Introduction
Human T-cell leukemia virus type 1 (HTLV-1) is
etiologically associated with two major diseases, adult T-cell
leukemia (ATL) and HTLV-1-associated myelopathy/tropical spastic
paraparesis (HAM/TSP) (1). ATL is
an aggressive malignant disease of CD4+ T cells that is
highly resistant to currently available chemotherapies, and HAM/TSP
presents with inflammatory symptoms and incomplete paralysis of the
limbs. Although various features of HTLV-1 biology have been
defined, the treatment of the aggressive subtypes of ATL and
HAM/TSP remains inadequate with minimal improvements.
Carotenoids have numerous bioactivities. In
particular, the marine carotenoid, fucoxanthin, and its metabolite,
fucoxanthinol, have multiple functions and studies from our
laboratories have shown that both carotenoids have considerable
potential for preventions and treatment of cancer (2–5). In
contrast, the bioactivities of peridinin, a carotenoid with a
structure similar to that of fucoxanthin (Fig. 1), have not been well studied.
Peridinin is cytotoxic to colorectal cancer cells in vitro
(6). However, so far, there is no
report on the use of peridinin in HTLV-1-associated diseases
including ATL. In this study, we tested the anti-ATL activity of
peridinin isolated from the gorgonian Isis hippuris both
in vitro and in vivo, and determined the underlying
molecular mechanisms of its anti-ATL effects. The results showed
the sensitivity of HTLV-1-infected T-cell lines to peridinin and
that peridinin is a potentially promising drug for
HTLV-1-associated diseases.
Materials and methods
Cell culture
HTLV-1-transformed T-cell lines, MT-2, MT-4,
HUT-102, C5/MJ and SLB-1, and ATL-derived T-cell lines, MT-1 and
ED-40515(-), were maintained in RPMI-1640 medium (cat. no.
30264-56, Nacalai Tesque, Inc., Kyoto, Japan) supplemented with 10%
fetal bovine serum (Biological Industries, Kibbutz Beit Haemek,
Israel) and 1% penicillin/streptomycin (cat. no. 09367-34, Nacalai
Tesque, Inc.)
Purification of peridinin
A hexane extract (totally 36.2 g) of the gorgonian
I. hippuris, collected July 1980, was subjected to
separation first on a silica gel column and then on Sephadex LH20
(cat. no. 17-0090-03, Pharmacia, Uppsala, Sweden) column to give a
crude peridinin fraction (591 mg), which was kept frozen for >30
years. Then, the fraction was passed through a Sephadex LH20 column
(MeOH-CH2Cl2, 1-1) twice followed by silica
(silica gel 60; cat. no. 107734, Merck, Darmstadt, Germany) flash
column and silica HPLC (cat. no. 38005-51, Cosmosil 5SL-II, 10×250
mm; Nacalai Tesque, Inc.) (hexane-EtOAc, 1-1) to yield 9.0 mg of
peridinin. Additional amount of peridinin was purified from crude
fractions of another specimen of the same gorgonian, with a final
total yield of 20.0 mg of peridinin. The 1H NMR spectrum
of the isolated peridinin in CDCl3 (cat. no. 139-18001,
Wako Pure Chemical Industries, Osaka, Japan) was identical to that
reported previously (7).
Measurement of cell proliferation and
cytotoxicity
Cell proliferation and cytotoxicity were measured by
the water-soluble tetrazolium (WST)-8 assay (cat. no. 07553-44,
Nacalai Tesque, Inc.). The WST-8 is taken up by viable cells and
reduced to the colored formazan product by mitochondrial
dehydrogenase (8). For the WST-8
assay, 100 μl (1×104) cells were seeded in 96-well
plates and treated with different concentrations of peridinin or
dimethyl sulfoxide (DMSO) (cat. no. 13407-45, Nacalai Tesque, Inc.)
for 24 h. Then, 10 μl of WST-8 was added to each well and incubated
for 6 h. Absorbance was measured at 450 and 620 nm by an iMark™
microplate absorbance reader (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). Cell viability was calculated by the percentage of
treated cells relative to that of solvent controls. All experiments
were performed in triplicates.
Cell cycle analysis
The cells were treated with different concentrations
of peridinin or DMSO for 24 h and stained with the CycleTEST Plus
DNA Reagent kit (cat. no. 340242, Becton-Dickinson Immunocytometry
Systems, San Jose, CA, USA) for analysis of changes in the cell
cycle. The stained cells were analyzed by an Epics XL flow
cytometer (Beckman Coulter, Inc., Brea, CA, USA). The MultiCycle
software (version 3.0) was used to calculate the percentage of
cells in each cell cycle phase.
Apoptosis assay
Apoptosis was assessed by the APO2.7 assay. Cells
were seeded in culture plates then treated with different
concentrations of peridinin or DMSO for 24 h, followed by analysis
by flow cytometry after staining with phycoerythrin-conjugated
APO2.7 antibody (cat. no. IM2088, Beckman Coulter, Marseille,
France), which specifically detects 7A6, a 38-kDa mitochondrial
membrane antigen expressed during apoptosis (9). For analysis of morphological changes
in nuclei, cells stained with DNA-specific Hoechst 33342 dye (cat.
no. 346-07951, Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) were analyzed using a Leica DMI6000 microscope (Leica
Microsystems, Wetzlar, Germany). In addition, apoptosis was also
assessed by monitoring cleavage of caspase-3, -8 and -9, as well as
poly(ADP-ribose) polymerase (PARP) by western blot analysis.
Protein extraction and western
blotting
Cells were harvested after treatment and lysed in a
lysis buffer containing 62.5 mM Tris-HCl (pH 6.8) (cat. no.
35434-21, Nacalai Tesque, Inc.), 2% sodium dodecyl sulfate (cat.
no. 31607-65, Nacalai Tesque, Inc.), 10% glycerol (cat. no.
17045-65, Nacalai Tesque, Inc.), 6% 2-mercaptoethanol (cat. no.
21438-82, Nacalai Tesque, Inc.) and 0.01% bromophenol blue (cat.
no. 021-02911, Wako Pure Chemical Industries). The lysates were
centrifuged and the supernatants were collected. The cell lysates
(20 μg) were separated on sodium dodecyl sulfate-polyacrylamide
gels followed by electroblotting to polyvinylidene difluoride
membranes (cat. no. IPVH00010EMD, Millipore, Darmstadt, Germany).
The following antibodies were used: cleaved caspase-3 (cat. no.
9664), -8 (cat. no. 9496) and -9 (cat. no. 9501), cleaved PARP
(cat. no. 9541), survivin (cat. no. 2808), Bak (cat. no. 3814),
Bcl-xL (cat. no. 2762), Akt (cat. no. 9272), phospho-Akt (Ser473)
(cat. no. 4060), phospho-Akt (Thr308) (cat. no. 13038),
phospho-IκBα (Ser32 and Ser36) (cat. no. 9246), phospho-IκB kinase
(IKK)α/β (Ser176 and Ser180) (cat. no. 2694), RelA (cat. no. 8242),
phospho-RelA (Ser536) (cat. no. 3033), 3-phosphoinositide-dependent
protein kinase 1 (PDK1) (cat. no. 3062), phospho-PDK1 (Ser241)
(cat. no. 3061), phospho-p70 S6 kinase (S6K) (Thr421 and Ser424)
(cat. no. 9204), phospho-phosphatase and tensin homologue (PTEN)
(Ser380) (cat. no. 9551) and phospho-extracellularly regulated
kinase (Erk)1/2 (Thr202 and Tyr204) (cat. no. 4370) were all from
Cell Signaling Technology, Inc. (Beverly, MA, USA); c-IAP2 (cat.
no. sc-7944), cyclin D2 (cat. no. sc-593) and IκBα (cat. no.
sc-371) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA); XIAP (cat. no. M044-3) and cyclin D1 (cat. no. K0062-3) were
from Medical & Biological Laboratories, Co. (Aichi, Japan);
Bcl-2 (cat. no. MS-597), CDK4 (cat. no. MS-299), CDK6 (cat. no.
MS-398), c-Myc (cat. no. MS-1054) and actin (cat. no. MS-1295) were
from Neomarkers, Inc. (Fremont, CA, USA).
Preparation of nuclear extracts and
electrophoretic mobility shift assay (EMSA)
Nuclear proteins were extracted and transcription
factors bound to specific DNA sequences were examined by EMSA, as
described previously (10). The
top strand sequences of the oligonucleotide probe or competitors
were as follows: for the nuclear factor-κB (NF-κB) element of the
interleukin-2 receptor α chain (IL-2Rα) gene,
5′-GATCCGGCAGGGGAATCTCCCTCTC-3′; and for the
activator protein-1 (AP-1) element of the interleukin-8
(IL-8) gene, 5′-GATCGTGATGACTCAGGTT-3′. The above
underlined sequences are the NF-κB and AP-1 binding sites,
respectively. In competition experiments, the nuclear extracts were
preincubated with 100-fold excess of unlabeled oligonucleotides for
15 min. To identify NF-κB proteins in the DNA-protein complex shown
by EMSA, antibodies specific for various NF-κB family proteins,
including p50 (cat. no. sc-114X), RelA (cat. no. sc-109X), c-Rel
(cat. no. sc-70X), p52 (cat. no. sc-298X) and RelB (cat. no. 226X)
(Santa Cruz Biotechnology Inc.) were used. These antibodies were
incubated with the nuclear extracts for 45 min at room temperature
before incubation with the radiolabeled probe.
Xenograft tumor model
Five-week-old female C.B-17/Icr-severe combined
immune deficiency (SCID) mice were obtained from Kyudo, Co. (Tosu,
Japan). The mice were kept in specific pathogen-free conditions and
housed in cages maintained in air-conditioned rooms (temperature,
24°C; humidity, 60%) set at 12-h light/12-h dark cycles. Mice were
provided with standard rodent diet (CE-2 from CLEA Japan, Inc.,
Tokyo, Japan) and water ad libitum. To induce malignancy,
1×107 HUT-102 cells suspended in 300 μl sterile
RPMI-1640 medium were inoculated subcutaneously into the
postauricular region of the SCID mice, which were then divided
randomly into two treatment groups (n=6/group). Peridinin was
solubilized in 5.2% polyethylene glycol 400 (cat. no. 161-09065,
Wako Pure Chemical Industries) and 5.2% Tween-80 (cat. no. 231181,
Becton-Dickinson, Franklin Lakes, NJ, USA) at a concentration of
0.56 mg/ml, and administered intraperitoneally for 5 days/week with
a 2-day rest, and the treatment was continued for 21 days,
beginning on the day subsequent to cell inoculation. The control
group received vehicle only, while the treated group received
peridinin at dose of 8.5 mg/kg. Tumor diameter was measured weekly
with a shifting caliper, and tumor volume was calculated. Mice were
weighed weekly. All mice were sacrificed on day 21 when tumors did
not reach the ethically allowed maximal size. Subsequently, tumors
were excised and their weight was measured. This study was
performed according to the Guidelines for Animal Experimentation of
the University of the Ryukyus (Nishihara, Japan), and was approved
by the Animal Care and Use Committee of the University of the
Ryukyus (reference no. 6042).
Morphological analysis of tumor tissues
and terminal deoxynucleotidyl transferase deoxyuridine triphosphate
nick end labeling (TUNEL) assay
Tumor specimens were collected from the control and
peridinin-treated groups, fixed in formalin (Wako Pure Chemical
Industries) solution, dehydrated through graded ethanol series
(Japan Alcohol Selling Co., Tokyo, Japan) and embedded in paraffin
(cat. no. 09620, Sakura Finetek Japan Co., Tokyo, Japan). The
paraffin-embedded specimens of ATL tumors were stained with
hematoxylin and eosin (H&E; cat. nos. 234-12 and 1159350025,
Merck) and examined histologically. Analysis of DNA fragmentation
by TUNEL assay was performed using a commercial kit (cat. no.
11684817910, Roche Applied Science, Penzberg, Germany). Cells were
examined under a light microscope (Axioskop 2 Plus) with an
Achroplan 40x/0.65 lens (both from Zeiss, Hallbergmoos, Germany).
Images were acquired with an AxioCam 503 color and AxioVision LE64
software (Zeiss).
Biomarker analysis
Serum concentrations of human soluble IL-2R (sIL-2R;
cat. no. DR2A00, R&D Systems, Inc., Minneapolis, MN, USA) and
human soluble cluster of differentiation 30 (sCD30; cat. no.
RBMS240R, BioVendor Inc., Brno, Czech Republic) were measured by
enzyme-linked immunosorbent assay (ELISA), according to the
protocol supplied by the manufacturer.
Statistical analysis
All values are expressed as the mean ± standard
deviation (SD). Differences between the control and treated groups
were tested for statistical significance by the unpaired t-test. A
P-value <0.05 denoted the presence of statistically significant
difference.
Results
Peridinin suppresses the survival of
HTLV-1-infected T-cell lines
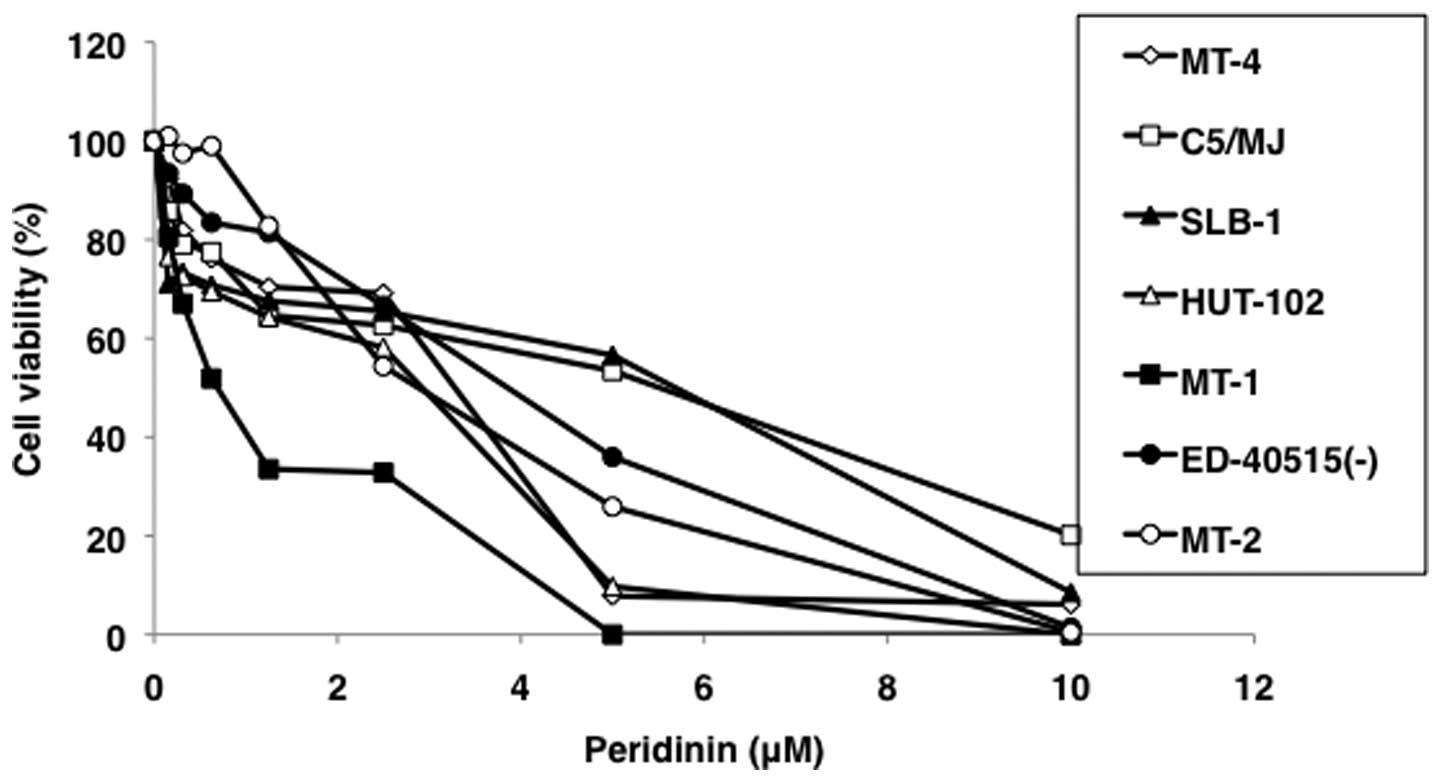
The suppressive effect of peridinin on the survival
of HTLV-1-transformed T-cell lines MT-2, MT-4, HUT-102, C5/MJ,
SLB-1, and patient-derived ATL cell lines MT-1 and ED-40515(-) was
confirmed by WST-8 assay. Furthermore, peridinin dose-dependently
suppressed the viability of HTLV-1-infected T-cell lines (Fig. 2). The IC50 values of
peridinin on HTLV-1-infected T-cell lines as estimated by the WST-8
assay were 0.71–5.38 μM (Table
I).
 | Table IThe IC50 values for
HTLV-1-infected T-cell lines. |
Table I
The IC50 values for
HTLV-1-infected T-cell lines.
| Cell line | IC50
(μM) |
|---|
|
HTLV-1-transformed | MT-2 | 2.23 |
|
HTLV-1-transformed | MT-4 | 3.06 |
|
HTLV-1-transformed | C5/MJ | 5.15 |
|
HTLV-1-transformed | SLB-1 | 5.38 |
|
HTLV-1-transformed | HUT-102 | 2.76 |
| ATL-derived | MT-1 | 0.71 |
| ATL-derived | ED-40515(-) | 3.53 |
Effects of peridinin on cell cycle
distribution
Disturbance of the cell cycle is one of therapeutic
targets for development of new anticancer drugs. The MT-2 and
HUT-102 cell lines were treated with different concentrations of
peridinin for 24 h and cell cycle was analyzed by propidium iodide
staining and DNA content analysis by flow cytometry. Low-dose
peridinin (1.25, 2.5 and 5.0 μM) significantly reduced the
proportion of cells in the S phase but increased those of cells in
the G1 phase population (Fig. 3A and B). These results suggest that
peridinin causes G1 cell cycle arrest in HTLV-1-infected
T-cell lines. The increase in G1 cell cycle arrest was
peridinin dose- and time-dependent (Fig. 3). On the other hand, the
percentages of cells in the sub-G1 phase of the
high-dose group (10 μM) (MT-2, 28.4%; HUT-102, 21.3%) were
significantly higher than those of low-dose groups and control
(MT-2, 3.1%; HUT-102, 6.9%) at 24 h (Fig. 3A). Furthermore, peridinin at high
dose markedly increased the percentage of apoptotic cells of
sub-G1 population and this effect was time-dependent
(Fig. 3C).
Effects of peridinin on apoptosis of
HTLV-1-infected T-cell lines
MT-2 and HUT-102 cells were treated with different
concentrations of peridinin for 24 h and apoptosis was assessed by
APO2.7 staining. There was no significant difference in rates of
apoptosis between the low-dose groups (1.25, 2.5 and 5.0 μM) and
negative control at 24 h, while high-dose peridinin (10 μM) induced
significant apoptosis of MT-2 and HUT-102 cells (Fig. 4A). As shown in Fig. 4B, by Hoechst-stained MT-2 cells
showed distinct morphological features of chromatin condensation
and fragmented nuclei in the presence of 10 μM peridinin. In
another method for apoptosis based on caspase-3, -8, -9 and PARP,
increases in cleaved caspase-3, -8, -9 and PARP expression levels
were detected in MT-2 cells treated with 10 μM peridinin (Fig. 4C).
Apoptosis is regulated by a balance between
pro-apoptotic and anti-apoptotic proteins. The Bcl-2 family members
play major roles in cell survival and apoptosis (11). For this reason, we measured the
effects of peridinin on the expression of Bcl-2, Bcl-xL and Bak by
western blot analysis. Peridinin inhibited the expression of Bcl-2
in a dose-dependent manner, but not that of Bcl-xL and Bak.
Inhibitor of apoptosis (IAP) proteins, including XIAP, c-IAP2 and
survivin directly bind to activated caspase-3, -7 and -9, and
inhibit their activities (12). As
shown in Fig. 5, peridinin reduced
the protein levels of survivin and XIAP, but not c-IAP2, in
cultured MT-2 cells and the effect was dose-dependent. These
results suggest that peridinin inhibits cell survival and induces
apoptosis through the regulation of Bcl-2 family members and
IAPs.
Cell cycle progression is governed by a family of
cyclins and CDKs, and cyclin D/CDK4/CDK6 is a critical determinant
of progression through G1 phase of the cell cycle
(13). We next evaluated the
effects of peridinin on cyclins and CDKs involved in cell cycle
arrest in MT-2 cells. As shown in Fig.
5, peridinin significantly decreased cyclin D1, cyclin D2, CDK4
and CDK6 levels in MT-2 cells.
Proto-oncogene c-myc encodes a basic
loop-helix-loop zipper transcription factor that plays crucial
roles in cell proliferation, apoptosis, differentiation and
metabolism (14,15). c-Myc can activate the expression of
many downstream cell cycle regulators, such as cyclin D1, cyclin D2
and CDK4 (14–16). Peridinin downregulated c-Myc
protein levels in MT-2 cells (Fig.
5), suggesting that c-Myc served as an upstream target in the
peridinin-mediated blockade of cell cycle progression.
Peridinin inhibits NF-κB
NF-κB encompases a family of transcription factors
that are involved in numerous biological processes including cell
growth and survival. The NF-κB proteins are usually sequestered in
the cytoplasm by a family of inhibitors including IκBα. Activation
of NF-κB occurs via phosphorylation of IκBα at Ser32 and Ser36.
This is followed by proteasome-mediated degradation resulting in
release and nuclear translocation of active NF-κB, where it
regulates the expression of several pro-survival proteins and cell
cycle regulatory molecules (17).
Interestingly, many genes downregulated by peridinin (cyclin D1,
cyclin D2, CDK4, CDK6, c-Myc, Bcl-2, XIAP and survivin) are also
regulated by NF-κB (18–25). Culture of MT-2 cells in the
presence of peridinin resulted in a significant dose-dependent
inhibition of IκBα protein phosphorylation (Fig. 6A). Pursuing further this line of
experiments, we examined, using the EMSA, the binding of NF-κB
family proteins to the NF-κB sequence of the IL-2Rα gene. To
validate the relevance of the visualized bands with regard to the
presence and specificity of the NF-κB-DNA complex, we incubated
nuclear extracts from MT-2 cells with antibodies specific for NF-κB
family proteins as well as to an excess of unlabeled competitors
NF-κB and AP-1 sequences. The band completely disappeared by excess
unlabeled NF-κB sequence, but not by AP-1 sequence, and was
affected by preincubation with antibodies specific for p50, RelA,
c-Rel, p52 and RelB, indicating that this band contains the
specific DNA-NF-κB subunit complex (Fig. 6B, right panel). Treatment with
peridinin completely abolished this binding process (Fig. 6B, left panel), indicating that
peridinin inhibits the NF-κB pathway. This inhibition can result
from upstream signaling events or from direct inhibition of one or
more stages of the NF-κB pathway. This latter possibility was
assessed by determining the types of proteins along the NF-κB
pathway that are direct targets for peridinin.
Attenuation of phosphorylation of IκBα (Fig. 6A) suggested that the upstream
kinase IKKβ was the likely site of the inhibitory action of
peridinin. To further explore this tenable conclusion, we examined
IKKβ phosphorylation. The results showed that peridinin suppressed
the phosphorylation of IKKβ (Fig.
6A).
Peridinin causes inhibition of
phosphorylation of Akt protein
RelA is phosphorylated at Ser536 by a variety of
kinases through various signaling pathways and such phosphorylation
enhances RelA transactivation potential (26). As shown in Fig. 6A, peridinin significantly inhibited
phosphorylation of RelA in MT-2 cells. Akt is a component of an
essential pathway for cell survival and growth during
carcinogenesis, and it can also phosphorylate RelA at Ser536
through an IKKα- or IKKβ-dependent mechanism (26). Peridinin induced dose-dependent
inhibition of Akt phosphorylation at Ser473 and Thr308 in MT-2
cells (Fig. 6C). Furthermore,
peridinin suppressed the phosphorylation of IKKα and IKKβ (Fig. 6A). Considered together, the above
results suggest that peridinin inhibits RelA phosphorylation by
inhibiting Akt and IKK activation.
Peridinin inhibits PDK1 protein
expression
PDK1 is an immediate downstream mediator of
phosphoinositide 3-kinase (PI3K) and is activated upon
phosphorylation at Ser241 (27).
Activated PDK1 controls cell proliferation and survival by further
activating its downstream cAMP-dependent, cGMP-dependent, protein
kinase C family of protein kinases, including Akt (28) and S6K (29). We next examined the effect of
peridinin on PDK1 protein. Peridinin reduced phosphorylation and
protein expression of PDK1 (Fig.
6C) and induced significant inhibition of S6K
phosphorylation.
The tumor suppressor PTEN directly dephosphorylates
phosphatidylinositol 3,4,5-triphosphate (PIP3) to PI(4,5)P2
and opposes the PI3K/Akt signaling (30). The oncogenicity of abnormal
PI3K/Akt signaling is emphasized by the observation that
deactivating mutations of the gene encoding PTEN are among the most
frequently occurring in human malignancy (30). However, ATL cells do not harbor
genetic alterations in PTEN but express high levels of PTEN
that is highly phosphorylated through loss of tumor suppressor
N-myc downstream-regulated gene 2 (31). It is believed that phosphorylation
keeps PTEN in an inactive form in the cytoplasm (32). To evaluate whether peridinin
increases PTEN activity by dephosphorylation, the phosphorylation
status of PTEN was examined. However, peridinin did not affect PTEN
phosphorylation (Fig. 6C). Erk
pathway is also involved in HTLV-1 transforming protein
Tax-mediated apoptosis protection (33), but peridinin did not inhibit Erk
phosphorylation (Fig. 6C).
Peridinin inhibits tumorigenesis in
vivo
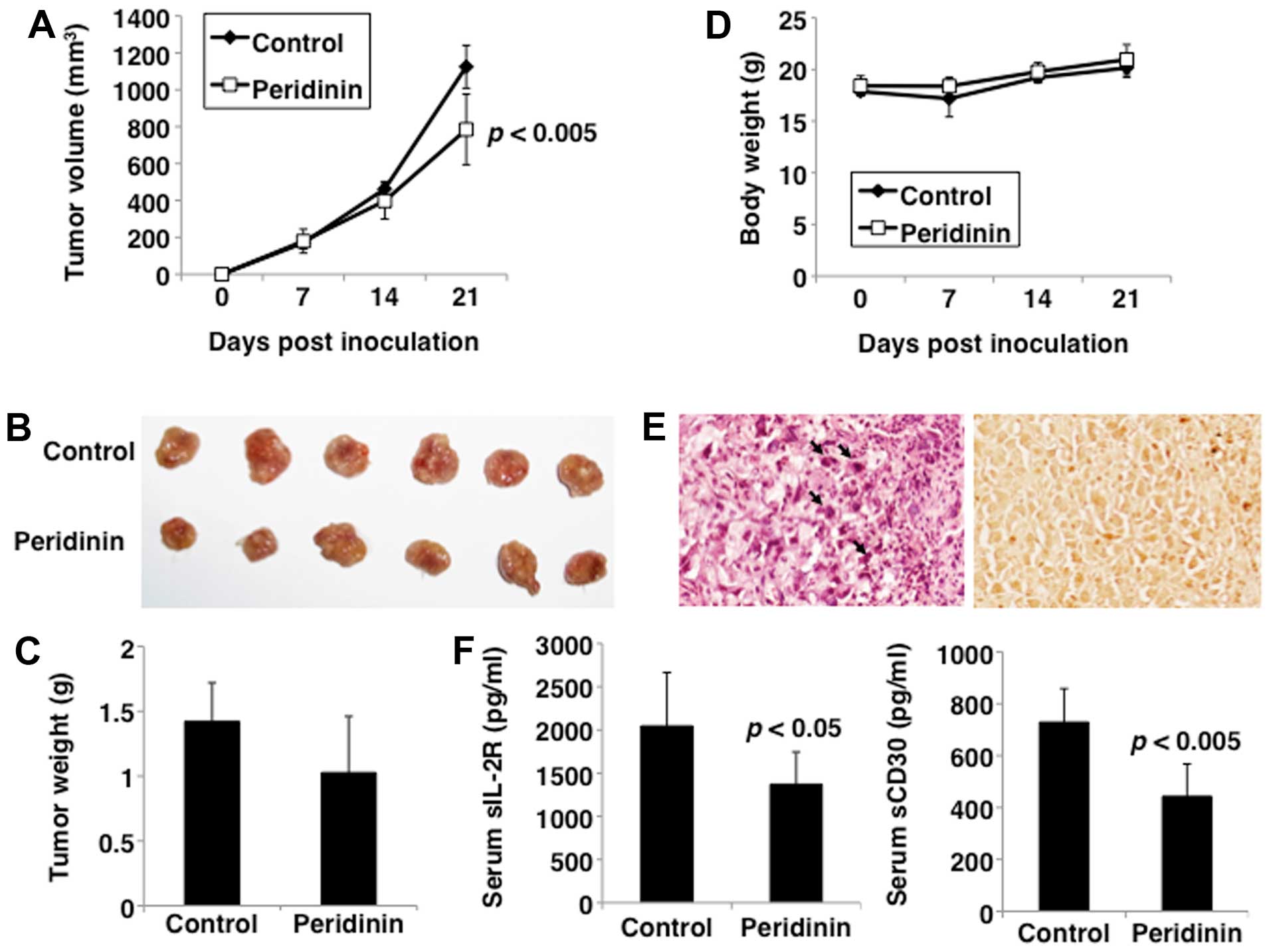
To determine the antitumor activity of peridinin
in vivo, HUT-102 cells were subcutaneously injected into the
postauricular region of the SCID mice. Mice were intraperitoneally
injected with vehicle or peridinin at 8.5 mg/kg 5 days a week over
a period of 21 days. Tumorigenesis was noted in all mice
transplanted with HUT-102 cells (Fig.
7A), but none showed signs of discomfort and/or suffering until
euthanasia. Mice treated with peridinin showed significantly
inhibited tumor growth compared with the control group
(P<0.005). Furthermore, the weight of the excised tumors was
lower after treatment with peridinin compared with those of
untreated mice, albeit statistically insignificant (Fig. 7B and C). In addition, mice seemed
to tolerate treatment with peridinin without overt signs of
toxicity or significant loss of body weight, similar to the
vehicle-treated group (Fig.
7D).
Tumor sections from the treated mice were further
examined by H&E staining. Apoptosis of tumor cells was noted in
the peridinin-treated group, which was characterized by cytoplasmic
condensation, chromatin hyperchromatism and condensation, and
nuclear fragmentation (Fig. 7E,
left panel). TUNEL staining also demonstrated the presence of
abundant apoptotic cells in the tumors of the peridinin-treated
group (Fig. 7E, right panel).
These findings suggest that the observed inhibition of tumor growth
in the peridinin-treated tumors in vivo was mainly due to
increased apoptosis.
Finally, to validate the results of the in
vivo xenograft model, the effect of peridinin on the surrogate
tumor markers sIL-2R (34) and
sCD30 (35) by ELISA was
investigated in serum samples. Compared with the control group,
treatment of mice with peridinin significantly reduced serum sIL-2R
and sCD30 levels (Fig. 7F).
Discussion
Our study showed that peridinin reduces cell
proliferation and viability of HTLV-1-infected T-cell lines. These
properties are similar to those described previously for
fucoxanthin (5). Both carotenoids
have similar structures, however, fucoxanthin is a
C40-skeletal carotenoid with an octaenone chromophore
whereas peridinin is a C37-skeletal butenolide
carotenoid (Fig. 1). These results
suggest no differences in the anti-ATL activities between
butenolide ring-structure and octaenone chromophore
carotenoids.
The goal of this study was to determine the effects
of peridinin on ATL and the mechanism of action. Peridinin potently
suppressed NF-κB activation and also downregulated NF-κB-regulated
gene products. The results also showed that peridinin inhibited
NF-κB activation by suppressing IKK activation, and the latter was
induced by suppressing Akt activation. In fact, Akt has been
reported to activate IKK (26).
While our results indicate that peridinin seems to inhibit IKK
activation through the suppression of Akt activation, we cannot
rule out direct inhibition of IKK activity by peridinin.
Our results also showed suppression of RelA
phosphorylation by peridinin. Both IKK and Akt are involved in RelA
phosphorylation (26). Thus,
peridinin-induced inhibition of RelA phosphorylation could be
mediated through the inhibition of both Akt and IKK. We also
investigated the effect of peridinin on the upstream event of Akt,
PDK1 and PTEN, and the results showed that peridinin inhibited the
phosphorylation of Akt at Thr308 through PDK1 repression, but did
not affect PTEN activity.
Our results showed that peridinin downregulated the
expression of NF-κB-regulated gene products involved in cellular
proliferation (cyclin D1, cyclin D2, CDK4, CDK6 and c-Myc) and
anti-apoptosis (XIAP, Bcl-2 and survivin). Akt also regulates cell
cycle and proliferation by indirectly modulating the levels of
cyclin D1 and cyclin D2 (36). Akt
interacts with and phosphorylates XIAP, a direct inhibitor of
caspase-3 and caspase-9, leading to inhibition of ubiquitination
and degradation of XIAP (37). In
addition, XIAP is known to prevent apoptosis through upregulation
of Akt cell survival signaling pathway (38). In addition, survivin and Bcl-2 have
been shown to be downstream targets of Akt signaling (39,40).
Thus, NF-κB and Akt can collaborate in ATL, depending on various
survival factors. The results indicate that peridinin downregulates
NF-κB-regulated gene products involved in cell proliferation and
cell survival through inactivation of IKK and Akt.
Our in vivo experiments demonstrated that at
a dose of 8.5 mg/kg body weight, peridinin markedly attenuated the
growth of subcutaneous ATL xenografts. Notably, examination of
tumors harvested from peridinin-treated mice showed increased
apoptosis. The results also showed that at that dose, the treated
mice showed no toxicity or any overt signs of ill health. These
results highlight the anti-ATL and safety properties of peridinin.
Further studies are needed to determine the long-term effects and
safety of peridinin against experimentally-induced ATL.
In conclusion, the anti-ATL activity of peridinin
was confirmed both in vitro and in vivo. Future
studies should focus on the efficacy of peridinin and characterize
its therapeutic potential against other human malignancies.
Acknowledgements
The authors thank Dr Taro Uchida for the excellent
assistance. We also thank Dr Michiyuki Maeda for providing
ED-40515(-) and the Fujisaki Cell Center, Hayashibara Biochemical
Laboratories, Inc. (Okayama, Japan) for providing C5/MJ, HUT-102
and MT-1. This study was supported in part by JSPS KAKENHI grant
numbers 15K18414 and 25461428.
References
|
1
|
Kannian P and Green PL: Human T
lymphotropic virus type 1 (HTLV-1): Molecular biology and
oncogenesis. Viruses. 2:2037–2077. 2010. View Article : Google Scholar
|
|
2
|
Rokkaku T, Kimura R, Ishikawa C, Yasumoto
T, Senba M, Kanaya F and Mori N: Anticancer effects of marine
carotenoids, fucoxanthin and its deacetylated product,
fucoxanthinol, on osteosarcoma. Int J Oncol. 43:1176–1186.
2013.PubMed/NCBI
|
|
3
|
Tafuku S, Ishikawa C, Yasumoto T and Mori
N: Anti-neoplastic effects of fucoxanthin and its deacetylated
product, fucoxanthinol, on Burkitt’s and Hodgkin’s lymphoma cells.
Oncol Rep. 28:1512–1518. 2012.PubMed/NCBI
|
|
4
|
Yamamoto K, Ishikawa C, Katano H, Yasumoto
T and Mori N: Fucoxanthin and its deacetylated product,
fucoxanthinol, induce apoptosis of primary effusion lymphomas.
Cancer Lett. 300:225–234. 2011. View Article : Google Scholar
|
|
5
|
Ishikawa C, Tafuku S, Kadekaru T, Sawada
S, Tomita M, Okudaira T, Nakazato T, Toda T, Uchihara JN, Taira N,
et al: Anti-adult T-cell leukemia effects of brown algae
fucoxanthin and its deacetylated product, fucoxanthinol. Int J
Cancer. 123:2702–2712. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sugawara T, Yamashita K, Sakai S, Asai A,
Nagao A, Shiraishi T, Imai I and Hirata T: Induction of apoptosis
in DLD-1 human colon cancer cells by peridinin isolated from the
dinoflagellate, Heterocapsa triquetra. Biosci Biotechnol Biochem.
71:1069–1072. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haugan JA, Englert G, Aakermann T, Glinz
E, Liaaen-Jensen S, Balzarini J, Fransson B, Ragnarsson U and
Francis GW: Algal carotenoids 58. Isomerization studies on
peridinin. Acta Chem Scand. 48:769–779. 1994. View Article : Google Scholar
|
|
8
|
Ishiyama M, Miyazono Y, Sasamoto K, Ohkura
Y and Ueno K: A highly water-soluble disulfonated tetrazolium salt
as a chromogenic indicator for NADH as well as cell viability.
Talanta. 44:1299–1305. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang C, Ao Z, Seth A and Schlossman SF: A
mitochondrial membrane protein defined by a novel monoclonal
antibody is preferentially detected in apoptotic cells. J Immunol.
157:3980–3987. 1996.PubMed/NCBI
|
|
10
|
Mori N and Prager D: Transactivation of
the interleukin-1alpha promoter by human T-cell leukemia virus type
I and type II Tax proteins. Blood. 87:3410–3417. 1996.PubMed/NCBI
|
|
11
|
Besbes S, Mirshahi M, Pocard M and Billard
C: New dimension in therapeutic targeting of BCL-2 family proteins.
Oncotarget. 6:12862–12871. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li G, Chang H, Zhai YP and Xu W: Targeted
silencing of inhibitors of apoptosis proteins with siRNAs: A
potential anti-cancer strategy for hepatocellular carcinoma. Asian
Pac J Cancer Prev. 14:4943–4952. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dang CV: c-Myc target genes involved in
cell growth, apoptosis, and metabolism. Mol Cell Biol. 19:1–11.
1999. View Article : Google Scholar
|
|
15
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pajic A, Spitkovsky D, Christoph B,
Kempkes B, Schuhmacher M, Staege MS, Brielmeier M, Ellwart J,
Kohlhuber F, Bornkamm GW, et al: Cell cycle activation by c-myc in
a Burkitt lymphoma model cell line. Int J Cancer. 87:787–793. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou J, Ching YQ and Chng WJ: Aberrant
nuclear factor-kappa B activity in acute myeloid leukemia: From
molecular pathogenesis to therapeutic target. Oncotarget.
6:5490–5500. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iwanaga R, Ohtani K, Hayashi T and
Nakamura M: Molecular mechanism of cell cycle progression induced
by the oncogene product Tax of human T-cell leukemia virus type I.
Oncogene. 20:2055–2067. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang Y, Ohtani K, Iwanaga R, Matsumura Y
and Nakamura M: Direct trans-activation of the human cyclin D2 gene
by the oncogene product Tax of human T-cell leukemia virus type I.
Oncogene. 20:1094–1102. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Iwanaga R, Ozono E, Fujisawa J, Ikeda MA,
Okamura N, Huang Y and Ohtani K: Activation of the cyclin D2 and
cdk6 genes through NF-kappaB is critical for cell-cycle progression
induced by HTLV-I Tax. Oncogene. 27:5635–5642. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duyao MP, Kessler DJ, Spicer DB,
Bartholomew C, Cleveland JL, Siekevitz M and Sonenshein GE:
Transactivation of the c-myc promoter by human T cell leukemia
virus type 1 tax is mediated by NF κB. J Biol Chem.
267:16288–16291. 1992.PubMed/NCBI
|
|
22
|
Kawakami A, Nakashima T, Sakai H, Urayama
S, Yamasaki S, Hida A, Tsuboi M, Nakamura H, Ida H, Migita K, et
al: Inhibition of caspase cascade by HTLV-I tax through induction
of NF-kappaB nuclear translocation. Blood. 94:3847–3854.
1999.PubMed/NCBI
|
|
23
|
Akita K, Kawata S and Shimotohno K:
p21WAF1 modulates NF-kappaB signaling and induces
anti-apoptotic protein Bcl-2 in Tax-expressing rat fibroblast.
Virology. 332:249–257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kawakami H, Tomita M, Matsuda T, Ohta T,
Tanaka Y, Fujii M, Hatano M, Tokuhisa T and Mori N: Transcriptional
activation of survivin through the NF-kappaB pathway by human
T-cell leukemia virus type I tax. Int J Cancer. 115:967–974. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hinz M, Krappmann D, Eichten A, Heder A,
Scheidereit C and Strauss M: NF-kappaB function in growth control:
Regulation of cyclin D1 expression and G0/G1-to-S-phase transition.
Mol Cell Biol. 19:2690–2698. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Viatour P, Merville MP, Bours V and
Chariot A: Phosphorylation of NF-kappaB and IkappaB proteins:
Implications in cancer and inflammation. Trends Biochem Sci.
30:43–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Casamayor A, Morrice NA and Alessi DR:
Phosphorylation of Ser-241 is essential for the activity of
3-phosphoinositide-dependent protein kinase-1: Identification of
five sites of phosphorylation in vivo. Biochem J. 342:287–292.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alessi DR, James SR, Downes CP, Holmes AB,
Gaffney PRJ, Reese CB and Cohen P: Characterization of a
3-phosphoinositide-dependent protein kinase which phosphorylates
and activates protein kinase Balpha. Curr Biol. 7:261–269. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pullen N, Dennis PB, Andjelkovic M, Dufner
A, Kozma SC, Hemmings BA and Thomas G: Phosphorylation and
activation of p70s6k by PDK1. Science. 279:707–710.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hollander MC, Blumenthal GM and Dennis PA:
PTEN loss in the continuum of common cancers, rare syndromes and
mouse models. Nat Rev Cancer. 11:289–301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakahata S, Ichikawa T, Maneesaay P, Saito
Y, Nagai K, Tamura T, Manachai N, Yamakawa N, Hamasaki M,
Kitabayashi I, et al: Loss of NDRG2 expression activates PI3K-AKT
signalling via PTEN phosphorylation in ATLL and other cancers. Nat
Commun. 5:33932014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rahdar M, Inoue T, Meyer T, Zhang J,
Vazquez F and Devreotes PN: A phosphorylation-dependent
intramolecular interaction regulates the membrane association and
activity of the tumor suppressor PTEN. Proc Natl Acad Sci USA.
106:480–485. 2009. View Article : Google Scholar :
|
|
33
|
Stoppa G, Rumiato E and Saggioro D: Ras
signaling contributes to survival of human T-cell leukemia/lymphoma
virus type 1 (HTLV-1) Tax-positive T-cells. Apoptosis. 17:219–228.
2012. View Article : Google Scholar :
|
|
34
|
Kamihira S, Atogami S, Sohda H, Momita S,
Yamada Y and Tomonaga M: Significance of soluble interleukin-2
receptor levels for evaluation of the progression of adult T-cell
leukemia. Cancer. 73:2753–2758. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nishioka C, Takemoto S, Kataoka S,
Yamanaka S, Moriki T, Shoda M, Watanabe T and Taguchi H: Serum
level of soluble CD30 correlates with the aggressiveness of adult
T-cell leukemia/lymphoma. Cancer Sci. 96:810–815. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dan HC, Sun M, Kaneko S, Feldman RI,
Nicosia SV, Wang HG, Tsang BK and Cheng JQ: Akt phosphorylation and
stabilization of X-linked inhibitor of apoptosis protein (XIAP). J
Biol Chem. 279:5405–5412. 2004. View Article : Google Scholar
|
|
38
|
Cheng JQ, Jiang X, Fraser M, Li M, Dan HC,
Sun M and Tsang BK: Role of X-linked inhibitor of apoptosis protein
in chemoresistance in ovarian cancer: Possible involvement of the
phosphoinositide-3 kinase/Akt pathway. Drug Resist Updat.
5:131–146. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liang Y-L, Wang L-Y, Wu H, Ma D-Z, Xu Z
and Zha X-L: PKB phosphorylation and survivin expression are
cooperatively regulated by disruption of microfilament
cytoskeleton. Mol Cell Biochem. 254:257–263. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pugazhenthi S, Nesterova A, Sable C,
Heidenreich KA, Boxer LM, Heasley LE and Reusch JE-B: Akt/protein
kinase B up-regulates Bcl-2 expression through cAMP-response
element-binding protein. J Biol Chem. 275:10761–10766. 2000.
View Article : Google Scholar
|