Introduction
Sialic acid-binding lectin is a lectin from Rana
catesbeiana oocytes (SBL). SBL preferentially binds to cancer
cells rather than normal cells (1)
because cancer cells often overexpress sialylated glycans on their
surface, which is usually associated with poor prognosis (2). SBL shows an agglutination activity
toward cancer cells by binding to the sialylated glycans on the
surface of cancer cells (1).
SBL also exhibits a prominent antitumor effect on
many types of cancer and tumor cells, such as breast, cervical,
oral cancer, glioblastoma, and T-cell leukemia, but not normal
cells, such as keratinocytes, fibroblasts, and lymphocytes
(3–6). Moreover, the treatment of cancer
cells with SBL ultimately leads to cell death (7). In mice with ascites tumor cells, the
injection of SBL inhibits tumor growth and prolongs the life span,
and sialidase protects cancer cells from SBL toxicity (8). Therefore, this selective antitumor
effect of SBL is due to the sialylated glycans on the surface of
tumors or cancer cells.
SBL is homologous with various members of the
ribonuclease (RNase) A superfamily and is also known as RC-RNase
(9,10). The RNase A superfamily exhibits
RNA-cleavage activity and has three catalytic amino acid residues.
Therefore, SBL also has RNase activity and the conserved catalytic
amino acid residues (His10, Lys35, and
His103). Huang et al demonstrated that the three
amino acid residues in the SBL molecule are required for inducing
cancer cell death as well as RNase activity using recombinant SBL
mutants with these amino acid residues replaced with alanine
residues (11).
The internalization of SBL into cancer cells causes
the degradation of ribosomal RNA, which leads to the inhibition of
protein synthesis and, in turn, induces cell death (8,12,13).
SBL-induced cell death is accompanied by mitochondrial dysfunction
(14), endoplasmic reticulum
stress (15), autophagocytosis
(16), and caspase activation
(3,5). Our previous studies showed that
mitogen-activated protein kinases (MAPKs) were phosphorylated in
two SBL-treated cell lines, human T-cell leukemia Jurkat cells and
malignant mesothelioma NCI-H28 cells (14,17).
However, it remains unclear whether MAPK activation is related to
SBL-induced cell death and how SBL activates MAPKs.
In this study, we found that SBL-induced cell death
and activation of p38 MAPK signaling in human breast cancer cell
lines. The analyses using p38 MAPK-specific inhibitors and short
interference RNA (siRNA) showed that p38 MAPK activation and
expression were associated with SBL-induced cell death.
Furthermore, RNase activity of SBL was required for the observed
SBL-induced cell death. SBL mutant lacking RNase activity indicated
that such RNase activity of SBL was important for SBL-induced p38
MAPK activation and subsequent caspase-3/7 activation. Together,
these data demonstrate that the RNA degradation by SBL triggers the
SBL-induced p38 MAPK activation that leads to cell death mediated
by caspase-3/7 activation.
Materials and methods
Antibodies and reagents
Mouse mAbs against p38 MAPK (no. 612168) and
phospho-p38 MAPK (no. 612280) were obtained from BD Biosciences. A
mouse mAb against β-actin (clone AC-74) was obtained from Sigma. A
rabbit polyclonal antibody against PARP was obtained from Roche
(no. 11835238001). A rabbit polyclonal SBL antibody was established
in our laboratory. Alexa Fluor 488-conjugated goat anti-rabbit IgG
(no. A11008) and Alexa Fluor 546-conjugated Phalloidin (no. A22283)
were obtained from Invitrogen. Native SBL was isolated by
sequential chromatography on Sephadex G-75, DEAE-cellulose,
hydroxyapatite, and SP-Sepharose as described previously (1). Two types of p38 MAPK inhibitor
(SB203580, no. 559389; and SB239063, no. 559404) and a JNK
inhibitor (SP600125, no. 420119) were from Calbiochem. A
pan-caspase inhibitor [zVAD-fmk,
benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone] was from
MBL. Signal Silence p38 MAPK siRNA (no. 6564) and Signal Silence
control siRNA (no. 6568) were from Cell Signaling Technology,
Inc.
Cell culture
Human breast cancer cell lines, SK-BR-3, MCF-7, and
MDA-MB231 were cultured in Dulbecco's modified Eagle's medium
supplemented with 10% fetal bovine serum, penicillin, and
streptomycin.
Cell viability
Cells were plated to each well of 96-well microtiter
plates before treatment with test samples. After the treatment at
37°C for indicated time, WST-8 (Dojindo) was added to each well to
detect viable cells. Color development was recorded at 450 nm using
the PowerScan HT (DS Pharma Biomedical).
Expression vector encoding an SBL
mutant
The full-length cDNA of SBL was prepared from a
bullfrog liver. To prepare the expression vector of the SBL mutant
lacking RNase activity, we replaced His103 of SBL with
Ala by a PCR method using the SBL cDNA in pET11d with a pel
B sequence of the pET22b vector as a template. The cycle
protocol for PCR was as follows: one cycle of 94°C for 2 min; 25
cycles of 94°C for 1 min, 51°C for 30 sec, and 68°C for 6 min; and
one cycle of 68°C for 4 min. The primer set for H103A was
5′-GTAGCGT TTGCTGGAATAGGACGA-3′ and 5′-GGGATATTGATT
CTCACATTTTAC-3′. The cDNA sequence was verified by DNA
sequencing.
Purification of recombinant SBL
E. coli BL21 (DE) pLysS was transformed with
SBL cDNA expression vector and then selected with carbenicillin and
chloramphenicol. The transformed cells were grown at 34°C in
Terrific Broth medium containing 50 μg/ml carbenicillin and 34
μg/ml chloramphenicol until the A600 reached 0.6 before isopropyl
1-thio-β-D-galactoside (IPTG) induction. After IPTG induction at a
final concentration of 0.4 mM for 2 days, the culture supernatant
was collected and then concentrated with 80% saturated ammonium
sulfate. Pellet containing proteins were dissolved in 20 mM HEPES
buffer (pH 7.0) and then dialyzed against the same buffer. The
crude sample was loaded onto a DEAE-Sepharose 6B column (GE
Healthcare), and the flow-through fraction was collected. The
fraction was loaded on a Heparin-Sepharose 6 column (GE Healthcare)
and eluted with 10 mM Tris-HCl (pH 7.5) containing 0.1–0.5 M NaCl.
The fractions containing H103A were loaded on a hydroxyapatite
column after a threefold dilution with 10 mM Tris-HCl (pH 7.5) and
then the flow-through fraction was collected. The purified protein
was resolved by SDS-PAGE and then stained with CBB. The purified
protein concentration was determined by DC protein assay (Bio-Rad
Laboratories).
Knockdown of p38 MAPK by siRNA
Double-stranded siRNAs were transfected into
MDA-MB231 cells using X-treme GENE siRNA Transfection Reagent
(Roche) according to the instruction manual. After transfection for
24 h, the cells were used for the following assays.
Analysis of RNA degradation
For analysis of intracellular RNA degradation, cells
(2×104 cells) were grown in the medium containing SBL.
After incubation for 24, 48, or 72 h, total RNA of the cells was
extracted with TRIzol reagent (Gibco BRL) according to the
instruction manual. A volume of 2 μg RNA was run on a 2% agarose
gel containing formaldehyde, and the RNA bands were visualized by
ethidium bromide.
Immunoblot analysis
Cells were washed twice with cold phosphate-buffered
saline (PBS) and then lysed with lysis buffer (1% Triton X-100, 20
mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA) containing protease
and phosphatase inhibitor cocktails (Roche). After incubation for
10 min on ice, the cell lysates were clarified by centrifugation at
20,400 g for 10 min at 4°C. The resultant supernatant was used in
the following experiments. The protein concentration was determined
using a Protein Quantification Kit-Rapid (Dojindo). For immunoblot
analysis, proteins resolved by SDS-PAGE were transferred to
nitrocellulose membranes. The blots were blocked with 5% fat-free
skim milk in TBS (20 mM Tris-HCl, 150 mM NaCl, pH 7.4) for 1 h.
Then the membranes were probed with each specific antibody.
Immunoreactive bands were detected using an ECL Western blotting
detection reagent (GE Healthcare).
Immunofluorescence analysis
Cells were cultured in growth medium containing test
samples. Cells were then fixed with 4% paraformaldehyde in PBS for
10 min and permeabilized with 0.2% Triton X-100 in PBS for 10 min.
Cells were blocked with 1.2% BSA in PBS before staining with
appropriate primary and secondary antibodies. After labeling with
the indicated antibodies, labeled slides were analyzed using a
fluorescent microscope, TIRFM (Olympus Corp.).
Caspase-3/7 activity
Caspase-3/7 activity was quantified using
SensoLyte® Homogeneous AMC Caspase-3/7 Assay kit
(AnaSpec Inc., no. 7118), according to the instruction manual.
Statistical analysis
Results are expressed as means ± SD and are
representative of at least two or three independent experiments
conducted in triplicate for each experiment. Statistical
comparisons between two groups were made using an unpaired
Student's t-test and among groups using one-way ANOVA followed by a
Tukey's multiple comparison test, with GraphPad Prism Version 5.0
software. A P-value of <0.05 was considered statistically
significant.
Results
SBL induces cell death through p38 MAPK
activation and expression in human breast cancer cell lines
SBL induces cell death in various types of human
cancer cells (7). To examine the
antitumor effect of SBL on breast cancer cells, we used one
estrogen-positive cell line (MCF-7) and two estrogen-negative cell
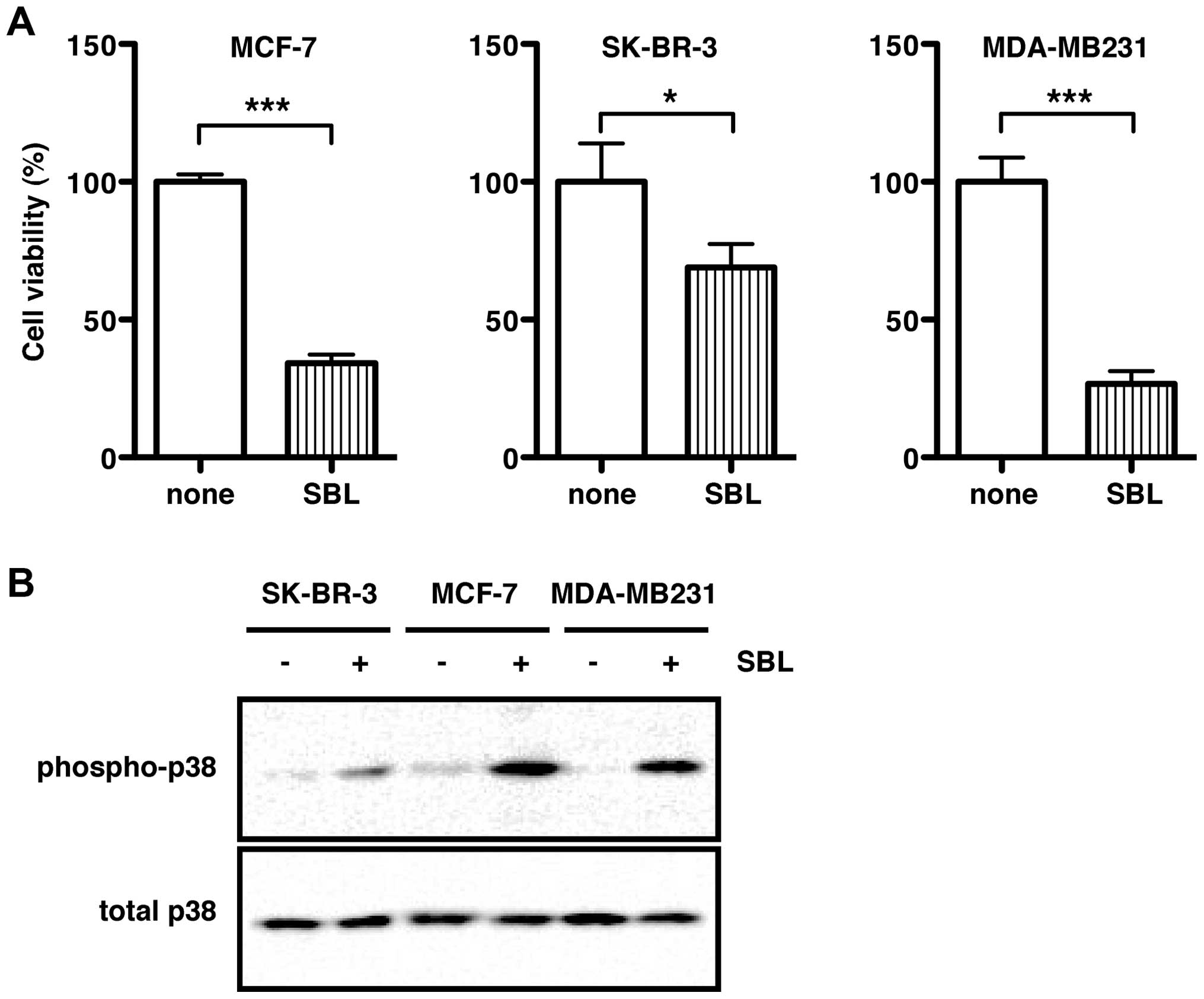
lines (SK-BR-3 and MDA-MB231) for a cell viability assay. After
treatment with SBL for 72 h, the cell viability of MCF-7, SK-BR-3,
and MDA-MB231 was significantly reduced to 30.4, 65.3 and 25.5% of
control levels (none, 100%), respectively (Fig. 1A). As shown in our previous study
using T-cell leukemia Jurkat cells and malignant mesothelioma
NCI-H28 cells (14,17), treatment with SBL markedly induced
phosphorylation of p38 MAPK in all the three breast cancer cell
lines, indicating that SBL activated the p38 MAPK signaling pathway
(Fig. 1B).
The p38 MAPK inhibitor SB203580, sufficiently
blocked SBL-induced phosphorylation of p38 MAPK (Fig. 2A, SB). In contrast, neither JNK
inhibitor, SP600125 (Fig. 2A, SP),
nor the pan-caspase inhibitor, VAD (Fig. 2A, VAD), had an effect on p38 MAPK
phosphorylation. To investigate whether SBL-induced cell death was
mediated by p38 MAPK activation, we tested the inhibitory effect of
the p38 MAPK inhibitor SB203580, on SBL-induced cell death using
MDA-MB231 cells. When treated with SBL, some MDA-MB231 cells shrank
and were dying or dead [Fig. 2B,
dimethyl sulfoxide (DMSO)]. The SBL-induced cell death was markedly
inhibited by the addition of SB203580 (Fig. 2B, SB). The p38 MAPK inhibitors,
SB203580 and SB239063 improved the cell viability in SBL-treated
cells from 36% (Fig. 2C, DMSO/SBL)
to 63% (Fig. 2C, SB/SBL) and 59%
(Fig. 2C, SB239063/SBL),
respectively.
To further investigate the relationship between
SBL-induced cell death and the p38 MAPK signaling pathway, we
performed a cell viability assay using a specific siRNA to p38 MAPK
(p38-siRNA). The expression of the endogenous p38 MAPK molecule was
assessed after 96 h transfection of p38-siRNA or scrambled control
siRNA (con-siRNA). When MDA-MB231 cells were treated with
p38-siRNA, the expression level of the p38 MAPK molecule was
suppressed to a half of that treated with con-siRNA (Fig. 2D). After treating with siRNA for 24
h, cells were further treated with SBL for 48 h. The cell viability
in the presence of SBL was partially rescued by the knockdown of
p38 MAPK molecule (Fig. 2E). These
results suggest that SBL induces cell death of the cancer cells
through activation of p38 MAPK.
RNase activity is required for
SBL-induced cell death
SBL exhibits RNase activity. When cells were treated
with SBL, 28S and 18S ribosomal RNA bands decreased in a
time-dependent manner, and some degraded RNA bands were detected
via formaldehyde-agarose gel electrophoresis (Fig. 3A). RNase activity has been shown to
be necessary for SBL to degrade RNA, as well as to cause cell death
of cancer cells (11). However, it
remains unclear how the SBL-induced RNA degradation leads to cell
death. To clarify the relationship between RNase activity and
SBL-induced cell death, we prepared a recombinant SBL mutant that
lacks RNase activity by replacing His103 (important for
its RNase and cell death-induced activities) with Ala (Fig. 3B, H103A). Escherichia coli
BL21 (DE) pLysS was transformed with H103A cDNA expression vector,
and then the recombinant protein was purified from the culture
medium of H103A-expressing BL21 (DE) pLysS using several
columns.
 | Figure 3RNase activity of SBL is required for
SBL-induced cell death. (A) RNA was extracted from MDA-MB231 cells
treated with or without SBL for the indicated times and then
analyzed on a formaldehyde-agarose gel. Asterisks indicate degraded
RNA. None, no SBL treatment for 72 h. SBL, 2 μM SBL treatment. (B)
For preparing an SBL mutant lacking RNase activity (H103A), the
histidine residue at position 103 essential for its catalytic
activity was replaced with an alanine residue (black dot). Numbers
indicate the number of amino acid residues. (C) Purified proteins
were run on 15% SDS-PAGE gel under reducing conditions and stained
with CBB (left panel). RNase activity of the purified proteins was
analyzed by RNA substrate zymography under non-reducing conditions
(right panel, Zymo). (D) MDA-MB231 cells were treated with 10 μM
SBL or 10 μM H103A for 72 h. None, no SBL treatment for 72 h. Total
RNA was analyzed by formaldehyde-agarose gel electrophoresis.
Asterisks indicate degraded RNA. (E) Cell viability after treatment
with 10 μM H103A for 72 h. The relative cell viability of cells
with no treatment with SBL (none) was set at 100%. Results are the
means ± SD for three independent experiments conducted in
triplicate. ns, not significant. (F) Cell morphology of MDA-MB231
cells after treatment with 2 μM SBL or 10 μM H103A for 72 h.
Arrowheads indicate dying or dead cells. (G) MDA-MB231 cells
treated with 4 μM SBL (middle), 4 μM H103A (right), or without SBL
(left, none) for 6 h were fixed with 4% paraformaldehyde,
permeabilized with 1% Triton X-100/PBS, and then stained with
anti-SBL antibody (green), phalloidin (red), and Hoechst 33342
(blue). |
The quality of the purified H103A protein was
assessed by Coomassie Brilliant Blue (CBB) staining, and its
molecular mass was estimated to be 13 kDa, which was the same as
that of native SBL protein (Fig.
3C, CBB). In a zymographic assay using an RNA-containing gel,
native SBL exhibited RNase activity, but H103A did not (Fig. 3C, Zymo). The cellular RNA from the
treated cells with native SBL was considerably degraded at 72 h
while H103A and no SBL treatment did not induce the cellular RNA
degradation (Fig. 3D).
Furthermore, H103A had no effect on cell viability (Fig. 3E). In agreement with the result of
cell viability, the H103A mutant did not initiate a cell death
phenotype as observed with the native SBL (Fig. 3F). These results indicate that
RNase activity is closely related to SBL-induced cell death.
The internalization of SBL into cells is required
for SBL to degrade cellular RNA (12). To exclude the possibility that
H103A is not capable of cellular internalization, the intracellular
localization of H103A was observed. H103A and native SBL were
localized to the perinuclear region in MDA-MB231 cells at 6 h
(Fig. 3G), indicating that H103A
is internalized.
As shown in Figs. 1
and 2, p38 MAPK activation was
associated with SBL-induced cell death. Therefore, it is likely
that the RNase activity of SBL may be of relevance to p38 MAPK
activation. We then examined the activation of p38 MAPK after
treatment with native SBL and the H103A mutant. Native SBL
increased the phosphorylation of p38 MAPK both at 48 and 72 h. In
contrast, H103A, as well as the untreated controls, did not promote
the phosphorylation of p38 MAPK even at 72 h (Fig. 4A). These results revealed that the
RNase activity of SBL is required for SBL-induced p38 MAPK
activation.
RNA degradation by SBL leads to
activation of caspase-3/7
Although SBL has been reported to activate
caspase-3/7 (3,5,18),
known as the major executioner caspases, the molecular mechanism
remains unclear. Indeed, the caspase-3/7 activity in SBL-treated
MDA-MB231 cells was 22-fold higher than that in the untreated cells
(Fig. 4B, none vs. SBL). This
upregulated caspase-3/7 activity by SBL was completely ablated in
the H103A mutant (Fig. 4B,
H103A).
Active caspase-3/7 cleaves a full-length poly
(ADP-ribose)-polymerase (PARP; 113 kDa) into two fragments: a) 89
kDa and b) 24 kDa. Therefore, we also examined SBL-induced
caspase-3/7 activation by checking the fragments of PARP. The
immunoblot analysis using an anti-PARP antibody showed that SBL
treatment caused an increase in the 89-kDa PARP fragment in a
time-dependent manner, whereas only a 113-kDa PARP band was
detected when H103A was treated for 72 h (Fig. 4C). These results indicate that
RNase activity of SBL is required for SBL-induced caspase-3/7
activation.
Activation of p38 MAPK signaling induced
by SBL leads to activation of caspase-3/7
Since RNA degradation by SBL remarkably induced p38
MAPK and caspase-3/7 activation, we speculated that SBL-induced p38
MAPK activation might be related to caspase-3/7 activation. To test
this hypothesis, we examined caspase-3/7 activity after treatment
with SBL in the presence of a p38 MAPK inhibitor. Caspase-3/7
activity after co-treatment of the p38 MAPK inhibitor, SB203580,
with SBL was suppressed by 41.8% compared with that after
co-treatment with the solvent, DMSO and SBL (Fig. 5A, SB). These results indicate that
the activation of p38 MAPK by SBL leads to caspase-3/7
activation.
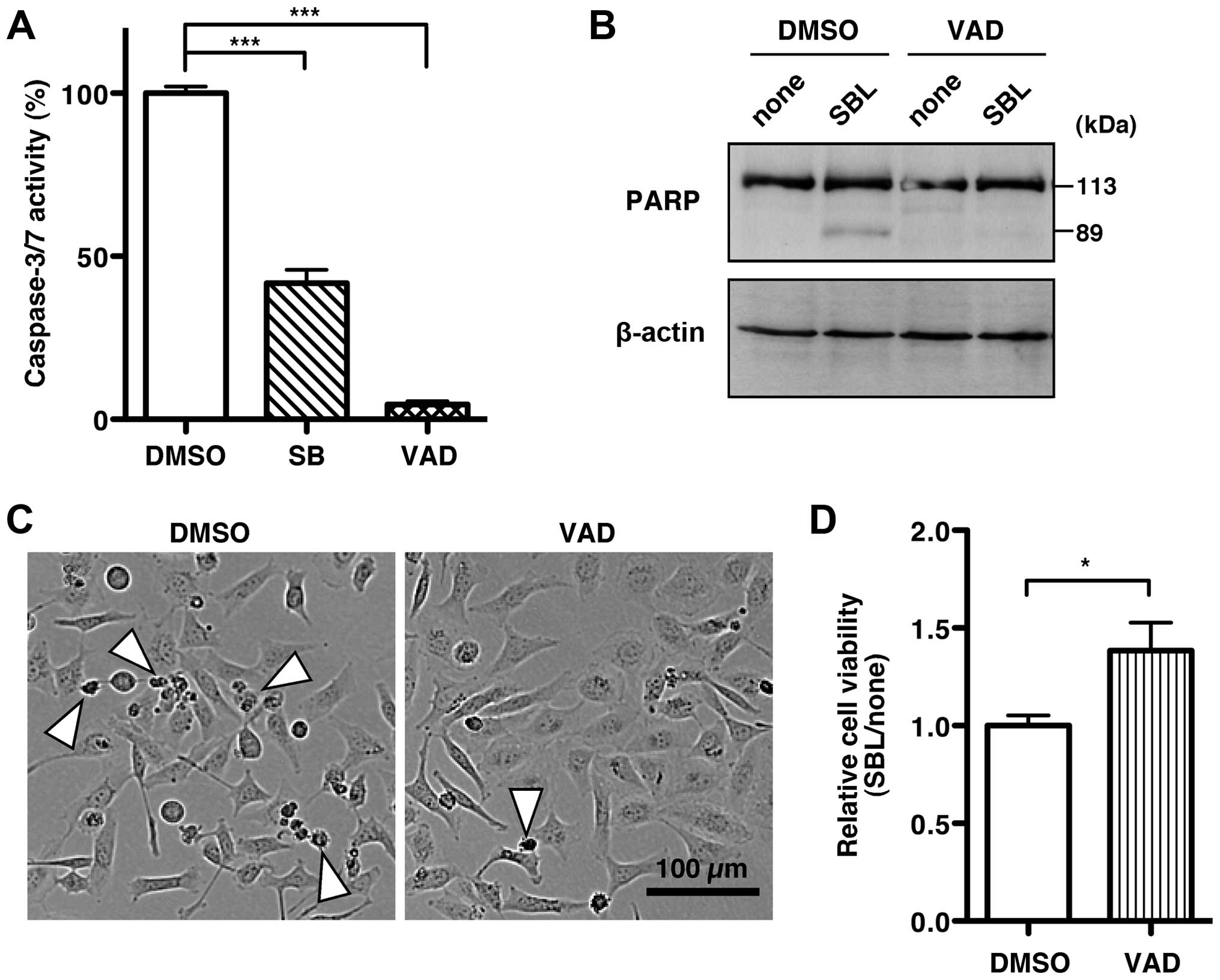
A pan-caspase inhibitor, zVAD-fmk, completely
inhibited the caspase-3/7 activation induced by SBL (Fig. 5A, VAD). Furthermore, zVAD-fmk
treatment inhibited the cleavage of PARP increased by SBL (Fig. 5B). Using zVAD-fmk, we then studied
how active caspase-3/7 contributes to SBL-induced cell death. In
the presence of zVAD-fmk, SBL-induced cell death decreased
(Fig. 5C). In addition, zVAD-fmk
increased the cell viability of SBL-treated MDA-MB231 cells
compared with the control (Fig.
5D). These results suggest that SBL induces cell death of
cancer cells through p38 MAPK-mediated activation of
caspase-3/7.
Discussion
Our previous study showed that SBL induced MAPK
phosphorylation. However, it remains unclear whether the MAPK
activation is related to SBL-induced cell death and RNase activity
of SBL. In this study, we demonstrate that SBL induces the
phosphorylation of p38 MAPK, due to RNA degradation by the RNase
activity of SBL. We also find that SBL-induced p38 MAPK activation
leads to caspase-3/7 activation and subsequent cell death.
In this study, the p38 MAPK inhibitor, SB inhibited
caspase-3/7 activity (Fig. 5A) but
caspase-3/7 inhibitor, VAD did not show any inhibitory effect on
p38 MAPK phosphorylation (Fig.
2A). This suggests that p38 MAPK is an upstream regulator of
caspase-3/7. However, SB did not completely inhibit caspase-3/7
activity (Fig. 5A). These results
indicate that caspase-3/7 may be also activated by a signaling
pathway other than p38 MAPK. Iordanov et al showed that both
JNK and p38 MAPK are activated by onconase, which is an RNase
isolated from Rana pipiens oocytes, and that JNK is related
to the cleavage of PARP and onconase-induced cell death (19). Since SBL also activates JNK
(14,17), JNK activation may also contribute
to capase-3/7 activation resulting in SBL-induced cell death.
Although the p38 MAPK inhibitor, SB significantly
inhibited SBL-induced cell death (Fig.
2C), approximately two-fifths of SB-treated cells still induced
SBL-dependent cell death. Given that H103A efficiently abrogated
SBL-induced cell death (Fig. 3E),
there could also be p38 MAPK-independent signaling pathways for
SBL-induced cell death. Inhibition of protein synthesis is most
likely related to this pathway because RNA damage leads to an
inhibition of protein synthesis in addition to p38 MAPK activation,
and inhibition of protein synthesis was observed in SBL-treated
cells (8).
SBL-induced cell death and p38 MAPK phosphorylation
in SK-BR-3 cells were much weaker than those of MCF-7 and MDA-MB231
cells. SBL requires at least three steps to induce cell death:
binding to cell surface via sialic acid, entering into cells,
followed by the activation of cell death-related signaling. From
the immunoblot analysis of cell lysate of SBL-treated cells, the
amount of SBL entered into the cells was lower in SK-BR-3 cells
than in MDA-MB231 and MCF-7 cells, suggesting that SBL
inefficiently enters into SK-BR-3 cells compared to MDA-MB231 and
MCF-7 cells (data not shown). Therefore, the weak reactivity
against the SBL-induced cell death and p38 MAPK phosphorylation in
SK-BR-3 cells might be due to the problem in the low binding of SBL
to cell surface of SK-BR-3 cells. Further studies such as
sialylation state and unidentified receptors in the cells are
required to address the question.
Shiga toxin, also known as verotoxin 1, acts as a
protein synthesis inhibitor within target cells. Shiga toxin
cleaves a specific adenine from 28S rRNA by its RNA
N-glycosidase activity, thereby inhibiting protein synthesis
(20,21). Moreover, Shiga toxin activates p38
MAPK or JNK (22,23). The ability of Shiga toxin to
activate p38 MAPK and JNK was lost by heat denaturation or
substituted mutation in the active site, indicating that the p38
MAPK and JNK activation by Shiga toxin depend on its RNA
N-glycosidase activity (22). Since an inhibitor of the p38 MAPK
was demonstrated to block the cytotoxicity of Shiga toxin (23), Shiga toxin induces cell death via
the p38 MAPK signaling pathway. Thus, the molecular mechanisms of
Shiga toxin-induced cell death are mediated in large part by the
ribotoxic stress response and are very similar to that of
SBL-induced cell death presented in this study. The B subunit of
Shiga toxin binds to globotriaosylceramide 3 (Gb3) on the host
cells, followed by the RNA N-glycosidase activity containing
A subunit internalizing into cells. Although the mechanism of the
internalization of SBL into cells is still unknown,
sialoglycoproteins may work as a receptor for SBL-internalization,
like Gb3 for the B subunit of Shiga toxin.
Onconase strongly induces cell death depending on
its RNase activity (24,25) by leading to the suppression of cell
cycle progression, followed by apoptosis (26). Onconase has also been shown to have
effective anticancer activity in animal models (27). Moreover, onconase has been studied
in advanced phase IIIb clinical trials against malignant
mesothelioma (28). Similar to
onconase, SBL also shows high toxicity against some cancer cells
and has antitumor effects in mouse models (3,8).
Moreover, SBL does not kill primary or normal cells because SBL
recognizes and selectively binds to sialoglycoproteins on tumor and
cancer cells (1,7,18).
Therefore, SBL may work as a potential antitumor drug as well as
onconase.
In conclusion, our findings demonstrate an important
role for RNase activity-dependent p38 MAPK activation and the
subsequent caspase-3/7 activation in SBL-induced cancer cell death.
Further studies regarding the molecular mechanism of SBL-induced
cancer cell death may be helpful to the development of anticancer
therapies in the future.
Acknowledgements
We appreciate Ms. Yuki Miura and Dr Kohta Takahashi
(Tohoku Pharmaceutical University, Sendai) for their assistance. We
also appreciate Dr Takashi Sugino (Shizuoka Cancer Center,
Shizuoka) for his kind gift of breast cancer cell lines. This study
was supported by the grant-in-aid for the ‘Academic Frontier’
Project (2006–2011) for Private Universities from the Ministry of
Education, Culture, Sports, Science and Technology of Japan; and a
Grant-in-Aid for Young Scientists (B) [grant no. 20790073 (to
Yukiko Kariya)].
Abbreviations:
|
CBB
|
Coomassie Brilliant Blue
|
|
DMSO
|
dimethyl sulfoxide
|
|
Gb3
|
globotriaosylceramide 3
|
|
IPTG
|
isopropyl 1-thio-β-D-galactoside
|
|
mAb
|
monoclonal antibody
|
|
MAPK
|
mitogen-activated protein kinase
|
|
PARP
|
poly (ADP-ribose)-polymerase
|
|
PBS
|
phosphate-buffered saline
|
|
RNase
|
ribonuclease
|
|
SBL
|
sialic acid-binding lectin
|
|
SD
|
standard deviation
|
|
siRNA
|
short interference RNA
|
References
|
1
|
Nitta K, Takayanagi G, Kawauchi H and
Hakomori S: Isolation and characterization of Rana catesbeiana
lectin and demonstration of the lectin-binding glycoprotein of
rodent and human tumor cell membranes. Cancer Res. 47:4877–4883.
1987.PubMed/NCBI
|
|
2
|
Dall'Olio F and Chiricolo M:
Sialyltransferases in cancer. Glycoconj J. 18:841–850. 2001.
View Article : Google Scholar
|
|
3
|
Hu CC, Tang CH and Wang JJ: Caspase
activation in response to cytotoxic Rana catesbeiana ribonuclease
in MCF-7 cells. FEBS Lett. 503:65–68. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liao YD, Huang HC, Chan HJ and Kuo SJ:
Large-scale preparation of a ribonuclease from Rana catesbeiana
(bullfrog) oocytes and characterization of its specific cytotoxic
activity against tumor cells. Protein Expr Purif. 7:194–202. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen JN, Yiang GT, Lin YF, Chou PL, Wu TK,
Chang WJ, Chen C and Yu YL: Rana catesbeiana ribonuclease induces
cell apoptosis via the caspase-9/-3 signaling pathway in human
glioblastoma DBTRG, GBM8901 and GBM8401 cell lines. Oncol Lett.
9:2471–2476. 2015.PubMed/NCBI
|
|
6
|
Ogawa Y, Sugawara S, Tatsuta T, Hosono M,
Nitta K, Fujii Y, Kobayashi H, Fujimura T, Taka H, Koide Y, et al:
Sialylglycoconjugates in cholesterol-rich microdomains of P388
cells are the triggers for apoptosis induced by Rana catesbeiana
oocyte ribonuclease. Glycoconj J. 31:171–184. 2014. View Article : Google Scholar
|
|
7
|
Tatsuta T, Sugawara S, Takahashi K, Ogawa
Y, Hosono M and Nitta K: Cancer-selective induction of apoptosis by
leczyme. Front Oncol. 4:1392014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nitta K, Ozaki K, Ishikawa M, Furusawa S,
Hosono M, Kawauchi H, Sasaki K, Takayanagi Y, Tsuiki S and Hakomori
S: Inhibition of cell proliferation by Rana catesbeiana and Rana
japonica lectins belonging to the ribonuclease superfamily. Cancer
Res. 54:920–927. 1994.PubMed/NCBI
|
|
9
|
Liao YD: A pyrimidine-guanine
sequence-specific ribonuclease from Rana catesbeiana (bullfrog)
oocytes. Nucleic Acids Res. 20:1371–1377. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Titani K, Takio K, Kuwada M, Nitta K,
Sakakibara F, Kawauchi H, Takayanagi G and Hakomori S: Amino acid
sequence of sialic acid binding lectin from frog (Rana catesbeiana)
eggs. Biochemistry. 26:2189–2194. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang HC, Wang SC, Leu YJ, Lu SC and Liao
YD: The Rana catesbeiana rcr gene encoding a cytotoxic
ribonuclease. Tissue distribution, cloning, purification,
cytotoxicity, and active residues for RNase activity. J Biol Chem.
273:6395–6401. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nitta K, Ozaki K, Tsukamoto Y, Furusawa S,
Ohkubo Y, Takimoto H, Murata R, Hosono M, Hikichi N, Sasaki K, et
al: Characterization of a Rana catesbeiana lectin-resistant mutant
of leukemia P388 cells. Cancer Res. 54:928–934. 1994.PubMed/NCBI
|
|
13
|
Nitta K, Ozaki K, Tsukamoto Y, Hosono M,
Ogawakonno Y, Kawauchi H, Takayanagi Y, Tsuiki S and Hakomori S:
Catalytic lectin (leczyme) from bullfrog (Rana catesbeiana) eggs.
Int J Oncol. 9:19–23. 1996.PubMed/NCBI
|
|
14
|
Tatsuta T, Hosono M, Sugawara S, Kariya Y,
Ogawa Y, Hakomori S and Nitta K: Sialic acid-binding lectin
(leczyme) induces caspase-dependent apoptosis-mediated
mitochondrial perturbation in Jurkat cells. Int J Oncol.
43:1402–1412. 2013.PubMed/NCBI
|
|
15
|
Tatsuta T, Hosono M, Miura Y, Sugawara S,
Kariya Y, Hakomori S and Nitta K: Involvement of ER stress in
apoptosis induced by sialic acid-binding lectin (leczyme) from
bullfrog eggs. Int J Oncol. 43:1799–1808. 2013.PubMed/NCBI
|
|
16
|
Yiang GT, Yu YL, Chou PL, Tsai HF, Chen
LA, Chen YH, Su KJ, Wang JJ, Bau DT and Wei CW: The cytotoxic
protein can induce autophagocytosis in addition to apoptosis in
MCF-7 human breast cancer cells. In Vivo. 26:403–409.
2012.PubMed/NCBI
|
|
17
|
Tatsuta T, Hosono M, Takahashi K, Omoto T,
Kariya Y, Sugawara S, Hakomori S and Nitta K: Sialic acid-binding
lectin (leczyme) induces apoptosis to malignant mesothelioma and
exerts synergistic antitumor effects with TRAIL. Int J Oncol.
44:377–384. 2014.
|
|
18
|
Tang CH, Hu CC, Wei CW and Wang JJ:
Synergism of Rana catesbeiana ribonuclease and IFN-gamma triggers
distinct death machineries in different human cancer cells. FEBS
Lett. 579:265–270. 2005. View Article : Google Scholar
|
|
19
|
Iordanov MS, Wong J, Newton DL, Rybak SM,
Bright RK, Flavell RA, Davis RJ and Magun BE: Differential
requirement for the stress-activated protein kinase/c-Jun
NH(2)-terminal kinase in RNAdamage-induced apoptosis in primary and
in immortalized fibroblasts. Mol Cell Biol Res Commun. 4:122–128.
2000. View Article : Google Scholar
|
|
20
|
Endo Y, Tsurugi K, Yutsudo T, Takeda Y,
Ogasawara T and Igarashi K: Site of action of a Vero toxin (VT2)
from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic
ribosomes. RNA N-glycosidase activity of the toxins. Eur J Biochem.
171:45–50. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saxena SK, O'Brien AD and Ackerman EJ:
Shiga toxin, Shiga-like toxin II variant, and ricin are all
single-site RNA N-glycosidases of 28 S RNA when microinjected into
Xenopus oocytes. J Biol Chem. 264:596–601. 1989.PubMed/NCBI
|
|
22
|
Smith WE, Kane AV, Campbell ST, Acheson
DW, Cochran BH and Thorpe CM: Shiga toxin 1 triggers a ribotoxic
stress response leading to p38 and JNK activation and induction of
apoptosis in intestinal epithelial cells. Infect Immun.
71:1497–1504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ikeda M, Gunji Y, Yamasaki S and Takeda Y:
Shiga toxin activates p38 MAP kinase through cellular Ca(2+)
increase in Vero cells. FEBS Lett. 485:94–98. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu Y, Mikulski SM, Ardelt W, Rybak SM and
Youle RJ: A cytotoxic ribonuclease. Study of the mechanism of
onconase cytotoxicity. J Biol Chem. 268:10686–10693.
1993.PubMed/NCBI
|
|
25
|
Lee JE and Raines RT: Contribution of
active-site residues to the function of onconase, a ribonuclease
with antitumoral activity. Biochemistry. 42:11443–11450. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Juan G, Ardelt B, Li X, Mikulski SM,
Shogen K, Ardelt W, Mittelman A and Darzynkiewicz Z: G1 arrest of
U937 cells by onconase is associated with suppression of cyclin D3
expression, induction of p16INK4A, p21WAF1/CIP1 and p27KIP and
decreased pRb phosphorylation. Leukemia. 12:1241–1248. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rybak SM, Pearson JW, Fogler WE, Volker K,
Spence SE, Newton DL, Mikulski SM, Ardelt W, Riggs CW, Kung HF, et
al: Enhancement of vincristine cytotoxicity in drug-resistant cells
by simultaneous treatment with onconase, an antitumor
ribo-nuclease. J Natl Cancer Inst. 88:747–753. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pavlakis N and Vogelzang NJ: Ranpirnase -
an antitumour ribo-nuclease: Its potential role in malignant
mesothelioma. Expert Opin Biol Ther. 6:391–399. 2006. View Article : Google Scholar : PubMed/NCBI
|