Introduction
Gastric cancer, also known as stomach cancer, is
cancer developing from the lining of the stomach (1). Cancer may spread from the stomach to
other parts of the body, particularly the liver, lungs, bones,
lining of the abdomen and lymph nodes (2). Globally stomach cancer is the fifth
leading cause of cancer and the third leading cause of death from
cancer making up 7% of cases and 9% of deaths. Gastric cancer is a
multifactorial disease (3,4). The pathogenesis of gastric cancer
represents a classic example of gene-environment interactions.
Cancer of the stomach is difficult to cure unless it is found at an
early stage before it has begun to spread. Because early stomach
cancer causes few symptoms, the disease is usually advanced when
the diagnosis is made (5,6). Thus, currently finding effective
target for therapies is necessary to patients.
MiRNAs are one family of small (about 22
nucleotides), non-coding RNAs which play an important role in
regulating diverse biological processes, including apoptosis,
proliferation, development, differentiation and metabolism
(7). Increasing evidence suggest
that some miRNAs perform as either tumor suppressors or oncogenes
in the tumorigenesis of many types of cancer, and have been
reported as potential biomarkers for tumor diagnosis, prognosis and
therapy (8,9). Altered miRNA expression in gastric
cancer has been found in many different studies. Several
deregulated miRNAs in gastric cancer such as miR-185, miR-107,
miR-210, miR-99, miR-155 and miR-95 have been shown to regulate
cell growth, apoptosis, migration and invasion (10–14).
These findings suggest that deregulation of miRNA may be associated
with tumorigenesis of gastric cancer. In addition, more extensive
investigations are needed to reveal the role of miRNAs in the
gastric cancer progression, and those miRNAs may be involved as
novel biomarkers for gastric cancer diagnosis, therapy and
prognosis. miR-224 is significantly deregulated in many types of
cancer, including lung, breast and colon cancer. Since deregulation
of miR-224 is common to a number of cancers, it has been supposed
that miR-224 may play an essential role in tumor development as
well as tumorigenesis (15,16).
However, the effects and function of miR-224 remains unclear,
especially in gastric cancer.
In the present study, we evaluated miR-224
expression levels in tumor tissues of patients with gastric cancer
and we found that miR-224 was significantly upregulated in gastric
cancer. We proved that miR-224 could promote gastric cancer cell
proliferation and downregulate apoptosis in vitro and in
vivo experiments. Furthermore, mTOR, which is usually
overexpressed in a variety of human cancers including lung, breast
and colon cancer, was identified as a direct target of miR-224. Our
results will be helpful for demonstrating the functions of miR-224
and its role in gastric cancer tumorigenesis.
Materials and methods
Tissue specimens and cell cultures
Human gastric cancer and the adjacent normal
non-tumor tissues were acquired from patients, undergoing surgical
resection in the Department of Gastric Cancer and Soft Tissue
Sarcomas, Nanjing Medical Cancer Center of Nanjing Medical
University, Nanjing, China, from January 2010 to December 2012 for
quantitative RT-PCR analysis. All tissue samples were immediately
frozen in the liquid nitrogen and then stored at −80°C for the
following studies.
Paraffin-embedded tumor tissues collected from
consecutive patients with gastric cancer between January 2010 and
December 2012 were performed for tissue assays. Clinical data
collection and postoperative follow-up procedures were done based
on a uniform guideline of the Nanjing Cancer Center of Nanjing
Medical University. Patient characteristics of clinicopathological
features are shown in Table I. All
samples were collected and analyzed with a prior written informed
consent, which was obtained from all patients, and the study was
approved by the Clinical Research Ethics Committee of Nanjing
Medical University Nanjing Cancer Center.
 | Table IClinicopathological characteristics
of gastric cancer patients. |
Table I
Clinicopathological characteristics
of gastric cancer patients.
| miR-224
expression | |
|---|
|
| |
|---|
| Low | High | |
|---|
|
|
| |
|---|
|
Characteristics | Patients n (%) | Patients n (%) | P-value |
|---|
| Total | 72 (45.0) | 88 (55.0) | |
| Age (years) | | | 0.136 |
| <60 | 45 (62.5) | 53 (60.2) | |
| ≥60 | 27 (37.5) | 35 (39.8) | |
| Gender | | | 0.121 |
| Female | 31 (43.1) | 18 (20.5) | |
| Male | 41 (56.9) | 70 (79.5) | |
| Site | | | 0.086 |
| Cardia | 12 (16.7) | 17 (19.3) | |
| Body | 36 (50.0) | 49 (55.7) | |
| Antrum | 0 (0.0) | 3 (3.4) | |
| Pylus | 5 (6.9) | 0 (0) | |
| Fundus | 19 (26.4) | 19 (21.6) | |
| Differentiatial
status | | | 0.534 |
|
Undifferentiated/poorly | 60 (83.3) | 79 (89.8) | |
| Moderate/well | 12 (16.7) | 9 (10.2) | |
| Nerve invasion | | | 0.121 |
| No | 39 (54.2) | 52 (59.1) | |
| Yes | 33 (45.8) | 36 (40.9) | |
| Vascular
invasion | | | 0.325 |
| No | 36 (50.0) | 54 (61.4) | |
| Yes | 36 (50.0) | 34 (38.6) | |
| T | | | 0.018 |
| T1/2/3 | 13 (18.1) | 20 (22.7) | |
| T4 | 59 (81.9) | 68 (77.3) | |
| N | | | 0.038 |
| N0/1/2 | 43 (59.7) | 55 (62.5) | |
| N3 | 27 (40.3) | 33 (37.5) | |
| M | | | 0.265 |
| M0 | 62 (86.1) | 77 (87.5) | |
| M1 | 10 (13.9) | 11 (12.5) | |
| Stage | | | 0.061 |
| I/II | 18 (25.0) | 30 (34.1) | |
| III/IV | 54 (75.0) | 58 (65.9) | |
Cell culture
Human gastric cancer cell lines, including GES-1,
HS-746T, MKN-45, HGC-27, SNU-5 and BGC-823, were purchased from the
American Type Culture Collection, the Cell Resource Center,
Shanghai Institute of Biochemistry and Cell Bank at the Chinese
Academy of Sciences. Cell lines were routinely authenticated by
DNA-fingerprinting and isoenzyme analyses and checked for
contamination by mycoplasma using Hoechst staining. All cell lines
were maintained in Roswell Park Memorial Institute (RPMI)-1640,
Dulbecco’s modified Eagle’s medium or Minimum Essential Medium,
containing 10% fetal bovine serum (FBS) and were incubated at 37°C
with 5% CO2.
RNA isolation, reverse transcription (RT)
and real-time PCR (RT-PCR)
Total RNA from tissue samples and cultured cells was
isolated through the mirVana miRNA Isolation kit (Ambion, Inc.,
Austin, TX, USA) based on the manufacturer’s instruction. Then the
cDNA was synthesized from total RNA with the TaqMan miRNA reverse
transcription kit (Applied Biosystems, Foster City, CA, USA).
Real-time PCR was conducted using the Applied Biosystems 7500
Sequence Detection system with iQ™ SYBR-Green Supermix (Bio-Rad
Laboratories, Hercules, CA, USA) containing 5 ng cDNA and 10 pM of
each primer. The data were normalized to the geometric mean of
housekeeping gene GAPDH or U6 small nuclear RNA expression and
calculated as 2−ΔΔCT method. Sequences of the primers
are summarized in Table II.
| Table IIPrimer sequences of RT-PCR
analysis. |
Table II
Primer sequences of RT-PCR
analysis.
| Gene | Forward primers
(5′-3′) | Reverse primers
(5′-3′) |
|---|
| GAPDH |
CATTCAAGACCGGACAGAGG |
ACATACTCAGCACCAGCATCACC |
| mTOR |
AUUGGAACCAAUGCAUGAC |
CACGACTTCGTCCTCCGGAC |
| Bcl-2 |
AGCACAGAAAGTGAGCTGC |
AGTCGTCCAGTTGGTGTA |
| Bax |
GATGTAGAGACAGGAACCC |
GGTCCATCGAGTGATGTTGTG |
| Caspase-9 |
GCAGCAGGTGAGTGGGCAGT |
ACTGTCGCCTGGTTCTCTGTGC |
| Caspase-3 |
CAGTTAGGAGACGACAG |
AGCGTCGCTGGATGTGTGA |
| PCNA |
CACCTTCTTGTCGACCGCCTA |
TCCGCGTCTGTTCGGCAT |
| p21 |
TCCTGGGACTCTTCTTATTTACCA |
TTGCCTGCTAAAGGCAATTACC |
| p27 |
GAGCAAGCAAGATTTACTCGA |
AGCCAGCTACATGGATCTAAA |
| Cyclin D1 |
GTCTGGTCGATTACCCTGG |
TCTTGTCTAAACAGA |
| Cyclin D2 |
GAGTGCCGGTGAGCGA |
TCCGCAAGAACCTGG |
| U6 |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT |
| mTOR (Mut) |
AUUGAGGUCAAUGCAUGACUA |
GACTCTTCTTATTTAACATG |
| miR-224 |
UUGCCUUGGUGGCGCC |
AUGACUGAACUGCAGGT |
Plasmids and transfection
In order to produce a miR-224 expression vector, a
281-bp genomic fragment which covers the region coding for
pri-miR-224 and its upstream and downstream regions was PCR
amplified and then cloned onto the pLVTHM vector (Addgene,
Cambridge, MA, USA). The full length of mTOR 3′-UTR is 4739 bp. The
miR-224 binding site in mTOR 3′-UTR is located at 4375 to 4381 bp.
The region of the human mTOR 3′-UTR from 4553 to 4761 bp was
generated by PCR amplification and subcloned into the sites of
pGL3-basic luciferase reporter plasmid (Promega, Madison, WI, USA).
The miR-224 mimics, negative control and anti-miR-224 inhibitors
were purchased from GeneCopoeia (Rockville, MD, USA) and then
transfected into gastric cancer cells with Lipofectamine 2000
reagent (Invitrogen, Carlsbad, CA, USA), based on the
manufacturer’s instructions. All the primers are listed in Table II.
Luciferase reporter assays
The cells, seeded in a 48-well plate, were
co-transfected with 50 nM single-stranded miRNA mimics,
anti-miR-224 inhibitors or negative control oligonucleotides, 50 ng
of firefly luciferase reporter and 10 ng of pRL-TK (Promega) with
the jetPRIME reagent. Cells were collected 36 h after the final
transfection, and then analyzed via Dual-luciferase reporter assay
system (Promega).
Apoptosis analysis by terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling
(TUNEL)
Apoptosis assay of samples was determined by TUNEL
using an In Situ Cell Death Detection kit, Fluorescein (Roche
Applied Science, Indianapolis, IN, USA) according to the
manufacturer’s protocol. The number of TUNEL-positive cells was
counted under a fluorescence microscope. The percentages of
apoptotic cells were calculated from the ratio of apoptotic cells
to total cells counted. Tissue sections were counterstained with
hematoxylin. Sections were mounted and observed by light
microscopy. The experiment was performed independently three times
for each cell line.
Western blot analysis
Proteins were extracted using T-PER Tissue Protein
Extraction reagent kit (Thermo Fisher Scientific, Waltham, MA, USA)
according to the manufacturer’s instructions. Protein
concentrations were determined by BCA protein assay kit, and equal
amounts of protein were loaded per well on a 10% sodium dodecyl
sulphate poly-acrylamide gel. Subsequently, proteins were
transferred onto polyvinylidene difluoride membrane. The resulting
membrane was blocked with Tris-buffered saline containing 0.05%
Tween-20 (TBS-T), supplemented with 5% skim milk (Sigma-Aldrich,
St. Louis, MO, USA) at room temperature for 2 h on a rotary shaker,
and followed by TBS-T washing. The specific primary antibody,
diluted in TBST, was incubated with the membrane at 4°C overnight.
Subsequently, the membrane was washed with TBS-T followed by
incubation with the peroxidase-conjugated secondary antibody at
room temperature for 1 h. The immunoactive proteins were detected
by using an enhanced chemiluminescence western blotting detection
kit. Western blot bands were observed using GE Healthcare ECL
Western blotting analysis system and exposed to X-ray film (Kodak).
The primary antibodies used were: mTOR, Bcl-2, Bax, caspase-9,
caspase-3, PCNA, p21, p27, cyclin D1, cyclin D2 and GAPDH.
Colony-forming assay
Gastric cancer cells were suspended in 0.9%
methylcellulose-based semisolid medium MethoCult H4100 (StemCell,
Beijing, China). After 14 days, individual primary clones (450
cells) were trypsinized and re-plated in the same conditions to
examine the secondary colony forming ability for self-renewal.
Migration and invasion assays
For the Transwell migration assays,
10×104 cells were planted in the top chamber with a
non-coated membrane. For the invasion assays, 2×105
cells were planted in the top chamber with a Matrigel-coated
membrane. For both assays, the cells were seeded in a serum-free
medium, and a medium with 10% serum was used as a chemoattractant
in the lower chamber. The cells were then incubated for 16 h at
37°C and 5% CO2 in a tissue culture incubator. After 16
h, the non-migrated/non-invading cells were removed from the upper
sides of the Transwell membrane filter inserts with cotton-tipped
swabs. The migrated/invaded cells on the lower sides of the inserts
were then stained with Giemsa, and finally the cells were
counted.
Establishment of xenograft tumor
models
The mouse experiments were conducted in the Animal
Laboratory Center. HS-746T cells (1×107 cells) treated
with miR-224 mimics, inhibitors or negative oligonucleotides were
suspended in 100 μl serum-free medium and injected subcutaneously
into the left flank of 4- to 6-week old male BALB/c nu/nu nude
mice. Tumor size was measured with digital caliper and calculated
every week. Tumor volume were measured every 7 days and at the end
of about 6 weeks after treatment, mice were sacrificed. Tumors were
excised, weighed, fixed in 10% neutral formalin, and embedded in
paraffin for histological analysis. All animal experiments were
performed after obtaining Ethics approval from the Endoscopy
Center, China-Japan Union Hospital, Jilin University, Changchun,
Jilin, P.R. China.
Immunohistochemistry
The xenograft tumors were performed for hematoxylin
and eosin (H&E) staining. In brief, fresh tissues were fixed in
paraffin. The sections were then deparaffinized and stained with
hematoxylin and eosin solution after being cut into 4 μm slices.
Then, the sliced sections were dehydrated and then mounted with
Permount. A microscope was used to analyze the results. For the
immunohistochemistry, the fresh tumor tissues were fixed in
formalin for 48 h. Then the tissue block was put in paraffin and
cut into the desired thickness with a microtome, and was then fixed
into a slide. After washing, the sections were prepared for
blocking and incubating with antibodies, including Ki-67 and mTOR,
which was diluted 1:100 in 5% horse serum with PBS at 4°C
overnight. Sections were then incubated with diluted
streptavidin-peroxidase HRP conjugates at room temperature by a
staining kit, based on the manufacturer’s instructions. The
sections were then stained with hematoxylin for 3 min and mounted
and analyzed under a phase-contrast microscope.
Immunofluorescence assays
After induction by conditioned culture medium, the
cells were fixed in 4% paraformaldehyde, permeabilized with 0.1%
Triton X-100 in PBS containing 0.5% BSA (PBS-BSA) for 30 min. The
cells were subsequently incubated with mTOR, caspase-3, cyclin D1
and CDK4 for 30 min, followed by labeling with Alexa Fluor
488-conjugated rabbit anti-mouse or goat anti-rabbit IgG antibody.
The cells were viewed under a fluorescent microscope.
Flow cytometry assays
Flow cytometric assay was used to clarify the cell
cycle arrest and cells of apoptosis. The cells were collected with
trypsinisation and then washed twice with PBS, and fixed in cold
80% ethanol and finally stored at 4°C overnight. The cells were
washed with PBS twice and RNase A (10 mg/ml) was administrated for
analysis. Propidium iodide was then added to tubes at a
concentration of 0.05 mg/ml and then incubated for 20 min at 4°C in
the dark. Cell cycle assays were examined with FACSCalibur flow
cytometer. FITC-labeled Annexin V/PI staining was applied based on
the manufacturer’s instructions (Nanjing KeyGen Biotech, Co., Ltd.,
Nanjing, China). In brief, 1×106 cells in each well were
suspended with buffer containing FITC-conjugated Annexin V/PI.
Samples were then analyzed via flow cytometry.
Statistical analysis
Data are expressed as means ± SEM. Treated cells,
tissues and the corresponding controls were compared using GraphPad
Prism (version 6.0; GraphPad Software, Inc., La Jolla, CA, USA) by
a one-way ANOVA with Dunn’s least significant difference tests.
Differences between groups were considered significant at
P<0.05.
Results
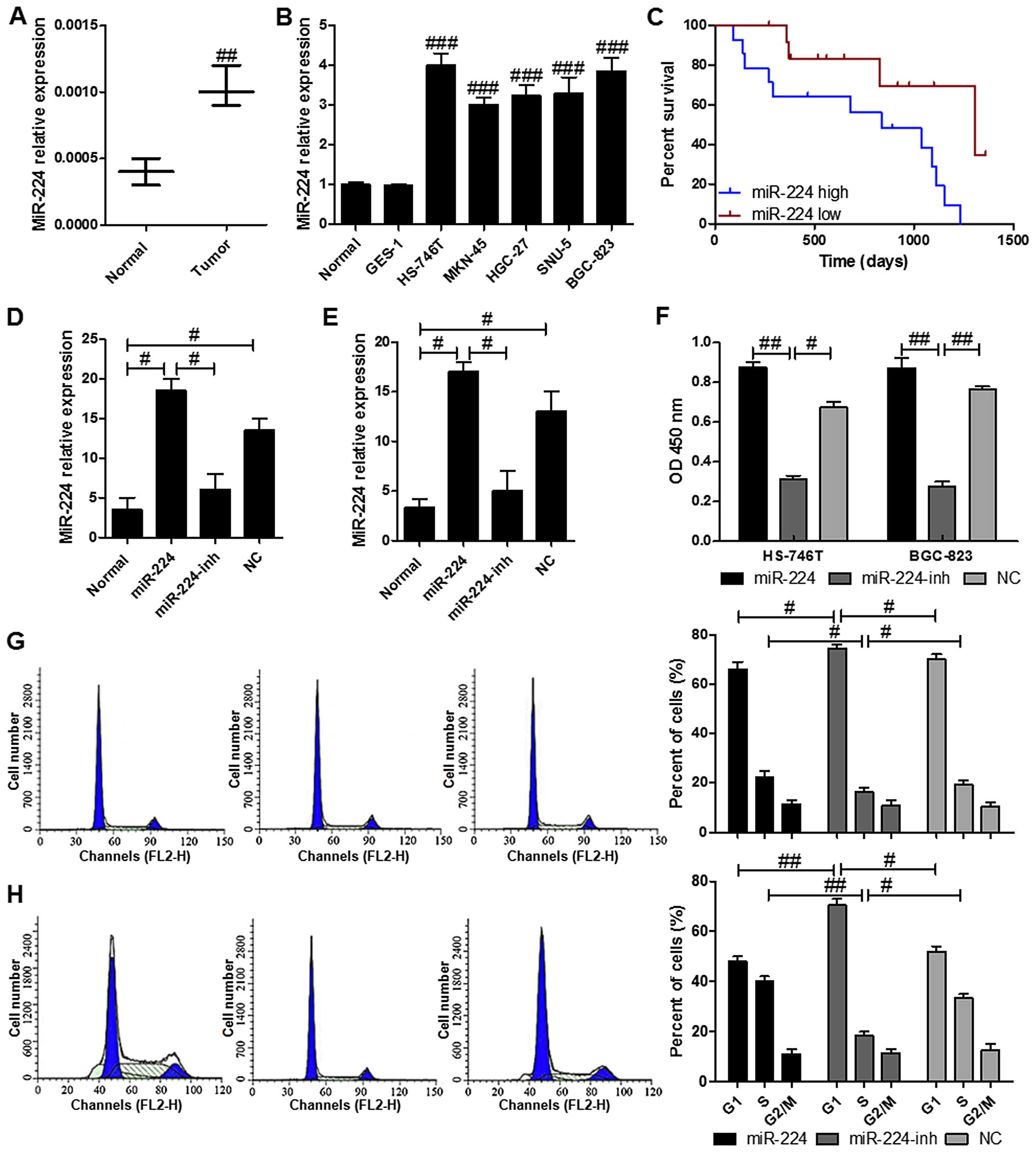
miR-224 is overexpressed in gastric
cancer tissues and cell lines as well as related to the poor
survival
We identified several miRNAs abnormally expressed in
gastric carcinoma using a GeneChip previously (Table III). Based on the result, we
predicted an upstream miRNA: miR-224. In order to investigate the
potential role of miR-224 in gastric cancer, the miR-224 levels was
evaluated. The results indicated a significantly higher mRNA levels
of miR-224 in the gastric cancer samples in comparison to the
normal ones with nontumor tissues (Fig. 1A). Also, as shown in Fig. 1B, no varied miR-224 levels were
observed in five different gastric cells of HS-746T, MKN-45,
HGC-27, SNU-5 and BGC-823, which were expressed markedly higher
than that in the normal tissues with non-tumor tissues and in the
normal gastric membrane cell line GES-1. Furthermore, we explored
the miR-224 expression in tumor tissues from patients (Fig. 1C). Similar to the findings, high
levels of miR-224 was associated with the cancer progression that
the high overall survival was linked to the lower levels of
miR-224. On the contrary, the patients with higher miR-224
displayed shorter survival. RT-PCR results determined that
transfection of miR-224 and its inhibitor restored and reduced its
expression in HS-746T and BGC-823 cell lines, repectively, compared
with the normal gastric cancer cell lines (Fig. 1D and E). As for cell proliferation,
restoration of miR-224 in HS-746T and BGC-823 cells resulted in
significant enhancement of cell proliferation, the proliferation
rate was suppressed in HS-746T and BGC-823 cells after transfection
with miR-224 inhibitor (Fig. 1F).
In addition, cell cycle assays were performed to determine if the
role of miR-224 in cell proliferation was due to the cell cycle
arrest. The results indicated that the cells in G1 phase was higher
in the HS-746T cells with miR-224 inhibitor compared to the gastric
cells treated with or without miR-224 mimics (Fig. 1G). Similar effects of miR-224 were
found in BGC-823 cells (Fig. 1H).
These results demonstrated that miR-224 regulated the gastric cell
proliferation.
| Table IIImiRNAs differentially expressed in
gastric cancer samples compared with adjacent normal samples. |
Table III
miRNAs differentially expressed in
gastric cancer samples compared with adjacent normal samples.
| Downregulated
(n=30) | Upregulated
(n=32) |
|---|
| miR-188 | miR-224 |
| miR-490 | miR-212 |
| miR-503 | miR-379 |
| miR-611 | miR-320 |
| miR-744 | miR-26b |
| miR-370 | miR-518b |
| miR-567 | |
| miR-353 | |
| miR-197 | |
| miR-575 | |
| miR-545 | |
| miR-433 | |
| miR-338 | |
| miR-19b | |
| miR-551 | |
| miR-21 | |
| miR-940 | |
| miR-847 | |
| miR-17-92 | |
| miR-222 | |
miR-244 targets and regulates mTOR
expression directly in gastric cancer cells
miRNAs mainly functions via its regulation of target
genes, and the target gene of miR-224 was further analyzed. After
checking the newly published CLASH data, about 457 genes were
targeted by miR-224 in HS-746T cells. Among the genes, mTOR, a key
regulator in apoptosis and proliferation, regulating cellular
processes, was focused on (17).
In order to confirm whether miR-224 could affect the expression of
mTOR, we performed luciferase reporter assays in HS-746T cells. We
then created luciferase reporter plasmid with wild-type (WT) or
mutant (Mut) targeting sequence of mTOR mRNA (Fig. 2A), which were cotransfected with
miR-224 mimics or the negative control (NC) oligonucleotides into
HS-746T cells for 48 h, and followed by determination of luciferase
activity in the transfected cancer cells. Our results indicated
that the reporter plasmid with WT targeting sequence of mTOR mRNA
caused a significant upregulation of luciferase activity in cells
transfected with miR-224, whereas reporter plasmid with mutant
sequence of mTOR produced no alteration of luciferase activity
(Fig. 2B). Next, we explored if
mTOR in gastric cancer cells was regulated similarly. HS-746T and
BGC-823 cells were transfected with miR-224 mimics, miR-224
inhibitor or negative control oligonucleotides, and mTOR mRNA and
protein levels were examined by RT-PCR and western blot analysis,
respectively. mTOR mRNA expressions were higher by miR-224 mimics
in HS-746T and BGC-823 cells, respectively (Fig. 2C and D). The level of mTOR protein
was consistently and substantially downregulated by miR-224
inhibitor in HS-746T and BGC-823 cells (Fig. 2E and F). Furthermore, we then
examined miR-224 expression after the transient transfection of
mTOR plasmid into HS-746T and BGC-823 cells. Western blot analysis
demonstrated that the transient transfection of mTOR into HS-746T
and BGC-823 cells increased mTOR expression at the protein levels.
mTOR induced miR-21 expression in the transient transfection
experiments in a dose-dependent manner (Fig. 2G and H). Taken together, our
results demonstrated that mTOR was a direct target of miR-224 in
gastric cancer cells.
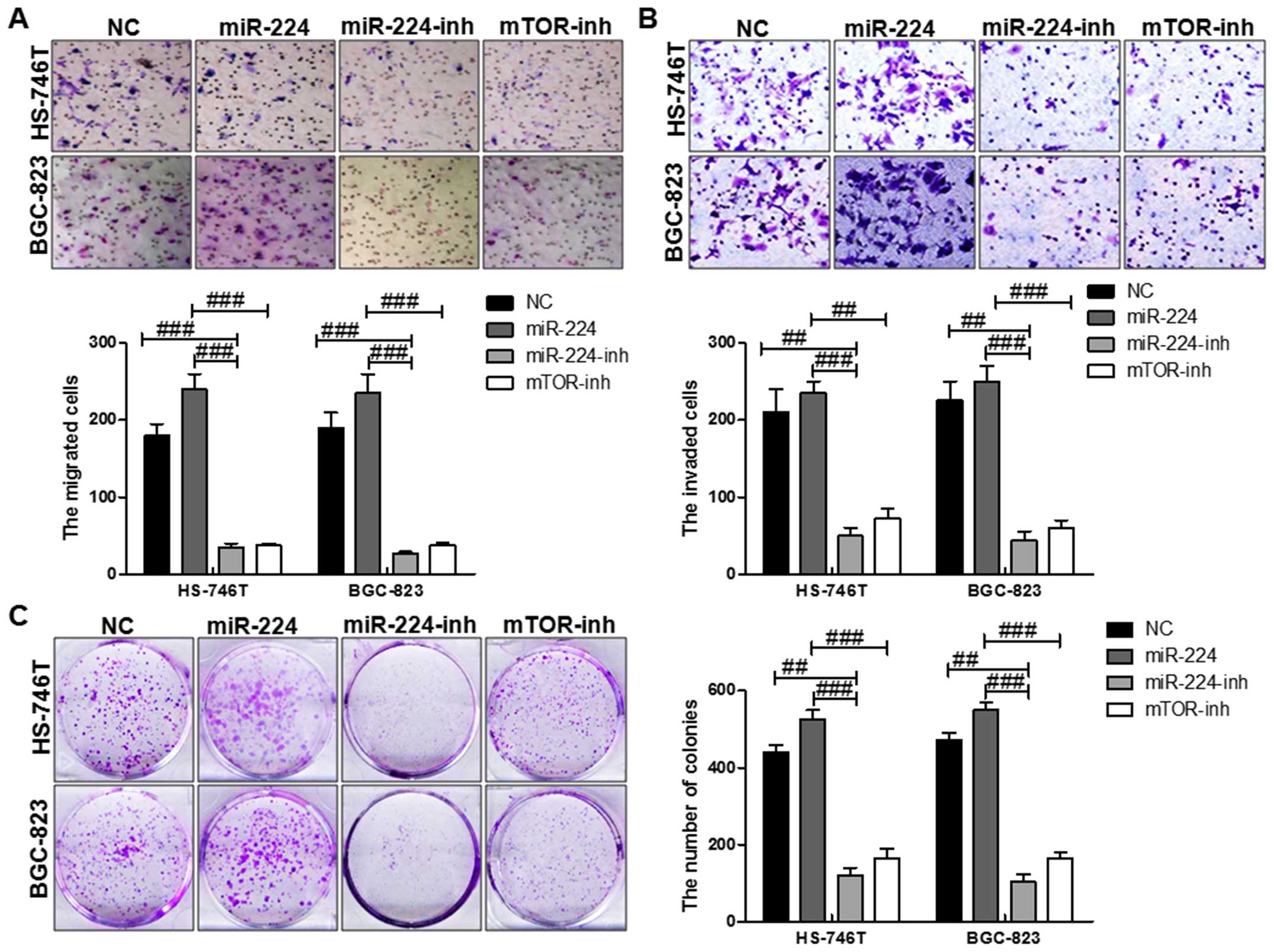
miR-224 accelerates gastric cancer cell
proliferation and migration partly through mTOR in vitro
mTOR was involved in many cellular processes,
including aoptosis, cell growth, diffrentiation and proliferation
(18). Thus, we further explored
the biological effects of miR-224 on mTOR. Cell function assays
showed that upregulation of miR-224 displayed higher proliferation,
migration and invasion capacity. At the same time, with miR-224
inhibitor and mTOR inhibition treatment, the migrated cells,
invaded cells as well as the colony formation were reversed
significantly, which further suggested that miR-224 could promote
gastric cancer progression via regulation with mTOR in both HS-746T
and BGC-823 cells (Fig. 3).
Together, the results showed inhibition of mTOR could partially
reverse the influence of miR-224 on cell growth migration,
proliferation and invasion.
miR-224 activates the mTOR signaling in
HS-746T and BGC-823 cells inhibiting apoptosis in vitro
Previous studies have suggested that miR-224 could
accelerate cell proliferation by Wnt/β-catenin signaling (19). In addition, we also observed here
that miR-224 could activate mTOR-regulated apoptic signaling
pathway via Bcl-2, Bax, caspase-9 and caspase-3 signal expression.
As shown in Fig. 4A, RT-PCR was
used to further confirm that miR-224 could activate mTOR mRNA
levels. Subsequently, it was notable that Bcl2, an important
anti-apoptotic factor, was found to be stimulated in miR-224 mimic
treatments, while inhibited with miR-224 inhibitor, the Bcl-2 was
downregulated significantly (Fig.
4B). Consistently, Bax, a pro-apoptotic factor, was
downregulated for miR-224 mimics, while being enhanced for miR-224
inhibitor treatment (Fig. 4C).
Next, the downstream signals of caspase-9/3, which are essential
regulators for apoptosis, were found to be inhibited in
miR-224-overexpressed gastric cell lines (Fig. 4D and E). Similarly, western blot
analysis displayed the role of miR-224 in suppressing apoptosis in
both HS-746T and BGC-823 cells via mTOR and Bcl-2 upregulation and
Bax, caspase-9/3 suppression, respectively (Fig. 4F–H). In this part, finally the
immunofluorescence assays were adopted to further confirm the role
of miR-224 in apoptosis inhibition in HS-746T gastric cells, which
showed higher mTOR expressive intensity while with lower caspase-3
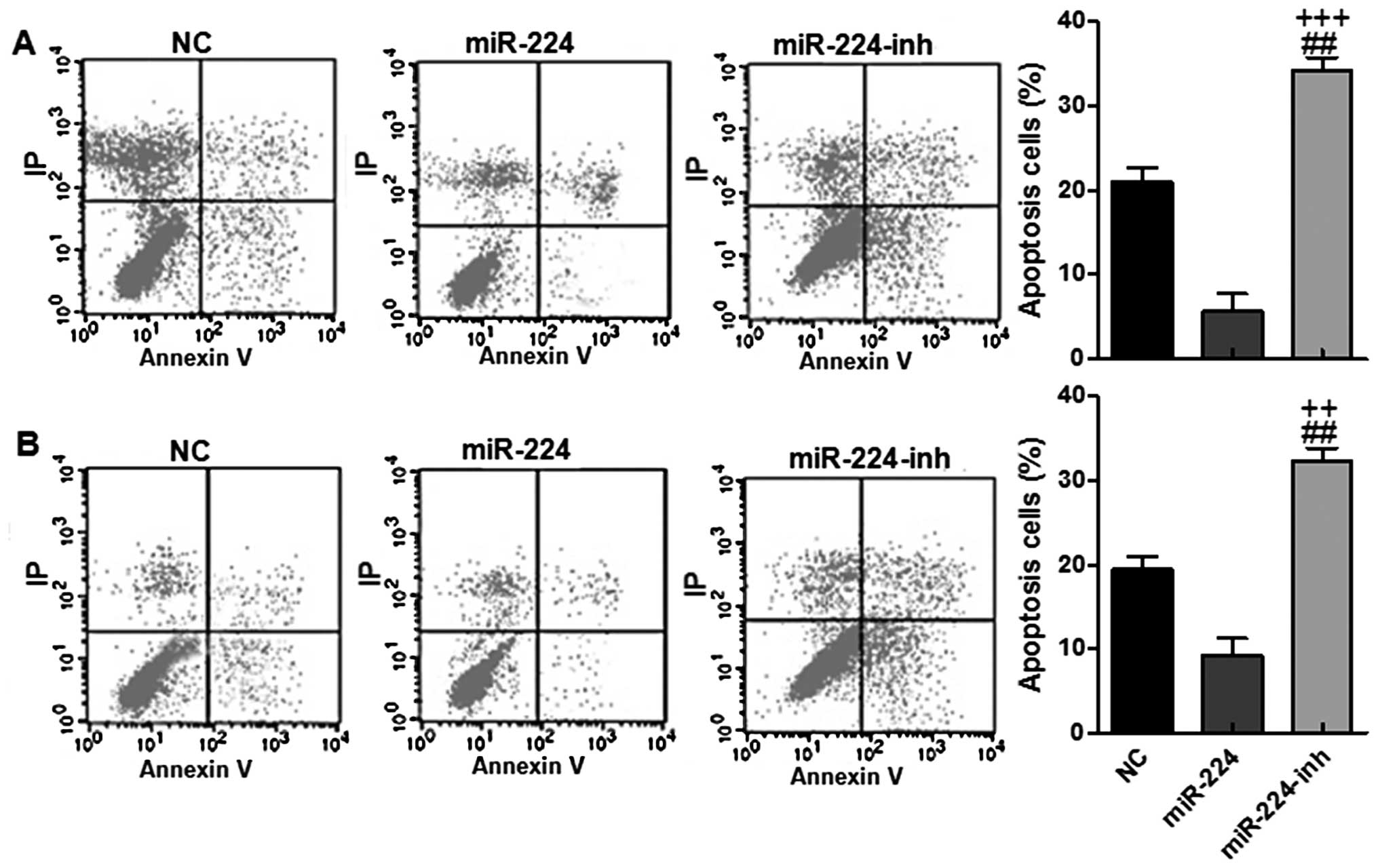
immunofluorescent intensity in the miR-224 groups (Fig. 4L). In addition, flow cytometry was
conducted to reveal that the apoptotic cells were lower in groups
with miR-224 high expression in both HS-746T and BGC-823 cell
lines, while after being treated with miR-224 inhibitor, the
apoptotic gastric cells were upregulated (Fig. 5A and B). Collectively, our results
above suggested that miR-224 could promote gastric cancer
progression via apoptosis suppression.
 | Figure 4The effects of miR-224 on apoptosis
in the gastric cancer cell lines via mTOR and its subsequent
signals. The relative mRNA levels of (A) mTOR, and its
down-streaming signals of (B) Bcl-2, (C) Bax, (D) caspase-9 and (E)
caspase-3 via RT-PCR assays. (F) The protein levels of mTOR, and
its downstream signals in both cell lines. The quantification of
(G) mTOR, (H) Bcl-2, (I) Bax, (J) caspase-9 and (K) caspase-3 in
both cell lines. (L) mTOR and caspase-3 expression were assessed by
immunofluorescence staining in the HS-746T gastric cancer cells
treated with the negative control miRNA or miR-224 mimics or
miR-224 inhibitors for 48 h. The values present mean ± SD; (n=6) of
the samples. #P<0.05, ##P<0.01 and
###P<0.001 vs. the NC group, and
+P<0.05, ++P<0.01 and
+++P<0.001 vs. the miR-224 group. |
The effects of miR-224 on proliferation
in the gastric cancer cell lines
In this regard, cell proliferation was determined to
clarify whether miR-224-regulated mTOR signaling pathway was
involved in gastric cancer progression. RT-PCR was conducted to
determine PCNA, p21, p27, cyclin D1 and cyclin D2 levels. As shown
in Fig. 6A, PCNA was upregulated
for miR-224 mimics, while being reduced in miR-224 inhibitor
treatment. Also, p21 and p27, as important factors inhibiting cell
proliferation, decrease cyclin D1 and cyclin D2 expression.
Fig. 6B–E shows downregulated p21
and p27 were observed in the miR-224 groups, which were reversed
for miR-224 inhibitor treatment in both gastric cancer cells.
Consistently, cyclin D1 and cyclin D2 were suppressed in the
miR-224-inh groups, suggesting that miR-224 inhibition might be a
potential therapeutic strategy for gastric cancer via cell
proliferation inhibition. In addition, western blot assays were
conducted to confirm that cyclin D1 and cyclin D2 were indeed
involved in miR-224-regulated mTOR signaling pathway. The results
were similar to mRNA levels. Fig.
6F–K shows PCNA, cyclin D1, and cyclin D2 protein levels were
upregulated in the miR-224 groups, while being downregulated in
miR-224-inh groups. Conversely, p21 and p27 were decreased for
miR-224 mimics and were increased due to miR-224 inhibitor usage.
Finally, immunofluorescence was used to confirm that cyclin D1 and
CDK4, as important cooperator of cyclin D1, were both stimulated
with high fluorescent intensity in miR-224 mimic groups, which were
weakened for miR-224 inhibitor treatment in the HS-746T gastric
cancer cells (Fig. 6L). Our
results above suggested that miR-224 could promote cell
proliferation, leading to gastric cancer progression.
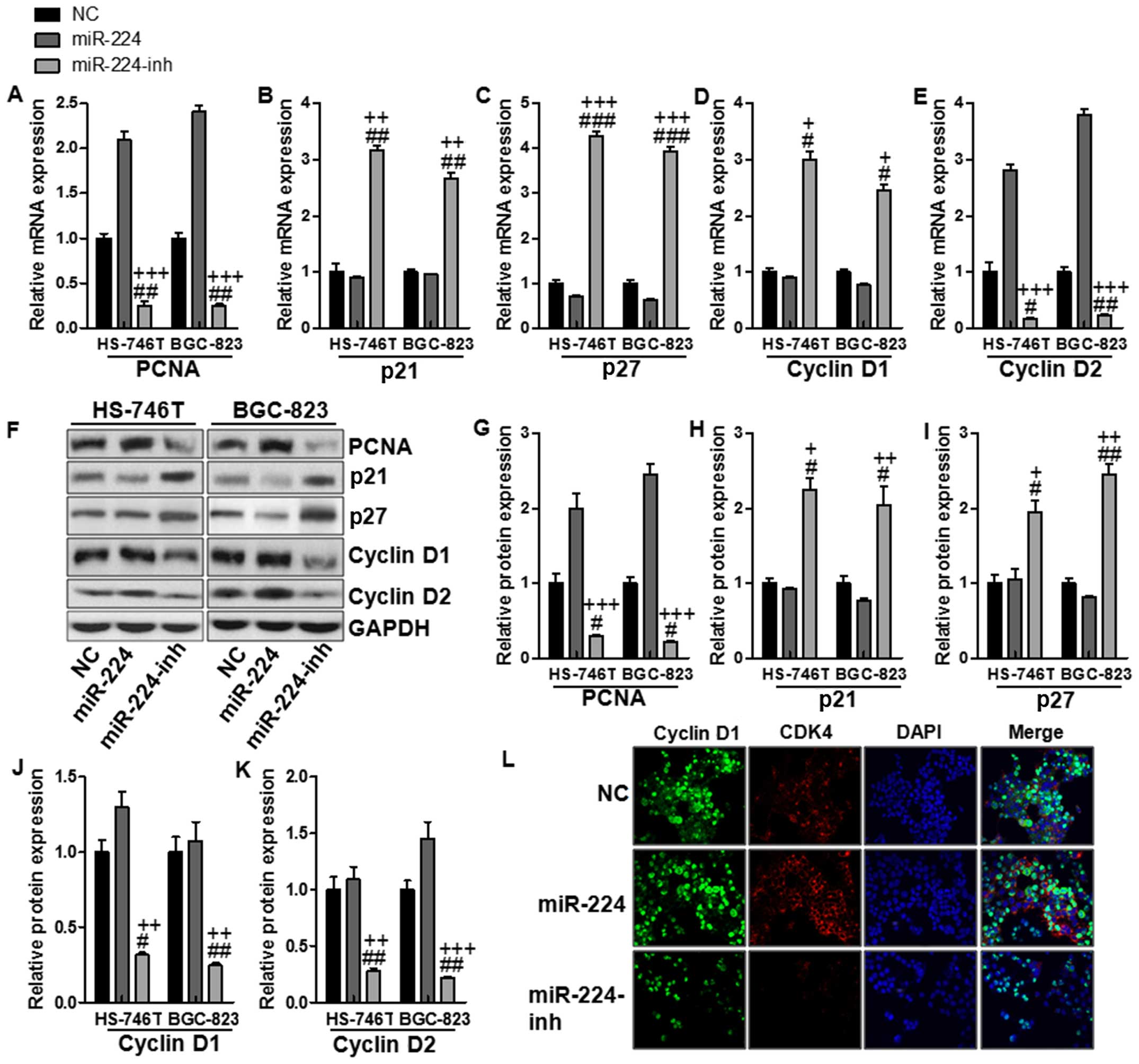 | Figure 6The effects of miR-224 on
proliferation in the gastric cancer cell lines. The relative mRNA
levels of (A) PCNA, (B) p21, (C) p27, (D) cyclin D1, and (E) cyclin
D2. (F) The protein levels of (G) PCNA, (H) p21, (I) p27, (J)
cyclin D1 and (K) cyclin D2. (L) Cyclin D1 and CDK4 expression were
assessed by immunofluorescence staining in the HS-746T gastric
cancer cells treated with the negative control miRNA or miR-224
mimics or miR-224 inhibitors for 48 h. The values present mean ± SD
(n=6) of the samples. #P<0.05, ##P<0.01
and ###P<0.001 vs. the NC group, and
+P<0.05, ++P<0.01 and
+++P<0.001 vs. the miR-224 group. |
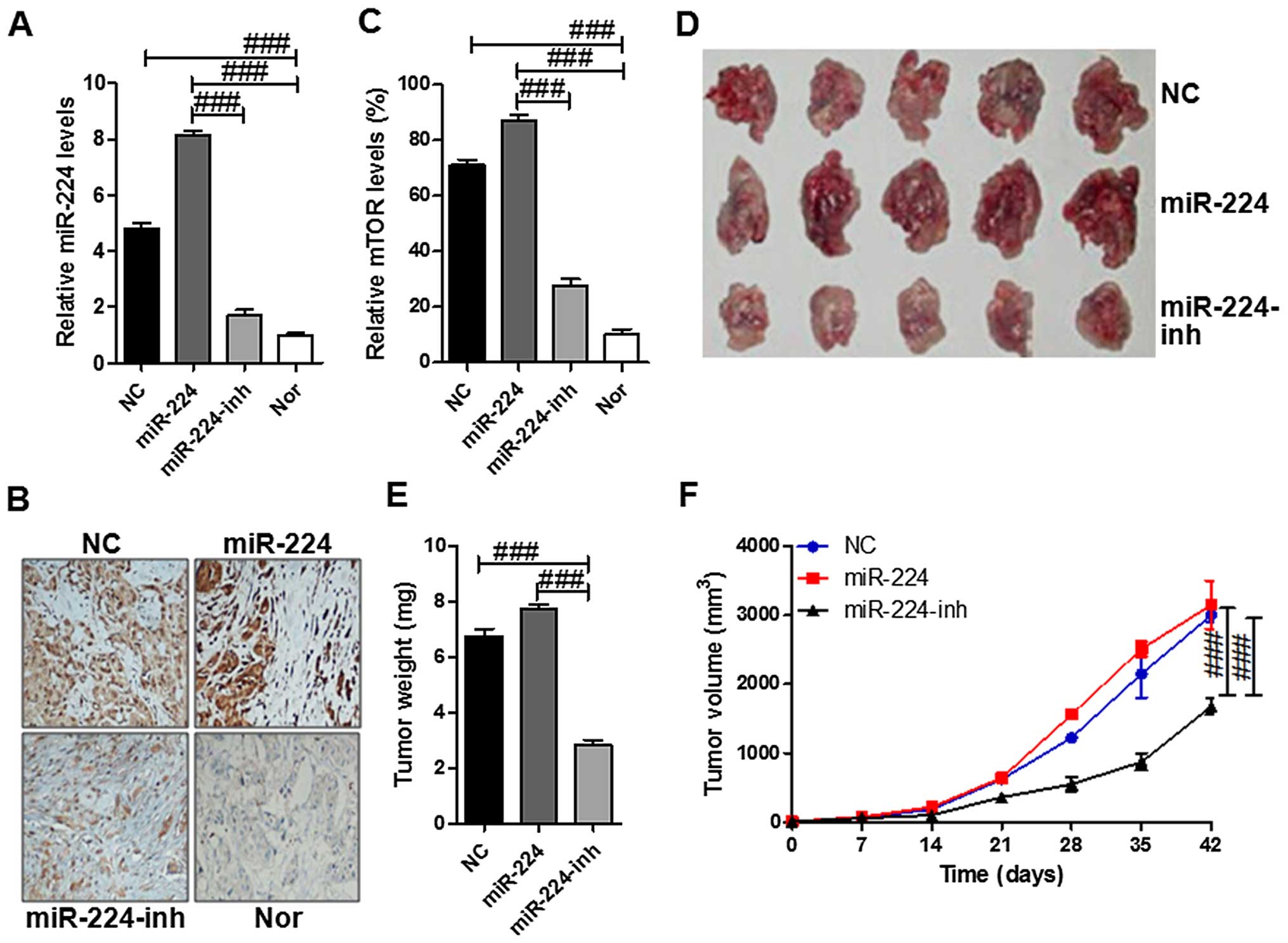
miR-224 promotes tumor growth by
targeting mTOR in vivo
To understand whether mTOR is involved in miR-224
mediated tumorigenesis in vivo, we engineered HS-746T cells
to stably overexpress miR-224. The control cells,
miR-224-overexpressing cells and the restored mTOR in
miR-224-overexpressing cells were subcutaneously inoculated into
nude mice, respectively. As shown in Fig. 7A, the miR-224 levels were higher in
the tissue samples with miR-224 mimics of HS-746T cells injection
compared to the one with miR-224-inhibitor. In addition, the mTOR
expressed levels displaying similar change with miR-224 via
immunohistochemical assays that mTOR was expressed highly in the
miR-224 group while being suppressed in the miR-224-inh groups
(Fig. 7B and C). Next, the tumor
incidence was evaluated biweekly, and tumors appeared in all the
mice. As shown in the Fig. 7D–F,
the tumors in the miR-224 group grew much more rapidly than the
other two tumor groups. Furthermore, the forced expression of
miR-224 significantly inhibited tumor growth in vivo. These
results indicated that miR-224 stimulated tumor growth and mTOR was
involved in the progression of tumor growth in vivo.
miR-224 inhibits apoptosis and enhanced
proliferation in gastric cancer in vivo
In this regard, we further determined the role of
miR-224 in the gastric cancer development in vivo. As shown
in Fig. 8A–C, immunohischemical
analysis was used to confirm that the tumors of the
miR-224-overexpressed group displayed lower TUNEL positive cells
while much higher Ki-67 indexes than the other two groups. In order
to further verify the above data and observations as well as the
possible molecular mechanisms, we assessed the mTOR-regulated
apoptotic signaling pathway and cyclin D1/2-associated cell
proliferation. Consistenly, in vivo experiments we found
that mTOR overexpressed in the miR-224 groups in comparison to the
other two groups (Fig. 8D). And
also, Bcl-2 was expressed highly, which was inhibited in miR-224
inhibitor use. Subsequently, caspase-9/3 were inactivated due to
miR-224 overexpression, which were activated for miR-224 inhibitor,
leading to apoptosis in the gastric cancer cells. Furthermore,
miR-224 could aggravate gastric cancer development via cell
proliferation upregulation of PCNA, cyclin D1/2, while
downregulated p21 and p27 (Fig.
8E). However, expression of these proteins could be reversed
for miR-224 inhibition, suggesting that suppressing miR-224 could
reduce gastric cancer progression. The above molecular link
provided a significant clue to the role of miR-224 in the process
of gastric cancer via mTOR-regulated signaling pathway.
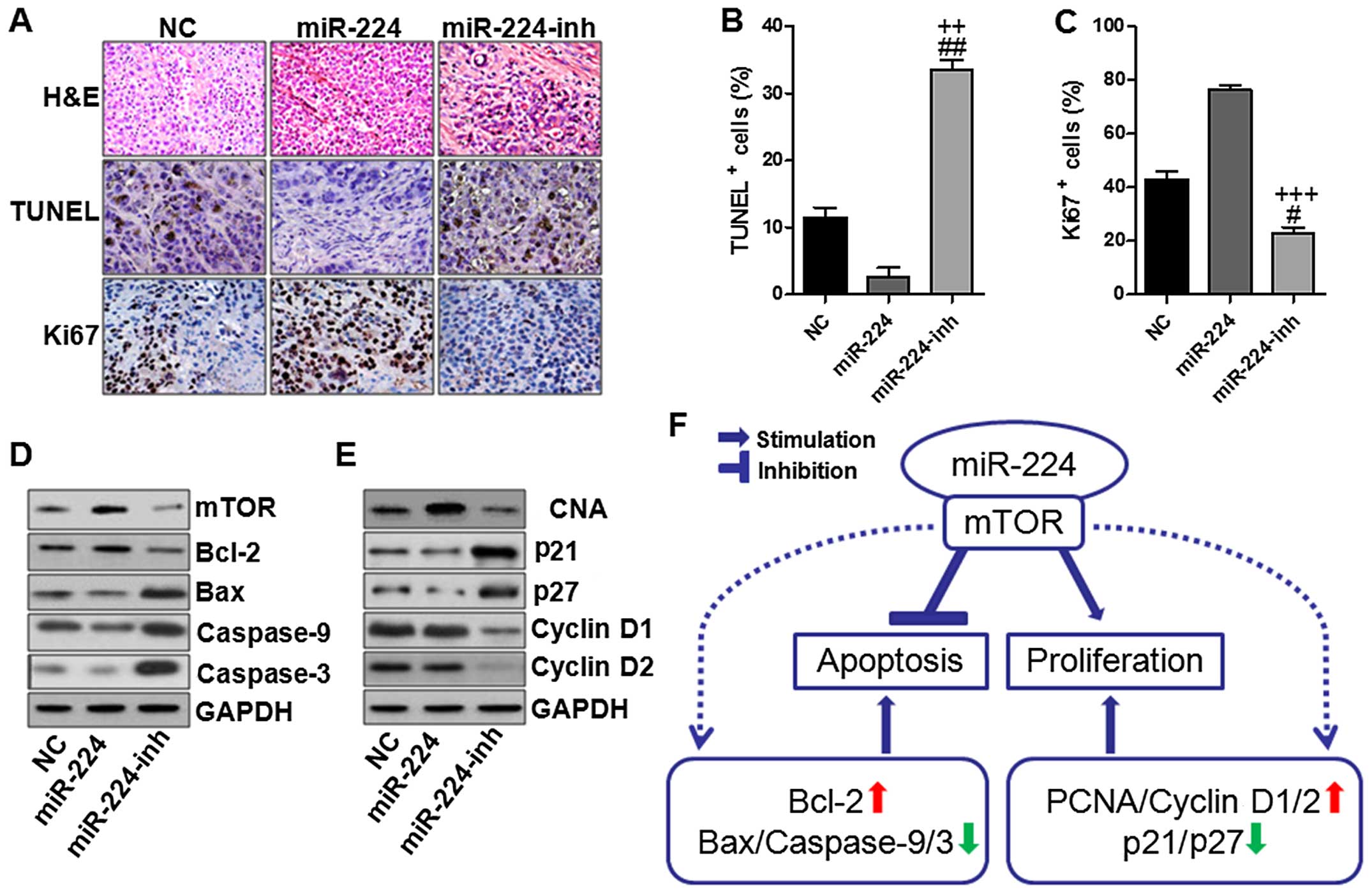 | Figure 8miR-224 inhibits apoptosis and
enhances proliferation in the gastric cancer. (A) Immunochemistry
assays were used to determine the pathological situation of tumor
tissues with H&E staining, TUNEL and Ki-67 quantification
assays. The TUNEL (B) and Ki-67 (C) positive cells were evaluated.
(D) Western blot analysis was conducted to analyze mTOR-regulated
apoptotic signaling pathway. (E) Western blot analysis was
conducted to analyze cell proliferation-associated signaling
pathway. (F) A proposed model for mTOR-related miR-224 upregulation
in the modulation of specific subsequent targets in gastric cancer
cells. miR-224 could operate with mTOR, leading to apoptosis
inhibition and cell proliferation in gastric cancer progression.
Once mTOR was activated for miR-224 high expression, Bcl-2 was
activated, causing Bax, caspase-9 and caspase-3 inactivation, thus
suppressing apoptosis. On the other hand, PCNA and cyclin D1/2 were
stimulated in gastric cancer cells, promoting the cancer cell
proliferation, thus, accelerating gastric cancer development. The
values present mean ± SD; (n=6) of the samples.
#P<0.05 and ##P<0.01 vs. the NC group,
and ++P<0.01 and +++P<0.001 vs. the
miR-224 group. |
Discussion
Presently, gastric cancer remains a major threat to
human health. Despite improvements in surgery and chemotherapy, the
outcomes in patients with advanced gastric cancer remain poor, with
a 5-year survival rate of less than 20% (20,21).
Over the past several years, targeted therapies have indeed
improved the outcome of a number of malignancies greatly, including
colorectal, lung and breast cancer (22,23).
However, less progress has been made regarding gastric cancer.
Molecular mechanism research on the different phases of this
disease will be useful and valuable for a better diagnosis and
development of therapeutic strategy.
MicroRNAs are small non-coding RNAs that regulate
the expression of target genes by inhibiting translation and/or
stability of mRNAs (24).
Accumulated evidence demonstrates that miRNAs play an important
role in many different biological processes, such as
differentiation, proliferation, survival, apoptosis, metabolism and
development (25,26). Numerous miRNAs act as either tumor
suppressors or oncogenes, and the aberrant miRNAs expression is
included in the initiation and progression of human tumors or
cancers (27). miR-224 has been
indicated to be increased in several solid tumors, including
colorectal cancer, hepatocellular carcinoma, lung and breast
cancer, and repressing many targets, such as SMAD4, API5, PHLPP1
and PHLPP2, promoting the migratory, invasive and proliferative
capacity of cancer cells (28,29).
However, further studies need to be done to clarify the biological
function of miR-224 in gastric cancer progression due to its
potential role in development of many other tumors.
In order to better understand the underlying
oncogenic role of miR-224 in gastric cancer, we attempted to find
other potential targets of miR-224. In the present study, we report
that mTOR is a novel direct target of miR-224 that could be
implicated in miR-224-induced gastric cancer cell proliferation and
invasion.
Mammalian target of rapamycin (mTOR) is a component
of the phosphatidylinositol 3-kinase (PI3K) cell survival pathway
that monitors the availability of nutrients, mitogenic signals,
cellular energy as well as oxygen levels, and thus is vital in the
regulation of cell growth and proliferation (30–32).
Abnormal activation of PI3K pathway is thought to be associated
with numerous cancers, and according to previous studies increased
activation of this pathway is often related to resistance to cancer
therapies (33). mTOR, as an
essential signal belonging to PI3K/AKT signaling pathway, operates
a key junction in the PI3K pathway-regulated cellular process
(34). Overall, the role of mTOR
in gastric cancer remains unclear. In the present study, we found
that the expression of mTOR is upregulated significantly in gastric
cancer cells or tissues compared to the normal ones, and higher
mTOR expression potentially contributes to the poor overall
survival of gastric cancer patients. We further demonstrated a
significant proportional correlation between miR-224 and mTOR in
gastric cancer cell samples. Moreover, inhibition of mTOR induced
same phenotypes as the suppression of miR-224 in gastric cancer
cells. These results suggest that mTOR and miR-224 have
collaborative interactions mediating gastric cancer progression. To
the best of our knowledge, there is no report regarding the role of
mTOR in cell proliferation in gastric cacner progression via
miR-224 regulation. The present study demonstrated that mTOR
positively regulates gastric cancer cell proliferation, invasion
and migration. However, further studies need to be done to reveal
the underlying mechanism of mTOR in the proliferation, invasion and
migration of gastric cancer.
In light of the significant upregulation of miR-224
in gastric cancer and the fact that mTOR is a direct target of
miR-224, upregulated mTOR might be attributable to the increased
miR-224 expression in gastric cancer. We found that mTOR expression
was significantly upregulated in gastric cancer. Next, we intended
to clarify the possible mechanism by which miR-224 and mTOR
regulated gastric cancer progression. We found that miR-224
significantly stimulated Bcl-2 expression via mTOR upregulation
in vivo and in vitro experiments, inducing Bax,
caspase-9 and caspase-3 down-regulation, inhibiting apoptosis in
gastric cancer development. Caspase-3 is known as an essential
activator for apoptosis development in tissues and cells via Bcl-2
and its family members, which are involved in cancer progression
based on previous studies (35–37).
Notably, miR-224 suppression could reverse expression of these
proteins with mTOR and Bcl-2 downregulation while with Bax,
caspase-9 and caspase-3 were upregulated, thus, leading to
apoptosis in gastric cancer and promoting cancer cell attenuating
gastric cancer progression. Collectively, the results above
elucidated that miR-224 might be linked to gastric cancer
progression via mTOR-regulated caspase-3 signaling pathway. In
addition, inhibiting miR-224, causing high expression of caspase-3,
could be effective in treating gastric cancer through apoptosis
activation.
mTOR exists as two separate complexes in the
cytoplasm (TORC1 and TORC2), which could regulate progression of
the cell cycle from the G1 to S phase through cyclin D. PNCA, p21,
and p27, cyclin D1 and cyclin D2 are known to be closely associated
with the cell cycle, leading to the alteration of cell
proliferation (38,39). G1/S progression is mainly regulated
by cyclin D1 and the cyclin-dependent kinase 4 (CDK4) (40). PCNA is an acidic nuclear protein,
being recognized as a histological marker for the G1/S phase in the
cell cycle (41). p21 is a CDK
inhibitor, which can bind to CDK-cyclin complexes and alter their
function in order to suppress cell proliferation (39). In the present study, we found that
PCNA and cyclin D1/2 were stimulated due to miR-224 upregulation
in vivo and in vitro experiments, promoting gastric
cancer cell proliferation, which was consistent with previous
studies. However, after miR-224 suppression, we found that
overexpressed PCNA, cyclin D1/2 were downregulated, while p21, and
p27 were upregulated, which might perform their role in suppressing
cell proliferation. Also, the immunofluorescent assays further
confirmed that higher levels of cyclin D1 with increased CDK4
expression in miR-224 groups indicated enhanced gastric cancer cell
proliferation due to miR-224. In addition, the weak fluorescent
intensity of cyclin D1 and CDK4 suggested that the cell
proliferation of gastric cancer was inhibited in miR-224
suppression. Our data above suggested that miR-224 could be a
target for gastric cancer suppression via mTOR-regulated apoptosis
and cell proliferation by caspase and cyclin D modulation.
The present study is the first to document the tumor
promoter role of miR-224 in gastric cancer progression. miR-224
could suppress apoptosis and enhance migratory, invasive and
proliferative behaviors by improving the expression of mTOR,
reducing apoptosis and enhancing cell proliferation (Fig. 8F). Our results illustrated that the
inhibition of the tumor promoter miR-224 might be useful and
efective in the treatment of gastric cancer.
References
|
1
|
JeJemal A, Bray F, Center MM, Ferlay J,
Ward E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Verdecchia A, Santaquilani M and Sant M:
Survival for cancer patients in Europe. Ann Ist Super Sanita.
45:315–324. 2009.PubMed/NCBI
|
|
3
|
Deng N, Goh LK, Wang H, Das K, Tao J, Tan
IB, Zhang S, Lee M, Wu J, Lim KH, et al: A comprehensive survey of
genomic alterations in gastric cancer reveals systematic patterns
of molecular exclusivity and co-occurrence among distinct
therapeutic targets. Gut. 61:673–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bang YJ, Van Cutsem E, Feyereislova A,
Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T,
et al; ToGA Trial Investigators. Trastuzumab in combination with
chemotherapy versus chemotherapy alone for treatment of
HER2-positive advanced gastric or gastro-oesophageal junction
cancer (ToGA): A phase 3, open-label, randomised controlled trial.
Lancet. 376:687–697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee J, Seo JW, Jun HJ, Ki CS, Park SH,
Park YS, Lim HY, Choi MG, Bae JM, Sohn TS, et al: Impact of MET
amplification on gastric cancer: Possible roles as a novel
prognostic marker and a potential therapeutic target. Oncol Rep.
25:1517–1524. 2011.PubMed/NCBI
|
|
6
|
Sasaki T, Kuniyasu H, Luo Y, Kitayoshi M,
Tanabe E, Kato D, Shinya S, Fujii K, Ohmori H and Yamashita Y: AKT
activation and telomerase reverse transcriptase expression are
concurrently associated with prognosis of gastric cancer.
Pathobiology. 81:36–41. 2014. View Article : Google Scholar
|
|
7
|
Jiang YW and Chen LA: microRNAs as tumor
inhibitors, oncogenes, biomarkers for drug efficacy and outcome
predictors in lung cancer (Review). Mol Med Rep. 5:890–894.
2012.PubMed/NCBI
|
|
8
|
Zhou C, Li G, Zhou J, Han N, Liu Z and Yin
J: miR-107 activates ATR/Chk1 pathway and suppress cervical cancer
invasion by targeting MCL1. PLoS One. 9:e1118602014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roldo C, Missiaglia E, Hagan JP, Falconi
M, Capelli P, Bersani S, Calin GA, Volinia S, Liu CG, Scarpa A, et
al: MicroRNA expression abnormalities in pancreatic endocrine and
acinar tumors are associated with distinctive pathologic features
and clinical behavior. J Clin Oncol. 24:4677–4684. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takahashi Y, Forrest AR, Maeno E,
Hashimoto T, Daub CO and Yasuda J: MiR-107 and MiR-185 can induce
cell cycle arrest in human non-small cell lung cancer cell lines.
PLoS One. 4:e66772009. View Article : Google Scholar
|
|
11
|
Zeng L, He X, Wang Y, Tang Y, Zheng C, Cai
H, Liu J, Wang Y, Fu Y and Yang GY: MicroRNA-210 overexpression
induces angiogenesis and neurogenesis in the normal adult mouse
brain. Gene Ther. 21:37–43. 2014. View Article : Google Scholar
|
|
12
|
Wang L, Chang L, Li Z, Gao Q, Cai D, Tian
Y, Zeng L and Li M: miR-99a and -99b inhibit cervical cancer cell
proliferation and invasion by targeting mTOR signaling pathway. Med
Oncol. 31:9342014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marcucci G, Maharry KS, Metzeler KH,
Volinia S, Wu YZ, Mrózek K, Nicolet D, Kohlschmidt J, Whitman SP,
Mendler JH, et al: Clinical role of microRNAs in cytogenetically
normal acute myeloid leukemia: miR-155 upregulation independently
identifies high-risk patients. J Clin Oncol. 31:2086–2093. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang X, Taeb S, Jahangiri S, Emmenegger
U, Tran E, Bruce J, Mesci A, Korpela E, Vesprini D, Wong CS, et al:
miRNA-95 mediates radioresistance in tumors by targeting the
sphingolipid phosphatase SGPP1. Cancer Res. 73:6972–6986. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mees ST, Mardin WA, Sielker S, Willscher
E, Senninger N, Schleicher C, Colombo-Benkmann M and Haier J:
Involvement of CD40 targeting miR-224 and miR-486 on the
progression of pancreatic ductal adenocarcinomas. Ann Surg Oncol.
16:2339–2350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang L, Dai T, Lin X, Zhao X, Chen X,
Wang C, Li X, Shen H and Wang X: MicroRNA-224 targets RKIP to
control cell invasion and expression of metastasis genes in human
breast cancer cells. Biochem Biophys Res Commun. 425:127–133. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee DF and Hung MC: All roads lead to
mTOR: Integrating inflammation and tumor angiogenesis. Cell Cycle.
6:3011–3014. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gridelli C, Maione P and Rossi A: The
potential role of mTOR inhibitors in non-small cell lung cancer.
Oncologist. 13:139–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li T, Lai Q, Wang S, Cai J, Xiao Z, Deng
D, He L, Jiao H, Ye Y, Liang L, et al: MicroRNA-224 sustains
Wnt/β-catenin signaling and promotes aggressive phenotype of
colorectal cancer. J Exp Clin Cancer Res. 35:21–27. 2016.
View Article : Google Scholar
|
|
20
|
Sasaki T, Kuniyasu H, Luo Y, Kitayoshi M,
Tanabe E, Kato D, Shinya S, Fujii K, Ohmori H and Yamashita Y:
Increased phosphorylation of AKT in high-risk gastric mucosa.
Anticancer Res. 33:3295–3300. 2013.PubMed/NCBI
|
|
21
|
Sukawa Y, Yamamoto H, Nosho K, Kunimoto H,
Suzuki H, Adachi Y, Nakazawa M, Nobuoka T, Kawayama M, Mikami M, et
al: Alterations in the human epidermal growth factor receptor
2-phosphatidylinositol 3-kinase-v-Akt pathway in gastric cancer.
World J Gastroenterol. 18:6577–6586. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Poplawski T, Tomaszewska K, Galicki M,
Morawiec Z and Blasiak J: Promoter methylation of cancer-related
genes in gastric carcinoma. Exp Oncol. 30:112–116. 2008.PubMed/NCBI
|
|
23
|
Oue N, Motoshita J, Yokozaki H, Hayashi K,
Tahara E, Taniyama K, Matsusaki K and Yasui W: Distinct promoter
hyper-methylation of p16INK4a, CDH1, and RAR-beta in intestinal,
diffuse-adherent, and diffuse-scattered type gastric carcinomas. J
Pathol. 198:55–59. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi XB, Xue L, Ma AH, Tepper CG, Kung HJ
and White RW: miR-125b promotes growth of prostate cancer xenograft
tumor through targeting pro-apoptotic genes. Prostate. 71:538–549.
2011. View Article : Google Scholar :
|
|
25
|
Szabó DR, Luconi M, Szabó PM, Tóth M,
Szücs N, Horányi J, Nagy Z, Mannelli M, Patócs A, Rácz K, et al:
Analysis of circulating microRNAs in adrenocortical tumors. Lab
Invest. 94:331–339. 2014. View Article : Google Scholar
|
|
26
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: Are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guo H, Liu H, Mitchelson K, Rao H, Luo M,
Xie L, Sun Y, Zhang L, Lu Y, Liu R, et al: MicroRNAs-372/373
promote the expression of hepatitis B virus through the targeting
of nuclear factor I/B. Hepatology. 54:808–819. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Prueitt RL, Yi M, Hudson RS, Wallace TA,
Howe TM, Yfantis HG, Lee DH, Stephens RM, Liu CG, Calin GA, et al:
Expression of microRNAs and protein-coding genes associated with
perineural invasion in prostate cancer. Prostate. 68:1152–1164.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Q, Wang G, Shan J-L, Yang ZX, Wang HZ,
Feng J, Zhen JJ, Chen C, Zhang ZM, Xu W, et al: MicroRNA-224 is
upregulated in HepG2 cells and involved in cellular migration and
invasion. J Gastroenterol Hepatol. 25:164–171. 2010. View Article : Google Scholar
|
|
30
|
Bhaskar PT and Hay N: The two TORCs and
Akt. Dev Cell. 12:487–502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tokunaga C, Yoshino K and Yonezawa K: mTOR
integrates amino acid- and energy-sensing pathways. Biochem Biophys
Res Commun. 313:443–446. 2004. View Article : Google Scholar
|
|
32
|
Betz C and Hall MN: Where is mTOR and what
is it doing there? J Cell Biol. 203:563–574. 2013. View Article : Google Scholar :
|
|
33
|
Frias MA, Thoreen CC, Jaffe JD, Schroder
W, Sculley T, Carr SA and Sabatini DM: mSin1 is necessary for
Akt/PKB phosphorylation, and its isoforms define three distinct
mTORC2s. Curr Biol. 16:1865–1870. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Betz C, Stracka D, Prescianotto-Baschong
C, Frieden M, Demaurex N and Hall MN: Feature Article: mTOR complex
2-Akt signaling at mitochondria-associated endoplasmic reticulum
membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad
Sci USA. 110:12526–12534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chang HY and Yang X: Proteases for cell
suicide: Functions and regulation of caspases. Microbiol Mol Biol
Rev. 64:821–846. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Beurel E and Jope RS: The paradoxical pro-
and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic
apoptosis signaling pathways. Prog Neurobiol. 79:173–189. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yo YT, Shieh GS, Hsu KF, Wu CL and Shiau
AL: Licorice and licochalcone-A induce autophagy in LNCaP prostate
cancer cells by suppression of Bcl-2 expression and the mTOR
pathway. J Agric Food Chem. 57:8266–8273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen Y, Robles AI, Martinez LA, Liu F,
Gimenez-Conti IB and Conti CJ: Expression of G1 cyclins,
cyclin-dependent kinases, and cyclin-dependent kinase inhibitors in
androgen-induced prostate proliferation in castrated rats. Cell
Growth Differ. 7:1571–1578. 1996.PubMed/NCBI
|
|
40
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhong W, Peng J, He H, Wu D, Han Z, Bi X
and Dai Q: Ki-67 and PCNA expression in prostate cancer and benign
prostatic hyperplasia. Clin Invest Med. 31:E8–E15. 2008.PubMed/NCBI
|