculated from the optical
densities of the HDACI-treated cells relative to the optical
density of DMSO-treated control cells with no HDACI exposure
(control value, 100%). The half-maximal inhibitory concentration
(IC50) values for romidepsin, TSA and SAHA on 24 and 72
h in 5637 cell line were calculated using GraphPad Prism version
6.01 software (GraphPad Software, Inc., La Jolla, CA, USA).
IC50 was considered as the drug concentration that
decreases the cell count by 50%. Non-linear regression curve
fitting was performed. The data were fitted to an exponential
first-order decay function.
Preparation of protein extraction,
separation of proteins and in-gel trypsin digestion
Total protein extraction from cell pellets was
prepared by the following method. In brief, cell pellets were lysed
in 0.4 ml lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM
Na2EDTA, 1 mM EGTA, 1% Triton X-100, protease inhibitor
cocktail pill). After cells were lysed, 50 μl of 10% SDS and 50 μl
of 1 M DTT were added into the mixture followed by incubation at
95°C for 10 min. The extraction was then sonicated and centrifuged
at 15,000 × g for 10 min. Supernatants were collected and stored at
−80°C for further analysis. The protein concentration of the
supernatants was determined by a BCA™ reducing reagent compatible
assay kit (Thermo Fisher Scientific, Rockford, IL, USA).
Equal amounts of protein (130 μg) from each sample
were fractioned by separation on a NuPAGE 4–12% Bis-Tris Gel (Life
Technologies, Grand Island, NY, USA). Sixteen gel fractions from
each lane representing one sample were treated with DTT for
reduction, then iodoacetamide for alkylation, and further digested
by trypsin in 25 mM NH4HCO3 solution. The
digested protein was extracted, and the extracted peptides were
dried and reconstituted in 20 μl of 0.1% formic acid before
nanospray LC/MS/MS analysis was performed.
Nanospray LC/MS/MS analysis
Sixteen tryptic peptide fractions from one cell
sample were analyzed sequentially using a Thermo Scientific Q
Exactive Hybrid Quadrupole-Orbitrap mass spectrometer equipped with
a Thermo Dionex UltiMate 3000 RSLCnano system. Tryptic peptide
samples were loaded onto a peptide trap cartridge at a flow rate of
5 μl/min. The trapped peptides were eluted onto a reversed-phase
25-cm C18 Picofrit column (New Objective, Woburn, MA, USA) using a
linear gradient of acetonitrile (3–36%) in 0.1% formic acid. The
elution duration was 110 min at a flow rate of 0.3 μl/min. Eluted
peptides from the Picofrit column were ionized and sprayed into the
mass spectrometer, using a Nanospray Flex Ion Source ES071 (Thermo
Fisher Scientific) under the following settings: spray voltage, 1.6
kV and capillary temperature, 250°C. The Q Exactive instrument was
operated in the data dependent mode to automatically switch between
full scan MS and MS/MS acquisition. Survey full scan MS spectra
(m/z 300–2000) was acquired in the Orbitrap with 70,000 resolution
(m/z 200) after accumulation of ions to a 3×106 target
value based on predictive AGC from the previous full scan. Dynamic
exclusion was set to 20 sec. The 12 most intense multiply-charged
ions (z ≥2) were sequentially isolated and fragmented in the Axial
higher energy collision-induced dissociation (HCD) cell using
normalized HCD collision energy at 25% with an AGC target 1e5 and a
maxima injection time of 100 ms at 17,500 resolution.
LC/MS/MS data analysis
The raw MS files were analyzed using the Thermo
Proteome Discoverer 1.4.1 platform (Thermo Fisher Scientific,
Bremen, GmbH) for peptide identification and protein assembly. For
each cell sample, 16 raw MS files obtained from 16 sequential LC/MS
analyses were grouped for a single database search against the
Human UniProtKB/Swiss-Prot human protein sequence databases (20597
entries, 12/20/2013) based on the SEQUEST and percolator algorithms
through the Proteome Discoverer 1.4.1 platform.
Carbamidomethylation of cysteines was set as a fixed modification.
The minimum peptide length was specified to be five amino acids.
The precursor mass tolerance was set to 15 ppm, whereas fragment
mass tolerance was set to 0.05 kDa. The maximum false peptide
discovery rate was specified as 0.01. The resulting Proteome
Discoverer Report contains all assembled proteins (a proteome
profile) with peptides sequences and matched spectrum counts. Three
proteome profiles were generated for the untreated control cells
and two HDACI-treated cell samples.
Protein quantification
Protein quantification used the normalized spectral
abundance factors (NSAFs) method to calculate the protein relative
abundance (20,21). To quantitatively describe the
relative abundance, the ppm (part per million) was chosen as the
unit and the 1,000,000 ppm value was assigned to each proteome
profile. A ppm value at the range of 0–1,000,000 ppm for each
identified protein in each proteome profile was calculated based on
its normalized NSAF.
The ppm was calculated as follows: RCN =
106 × NSAFN, where RCN is the
relative concentration of protein N in the proteome of test sample,
NSAFN is the protein's normalized spectral abundance
factor and N is the protein index.
NSAFs were calculated as follows: NSAFN =
(SN/LN)/(∑ni=1Si/Li),
where N is the protein index, SN is the number of
peptide spectra matched to the protein, LN is the length
of protein N (number of amino acid residues), and n is the total
number of proteins in the input database (proteome profile for one
cell sample). The ratio of HDACI treated vs. untreated control was
defined as 1,000 if the protein was not identified in untreated
control, or as 0.001 if the protein was not identified in
HDACI-treated sample.
Signaling pathway analysis
The cell functions are executed and regulated by the
entire sets of proteins (the proteome). The regulation of different
cellular functions has been categorized into a number of pathways,
such as cell cycle and apoptosis signaling pathways. In each
pathway, the components according to their functions are generally
named as ligands, receptors, activating regulators, inhibitory
regulators and effectors. In order to measure the activation
strength of a pathway, the protein molecules that belong to
ligands, receptors, activating regulators, inhibitory regulators,
or effectors were grouped and their relative abundances (ppm) were
summed. The protein list for all analyzed pathways and processes
was obtained from the Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway database (http://www.genome.jp/kegg/pathway.html), and their
functional annotations were manually confirmed using the UniProtKB
protein database and the NCBI protein database or available
publications.
Statistical data analysis
All quantitative values are presented as means ± SD.
Data were statistically analyzed using two-way analysis of variance
(ANOVA) for comparison among groups. Student's t-test was used to
analyze the statistical significance of differences between
untreated controls and HDACI-treated groups. All P-values were
determined using a two-sided test, and P-values <0.05 were
considered to indicate significance.
Results
HDACIs inhibit cell proliferation and
induce cytotoxicity in human bladder cancer cells
To investigate the effect of HDACIs on bladder
cancer cell growth and proliferation, we selected human bladder
cancer 5637 cells, a cell line commonly used as a model for
studying bladder carcinoma. The dose-response of romidepsin, TSA
and SAHA inhibition of the growth of 5637 cell line was
characterized in vitro using the MTS assay. Romidepsin, TSA
or SAHA at concentrations of 0.1 nM to 100 μM caused dose-dependent
inhibition of the proliferation of 5637 cells at 72 h (Fig. 1A). The half-maximal inhibitory
concentration (IC50) values of romidepsin, TSA and SAHA
at 72 h in this line were 1.0±0.1 nM, 100±3.5nM and 1.9±0.1 μM,
respectively. These results indicate that HDACIs can potently
inhibit cell proliferation and induce cell toxicity in bladder
cancer cells.
Previous study has demonstrated that HDACIs increase
histone acetylation levels in human bladder cancer cells and that
these levels peak at 24 h and decrease gradually over 48–72 h
(22). Therefore, we chose 24-h
treatment with HDACIs for this in vitro study. To establish
the appropriate HDACI treatment concentration for our proteomic
studies, we performed cytotoxicity assays in 5637 cells in response
to romidepsin, TSA or SAHA treatment at different concentrations.
As shown in Fig. 1B, with
dose-increased HDACI treatment for 24 h, the viability of 5637
cells correspondingly decreased, and the romidepsin, TSA and SAHA
working concentrations resulting in 50% cell viability were 50±3.5
nM, 200±20 nM and 7.5±0.5 μM, respectively. Since the activity of
romidepsin and TSA was much more potent than SAHA in cytotoxicity
in 5637 cells (Fig. 1), we
therefore, finally used the working concentrations of 50 and 200 nM
for 24-h treatment for romidepsin and TSA, respectively, for the
following proteomic experiments.
Quantitative proteomic analysis of
bladder cancer cells following HDACI treatment
To analyze the mechanisms responsible for the effect
of HDACIs on cell proliferation and cytotoxicity in bladder cancer
cells, the whole cell proteome profiles of the HDACI-treated and
untreated 5637 cells were compared using quantitative proteomic
studies. Differentially expressed proteins were identified and
quantified by nanospray LC/MS/MS mass spectrometry. The selection
criteria for deregulation were the same for all the samples:
identification based on at least two unique peptides and fold
difference >2.0 or <−2.0.
Using the nanospray LC/MS/MS analysis, a total of
6003 non-redundant proteins were identified in both HDACI treated
and untreated 5637 cells. Of these, 4865, 4618 and 4674 were
quantified in romidepsin-treated, TSA-treated and untreated cells,
respectively. A total of 3518 proteins were common to the two
HDACI-treated cells and untreated cells.
Compared with the untreated control, there were 5698
differentially expressed proteins in romidepsin-treated 5637 cells,
including 2969 upregulated proteins (1845 ≥2-fold upregulated
proteins) and 2729 downregulated proteins (1626 ≥2-fold down
regulated proteins). The fold changes ranged from 45.51 to -35.99
and 1979 of these proteins (both upregulated and downregulated
proteins) showed >10-fold deregulation. For the TSA-treated 5637
cells, a total of 5497 proteins were differentially regulated; 2808
were upregulated (1709 ≥2-fold upregulated) and 2689 downregulated
(1563 ≥2-fold down-regulated). The fold changes ranged from 36.18
to −26.83 and 1826 of these proteins (both upregulated and
downregulated proteins) showed more than 10-fold deregulation. A
total of 1082 ≥2-fold upregulated proteins and 1140 ≥2-fold
down-regulated proteins were common to both romidepsin-treated and
TSA-treated 5637 cells.
Functional classification of
differentially expressed proteins in HDACI-treated bladder cancer
cells
To gain an initial understanding of the role and
function of the identified proteins between the HDACI treated and
untreated 5637 bladder cancer cells, we merged the protein datasets
and used pathway software to provide a descriptive analysis. The
functional correlation analysis of the differentially regulated
proteins was done by database search using UniProt, Swiss-Prot and
PANTHER. The categorization of differentially expressed proteins
(≥2-fold upregulated and downregulated proteins) according to their
molecular function, biological process and cellular component is
shown in Fig. 2. These data are
based on a compilation of proteins from the romidepsin-treated cell
samples and are presented to demonstrate the range of molecular
functions (Fig. 2A) and biological
processes (Fig. 2B) represented by
the identified proteins, and the cellular component (Fig. 2C) to which the proteins belong.
According to cellular component, the analysis revealed a high
percentage of proteins corresponding to the cell part, organelle,
macromolecular complex, extracellular region and extracellular
matrix (Fig. 2C). Based on
molecular function, the most general categories of proteins were
catalytic activity, binding activity, structural molecule activity,
nucleic acid transcription factor activity, enzyme regulator
activity, transporter activity and receptor activity (Fig. 2A). Differentially expressed
proteins related to 13 biological processes, including metabolic
process, cellular process, localization, biological regulation,
developmental process, cellular component organization or
biogenesis, response to stimulus and apoptotic process (Fig. 2B).
A majority of the molecular functions and biological
processes were affected in both romidepsin-treated and TSA-treated
bladder cancer cells. Although romidepsin caused more
differentially expressed proteins (3471 ≥2-fold upregulated and
downregulated) than those caused by TSA (3272 ≥2-fold upregulated
and downregulated proteins), the percentages of proteins in each
category of the molecular function and biological process were
similar between the romidepsin-treated (Fig. 2A and B) and TSA-treated (data not
shown) 5637 cells. There were 1845 and 1709 ≥2-fold upregulated
proteins and 1626 and 1563 ≥2-fold downregulated proteins in
romidepsin-treated and TSA-treated cell samples, respectively. We
also compared and showed that in either the upregulated proteins or
the downregulated proteins, there was no significant difference for
the percentages of proteins in each category of the molecular
function and biological process between the romidepsin-treated
cells and TSA-treated cells (data not shown), suggesting that both
romidepsin and TSA exert the same or similar actions on functional
categories in our cell model of bladder cancer.
Biological pathway analysis according to
the Kyoto Encyclopedia of Genes and Genomes (KEGG)
Next, we used KEGG pathway analysis to identify the
biological pathways of the proteins that were significantly
differentially expressed (≥2-fold upregulated and downregulated) in
the HDACI-treated 5637 bladder cancer cells. Pathway analysis using
KEGG database by DAVID bioinformatics resources tool (DAVID v6.7,
the Database for Annotation, Visualization and Integrated
Discovery) showed that the downregulated proteins were associated
with multiple pathways, such as cell cycle, bladder cancer, lysine
degradation, valine, leucine, and isoleucine degradation, and all
major annotated lipid metabolism pathways including
glycerophospholipid metabolism, steroid biosynthesis, glycerolipid
metabolism, sphingolipid metabolism and ether lipid metabolism
(Table I). We also performed the
same analysis for HDACI upregulated proteins, which were enriched
in the DNA replication and nucleotide related processes, including
ribosome, amino sugar and nucleotide sugar metabolism, mismatch
repair, basal transcription factors, nucleotide excision repair,
purine metabolism, pyrimidine metabolism and RNA polymerase
(Table I).
 | Table IMain metabolic and enzymatic pathways
associated with the upregulated and downregulated proteins in
romidepsin-treated 5637 cells as analyzed by the Kyoto Encyclopedia
of Genes and Genomes.a |
Table I
Main metabolic and enzymatic pathways
associated with the upregulated and downregulated proteins in
romidepsin-treated 5637 cells as analyzed by the Kyoto Encyclopedia
of Genes and Genomes.a
| Biological
pathway | % | P-value | Benjamini |
|---|
| 1845 ≥2-fold
upregulated proteins |
| Ribosome | 1.8 |
4.0×10−9 |
7.1×10−7 |
| Oxidative
phosphorylation | 2.1 |
5.8×10−7 |
3.4×10−5 |
| Ubiquitin mediated
proteolysis | 2.0 |
6.1×10−6 |
2.1×10−4 |
| Lysosome | 1.5 |
7.1×10−4 |
1.5×10−2 |
| Amino sugar and
nucleotide sugar metabolism | 0.8 |
1.3×10−3 |
2.2×10−2 |
| Mismatch
repair | 0.5 |
6.8×10−3 |
8.8×10−2 |
| Basal
transcription factors | 0.6 |
7.6×10−3 |
9.1×10−2 |
| DNA
replication | 0.6 |
9.3×10−3 |
1.0×10−2 |
| Nucleotide
excision repair | 0.6 |
3.3×10−2 |
2.8×10−2 |
| Purine
metabolism | 1.5 |
4.1×10−2 |
3.2×10−2 |
| Pyrimidine
metabolism | 1.0 |
6.2×10−2 |
4.0×10−2 |
| RNA
polymerase | 0.4 |
6.2×10−2 |
3.9×10−2 |
| 1626 ≥2-fold
downregulated proteins |
| N- and O-Glycan
biosynthesis | 1.3 |
5.4×10−5 |
1.2×10−4 |
|
Glycerophospholipid metabolism | 1.0 |
4.7×10−3 |
1.5×10−2 |
| Cell cycle | 1.4 |
4.8×10−3 |
1.3×10−2 |
| Lysine
degradation | 0.7 |
1.1×10−2 |
2.0×10−2 |
| Valine, leucine
and isoleucine degradation | 0.6 |
3.2×10−2 |
3.8×10−2 |
| Glycerolipid
metabolism | 0.6 |
3.6×10−2 |
3.9×10−2 |
| Sphingolipid
metabolism | 0.5 |
4.7×10−2 |
4.1×10−2 |
| Biosynthesis of
unsaturated fatty acids | 0.4 |
3.2×10−2 |
3.1×10−2 |
| Steroid
biosynthesis | 0.3 |
5.3×10−2 |
4.0×10−2 |
| Bladder
cancer | 0.5 |
6.6×10−2 |
4.1×10−2 |
| Ether lipid
metabolism | 0.5 |
7.6×10−2 |
4.3×10−2 |
| Methane
metabolism | 0.2 |
8.8×10−2 |
4.7×10−2 |
HDACI-induced cell death in bladder
cancer cells is mediated via modulating cell cycle progression,
apoptosis and DNA damage repair
Given that HDACIs have been shown to exert a variety
of anticancer activities in different types of tumors and that both
romidepsin and TSA induced cell growth inhibition and cell death in
our bladder cancer cells (Fig. 1),
we elucidated the mechanism underlying the effect of HDACIs on cell
proliferation and cytotoxicity in this model system. We performed
pathway-clustering analyses of the HDACI-responsive proteome for
pathways involved in cell death. The results showed that cell
cycle, apoptosis, oxidative stress, autophagy, and DNA damage
repair were the most prominent pathways enriched with altered
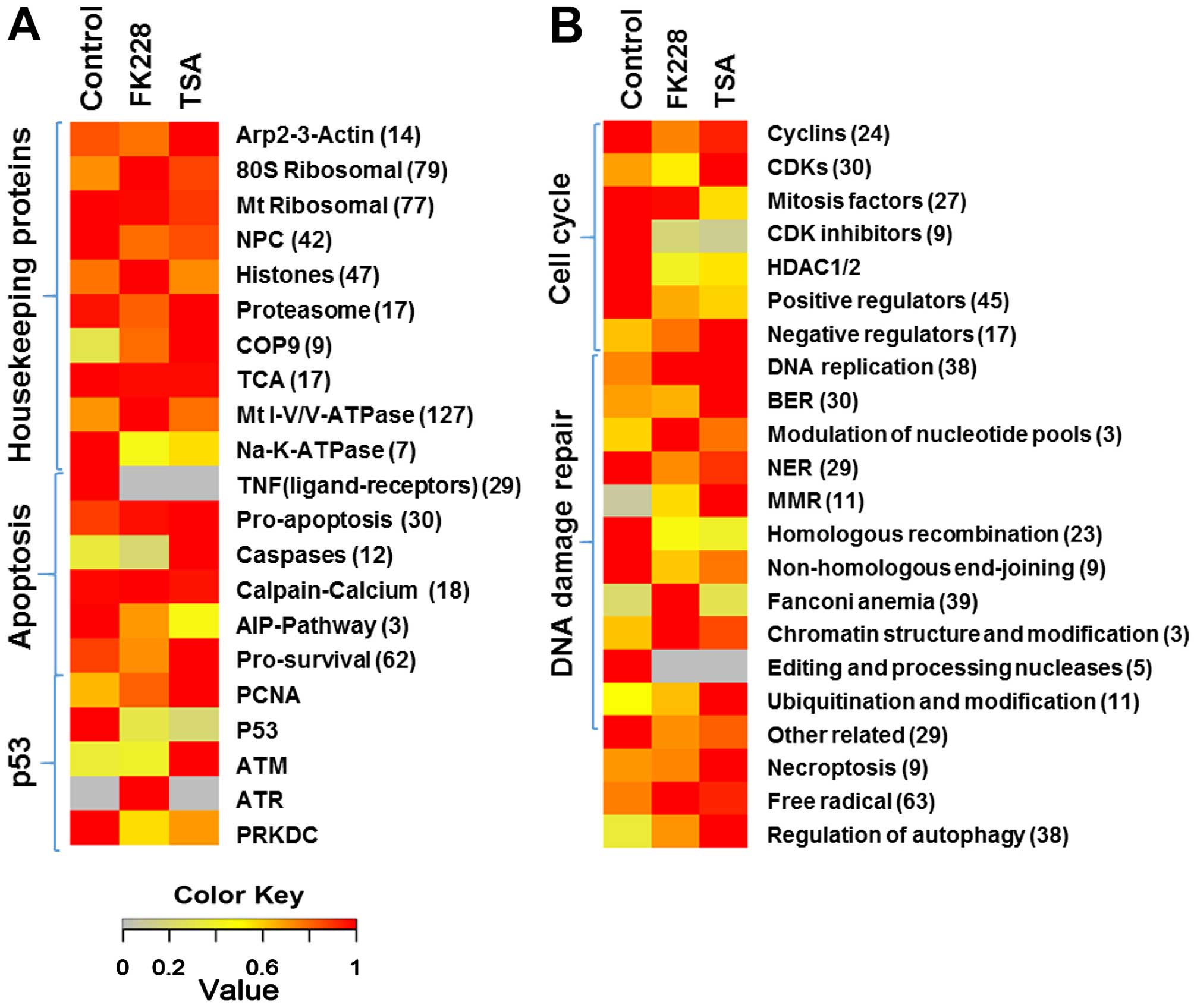
protein levels in HDACI-treated cells (Fig. 3), suggesting that these pathways
are involved in HDACI-induced cell death in 5637 bladder cancer
cells.
To understand more about the mechanisms of
HDACI-induced cell death in our bladder cancer cell model, we
identified the differentially expressed proteins related to cell
death in these pathways in response to HDACI treatment. Table II shows part of the differentially
expressed proteins involved in cell death in both romidepsin and
TSA treated cells. These include 37 proteins involved in cell cycle
progression, 19 proteins associated with apoptosis process, 30
proteins in various DNA damage repair pathways and 11 proteins
involved in ROS generation and autophagy regulation. The functions
and levels of the proteins in each pathway are listed in the
table.
| Table IIAlterations in the levels of the
proteins associated with cell death in bladder cancer cells in
response to romidepsin or trichostatin A (TSA) treatment. |
Table II
Alterations in the levels of the
proteins associated with cell death in bladder cancer cells in
response to romidepsin or trichostatin A (TSA) treatment.
| | | | Protein level
(ppm) |
|---|
| | | |
|
|---|
| Accession no. | Protein name | Symbol | Protein
function | Untreated | Romidepsin | TSA |
|---|
| Regulation of cell
cycle |
| 116176 | G2/mitotic-specific
cyclin-B1 | CCNB1 | Cyclin | 7.19 | 0 | 0 |
| 5921731 | G2/mitotic-specific
cyclin-B2 | CCNB2 | Cyclin | 7.83 | 0 | 0 |
| 218511966 | Cyclin-K | CCNK | Cyclin | 37.57 | 23.28 | 7.47 |
| 74753368 | Cyclin-L1 | CCNL1 | Cyclin | 11.84 | 6.41 | 0 |
| 9296942 | Cyclin-T1 | CCNT1 | Cyclin | 12.87 | 0 | 0 |
| 6226784 | Cyclin-dependent
kinase 10 | CDK10 | CDK | 25.95 | 0 | 0 |
| 205371737 | Anaphase-promoting
complex subunit 4 | APC4 | Mitosis factor | 15.42 | 8.36 | 5.36 |
| 37537861 | Anaphase-promoting
complex subunit 5 | APC5 | Mitosis factor | 4.12 | 0 | 0 |
| 37537762 | Cell division cycle
protein 20 homolog | CDC20 | Mitosis factor | 12.48 | 0 | 0 |
| 37537763 | Cell division cycle
protein 16 homolog | CDC16 | Mitosis factor | 20.09 | 16.33 | 0 |
| 254763423 | Cell division cycle
protein 23 homolog | CDC23 | Mitosis factor | 15.65 | 0 | 7.26 |
| 12644198 | Cell division cycle
protein 27 homolog | CDC27 | Mitosis factor | 7.56 | 0 | 0 |
| 12230256 | Mitotic spindle
assembly checkpoint protein MAD2A | MD2L1 | Mitosis factor | 45.58 | 0 | 21.14 |
| 729143 | Cyclin-dependent
kinase inhibitor 1 | CDN1A | CDK inhibitor | 0 | 20.58 | 0 |
| 3041660 | Cyclin-dependent
kinase inhibitor 2A | CD2A1 | CDK inhibitor | 59.89 | 173.11 | 55.56 |
| 172047302 | Cyclin-D1-binding
protein 1 | CCNDBP1 | CDK inhibitor | 0 | 9.38 | 12.04 |
| 1709658 |
Serine/threonine-protein kinase PLK1 | PLK1 | Positive
regulator | 30.99 | 5.60 | 0 |
| 68571766 | DNA replication
licensing factor MCM4 | MCM4 | Positive
regulator | 21.65 | 7.82 | 15.06 |
| 19858646 | DNA replication
licensing factor MCM5 | MCM5 | Positive
regulator | 67.89 | 59.79 | 59.04 |
| 76803807 | Origin recognition
complex subunit 1 | ORC1 | Positive
regulator | 7.23 | 3.92 | 5.03 |
| 6174924 | Origin recognition
complex subunit 5 | ORC5 | Positive
regulator | 21.48 | 7.76 | 19.92 |
| 25091097 | Double-strand-break
repair protein rad21 homolog | RAD21 | Positive
regulator | 167.81 | 32.10 | 27.47 |
| 13633914 | Mothers against
decapentaplegic homolog 2 | SMAD2 | Positive
regulator | 13.34 | 7.23 | 9.28 |
| 51338669 | Mothers against
decapentaplegic homolog 3 | SMAD3 | Positive
regulator | 51.29 | 7.94 | 10.20 |
| 29336622 | Structural
maintenance of hromosomes protein 1A | SMC1A | Positive
regulator | 434.44 | 177.95 | 207.35 |
| 29337005 | Structural
maintenance of chromosomes protein 3 | SMC3 | Positive
regulator | 442.71 | 183.07 | 188.71 |
| 209572720 | Cohesin subunit
SA-1 | STAG1 | Positive
regulator | 17.33 | 10.73 | 3.44 |
| 73621291 | Cohesin subunit
SA-2 | STAG2 | Positive
regulator | 43.01 | 10.97 | 0 |
| 135674 | Transforming growth
factor β-1 | TGFB1 | Positive
regulator | 23.96 | 0 | 0 |
| 132164 |
Retinoblastoma-associated protein | RB | Positive
regulator | 3.36 | 0 | 0 |
| 1345590 | 14-3-3 protein
β/α | YWHAB | Negative
regulator | 468.41 | 644.94 | 827.91 |
| 51702210 | 14-3-3 protein
ɛ | YWHAE | Negative
regulator | 708.35 | 1006.08 | 1053.59 |
| 1345593 | 14-3-3 protein
η | YWHAH | Negative
regulator | 215.22 | 343.05 | 563.68 |
| 48428721 | 14-3-3 protein
γ | YWHAG | Negative
regulator | 428.69 | 464.67 | 719.29 |
| 112690 | 14-3-3 protein
θ | YWHAQ | Negative
regulator | 483.03 | 771.58 | 937.41 |
| 52000887 | 14-3-3 protein
ζ/δ | YWHAZ | Negative
regulator | 699.13 | 771.58 | 1025.84 |
| 398953 | 14-3-3 protein
σ | SFN | Negative
regulator | 452.08 | 476.40 | 698.92 |
| Regulation of
apoptosis |
| 6094511 | Tumor necrosis
factor receptor type 1-associated DEATH domain protein | TRADD | Pro-apoptosis | 0 | 10.82 | 0 |
| 20141188 | Apoptotic
protease-activating factor 1 | APAF | Pro-apoptosis | 0 | 5.41 | 0 |
| 18203316 | Diablo homolog,
mitochondrial | DBLOH | Pro-apoptosis | 195.46 | 211.86 | 253.83 |
| 17376879 | Serine protease
HTRA2, mitochondrial | HTRA2 | Pro-apoptosis | 54.40 | 73.70 | 75.69 |
| 728945 | Apoptosis regulator
BAX | BAX | Pro-apoptosis | 97.32 | 351.63 | 203.12 |
| 2493274 | Bcl-2 homologous
antagonist/killer | BAK | Pro-apoptosis | 29.52 | 63.99 | 41.07 |
| 2493285 | BH3-interacting
domain death agonist | BID | Pro-apoptosis | 31.94 | 207.73 | 88.89 |
| 23396740 | Bcl-2-like protein
13 | B2L13 | Pro-apoptosis | 12.84 | 20.88 | 26.80 |
| 2810997 | DNA fragmentation
factor subunit α | DFFA | Pro-apoptosis | 9.41 | 40.79 | 39.27 |
| 575773389 | Serine-protein
kinase ATM | ATM | Pro-apoptosis | 1.02 | 1.10 | 2.84 |
| 77416852 | Caspase-3 | CASP3 | Caspase | 11.24 | 24.37 | 46.93 |
| 115612 | Calpain small
subunit 1 | CPNS1 |
Calpain-calcium | 81.34 | 151.15 | 129.35 |
| 62906858 | Interleukin-1
β | IL1B | Pro-survival | 46.31 | 0 | 16.11 |
| 125987833 | Interleukin-1
receptor-associated kinase-like 2 | IRAK2 | Pro-survival | 14.95 | 0 | 0 |
| 18202671 | Myeloid
differentiation primary response protein MyD88 | MYD88 | Pro-survival | 10.52 | 0 | 0 |
| 21542418 | Nuclear factor
NF-kappa-B p105 subunit | NFKB1 | Pro-survival | 9.65 | 0 | 0 |
| 125193 | cAMP-dependent
protein kinase type I-α regulatory subunit | PRKAR1A | Pro-survival | 147.13 | 8.86 | 34.12 |
| 229463042 | cAMP-dependent
protein kinase type I-β regulatory subunit | PRKAR1B | Pro-survival | 32.70 | 0 | 0 |
| 125198 | cAMP-dependent
protein kinase type II-α regulatory subunit | PRKAR2A | Pro-survival | 92.50 | 75.20 | 85.81 |
| Regulation of DNA
damage repair |
| 73921676 | DNA-(apurinic or
apyrimidinic site) lyase 2 | APEX2 | Base excision
repair | 12.02 | 0 | 0 |
| 251757259 | DNA ligase 3 | LIG3 | Base excision
repair | 37.04 | 23.42 | 17.18 |
| 317373290 | DNA repair protein
XRCC1 | XRCC1 | Base excision
repair | 49.20 | 16.00 | 41.07 |
| 130781 | Poly [ADP-ribose]
polymerase 1 | PARP1 | Base excision
repair | 380.84 | 236.36 | 311.96 |
| 17380230 | Poly [ADP-ribose]
polymerase 2 | PARP2 | Base excision
repair | 26.71 | 5.79 | 0 |
| 296453081 | DNA repair protein
complementing XP-C cells | XPC | Nucleotide excision
repair | 3.31 | 0 | 0 |
| 12643730 | DNA damage-binding
protein 1 | DDB1 | Nucleotide excision
repair | 188.50 | 109.56 | 148.24 |
| 12230033 | DNA damage-binding
protein 2 | DDB2 | Nucleotide excision
repair | 29.17 | 0 | 10.15 |
| 119541 | TFIIH basal
transcription factor complex helicase XPB subunit | ERCC3 | Nucleotide excision
repair | 31.86 | 17.27 | 11.08 |
| 17380326 | General
transcription factor IIH subunit 2 | GTF2H2 | Nucleotide excision
repair | 31.54 | 8.55 | 0 |
| 50403772 | General
transcription factor IIH subunit 3 | GTF2H3 | Nucleotide excision
repair | 30.33 | 21.92 | 14.07 |
| 17380328 | General
transcription factor IIH subunit 4 | GTF2H4 | Nucleotide excision
repair | 74.15 | 7.31 | 56.28 |
| 1706232 | Cyclin-H | CCNH | Nucleotide excision
repair | 28.93 | 10.45 | 26.83 |
| 25091548 | Pre-mRNA-splicing
factor SYF1 | XAB2 | Nucleotide excision
repair | 87.42 | 51.33 | 35.48 |
| 108936013 | Cullin-4A | CUL4A | Nucleotide excision
repair | 36.93 | 22.24 | 22.84 |
| 60392986 | DNA repair protein
RAD50 | RAD50 | Homologous
recombination | 71.21 | 51.46 | 36.33 |
| 17380137 | Double-strand break
repair protein MRE11A | MRE11A | Homologous
recombination | 39.59 | 4.77 | 24.48 |
| 74762960 | Nibrin | NBN | Homologous
recombination | 41.30 | 13.4 | 5.75 |
| 116242745 | DNA endonuclease
RBBP8 | RBBP8 | Homologous
recombination | 3.47 | 0 | 0 |
| 166898077 | Crossover junction
endonuclease MUS81 | MUS81 | Homologous
recombination | 5.65 | 0 | 0 |
| 2501242 | DNA topoisomerase
3-α | TOP3A | Homologous
recombination | 3.11 | 0 | 0 |
| 38258929 | DNA-dependent
protein kinase catalytic subunit | PRKDC | Non-homologous
end-joining | 521.31 | 295.21 | 363.21 |
| 125731 | X-ray repair
cross-complementing protein 5 | XRCC5 | Non-homologous
end-joining | 582.87 | 387.37 | 443.99 |
| 125729 | X-ray repair
cross-complementing protein 6 | XRCC6 | Non-homologous
end-joining | 772.18 | 454.52 | 619.04 |
| 74760390 | WD
repeat-containing protein 48 | WDR48 | Fanconi anemia | 4.60 | 0 | 0 |
| 48428038 | Aprataxin | APTX | Editing and
processing nuclease | 43.74 | 0 | 0 |
| 146325723 | E3
ubiquitin-protein ligase SHPRH | SHPRH | Ubiquitination and
modification | 1.85 | 0 | 0 |
| 68565701 | Telomere-associated
protein RIF1 | RIF1 | Other related | 51.65 | 15.02 | 8.76 |
| 1705919 | Dual specificity
protein kinase CLK2 | CLK2 | Other related | 12.48 | 0 | 0 |
| 55976619 | Pre-mRNA-processing
factor 19 | PRPF19 | Other related | 463.44 | 241.12 | 335.32 |
| ROS generation |
| 14916998 | Glutathione
reductase | GSHR | Reductase | 17.90 | 6.47 | 8.30 |
| 182705230 | Thioredoxin
reductase 2 | TRXR2 | Reductase | 17.83 | 0 | 0 |
| 2506326 | Xanthine
dehydrogenase/oxidase | XDH | Oxidase | 0 | 2.67 | 2.53 |
| Regulation of
autophagy |
| 254763436 | Protein kinase,
AMP-activated, α 1 catalytic subunit | PRKAA1 | Autophagy | 22.28 | 36.23 | 46.51 |
| 20178289 | Interferon, α
21 | IFNA21 | Autophagy | 0 | 17.86 | 0 |
| 74762700 |
Phosphoinositide-3-kinase, regulatory
subunit 4 | PIK3R4 | Autophagy | 0 | 4.97 | 3.19 |
| 74730233 |
Phosphatidylinositol 3-kinase, catalytic
subunit type 3 | PIK3C3 | Autophagy | 0 | 3.81 | 4.89 |
| 62286592 | Autophagy related
7 | ATG | Autophagy | 22.15 | 62.42 | 12.33 |
| 62510482 | Autophagy related
16-like 1 (S. cerevisiae) | ATG16L1 | Autophagy | 5.13 | 11.12 | 0 |
| 44888808 | GABA(A)
receptor-associated protein-like 2 | GABARAPL | Autophagy | 0 | 28.85 | 111.11 |
| 61212142 | Autophagy related
3 | ATG3 | Autophagy | 0 | 10.75 | 55.20 |
For example, our data showed that multiple
autophagy-associated proteins, such as ATG3, PRKAA1, GABARAPL and
ATG7, were highly upregulated (Table
II), suggesting that these proteins might have important roles
in HDACI-induced autophagy in bladder carcinoma.
HDACIs enhance global histone and
non-histone protein acetylation levels and induce deregulation of
chromatin modification proteins
Since both romidepsin and TSA are HDACIs, we
assessed the effect of the two HDACIs on lysine acetylation in 5637
cells. We first verified whether inhibition of histone
deacetylation by the HDACIs altered global acetylation in our model
system. We searched the whole cell proteome and identified the
non-redundant peptides containing the acetylated lysine residues.
As shown in Table III, both
romidepsin and TSA significantly increased global histone and
non-histone lysine acetylation levels compared to the untreated
control. Romidepsin induced ~2.5-fold and 2-fold increases in
histone and non-histone protein acetylation levels, respectively
(P<0.01), while TSA increased global lysine acetylation levels
63 and 50% in histone and non-histone proteins, respectively
(P<0.05), indicating that romidepsin exerts a more potent effect
than TSA on the lysine acetylated profile of both non-histone
substrates and core histones in 5637 cells.
| Table IIIHistone deacetylase inhibitors induce
enhanced global lysine acetylation in histones and non-histone
proteins in 5637 bladder cancer cells as determined by proteomic
analysis. |
Table III
Histone deacetylase inhibitors induce
enhanced global lysine acetylation in histones and non-histone
proteins in 5637 bladder cancer cells as determined by proteomic
analysis.
| Treatment | Histone
protein | Non-histone
protein |
|---|
| Untreated | 172 | 426 |
| Romidepsin | 422 | 841 |
| Trichostatin A | 280 | 638 |
Next, the overall increased lysine acetylation
levels in histone proteins prompted us to further investigate the
impact of HDACIs on site-specific histone lysine acetylation. To
this end, we applied the quantitative proteomics to profile histone
lysine acetylation in 5637 cells after romidepsin or TSA treatment,
followed by protein sequence database search for peptide
identification and post-translational modification site mapping.
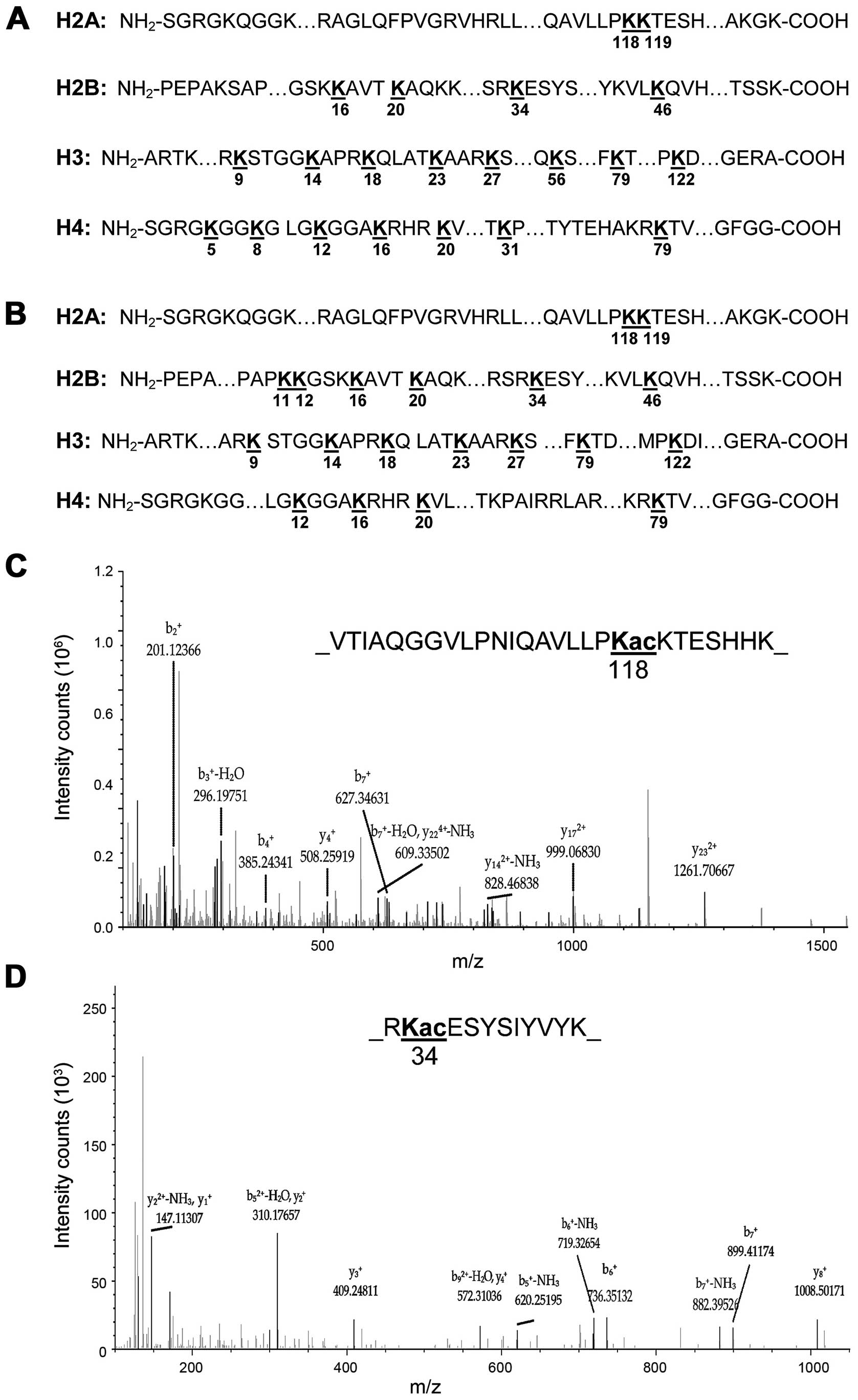
The diagram of Fig. 4 shows that a
total of 23 lysine acetylation (Kac) sites in core histones were
identified, in most of them Kac sites were previously reported in
core histones in mammalian cells. Importantly, we identified two
new histone marks, including H2AK118ac and H2BK34ac in both
romidepsin and TSA treated 5637 cells (Fig. 4A and B). The representative spectra
of histone lysine acetylated peptides are shown in Fig. 4C and D, including the spectra for
peptides of H2AK118ac and H2BK34ac. In addition, the sequences of
the identified lysine acetylated peptides in core histones in
romidepsin and TSA treated cells are listed in Table IV.
| Table IVThe identified lysine acetylation
(Kac) sites in core histones and the lysine-acetylated peptide
sequences in histone deacetylase inhibitor-treated bladder cancer
5637 cells. |
Table IV
The identified lysine acetylation
(Kac) sites in core histones and the lysine-acetylated peptide
sequences in histone deacetylase inhibitor-treated bladder cancer
5637 cells.
| Modified histone
site | Modified peptide
sequence |
|---|
| Romidepsin
treatment |
| H2AK118ac |
_VTIAQGGVLPNIQAVLLPK(ac)K(ac)TESHHK_ |
| H2AK119ac |
_VTIAQGGVLPNIQAVLLPK(ac)K(ac)_ |
| H2BK16ac |
_K(ac)AVTK(ac)AQK_ |
| H2BK20ac |
_K(ac)AVTK(ac)AQK_ |
| H2BK34ac |
_K(ac)ESYSIYVYK_ |
| H2BK46ac |
_VLK(ac)QVHPDTGISSK_ |
| H3K9ac |
_K(ac)STGGK(ac)APR_ |
| H3K14ac |
_K(ac)STGGK(ac)APR_ |
| H3K18ac |
_K(ac)QLATK(ac)AAR_ |
| H3K23ac |
_K(ac)QLATK(ac)AAR_ |
| H3K27ac |
_K(ac)SAPATGGVKKPHR_ |
| H3K56ac |
_YQK(ac)STELLIR_ |
| H3K79ac |
_EIAQDFK(ac)TDLR_ |
| H3K122ac |
_VTIMPK(ac)DIQLAR_ |
| H4K5ac |
_GK(ac)GGK(ac)GLGK_ |
| H4K8ac |
_GK(ac)GGK(ac)GLGK_ |
| H4K12ac |
_GLGK(ac)GGAK(ac)R_ |
| H4K16ac |
_GLGK(ac)GGAK(ac)R_ |
| H4K20ac |
_K(ac)VLRDNIQGITKPAIR_ |
| H4K31ac |
_DNIQGITK(ac)PAIR_ |
| H4K79ac |
_K(ac)TVTAMDVVYALKR_ |
| Trichostatin A
treatment |
| H2AK118ac |
_VTIAQGGVLPNIQAVLLPK(ac)K(ac)TESHHK_ |
| H2AK119ac |
_VTIAQGGVLPNIQAVLLPK(ac)K(ac)_ |
| H2BK11ac |
_SAPAPK(ac)K(ac)GSK_ |
| H2BK12ac |
_SAPAPK(ac)K(ac)GSK_ |
| H2BK16ac |
_K(ac)AVTK(ac)AQK_ |
| H2BK20ac |
_K(ac)AVTK(ac)AQK_ |
| H2BK34ac |
_K(ac)ESYSIYVYK_ |
| H2BK46ac |
_VLK(ac)QVHPDTGISSK_ |
| H3K9ac |
_K(ac)STGGK(ac)APR_ |
| H3K14ac |
_K(ac)STGGK(ac)APR_ |
| H3K18ac |
_K(ac)QLATK(ac)AAR_ |
| H3K23ac |
_K(ac)QLATK(ac)AAR_ |
| H3K27ac |
_K(ac)SAPATGGVKKPHR_ |
| H3K79ac |
_EIAQDFK(ac)TDLR_ |
| H3K122ac |
_VTIMPK(ac)DIQLAR_ |
| H4K12ac |
_GLGK(ac)GGAK(ac)R_ |
| H4K16ac |
_GLGK(ac)GGAK(ac)R_ |
| H4K20ac |
_K(ac)VLRDNIQGITKPAIR_ |
| H4K79ac |
_K(ac)TVTAMDVVYALKR_ |
Finally, we quantified dynamic change in global
protein abundance of the chromatin modifying proteins in
HDACI-induced bladder cancer cells. Unexpectedly, we found that the
protein levels of HDAC1, HDAC2 and HDAC3 in the deacetylation
complexes of Mi-2/NuRD, CoREST, NcoR, SMRT and Sin3 were all
downregulated in both romidepsin and TSA treated cells. As seen in
Table V, treatment with romidepsin
and TSA induced 2- and 1.7-fold downregulation for HDAC1, 3.2- and
2-fold downregulation for HDAC2, and 5.5- and 2.2-fold
downregulation for HDAC3, respectively. The levels of Sin3 histone
deacetylase corepressor complex component SDS3, a regulatory
protein that augments histone deacetylase activity of HDAC1, were
also reduced in response to exposure to romidepsin or TSA (Table V). Additionally, romidepsin and TSA
decreased the levels of histone deacetylase complex subunit SAP130
by 2.8- and 2.2-fold, respectively, in the 5637 cells. In contrast,
the levels of the lysine acetyltransferase KAT6A and histone
acetyltransferase type B catalytic subunit, the latter acetylates
histone H4 at H4K5ac and H4K12ac, were both elevated following the
HDACI induction (Table V). These
data suggest that romidepsin and TSA induced global acetylation in
core histones and non-histone proteins are mediated partly through
the elevated levels of HATs and reduced levels of HDACs in 5637
bladder cancer cells.
| Table VThe differentially expressed
chromatin modifying proteins in response to histone deacetylase
inhibitor treatment in bladder cancer 5637 cells. |
Table V
The differentially expressed
chromatin modifying proteins in response to histone deacetylase
inhibitor treatment in bladder cancer 5637 cells.
| Accession no. | Protein name | Symbol | Complex | Protein
function | Protein level
(ppm) |
|---|
|
|---|
| Untreated | Romidepsin | TSA |
|---|
| 2498443 | Histone deacetylase
1 | HDAC1 | Mi-2/NuRD; CoREST;
Sin 3 | Lysine
deacetylase | 374.75 | 189.09 | 224.75 |
| 68068066 | Histone deacetylase
2 | HDAC2 | Mi-2/NuRD; CoREST;
Sin 3 | Lysine
deacetylase | 421.20 | 131.42 | 213.11 |
| 3334210 | Histone deacetylase
3 | HDAC3 | Mi-2/NuRD;
NcoR/SMRT | Lysine
deacetylase | 87.32 | 15.77 | 40.49 |
| 74717977 | Histone deacetylase
complex subunit SAP130 | SP130 | Sin 3 | Repressor | 17.83 | 6.44 | 8.26 |
| 68053233 | Sin3 histone
deacetylase corepressor complex component SDS3 | SDS3 | Sin 3 | Corepressor | 37.98 | 0 | 26.42 |
| 3334209 | Histone
acetyltransferase type B catalytic subunit | HAT1 | KATs | Lysine
acetyltransferase | 52.02 | 48.34 | 103.42 |
| 215274095 | Histone
acetyltransferase KAT6A | KAT6A | KATs | Lysine
acetyltransferase | 1.55 | 3.37 | 2.16 |
Discussion
Although HDACIs such as romidepsin and vorinostat
(SAHA) have been approved for the treatment of CTCL, there is no
currently approved HDACI for any solid tumor indication; therefore,
we explored the potential for the development of HDACI as a novel
therapeutic for bladder urothelial carcinoma. In the present study,
we have demonstrated that romidepsin, SAHA and TSA suppressed cell
growth and caused cell death in 5637 bladder cancer cells in
vitro. Furthermore, our quantitative proteomic studies showed
that 2472 proteins were 2-fold upregulated and 2049 proteins were
2-fold downregulated in this model in response to romidepsin and
TSA exposure, among them 1082 ≥2-fold upregulated proteins and 1140
≥2-fold downregulated proteins were common to both romidepsin and
TSA treatment, as compared to the untreated controls (P<0.05).
The subsequent bioinformatic analysis revealed that those
differentially expressed proteins were mainly involved in
biological and metabolic functions and cell death associated
pathways. HDACI exposure also enhanced global acetylation levels in
both histone and non-histone proteins. Twenty-three lysine
acetylation marks were detected on core histones in HDACI-treated
bladder cancer cells including two newly identified histone Kac
sites (H2AK118ac and H2BK34ac). These data suggest that
HDACI-induced alterations in protein expression is mediated, at
least in part, through histone modification, leading to changes in
biological and metabolic functions and cell death in bladder cancer
cells. By establishing the link between histone modification and
whole proteome in response to HDACI treatment, this study may
deepen our understanding of HDACI-mediated therapeutics in bladder
cancer.
A major goal of the chemotherapy of human
malignancies is the inhibition of cell proliferation, and
drug-induced cancer cell growth arrest is mediated partly by
blocking cell cycle progression (23). The eukaryotic cell cycle is
regulated via the sequential activation and inactivation of
cyclin-dependent kinases (CDKs) that drive cell cycle progression
through the phosphorylation and dephosphorylation of regulatory
proteins (24–26). The activities of CDKs are
positively regulated by cyclins and negatively regulated by CDK
inhibitors (CKIs). Thus, the cell cycle is regulated by cyclins,
CDKs and CKIs. Changes in the expression of specific CDKs or their
regulatory proteins such as cyclins and CKIs can lead to
uncontrolled cell proliferation and eventually to carcinogenesis
(25,27). Whereas, downregulating the levels
of cyclins or upregulating CKIs lead to blockade of cell cycle
progression.
In this study, we showed that romidepsin and TSA
downregulated the protein expression of cyclins B1/B2 and
upregulated the expression of anaphase promoting complex-1 (APC1)
and 14-3-3 proteins in 5637 cells. Cyclin B binds to and activates
CDK1. The complex of cyclin B and CDK1 is responsible for the
control of G2/M checkpoint, while APC1 acts by mediating
ubiquitination and degradation of cyclin B and subsequent
inactivation of CDK1. On the other hand, CDK1 activity is
suppressed via phosphorylation of Thr-14 and Tyr-15 by the Wee-1
protein kinase (28) and is
activated by CDC25 protein phosphatases, which function to remove
the inhibitory phosphates from CDK1 (29). 14-3-3 proteins are involved in the
regulation of G2/M checkpoint by 14-3-3-mediated CDC25 inactivation
and Wee-1 activation. Romidepsin and TSA caused reduced levels of
cyclin B and elevated levels of APC1 and 14-3-3 proteins in 5637
cells, suggesting that romidepsin and TSA suppress bladder cancer
cell proliferation through cell cycle blockade at the G2/M phase,
and that this occurs via the HDACI downregulation of cyclin B and
upregulation of APC1 and 14-3-3 proteins, leading to cell cycle
arrest and cell growth suppression in bladder cancer cells.
The other goal of cancer chemotherapy is to commit
tumor cells to death or apoptosis following exposure to anticancer
agents. Apoptosis is a highly regulated cellular process between
cell proliferation and cell death and drug-induced cell death is
mediated, at least in part, by apoptotic cell death (30).
In the present study, we found that the levels of
caspase-3 were significantly increased in 5637 cells following
treatment with romidepsin or TSA. In addition, both romidepsin and
TSA enhanced Bax and Bak expression and triggered phosphorylation
of Bcl-2 at Ser-70. It is known that the expression of
pro-apoptotic proteins is mediated through p53-dependent and
-independent pathways. In this study, we showed that the levels of
p53 protein as well as other p53-pathway proteins, such as
DNA-dependent protein kinase (PRKDC), were not elevated in response
to HDACI exposure (Fig. 3A), and
we confirmed by DNA sequencing that p53 gene is mutated in this
cell line (data not shown), suggesting that the increased
expression of apoptosis-associated proteins is not under direct
control by p53 in 5637 bladder carcinoma cells. Additionally, it
has been shown that phosphorylation of Bcl-2 is induced by several
drugs in a panel of cancer cell lines derived from leukemia,
lymphoma, and breast and prostate cancer (31–35).
Phosphorylation of Bcl-2 is cell cycle-dependent, occurs at G2/M
(34) and results in concomitant
apoptosis (34,36). Interestingly, treatment with HDACIs
was found to induce Bcl-2 Ser-70 phosphorylation at the G2/M phase
of the cell cycle, with concomitant apoptosis in bladder cancer
cells. This is consistent with the literature reporting that Bcl-2
phosphorylation at Ser-70 and loss of anti-apoptotic function in
response to antitumor drugs and subsequent elimination of tumor
cells via apoptosis (35). These
results suggest that a similar mechanism (Bcl-2 phosphorylation at
G2/M) may be involved with induction of apoptosis by HDACIs in this
model system. Taken together, these data strongly support the
involvement of Bcl-2 family proteins in HDACI-induced apoptosis,
possibly acting through a p53-independent, mitochondria-dependent
intrinsic apoptotic pathway in human bladder cancer 5637 cells.
A wide range of DNA damage can be inflicted, both
from extracellular agents including some antitumor drugs and via
endogenous mechanisms (37).
Genotoxic cancer therapeutics such as cisplatin and mitomycin C
bind to DNA, forming adducts that in turn can be repaired by the
DNA repair machinery or lead to permanent DNA damage. Anticancer
agent-induced DNA damage leads to transient arrest in the G1, S, G2
and M phases of the cell cycle, allowing cells to have sufficient
time to repair damaged DNA before resuming cell cycle progression.
However, severe DNA injury that is too extensive for intracellular
repair mechanisms will lead to activation of intrinsic apoptosis
pathway and cell death. Although DNA damage also affects normal
cells, tumor cells are often more vulnerable because of defects in
DNA repair pathways or critical cell cycle checkpoints.
Five main mechanisms are involved in DNA repair: i)
base excision repair, which corrects non-bulky damage; ii)
nucleotide excision repair, which corrects lesions that disrupt the
double helical structure of DNA; iii) mismatch repair, which
corrects replication errors; iv) double-strand break repair, which
corrects double-strand breaks through two different pathways,
homologous recombination and non-homologous end-joining; and v)
direct repair, which corrects methylated or alkylated bases
(38). Although the DNA lesions
induced directly by HDACIs or indirectly via endogenous mechanisms
such as the generation of free radicals, as well as the relevant
DNA repair mechanisms responsible for the removal of those lesions
in 5637 cells are still not known, our proteomic analysis revealed
that the levels of multiple DNA repair proteins in multiple repair
mechanisms were decreased in HDACI-treated bladder cancer cells.
For example, the protein expression of XRCC1 and PARP1/2 in base
excision repair, XPC and ERCC3 in nucleotide excision repair, RAD50
and MRE11A in homologous recombination, and XRCC5 and XRCC6 in
non-homologous end-joining were all reduced after romidepsin and
TSA treatment. The downregulation of DNA repair protein expression
by HDACIs significantly impairs cellular DNA repair activity and
DNA damage response, which in turn results in inhibition of
transcription, replication, and chromosome segregation leading to
blockade of cell cycle progression or apoptosis in bladder
carcinoma cells.
Studies suggest oxidative stress as a mechanism to
the primary modes of action of antitumor agents. Oxidative stress
is a redox (reduction-oxidation) disequilibrium state, in which the
generation of ROS overwhelms the antioxidant defense mechanisms
(39). ROS such as superoxide and
hydroxyl radicals are highly toxic, as a result of their actions as
oxidizing agents and can have damaging effects on cell physiology.
Under conditions that can cause oxidative stress, cells are exposed
to excessive ROS that can oxidize membrane fatty acids, initiating
lipid peroxidation, oxidize proteins (40) and cause DNA damage (41). At high level, excessive ROS may
cause severe damage to cells, including necrosis and apoptosis
(42).
Redox state in the cell is regulated by redox
proteins. In this study, we found that romidepsin and TSA
downregulated the expression of glutathione reductase (GSHR) and
thioredoxin reductase 2 (TRXR2) and upregulated the expression of
xanthine dehydrogenase/oxidase (XDH), which lead to ROS formation
in 5637 cells. These findings suggest that oxidative stress is
involved in the antitumor effects of HDACIs in bladder cancer and
that exposure to HDACIs may alter the antioxidant defense system
and redox mechanisms in cells. This notion is supported by the
reports from other researchers demonstrating that HDACIs induce
cell death through ROS production (43).
Although the mechanism for the link between
HDACI-induced oxidative stress and cell death is not well
understood, several lines of evidence suggest that HDACIs induce
cell death via ROS generation by the following mechanisms: i)
excess of ROS may facilitate the detachment of cytochrome c
from the mitochondrial membrane and increases the mobilized pool of
cytochrome c, which is a prerequisite for its release into
the cytoplasm through the pores created by pro-apoptotic Bcl-2
family members such as Bax and Bak; ii) ROS may also directly
damage mitochondrial membrane and induce membrane potential loss
that favors cytochrome c release; iii) death receptor
aggregation may also result from ROS production and induce cell
death through a different pathway; iv) downregulation of
anti-apoptotic molecules and/or upregulation of pro-apoptotic
signals are involved in ROS-induced cell death (44); and v) HDACI-induced ROS causes
oxidative DNA damage (43), as
evidenced by the levels of phosphorylated histone H2AX (γ-H2AX) and
ataxia telangiectasia mutated (ATM), early markers of DNA damage,
significantly increase after the administration of HDACIs (45,46).
Cellular oxidative DNA damage induced by endogenous ROS production
via HDACI treatment can lead to bladder cancer cell death (47).
Combining our results discussed above, we propose a
possible mechanism by which HDACIs cause bladder cancer cell growth
arrest and cell death as shown in Fig.
5. In this model, HDACIs alter changes in the levels and
activities of proteins involved in the signaling pathways of cell
cycle progression, apoptotic cell death, DNA damage repair, ROS
generation, endoplasmic reticulum (ER) stress (48–50)
and autophagy regulation (51),
which are associated with cell death. In the proposed pathways
depicted here (Fig. 5), romidepsin
and TSA induce cell cycle arrest and apoptotic cancer cell death;
cell cycle blockade not only causes cancer cell growth arrest, but
prolonged cell cycle arrest also triggers cell suicide, usually in
the form of apoptosis. In addition, the HDACIs increase DNA damage
directly or indirectly through ROS production, which in turn
promotes apoptosis. On the other hand, romidepsin and TSA mediate
cancer cell death via inducing ER stress and autophagy. Because
HDACIs target cell survival and cell death through multiple closely
related but distinct mechanisms, they may act collaboratively or
synergistically to promote apoptotic death of bladder cancer cells
through these signaling pathways and their downstream molecular
events.
Finally, our data indicate that dysregulation of
protein expression in HDACI-treated 5637 cells was associated with
enhanced lysine acetylation in histone and non-histone proteins as
well as alterations in the levels of chromatin modifying proteins,
suggesting a role for epigenetic modification.
The three main epigenetic mechanisms (DNA
methyltion, histone modifications and RNA-mediated gene silencing)
have been studied primarily in the context of gene expression
(52,53). The second epigenetic mechanism
encompasses various histone modifications, including acetylation,
glycosylation, methylation, phosphorylation and ubiquitination of
specific residues in the N-terminal tails of histones (54). Histone modifications are
post-translational alterations of histone proteins that interact
with DNA to form a complex known as chromatin (54). Besides regulating several cellular
processes including gene transcription, proliferation, and
autophagy, histone modifications also affect many other
chromatin-based processes such as DNA repair, replication and
recombination (54).
The best-studied histone modification is lysine
acetylation. The acetylation of histone modulates transcription by
altering the accessibility of DNA to proteins, such as
transcriptional regulators (transcriptional activators and
repressors), and binding of regulatory proteins (transcription
factors or repressors) to the promoter sequence of a gene resulting
in activation or blocking of transcription. Furthermore, the
activity of non-histone proteins, such as transcription factors and
repressors, can also be modulated by post-translational protein
modifications (e.g., acetylation, phosphorylation or
glycosylation), and these modifications could change protein
conformation and lead to changes in activity.
As the variety of gene expression profiles is
determined by distinct sets of transcriptional regulators
(transcription factors or repressors) that control and determine
which genes are switched on or off, HDACIs may upregulate or
downregulate gene expression via altering the activity of
transcription factors or repressors by post-translational
modifications (PTMs) in our bladder tumor cells. Additionally,
since the majority of cellular functions are carried out by
proteins, HDACIs may modulate biological changes not only through
alterations at the protein level but also by PTMs in bladder
carcinoma cells. However, the role of the two newly identified
histone markers (H2AK118ac and H2BK34ac) in the antitumor activity
of HDACIs in bladder cancer, as well as the precise mechanism for
how HDACIs upregulate or downregulate specific gene and protein
expression through histone modifications and PTMs will require
further investigation.
In summary, we have profiled the antitumor activity
of HDACIs in association with enhanced lysine acetylation in
histone and non-histone proteins as well as altered levels of
chromatin modifying proteins in bladder cancer cells. Proteomic
data analysis further revealed dysregulation of protein expression
involved in multiple biological functions and cell death associated
pathways in romidepsin and TSA treated 5637 cells. These results
suggest that the antitumor effect of HDACIs in bladder carcinoma is
mediated through modulation of these pathways by histone
modifications and PTMs, leading to cancer cell growth arrest and
cell death. Our findings may be helpful for developing HDCAIs in
combination with other therapeutics targeted at modulating relevant
cell death pathways or at inhibiting cell proliferation in tumors.
Further studies are needed to investigate the anticancer activity
of HDACIs in bladder cancer via the modulation of signaling
pathways (e.g., PI3K-PTEN-mTOR pathway) (55) or the inhibition of regulatory
enzymes in histone modifications and PTMs (56) influencing cell survival and
death.
Acknowledgements
The present study was supported by the Intramural
Research Program of the U.S. National Cancer Institute, the
National Institutes of Health.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaufman DS, Shipley WU and Feldman AS:
Bladder cancer. Lancet. 374:239–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weintraub MD, Li QQ and Agarwal PK:
Advances in intravesical therapy for the treatment of non-muscle
invasive bladder cancer (Review). Mol Clin Oncol. 2:656–660.
2014.PubMed/NCBI
|
|
4
|
Stenzl A, Cowan NC, De Santis M, Kuczyk
MA, Merseburger AS, Ribal MJ, Sherif A and Witjes JA; European
Association of Urology (EAU). Treatment of muscle-invasive and
metastatic bladder cancer: Update of the EAU guidelines. Eur Urol.
59:1009–1018. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marks P, Rifkind RA, Richon VM, Breslow R,
Miller T and Kelly WK: Histone deacetylases and cancer: Causes and
therapies. Nat Rev Cancer. 1:194–202. 2001. View Article : Google Scholar
|
|
6
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: Molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marks PA and Xu WS: Histone deacetylase
inhibitors: Potential in cancer therapy. J Cell Biochem.
107:600–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schrump DS: Cytotoxicity mediated by
histone deacetylase inhibitors in cancer cells: Mechanisms and
potential clinical implications. Clin Cancer Res. 15:3947–3957.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakagawa M, Oda Y, Eguchi T, Aishima S,
Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, et al:
Expression profile of class I histone deacetylases in human cancer
tissues. Oncol Rep. 18:769–774. 2007.PubMed/NCBI
|
|
10
|
Zhang Z, Yamashita H, Toyama T, Sugiura H,
Ando Y, Mita K, Hamaguchi M, Hara Y, Kobayashi S and Iwase H:
Quantitation of HDAC1 mRNA expression in invasive carcinoma of the
breast. Breast Cancer Res Treat. 94:11–16. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marquard L, Poulsen CB, Gjerdrum LM, de
Nully Brown P, Christensen IJ, Jensen PB, Sehested M, Johansen P
and Ralfkiaer E: Histone deacetylase 1, 2, 6 and acetylated histone
H4 in B- and T-cell lymphomas. Histopathology. 54:688–698. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Witt O, Deubzer HE, Milde T and Oehme I:
HDAC family: What are the cancer relevant targets? Cancer Lett.
277:8–21. 2009. View Article : Google Scholar
|
|
13
|
Ozawa A, Tanji N, Kikugawa T, Sasaki T,
Yanagihara Y, Miura N and Yokoyama M: Inhibition of bladder tumour
growth by histone deacetylase inhibitor. BJU Int. 105:1181–1186.
2010. View Article : Google Scholar
|
|
14
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoshida M, Kijima M, Akita M and Beppu T:
Potent and specific inhibition of mammalian histone deacetylase
both in vivo and in vitro by trichostatin A. J Biol Chem.
265:17174–17179. 1990.PubMed/NCBI
|
|
17
|
Tan J, Cang S, Ma Y, Petrillo RL and Liu
D: Novel histone deacetylase inhibitors in clinical trials as
anti-cancer agents. J Hematol Oncol. 3:52010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McGraw AL: Romidepsin for the treatment of
T-cell lymphomas. Am J Health Syst Pharm. 70:1115–1122. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li QQ, Wang G, Liang H, Li JM, Huang F,
Agarwal PK, Zhong Y and Reed E: β-Elemene promotes
cisplatin-induced cell death in human bladder cancer and other
carcinomas. Anticancer Res. 33:1421–1428. 2013.PubMed/NCBI
|
|
20
|
Florens L, Carozza MJ, Swanson SK,
Fournier M, Coleman MK, Workman JL and Washburn MP: Analyzing
chromatin remodeling complexes using shotgun proteomics and
normalized spectral abundance factors. Methods. 40:303–311. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Paoletti AC, Parmely TJ, Tomomori-Sato C,
Sato S, Zhu D, Conaway RC, Conaway JW, Florens L and Washburn MP:
Quantitative proteomic analysis of distinct mammalian Mediator
complexes using normalized spectral abundance factors. Proc Natl
Acad Sci USA. 103:18928–18933. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ou JN, Torrisani J, Unterberger A,
Provençal N, Shikimi K, Karimi M, Ekström TJ and Szyf M: Histone
deacetylase inhibitor Trichostatin A induces global and
gene-specific DNA demethylation in human cancer cell lines. Biochem
Pharmacol. 73:1297–1307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Swanton C: Cell-cycle targeted therapies.
Lancet Oncol. 5:27–36. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dynlacht BD: Regulation of transcription
by proteins that control the cell cycle. Nature. 389:149–152. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wesierska-Gadek J, Gueorguieva M and Horky
M: Dual action of cyclin-dependent kinase inhibitors: Induction of
cell cycle arrest and apoptosis. A comparison of the effects
exerted by roscovitine and cisplatin. Pol J Pharmacol. 55:895–902.
2003.
|
|
28
|
Parker LL, Sylvestre PJ, Byrnes MJ III,
Liu F and Piwnica-Worms H: Identification of a 95-kDa WEE1-like
tyrosine kinase in HeLa cells. Proc Natl Acad Sci USA.
92:9638–9642. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mitra J and Enders GH: Cyclin A/Cdk2
complexes regulate activation of Cdk1 and Cdc25 phosphatases in
human cells. Oncogene. 23:3361–3367. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fisher DE: Apoptosis in cancer therapy:
Crossing the threshold. Cell. 78:539–542. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Haldar S, Chintapalli J and Croce CM:
Taxol induces bcl-2 phosphorylation and death of prostate cancer
cells. Cancer Res. 56:1253–1255. 1996.PubMed/NCBI
|
|
32
|
Blagosklonny MV, Schulte T, Nguyen P,
Trepel J and Neckers LM: Taxol-induced apoptosis and
phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a
novel c-Raf-1 signal transduction pathway. Cancer Res.
56:1851–1854. 1996.PubMed/NCBI
|
|
33
|
Blagosklonny MV, Giannakakou P, el-Deiry
WS, Kingston DG, Higgs PI, Neckers L and Fojo T: Raf-1/bcl-2
phosphorylation: A step from microtubule damage to cell death.
Cancer Res. 57:130–135. 1997.PubMed/NCBI
|
|
34
|
Haldar S, Basu A and Croce CM: Bcl2 is the
guardian of microtubule integrity. Cancer Res. 57:229–233.
1997.PubMed/NCBI
|
|
35
|
Haldar S, Basu A and Croce CM: Serine-70
is one of the critical sites for drug-induced Bcl2 phosphorylation
in cancer cells. Cancer Res. 58:1609–1615. 1998.PubMed/NCBI
|
|
36
|
Chang BS, Minn AJ, Muchmore SW, Fesik SW
and Thompson CB: Identification of a novel regulatory domain in
Bcl-X(L) and Bcl-2. EMBO J. 16:968–977. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Roos WP, Thomas AD and Kaina B: DNA damage
and the balance between survival and death in cancer biology. Nat
Rev Cancer. 16:20–33. 2016. View Article : Google Scholar
|
|
38
|
Friedberg EC, Walker GC, Siede W and
Schultz RA: DNA Repair and Mutagenesis. 2nd edition. ASM Press;
Washington, DC: 2006
|
|
39
|
Halliwell B: Free radicals, antioxidants,
and human disease: Curiosity, cause, or consequence? Lancet.
344:721–724. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brot N, Weissbach L, Werth J and Weissbach
H: Enzymatic reduction of protein-bound methionine sulfoxide. Proc
Natl Acad Sci USA. 78:2155–2158. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Demple B and Linn S: 5,6-Saturated thymine
lesions in DNA: Production by ultraviolet light or hydrogen
peroxide. Nucleic Acids Res. 10:3781–3789. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xia T, Kovochich M, Brant J, Hotze M,
Sempf J, Oberley T, Sioutas C, Yeh JI, Wiesner MR and Nel AE:
Comparison of the abilities of ambient and manufactured
nanoparticles to induce cellular toxicity according to an oxidative
stress paradigm. Nano Lett. 6:1794–1807. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Feng R, Oton A, Mapara MY, Anderson G,
Belani C and Lentzsch S: The histone deacetylase inhibitor, PXD101,
potentiates bortezomib-induced anti-multiple myeloma effect by
induction of oxidative stress and DNA damage. Br J Haematol.
139:385–397. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rosato RR, Maggio SC, Almenara JA, Payne
SG, Atadja P, Spiegel S, Dent P and Grant S: The histone
deacetylase inhibitor LAQ824 induces human leukemia cell death
through a process involving XIAP down-regulation, oxidative injury,
and the acid sphingomyelinase-dependent generation of ceramide. Mol
Pharmacol. 69:216–225. 2006.
|
|
45
|
Feng R, Ma H, Hassig CA, Payne JE, Smith
ND, Mapara MY, Hager JH and Lentzsch S: KD5170, a novel
mercaptoketone-based histone deacetylase inhibitor, exerts
antimyeloma effects by DNA damage and mitochondrial signaling. Mol
Cancer Ther. 7:1494–1505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rosato RR, Almenara JA, Maggio SC, Coe S,
Atadja P, Dent P and Grant S: Role of histone deacetylase
inhibitor-induced reactive oxygen species and DNA damage in
LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther.
7:3285–3297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Mitochondrial oxidative stress: Implications for cell death. Annu
Rev Pharmacol Toxicol. 47:143–183. 2007. View Article : Google Scholar
|
|
48
|
Rao R, Nalluri S, Fiskus W, Savoie A,
Buckley KM, Ha K, Balusu R, Joshi A, Coothankandaswamy V, Tao J, et
al: Role of CAAT/enhancer binding protein homologous protein in
panobinostat-mediated potentiation of bortezomib-induced lethal
endoplasmic reticulum stress in mantle cell lymphoma cells. Clin
Cancer Res. 16:4742–4754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rao R, Nalluri S, Kolhe R, Yang Y, Fiskus
W, Chen J, Ha K, Buckley KM, Balusu R, Coothankandaswamy V, et al:
Treatment with panobinostat induces glucose-regulated protein 78
acetylation and endoplasmic reticulum stress in breast cancer
cells. Mol Cancer Ther. 9:942–952. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kahali S, Sarcar B, Prabhu A, Seto E and
Chinnaiyan P: Class I histone deacetylases localize to the
endoplasmic reticulum and modulate the unfolded protein response.
FASEB J. 26:2437–2445. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J,
Ma C, Sun Y, Zhang S, Feng W, et al: Function and molecular
mechanism of acetylation in autophagy regulation. Science.
336:474–477. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hassler MR and Egger G: Epigenomics of
cancer - emerging new concepts. Biochimie. 94:2219–2230. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jerónimo C and Henrique R: Epigenetic
biomarkers in urological tumors: A systematic review. Cancer Lett.
342:264–274. 2014. View Article : Google Scholar
|
|
54
|
Bannister AJ and Kouzarides T: Regulation
of chromatin by histone modifications. Cell Res. 21:381–395. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Abbosh PH, McConkey DJ and Plimack ER:
Targeting signaling transduction pathways in bladder cancer. Curr
Oncol Rep. 17:582015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
O'Rourke CJ, Knabben V, Bolton E, Moran D,
Lynch T, Hollywood D and Perry AS: Manipulating the epigenome for
the treatment of urological malignancies. Pharmacol Ther.
138:185–196. 2013. View Article : Google Scholar : PubMed/NCBI
|