Introduction
Kidney cancer, one of the most common malignancies
in genitourinary system, affects approximately 208,500 people all
around the world each year. In addition, the global incidence of
kidney cancer is continuously increasing (1). Renal cell carcinoma (RCC) is
identified as the most common type of kidney cancer, which is
responsible for approximately 90–95% of primary kidney cancer cases
(2). RCC starts in the cells of
the proximal renal tubular epithelium, with classic symptoms
including haematuria, flank pain and an abdominal mass. Although
smoking, NSAIDs medication and family history are suggested as risk
factors, the exact pathogenies of RCC remain poorly understood
(3,4). Besides, the therapy for RCC is still
limited. Surgery is applied primarily in RCC treatment, as RCC is
often insensitive to chemotherapy and radiotherapy. Whereas,
surgery is not always efficient when the cancer has spread around
the body. In recent years, target therapy has improved the
treatment of RCC (5).
Neutralization of vascular endothelial growth factor is proved to
prolong the time to progression of disease in RCC patients. Thus,
the research on potential targeting factors may light up the
prospect for RCC treatment.
The transmembrane glycoproteins of triggering
receptor expressed on myeloid cells (TREM) belong to the single
immunoglobulin variable (IgV) domain receptor family (6). TREMs map to human chromosome 6p21.1
and encode TREM-1, TREM-2, TREM-4, TREM-5 and TREM-like genes in
human. TREM-2 is encoded by a 1041-nucleotide long cDNA. This
receptor consists of an extracellular domain, a transmembrane
region and a short cytoplasmic tail (7,8).
TREM-2 is involved in many biological processes (9). In bone remodeling, TREM-2 has been
suggested to favor osteoclast differentiation and morphology
(10). In immune responses, TREM-2
is associated with the upregulation of CD40, CD86, MHC class II in
dendritic cells and the maturation of dendritic cells (11,12).
TREM-2 also plays a role in suppressing the production of TNF and
IL-6 and TLR signaling in macrophages (13,14).
Moreover, TREM-2 is also involved in the pathology of some
diseases. Related research proved that the defects in TREM-2 may be
a cause of polycystic lipomembranous osteodysplasia with sclerosing
leukeoncephalopathy (PLOSL) (15),
and a rare missense mutation (rs75932628-T) in TREM-2 may confer an
obvious risk of Alzheimer’s disease (16). Recently, TREM-2 is suggested
functioning in human malignancies. Wang et al (17) have indicated that highly expressed
TREM-2 promotes cell proliferation and invasion in glioma cells.
Other studies also suggest that abnormally expressed TREM-2 may be
associated with tumor immune evasion in lung cancer (18). Whereas, the effects of TREM-2 on
RCC are still less known.
In the present study, we revealed the biological
effects of TREM-2 on RCC cells for the first time. We found that
the expression of TREM-2 is abnormally elevated in RCC tumor
tissues. TREM-2 functioned as an oncogene in both RCC cell lines
and tumor-bearing mouse model in vivo. The effect of TREM2
on RCC progression might be related to the regulation of apoptotic
proteins and PTEN-PI3K/Akt pathway. Therefore, TREM-2 may provide a
novel approach to the therapy for RCC.
Materials and methods
Tissue samples
Renal tumor tissues and adjacent normal tissues were
collected from 40 patients with RCC treated at the Huadong
Hospital, Fudan University. The tissues were stored at −80°C until
being used. The study was approved by the Ethics Committee of
Huadong Hospital, Fudan University. Informed and written consent
were obtained from all patients according to the guidelines of the
ethics committee.
Cell culture
Five RCC cell lines, Caki-1, Caki-2, ACHN, 786-0 and
OS-RC-2 were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA). Cells were cultured in RMPI-1640 medium
supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U/ml
penicillin and 100 μg/ml streptomycin. Cell culture was maintained
at 37°C in a humidified 5% CO2 atmosphere.
RNA interference
To knock down the expression of TREM-2 in RCC cells,
siRNA transfection was performed. siRNAs targeting three positions
of human TREM-2 mRNA (NM_001271821.1; siRNA1: 125-147UCUUACUCUUUGUC
ACAGA, siRNA1: 386–408 UUACGCUGCGGAAUCUACA and siRNA3: 591–613
GAGACACGUGAAGGAAGAU) were synthesized. A non-specific scramble
siRNA sequence served as negative control (NC). siRNAs were
transfected into RCC cells using Lipofectamine 2000 (Invitrogen)
according to the manufacturer’s instructions. The following assays
were performed at 48 h post-RNA interference.
CCK-8 assay
To analyze the cell proliferation, Caki-2 and ACHN
cells (5×103) were seeded into 96-well plates and
examined at 0, 24, 48 and 72 h after siRNA transfection using
commercial Cell Counting kit (7seabitech) per the instructions of
the manufacturer. Absorbance excited at 450 nm of reacted cells
were detected to valuate cell growth.
Cell cycle assay
At 48 h after siRNA transfection, Caki-2 and ACHN
cells were collected and fixed by 70% ethanol at −20°C for 2 h.
After being washed with phosphate-buffered saline (PBS), the cells
were incubated with propidudium iodide (PI; 0.05 mg/ml;
Sigma-Aldrich) in the dark for 30 min. In addition, cell cycle
assay was performed using flow cytometer (BD Biosciences, San Jose,
CA, USA) and analyzed using FlowJo cell cycle analysis
software.
Cell apoptosis assay
Caki-2 and ACHN cells were harvested at 48 h
post-RNA interference. Cell apoptosis assay was performed using
Annexin V apoptosis detection kit APC (eBioscience, San Diego, CA,
USA). The cells double stained with Annexin V-fluorescein
isothiocyanate (FITC) and PI were then examined by flow cytometer
(BD Biosciences). At least 10,000 cells were obtained for each
experiment.
Reverse transcription and real-time PCR
(qRT-PCR)
Total RNA was extracted from RCC cells and tissue
samples using TRIzol reagent (Invitrogen). Reverse transcription
was performed via cDNA Synthesis kit (Fermentas, Waltham, MA, USA).
qRT-PCR was processed using a standard SYBR-Green PCR kit
(Fermentas). All the procedures were performed according to the
manufacturer’s instructions. The cycle conditions were 10 min at
95°C, 40 cycles of 15 sec at 95°C and 45 sec at 60°C, 15 sec at
95°C, 1 min at 60°C followed by 15 sec at 95°C and 15 sec at 60°C.
GAPDH served as internal control. The primer sequences were the
following: TREM-2 NM_001271821.1): primer F,
5′-TGGCACTCTCACCATTACG-3′ and primer R,
5′-CCTCCCATCATCTTCCTTCAC-3′; Bax (NM_004324.3): primer F,
5′-AGCTGAGCGAGTGTCTCAAG-3′ and primer R,
5′-TGTCCAGCCCATGATGGTTC-3′; Bcl2 (NM_000633.2): primer F,
5′-AGACCGAA GTCCGCAGAACC-3′ and primer R, 5′-GAGACCACACTGC
CCTGTTG-3′; PCNA (NM_002592.2): primer F,
5′-GCCTGACAAATGCTTGCTGAC-3′ and primer R,
5′-TTGAGTGCCTCCAACACCTTC-3′; caspase-3 (NM_004346.3): primer F,
5′-AACTGGACGTGGCATTGAG-3′ and primer R, 5′-ACAAAGCGACTGGATGAACC-3′;
GAPDH (NM_001256799.1): primer F, 5′-CACCCACTCCTCCACCTTTG-3′ and
primer R, 5′-CCACCACCCTGTTGCTGTAG-3′.
Western blotting
Tissue samples were collected and put into
homogenizer to grind into tissue homogenate. Treated and untreated
RCC cells were harvested and washed twice with PBS. Then, tissue
homogenate and cells were disrupted in a radio-immunoprecipitation
assay lysis buffer. After protein normalization, tissue and cell
samples were separated in SDS-PAGE and transferred to a
nitrocellulose membrane. The blots were then incubated with
appropriate primary and secondary antibodies following blocked with
5% skim milk. Visualization was performed using the enhanced
chemiluminescence (ECL; Millipore, Billerica, MA, USA). The
antibody list was as follows: TREM-2 (1:800, Ab86491; Abcam,
Cambridge, MA, USA), PCNA (1:1,000, #13110; Cell Signaling
Technology Danvers, MA, USA), Bax (1:300, Sc-493; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), Bcl2 (1:300, Sc-492; Santa
Cruz Biotechnology), caspase-3 (1:500, Ab44976; Abcam), PTEN
(1:1,000, #9188; Cell Signaling Technology), PI3K (1:1,000,
Ab189403; Abcam), p-PI3K (1:1,000, Ab182651; Abcam), Akt (1:1,000,
#9272; Cell Signaling Technology), p-Akt (1:1,000, #9271, Cell
Signaling Technology), GAPDH (1:1,500, #5174; Cell Signaling
Technology) and HRP-labeled secondary antibodies (1:1,000, A0208,
A0181, A0216; Beyotime Institute of Biotechnolgy, Haimen,
China).
Nude mouse xenograft model
Animal experiments were approved and performed per
the guidelines of Animal Care and Use Committee of Huadong
Hospital, Fudan University (Shanghai, China). Twelve BALB/c nude
mice aged 4-weeks (SLAC animal) were maintained under specific
pathogen-free conditions using a laminar air-flow rack. All the
mice were fed with sterilized food and autoclaved water. Injection
was performed as previously described (19). Briefly, after one week of
acclimatization, 12 nude mice were divided into two groups, NC
group and siRNA group (n=6/group) randomly, and subcutaneously
injected with ACHN cells transfected with NC-siRNA and TREM-2-siRNA
(2×106) to the armpit of nude mouse, respectively. After
1 week of tumorigenesis, the shortest and longest diameter of the
tumor were measured with calipers every 3 days, and tumor volume
(mm3) was calculated using the following standard
formula: (the shortest diameter)2 × (the longest
diameter) × 0.5. On day 34 post-injection, the mice were sacrificed
by cervical dislocation and the tumors were harvested. The wet
weights of each tumor were examined. During the experimental
procedure, all mice were monitored every 2 days. None of the mice
died prior to the experimental endpoint.
Statistical analysis
At least 3 independent experiments were performed in
every assay. Data analysis used GraphPad Prism software. Data are
expressed as mean (± SD) and analyzed with t-test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant result.
Results
TREM-2 expression is elevated in RCC
tissues
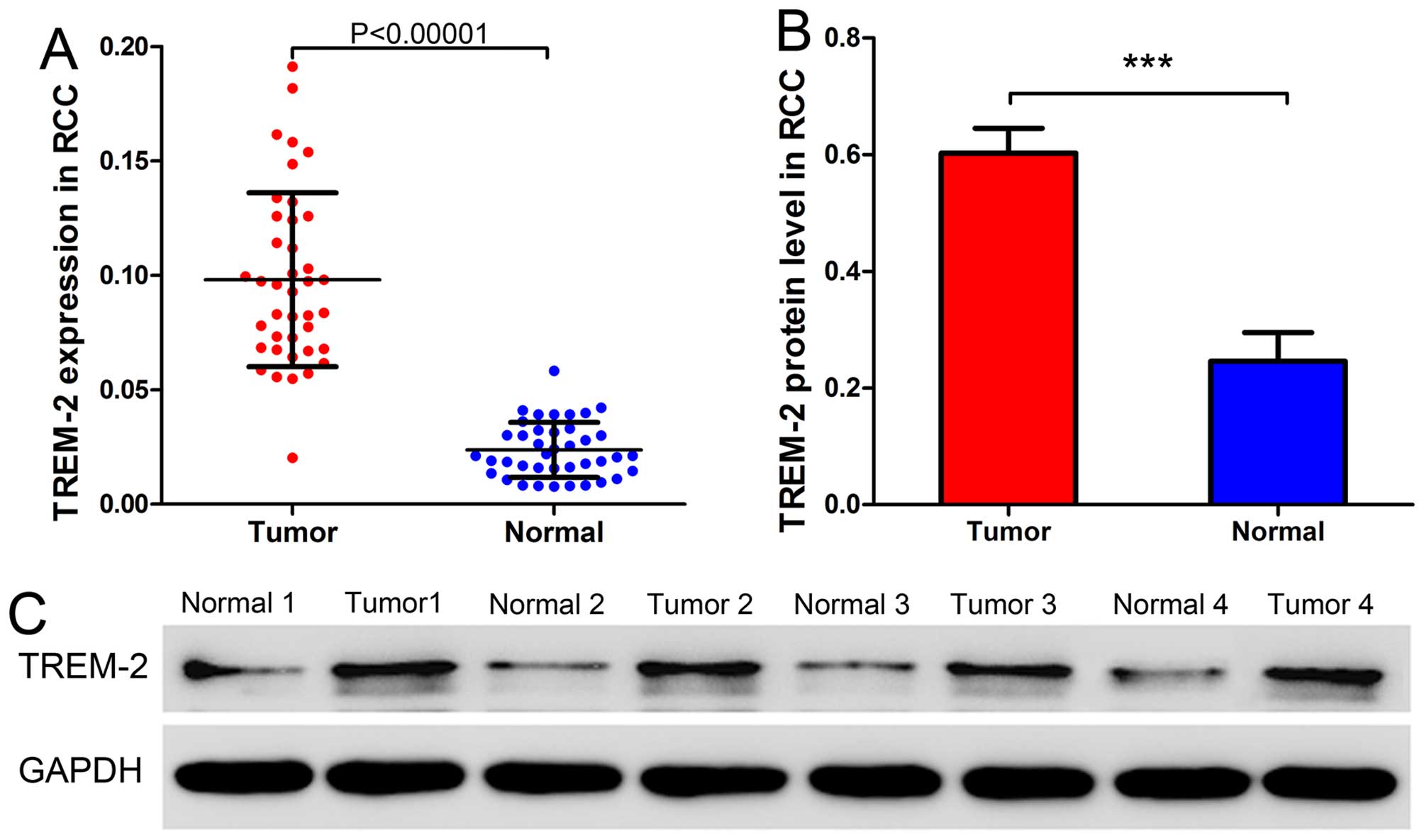
To profile the expression pattern of TREM-2 in RCC,
we analyzed the mRNA and protein expression of TREM-2 in tumor and
adjacent normal tissues from 40 patients with RCC using qRT-PCR and
western blot analysis. As Fig. 1
shows, both mRNA and protein levels of TREM2 were significantly
higher in tumor tissues than adjacent normal tissues. These data
indicated that the expression of TREM-2 was abnormally elevated in
RCC tumor tissues.
TREM-2 knockdown via RNA
interference
We then examined TREM-2 expression level in five RCC
cell lines, including Caki-1, Caki-2, ACHN, 786-0 and OS-RC-2,
using qRT-PCR and western blot analysis. We found that TREM2 was
highly expressed in ACHN and Caki-2 cell lines as comparing to
other three cell lines (Fig. 2A and
B). In addition, we employed these two RCC cell lines to carry
out the following experiments. To further analyzed the biological
functions of TREM-2 in RCC, TREM-2 expression was silenced in ACHN
and Caki-2 cells using RNA interference. The results of qRT-PCR
showed that, the mRNA level of TREM-2 was significantly suppressed
after siRNA transfection. In addition, the siRNA2 (targeting
position 386–408: UUACGCUGCGGAAUCUACA) silenced TREM-2 expression
more effectively as comparing to the other two siRNAs. Thus, siRNA2
was selected as TREM-2-siRNA transfected into cancer cells for the
following assays. The western blot data also confirmed that siRNA2
decreased the protein level of TREM-2 in two RCC cell lines
(Fig. 2C and D).
Depletion of TREM-2 inhibits cell
proliferation in RCC cell lines
We analyzed the cell growth of ACHN and Caki-2 at 0,
24, 48 and 72 h post-RNA interference using CCK-8 assay. As shown
in Fig. 3, obvious decrease of
cell growth was detected in both ACHN and Caki-2 cells at 48 and 72
h after siRNA transfection. Whereas, the cell proliferation was not
affected in the NC group. These data suggested that knockdown of
TREM-2 might suppress cell growth in RCC progression.
TREM-2 knockdown induces G1 phase arrest
in RCC cell lines
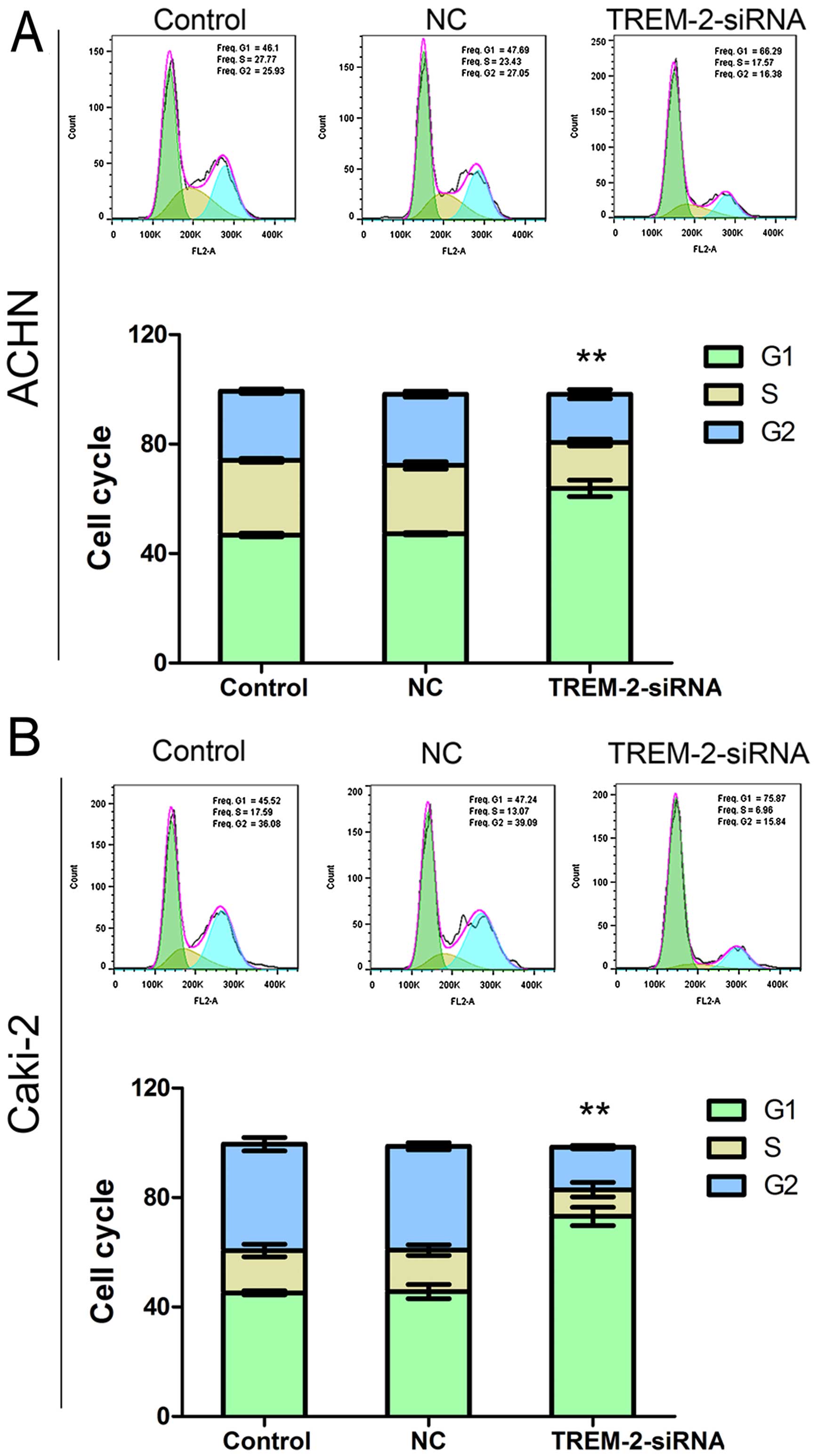
As silencing TREM2 inhibited cell growth in RCC, we
analyzed the effects of TREM-2 on the cell cycle progress in ACHN
and Caki-2 cells sequentially. The RCC cells were stained by PI at
48 h post-RNA interference, and the cell cycle was examined by flow
cytometry. As shown in Fig. 4,
proportion of G1 phase was remarkably increased in siRNA-TREM-2
group than in the NC group, with increased ratios: 35.13 and 60.30%
in ACHN and Caki-2, respectively. These results indicated that,
silencing the expression of TREM-2 might inhibit cell cycle
progress via inducing THE G1 phase arrest.
Silencing TREM-2 causes cell apoptosis in
RCC cell lines
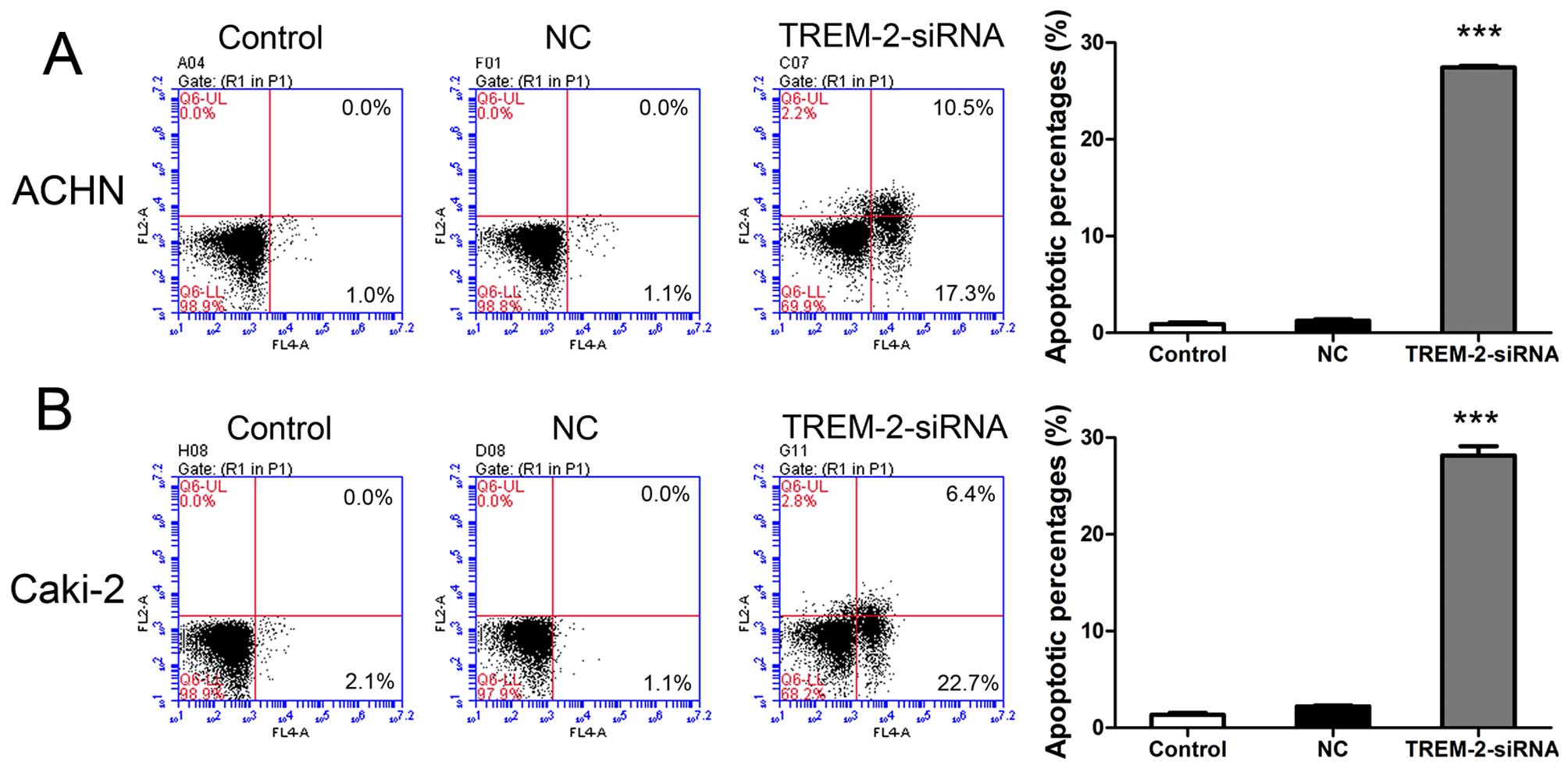
We analyzed the effects of TREM-2 on cell apoptosis
in ACHN and Caki-2 cells at 48 h after siRNA transfection. The
cells were double stained by Annexin V-FITC and PI, and examined
using flow cytometry. As illustrated in Fig. 5, ~22-fold increase and 13-fold
increase of cell apoptosis was examined in ACHN and Caki-2 cells of
siRNA groups as comparing to NC groups. The data showed that
knockdown of TREM-2 induced significant cell apoptosis in RCC cell
lines.
Silencing TREM-2 inhibits xenografted
ACHN tumor development in nude mice
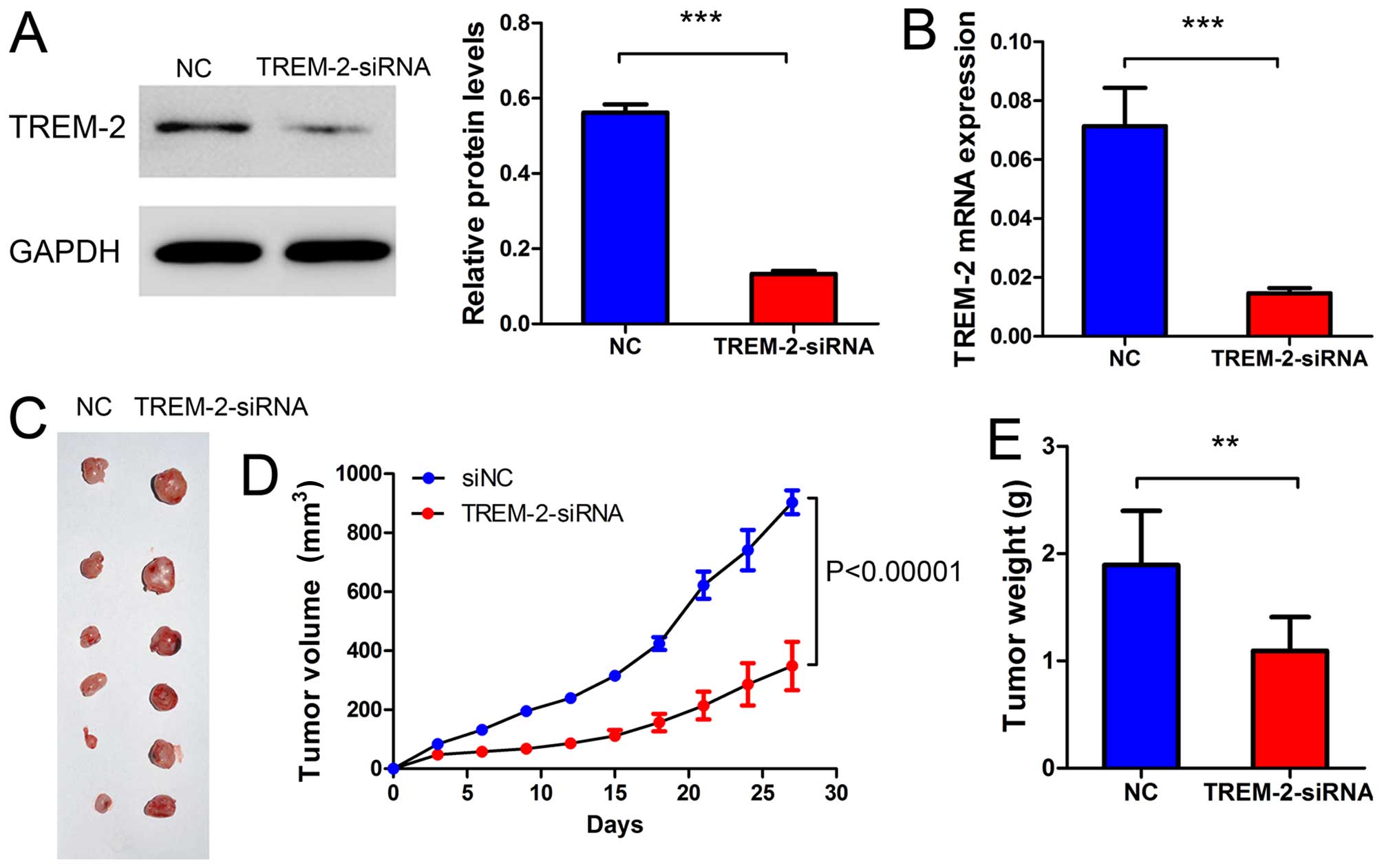
We then examined whether TREM-2 silenced in RCC
cells could inhibit tumor growth in vivo. The ACHN cells
transfected with NC-siRNA or TREM-2-siRNa were subcutaneously
injected into nude mice, respectively. Tumor volumes were measured
for 27 days. The weight of tumors were measured on day 34. As shown
in Fig. 6A–C, the mRNA expression
and protein level of TREM-2 were significantly decreased in
TREM-2-siRNA group compared with NC group. As shown in Fig. 6C–E, both the volume and the weight
of RCC tumors were obviously decreased after TREM-2-siRNA
transfection. These results implicated that TREM-2 knockdown might
inhibit the tumor development of RCC in vivo.
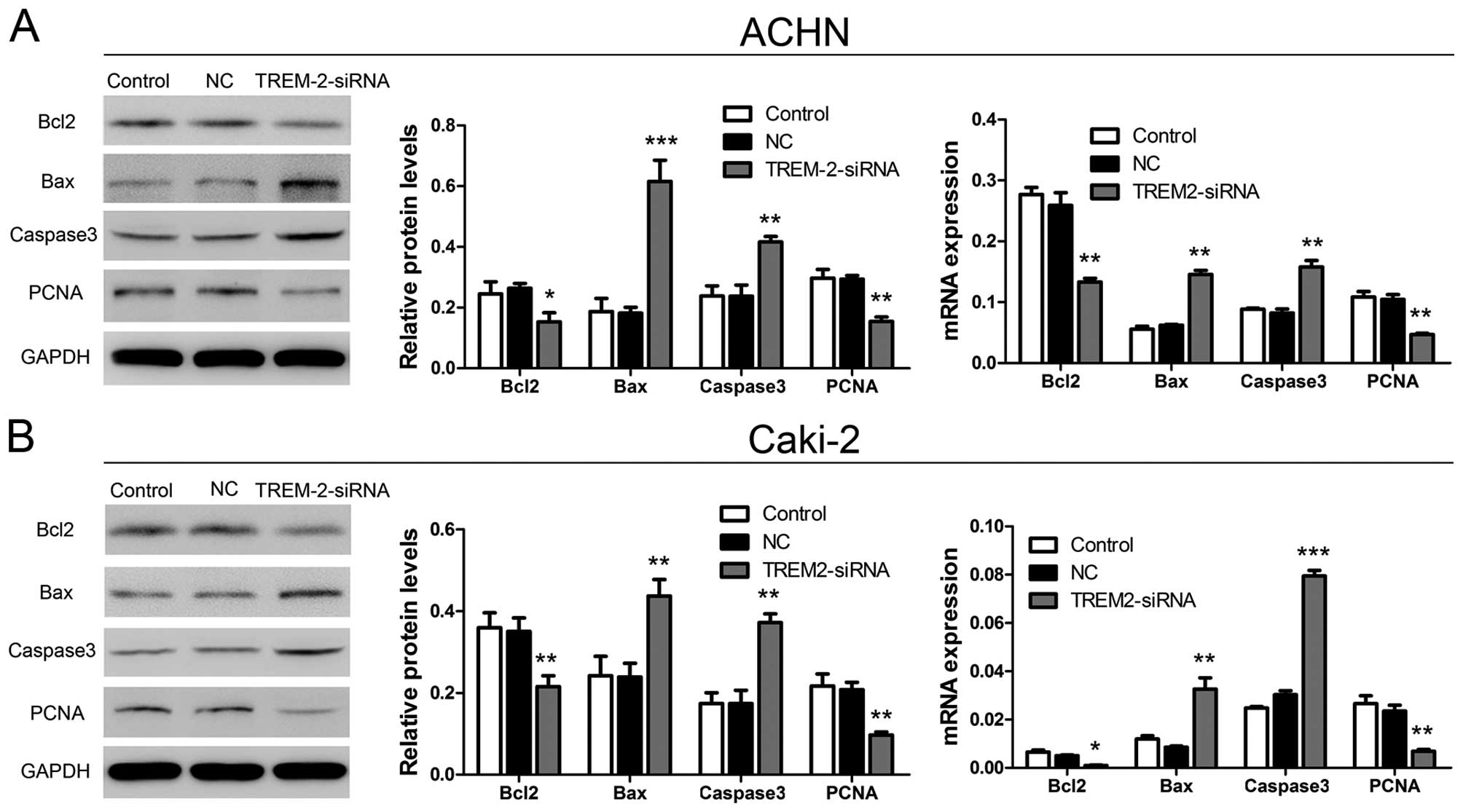
Silencing TREM-2 altered endogenous
expression of proteins related to apoptosis and the cell cycle in
RCC cell lines
As described above, TREM-2 might act as a promoter
in cell proliferation through inducing cell cycle progress and
inhibiting cell apoptosis. We then examined four related protein
levels, including Bcl2, Bax, caspase-3 and PCNA, using qRT-PCR and
western blot analysis at 48 h post-RNA interference. As shown in
Fig. 7, mRNA expression of Bcl2
(decreased ratios: ACHN, 48.55% and Caki-2, 80.47%) and PCNA
(decreased ratios: ACHN, 55.11% and Caki-2, 69.58%) were obviously
decreased after TREM-2 knockdown. Whereas, the mRNA expression of
Bax (increased ratios: ACHN, 134.16% and Caki-2, 172.83%) and
caspase-3 (increase ratios: ACHN, 91.77% and Caki-2, 162.50%) were
significantly increased in siRNA-TREM-2 groups compared to NC
groups. The same results were also examined by western blot
analysis. The protein levels of Bcl-2 and PCNA were reduced, while
the protein levels of Bax and caspase-3 were elevated after siRNA
transfection. The decreased ratios of Bcl2 and PCNA protein levels
were 41.94 and 47.39% in ACHN, 38.54 and 53.59% in Caki-2,
respectively. The increased ratios of Bax and caspase-3 protein
levels were 239.18 and 75.45% in ACHN, and 82.99 and 113.98% in
Caki-2, respectively.
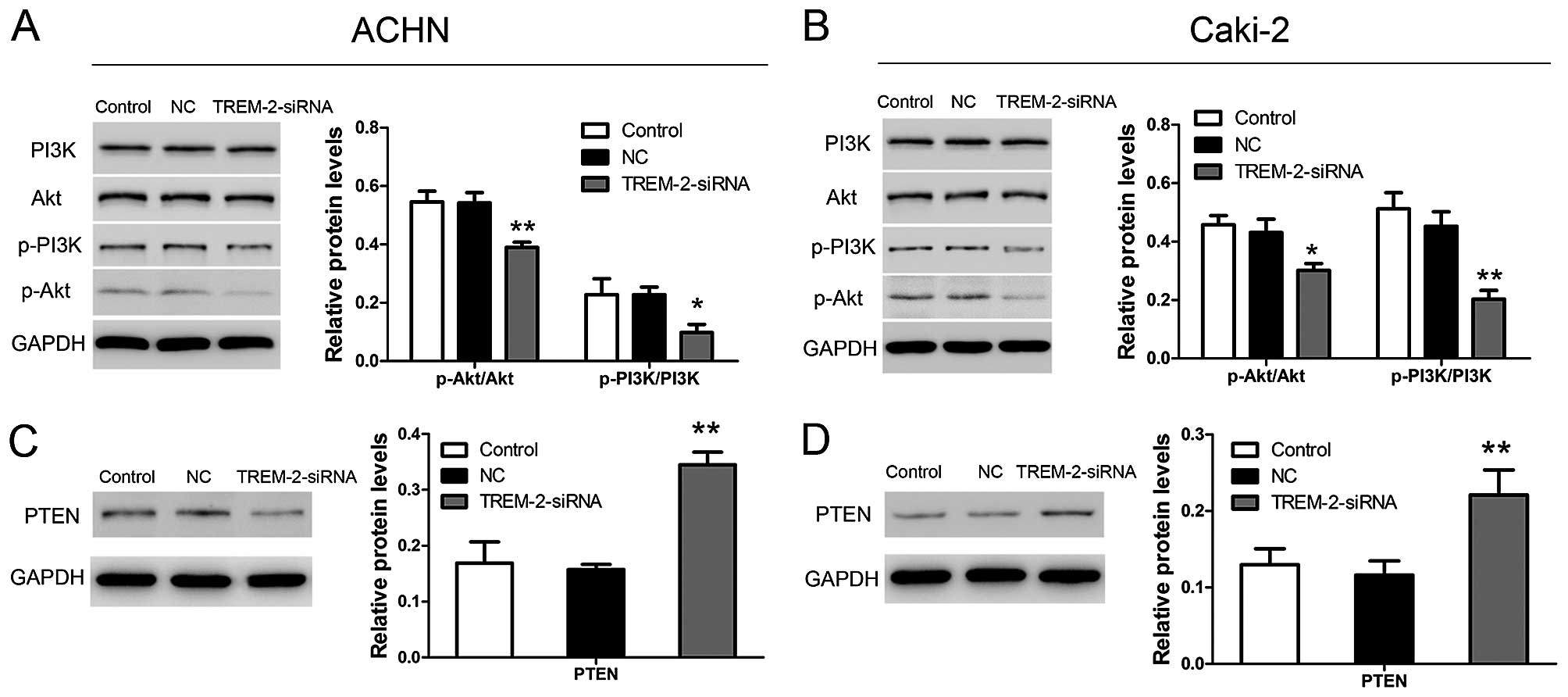
Depletion of TREM-2 inactivates
PTEN-PI3K/Akt signaling pathway in RCC cell lines
To reveal the functional mechanisms of TREM-2 in
cell growth, we analyzed the PTEN-PI3K/Akt signaling pathway using
western blot analysis. As Fig. 8A and
B show, TREM-2 knockdown significantly increased the protein
level of PTEN in ACHN and Caki-2 cells. Depletion of TREM-2
inhibited phoshoprylation of PI3K and Akt in both cell lines
(Fig. 8C and D). These data
suggested that TREM-2 knockdown inhibited the activation of
PI3K/AKT pathway via upregulating the PTEN level in RCC cell
lines.
Discussion
Immunotherapy and targeted therapy have recently
provided new insights into the treatment of RCC. Research on novel
targeting factors still remains urgent. TREM-1, a member of TREM
family, has been suggested to be involved in progression of certain
human malignancies. Liao et al (20) has proved that TREM-1 is related to
the aggressive tumor behavior and has potential value as a
prognostic factor for hepatocellular carcinoma. TREM-1 is
upregulated in macrophages and is associated with cancer recurrence
and poor survival of patients with lung cancer (21,22).
TREM-2 is also suggested to act as an oncogene in glioma (17). Moreover, TREM-2 is proven to
promote tumor immune evasion in lung cancer cells (18). Inspired by the association between
TREMs and human cancers, we sought to reveal the role of TREM-2 in
RCC. We found that the expression of TREM-2 is remarkably
facilitated in tumor tissues compared with the adjacent normal
tissues of patients with RCC. It indicated that elevated TREM-2
might play a role in the RCC progression.
Then, we knocked down the TREM-2 expression in
selected RCC cancer cell lines via RNA interference. In addition,
we examined effects of TREM-2 depletion on cell proliferation,
apoptosis and cell cycle in ACHN and Caki-2. The data showed that
TREM-2 knockdown significantly inhibited cell growth, and induced
cell apoptosis in two RCC cell lines. Similar results were also
found in research by Wang et al (17). In their previous study, silencing
TREM-2 suppressed cell proliferation and promotes cell apoptosis in
glioma cells. In cell cycle assay, we found that depletion of
TREM-2 induced arrest in G1 phase of cell cycle in RCC cells. In
vivo, the data affirmed that knockdown of TREM-2 inhibited
tumorigenesis of RCC cells. The above results illustrated that
TREM-2 might function as an oncogene in RCC development.
To confirm the functional mechanisms of TREM-2 in
RCC cells, we analyzed the protein levels and mRNA expression of
factors related to apoptosis and cell cycle using western blot and
qRT-PCR analysis. We found that in ACHN and Caki-2 cells, both mRNA
expression and protein levels of Bcl2 and PCNA were obviously
decreased post-RNA interference. Whereas, expression of Bax and
caspase-3 were elevated in TREM-2-siRNA groups as comparing to NC
groups in two RCC cell lines. Bcl2 belongs to the antiapoptotic
Bcl2 family, which acts to prevent or delay cell death. Previous
studies indicate that increased expression of Bcl2 in RCC may
decrease the levels of apoptosis and promote resistant to treatment
(23,24). PCNA, a progressivity factor for DNA
polymerase δ, is essential for DNA replication (25,26).
In addition, suppression of PCNA may cause G1 phase arrest in cell
cycle and promote cell death in cancer cells (27). Bax and caspase-3 act as promoters
in cell apoptosis. Especially, activation of caspase-3 plays a
central role in the execution-phase of cell apoptosis (28). Thus, the results of this study
implicated that TREM-2 affects cell growth, apoptosis and cell
cycle through regulating the mRNA expression and intracellular
levels of related proteins.
Finally, we analyzed the effects of TREM-2 on
PTEN-PI3K/Akt pathway. PI3K is a lipid kinase and generated
PI(3,4,5)P3, which is an essential second messenger in
translocation of Akt to plasma membrane. Activated Akt plays a
pivotal role in fundamental cellular functions (29,30).
The activation of PI3K/Akt pathway has been suggested involved in
many kinds of human tumors, such as breast, lung cancer and
leukemia (31–33). PI3K/Akt pathway also plays a key
role in RCC. Activation of PI3K/Akt is associated with the decrease
of the survival rate in RCC (34).
Research has proven that certain anticancer agents, such as Klotho
and β-elemene, inhibit the tumor progression through suppressing
PI3K/Akt signaling in RCC (35–38).
TREM-2 has been proven as an activator in PI3K/Akt signaling
pathway. Elevated TREM-2 can mediate bacterial killing via
activating PI3K/Akt pathway (39,40).
PTEN is identified as a tumor suppressor that is mutated in a large
number of cancers at high frequency (41). PTEN acts as a key negative
regulator in the alternations of PI3K/Akt through decreasing the
intracellular level of PI(3,4,5)P3 in cells (42). In addition, the disturbed
expression and function of PTEN have been examined in both human
cancer cell lines and human malignancies (43–45).
Research has implicated that inhibition of PTEN may cause permanent
activation of the PI3K/Akt pathway (46). In the present study, we found that
TREM-2 knockdown significantly inactivated the PI3K/Akt pathway by
downregulating the intracellular protein levels of p-PI3K and
p-Akt, and increased the intracellular protein level of PTEN in two
RCC cell lines. Therefore, the data suggested that TREM-2 might act
as a regulator of PI3K/Akt through altering the PTEN level in
RCC.
In conclusion, this study first revealed the role of
TREM-2 in RCC progression. We found that TREM-2 is abnormally
elevated in RCC tissues. Knockdown of TREM-2 obviously inhibited
cell proliferation, induced cell apoptosis and G1 phase arrest in
RCC cells. The in vivo experiments also confirmed that
depletion of TREM-2 suppressed the tumor development of RCC.
Moreover, the oncogene functional effects of TREM-2 might be caused
by its regulation of related proteins and PTEN-PI3K/Akt pathway.
Therefore, TREM-2 may be considered as a potential therapeutic
target in the treatment of RCC.
References
|
1
|
Yi Z, Fu Y, Zhao S, Zhang X and Ma C:
Differential expression of miRNA patterns in renal cell carcinoma
and nontumorous tissues. J Cancer Res Clin Oncol. 136:855–862.
2010. View Article : Google Scholar
|
|
2
|
Rini BI, Campbell SC and Escudier B: Renal
cell carcinoma. Lancet. 373:1119–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheungpasitporn W, Thongprayoon C,
O’Corragain OA, Edmonds PJ, Ungprasert P, Kittanamongkolchai W and
Erickson SB: The risk of kidney cancer in patients with kidney
stones: A systematic review and meta-analysis. QJM. 108:205–212.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ljungberg B, Campbell SC, Choi HY, Jacqmin
D, Lee JE, Weikert S and Kiemeney LA: The epidemiology of renal
cell carcinoma. Eur Urol. 60:615–621. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singer EA, Gupta GN, Marchalik D and
Srinivasan R: Evolving therapeutic targets in renal cell carcinoma.
Curr Opin Oncol. 25:273–280. 2013.PubMed/NCBI
|
|
6
|
Allcock RJ, Barrow AD, Forbes S, Beck S
and Trowsdale J: The human TREM gene cluster at 6p21.1 encodes both
activating and inhibitory single IgV domain receptors and includes
NKp44. Eur J Immunol. 33:567–577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klesney-Tait J, Turnbull IR and Colonna M:
The TREM receptor family and signal integration. Nat Immunol.
7:1266–1273. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Whittaker GC, Orr SJ, Quigley L, Hughes L,
Francischetti IM, Zhang W and McVicar DW: The linker for activation
of B cells (LAB)/non-T cell activation linker (NTAL) regulates
triggering receptor expressed on myeloid cells (TREM)-2 signaling
and macrophage inflammatory responses independently of the linker
for activation of T cells. J Biol Chem. 285:2976–2985. 2010.
View Article : Google Scholar :
|
|
9
|
Paradowska-Gorycka A and Jurkowska M:
Structure, expression pattern and biological activity of molecular
complex TREM-2/DAP12. Hum Immunol. 74:730–737. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Filbert EA: Investigations of mechanisms
involved in LPS-stimulated osteoclastogenesis. University of
Connecticut, School of Dental Medicine; SoDM Masters Theses, Paper
155. 2007
|
|
11
|
Sharif O and Knapp S: From expression to
signaling: Roles of TREM-1 and TREM-2 in innate immunity and
bacterial infection. Immunobiology. 213:701–713. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tomasello E, Desmoulins P-O, Chemin K,
Guia S, Cremer H, Ortaldo J, Love P, Kaiserlian D and Vivier E:
Combined natural killer cell and dendritic cell functional
deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity.
13:355–364. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Helming L, Tomasello E, Kyriakides TR,
Martinez FO, Takai T, Gordon S and Vivier E: Essential role of
DAP12 signaling in macrophage programming into a fusion-competent
state. Sci Signal. 1:ra112008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Turnbull IR, Gilfillan S, Cella M, Aoshi
T, Miller M, Piccio L, Hernandez M and Colonna M: Cutting edge:
TREM-2 attenuates macrophage activation. J Immunol. 177:3520–3524.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Paloneva J, Manninen T, Christman G,
Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R,
Salmaggi A, et al: Mutations in two genes encoding different
subunits of a receptor signaling complex result in an identical
disease phenotype. Am J Hum Genet. 71:656–662. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guerreiro R, Wojtas A, Bras J,
Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe
JS, Younkin S, et al; Alzheimer Genetic Analysis Group. TREM2
variants in Alzheimer’s disease. N Engl J Med. 368:117–127. 2013.
View Article : Google Scholar
|
|
17
|
Wang XQ, Tao BB, Li B, Wang XH, Zhang WC,
Wan L, Hua XM and Li ST: Overexpression of TREM2 enhances glioma
cell proliferation and invasion: A therapeutic target in human
glioma. Oncotarget. 7:2354–2366. 2016.
|
|
18
|
Yao Y, Li H, Wang Y and Zhou J: Triggering
receptor expressed on myeloid cells-2 (TREM-2) elicited by lung
cancer cells to facilitate tumor immune evasion. J Clin Oncol.
31(Suppl): 220542013.
|
|
19
|
Chen Y, Guo Y, Yang H, Shi G, Xu G, Shi J,
Yin N and Chen D: TRIM66 overexpresssion contributes to
osteosarcoma carcinogenesis and indicates poor survival outcome.
Oncotarget. 6:23708–23719. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liao R, Sun TW, Yi Y, Wu H, Li YW, Wang
JX, Zhou J, Shi YH, Cheng YF, Qiu SJ, et al: Expression of TREM-1
in hepatic stellate cells and prognostic value in hepatitis
B-related hepatocellular carcinoma. Cancer Sci. 103:984–992. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ho CC, Liao WY, Wang CY, Lu YH, Huang HY,
Chen HY, Chan WK, Chen HW and Yang PC: TREM-1 expression in
tumor-associated macrophages and clinical outcome in lung cancer.
Am J Respir Crit Care Med. 177:763–770. 2008. View Article : Google Scholar
|
|
22
|
Yuan Z, Mehta HJ, Mohammed K, Nasreen N,
Roman R, Brantly M and Sadikot RT: TREM-1 is induced in tumor
associated macrophages by cyclo-oxygenase pathway in human
non-small cell lung cancer. PLoS One. 9:e942412014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gobé G, Rubin M, Williams G, Sawczuk I and
Buttyan R: Apoptosis and expression of Bcl-2, Bcl-XL, and Bax in
renal cell carcinomas. Cancer Invest. 20:324–332. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sejima T and Miyagawa I: Expression of
bcl-2, p53 oncoprotein, and proliferating cell nuclear antigen in
renal cell carcinoma. Eur Urol. 35:242–248. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fisher PA, Moutsiakis DL, McConnell M,
Miller H and Mozzherin DJ: A single amino acid change (E85K) in
human PCNA that leads, relative to wild type, to enhanced DNA
synthesis by DNA polymerase δ past nucleotide base lesions (TLS) as
well as on unmodified templates. Biochemistry. 43:15915–15921.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang P, Sun Y, Hsu H, Zhang L, Zhang Y
and Lee MY: The interdomain connector loop of human PCNA is
involved in a direct interaction with human polymerase δ. J Biol
Chem. 273:713–719. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pleschke JM, Kleczkowska HE, Strohm M and
Althaus FR: Poly(ADP-ribose) binds to specific domains in DNA
damage checkpoint proteins. J Biol Chem. 275:40974–40980. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cohen GM: Caspases: The executioners of
apoptosis. Biochem J. 326:1–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fry MJ: Phosphoinositide 3-kinase
signalling in breast cancer: How big a role might it play? Breast
Cancer Res. 3:304–312. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin X, Böhle AS, Dohrmann P, Leuschner I,
Schulz A, Kremer B and Fändrich F: Overexpression of
phosphatidylinositol 3-kinase in human lung cancer. Langenbecks
Arch Surg. 386:293–301. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Martínez-Lorenzo MJ, Anel A, Monleón I,
Sierra JJ, Piñeiro A, Naval J and Alava MA: Tyrosine
phosphorylation of the p85 subunit of phosphatidylinositol 3-kinase
correlates with high proliferation rates in sublines derived from
the Jurkat leukemia. Int J Biochem Cell Biol. 32:435–445. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Merseburger AS, Hennenlotter J, Kuehs U,
Simon P, Kruck S, Koch E, Stenzl A and Kuczyk MA: Activation of
PI3K is associated with reduced survival in renal cell carcinoma.
Urol Int. 80:372–377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhu Y, Xu L, Zhang J, Xu W, Liu Y, Yin H,
Lv T, An H, Liu L, He H, et al: Klotho suppresses tumor progression
via inhibiting PI3K/Akt/GSK3β/Snail signaling in renal cell
carcinoma. Cancer Sci. 104:663–671. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhan YH, Liu J, Qu XJ, Hou KZ, Wang KF,
Liu YP and Wu B: β-Elemene induces apoptosis in human renal-cell
carcinoma 786–0 cells through inhibition of MAPK/ERK and
PI3K/Akt/mTOR signalling pathways. Asian Pac J Cancer Prev.
13:2739–2744. 2012. View Article : Google Scholar
|
|
37
|
Li H, Zeng J and Shen K: PI3K/AKT/mTOR
signaling pathway as a therapeutic target for ovarian cancer. Arch
Gynecol Obstet. 290:1067–1078. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Roy SK, Srivastava RK and Shankar S:
Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of
FOXO transcription factor, leading to cell cycle arrest and
apoptosis in pancreatic cancer. J Mol Signal. 5:102010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun M, Zhu M, Chen K, Nie X, Deng Q,
Hazlett LD, Wu Y, Li M, Wu M and Huang X: TREM-2 promotes host
resistance against Pseudomonas aeruginosa infection by suppressing
corneal inflammation via a PI3K/Akt signaling pathway. Invest
Ophthalmol Vis Sci. 54:3451–3462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu M, Li D, Wu Y, Huang X and Wu M:
TREM-2 promotes macrophage-mediated eradication of Pseudomonas
aeruginosa via a PI3K/Akt pathway. Scand J Immunol. 79:187–196.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Myers MP and Tonks NK: PTEN: Sometimes
taking it off can be better than putting it on. Am J Hum Genet.
61:1234–1238. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Eng C: PTEN: One gene, many syndromes. Hum
Mutat. 22:183–198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Muñoz J, Lázcoz P, Inda MM, Nistal M,
Pestaña A, Encío IJ and Castresana JS: Homozygous deletion and
expression of PTEN and DMBT1 in human primary neuroblastoma and
cell lines. Int J Cancer. 109:673–679. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nassif NT, Lobo GP, Wu X, Henderson CJ,
Morrison CD, Eng C, Jalaludin B and Segelov E: PTEN mutations are
common in sporadic microsatellite stable colorectal cancer.
Oncogene. 23:617–628. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Osaki M, Oshimura M and Ito H: PI3K-Akt
pathway: Its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|