Introduction
Ovarian cancer is associated with the highest
mortality rate among women of all other gynecological cancers in
the world (1). Surgical removal of
the tumor with following platinum-based chemotherapy are the
standard methods used to treat the disease (1). However, with increasing
chemoresistance level of the tumor, discovery of new effectual
alternative approaches is becoming an urgent need.
Flavonoids are a group of the most abundant
polyphenols in our daily diet and display a wide range of
pharmacological properties (2).
Quercetin-3-O-β-D-galactopyranoside, also known as
hyperoside, is a flavonol glycoside mainly found in plants of the
genera Hypericum and Crataegus (3). Previous studies have shown that
hyperoside has anti-oxidant (3,4),
anti-inflammatory (5,6) and anticancer activities (7,8).
Furthermore, hyperoside produced anticancer effects through
inducing apoptosis (8). However,
the effect of hyperoside on ovarian cancer cells, especially
whether it induces apoptosis, and the underlying mechanisms are all
still unknown.
Anticancer drugs induce programmed cell death (PCD).
Apoptosis, also known as type I cell death, is regarded as the
principal cell death mechanism (9). However, cancer cells trigger multiple
pathways to escape from apoptosis (10,11).
One of the most important mechanism by which ovarian cancer cells
avoid cisplatin-induced apoptosis is that progesterone receptor
membrane component (PGRMC) 1 overexpressing in these cells
simultaneously induce epithelial grow factor receptor (EGFR)
stabilization to promote cell survival and induce cytochrome p450
activation to accelerate drug efflux (12–14).
Moreover, the type II PCD, autophagy, which shows a biphasic effect
on cell viability, possibly is also responsible for chemoresistance
(15). Accordingly, PD168393, an
EGFR-TKI, induce autophagy as a cytostatic, but not a cytotoxic,
response in malignant peripheral nerve sheath tumor (MPNST) cells
that is accompanied by suppression of Akt and mTOR activation
(16). In addition to EGFR,
genetic and pharmacologic autophagy blockade via PI3K/mTOR
inhibition reverses apoptotic resistance and result in significant
cell apoptosis (17). Besides, p53
(18), VEGF (19), MAPK14/p38α signaling (20) and microRNAs (21,22)
are also engaged in autophagy-mediated cancer cell chemoresistance.
Nevertheless, other studies support a role of autophagy for tumor
suppression. Patients with low levels of autophagy-related protein
(ATG) 5 in their tumors had reduced progression-free survival
(23). Ursolic acid promotes
cancer cell death by inducing ATG 5-dependent autophagy (24). Thus, molecules leading to cytotoxic
autophagy may circumvent apoptotic resistance and benefit cancer
treatment.
Notably, a linkage between autophagy and PGRMC1 has
been identified (25), PGRMC1 not
only induces chemoresistance, but also promotes autophagy. PGRMC1
inhibitor or PGRMC1-knockdown leads to autophagic flux
inhibition (25). In addition,
PGRMC1-knockdown cells had increased levels of ubiquitinated
proteins, a substrate of autophagy (25). The roles of PGRMC1 and autophagy in
the presence of hyperoside in survival of ovarian cancer cells were
investigated. We focused on the effect of hyperoside on cell
viability, apoptosis and autophagy of ovarian cancer cells,
additionally, detailed mechanisms involved including PGRMC1/AKT
signaling and Bcl-2 family were thoroughly investigated in this
study.
Materials and methods
Cells, plasmids, transfection and
reagents
The ovarian cancer cell lines SKOV-3 and HO-8910
were obtained from the American Type Culture Collection (ATCC,
Rockville, MD, USA). SKOV-3 and HO-8910 cells were sustained in
Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) with 10%
(v/v) fetal bovine serum (FBS; Gibco), 100 IU/ml penicillin and 100
ng/ml streptomycin (PAA Laboratories GmbH, Pasching, Austria) at
37°C, in 5% CO2 humidified atmosphere. A 603-bp fragment
of PGRMC1 complementary DNA was amplified from SKOV-3 cells
by RT-PCR and inserted into the secretory vector, pSecTag2B (a kind
gift of Professor C. Lu, Department of Molecular Virology, Nanjing
Medical University, China), to generate recombinant pPGRMC1. Short
hairpin RNAs for PGRMC1 knockdown were purchased from
Shanghai GenePharma Co. (Shanghai, China) and an optimized shPGRMC1
was determined by western blotting. Transfections of SKOV-3 cells
were performed with the Lipofectamine 2000 reagent (Invitrogen
Inc., Carlsbad, CA, USA) according to the manufacturer's
instructions. Hyperoside (MW: 464.38, HPLC ≥98%), 3-methyladenine
(3-MA) and monodansylcadaverine (MDC) were purchased from
Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Hyperoside was
dissolved in aqueous DMSO and delivered to cells in media
containing this solvent at a final concentration of 0.1% (v/v).
Cisplatin was a kind gift of Dr Wei Zhu (26).
Cell proliferation assay
Cell viability in the treated cells was determined
by using Cell Counting Kit-8 (CCK-8) kit (Dojindo Laboratories,
Kumamoto, Japan) according to the manufacturer's instructions and
as previously described by us (27). Briefly, cells were plated at a
density of 3×103 cells/well with 100 µl of medium
in 96-well plates with increasing doses of hyperoside (0, 50 and
100 µM, dissolved in DMSO). After treatment, CCK-8 solution
(10 µl) was added to each well and the plates were incubated
at 37°C for 90 min. The absorbance of the cell suspension was
measured with a microplate reader at a wavelength of 490 nm. The
highest concentration of hyperoside does not interfere with the
CCK-8 assay reagents in the absence of cells (data not shown).
Medium containing 10% CCK-8 served as a control.
Plate colony formation assay
To evaluate the ability of cell proliferation, the
plate colony formation assay was performed as described previously
(28). Briefly, cells
(~2×102) were seeded in each well of 12-well plates with
complete medium containing hyperoside with increasing concentration
from 0 to 100 µM. Cultures were supplemented with
conditional complete medium per week. Cells were then fixed in
methanol/glacial acetic acid (7:1), washed with water and stained
with crystal violet (0.2 g/l). Colonies were scored 14–21 days
after seeding the cells.
Cell apoptosis assay
The percentage of cells actively undergoing
apoptosis was determined by flow cytometry using an Annexin V assay
kit according to the manufacturer's instructions and as previously
described (27). Briefly, cells
were incubated with increasing doses of hyperoside (vehicle, 50 and
100 µM) for 72 h, and then the cells were harvested with
trypsin, washed in phosphate-buffered saline (PBS), and counted.
After counting, 1×105 cells were resuspended in binding
buffer at a concentration of 1×106 cells/ml. Next, 10
µl of Annexin V and 5 µl of PI were added, and the
cells were incubated at room temperature for ≥15 min in the dark.
After incubation, the percentage of apoptotic cells was analyzed by
flow cytometry (FACScan; BD Biosciences, USA).
Flow cytometry
To detect the expression of LC3B in ovarian cancer
cells, 1×106 cells were collected and suspended in cold
PBS, fixed with 80% methanol (5 min) and then permeabilized with
0.1% PBS-Tween for 20 min. The cells were then incubated in 1X
PBS/10% normal goat serum/0.3 M glycine to block non-specific
protein-protein interactions followed by the LC3B mAb (Abcam,
ab213934) for 30 min at 22°C. The secondary antibody used was Alexa
Fluor® 488 goat anti-mouse IgG (H+L) (ab150117) at
1:2,000 dilution for 30 min at 22°C. Acquisition of >10,000
events were collected and analyzed with the FACScan (BD
Biosciences).
MDC staining of autophagic vacuoles
MDC staining of autophagic vacuoles was performed
for autophagy analysis. Briefly, SKOV-3 cells or HO-8910 cells were
seeded onto cover slips placed onto a 6-well plate at a density of
1×105 cells/ml 24 h before treatment and were incubated
overnight at 37°C. After a 48 h treatment with hyperoside (100
µM), the cells were incubated for 20 min with MDC (0.05
mmol/l) at 37°C and were then washed four times with PBS.
Autophagic vacuoles were observed under a fluorescence
microscope.
Immunofluorescence staining
For immunofluorescence, cells were fixed in 3:1
methanol:acetone at 4°C for 45 min as recommended, and then washed,
blocked with 10% normal goat serum and probed with mouse anti-LC3B
(Abcam, ab213934) and, where indicated, rabbit anti-PGRMC1 (Abcam,
ab88381). Fluorescein isothiocyanate (FITC) conjugated goat
anti-rabbit IgG (H+L) and Cy3® conjugated goat
anti-mouse IgG (H+L) were used as secondary antibodies. All other
procedures for staining were according to the manufacturer's
instructions. Images were observed and recorded with a Zeiss
Axiovert 200 M epifluorescence microscope (Carl Zeiss Inc.,
Thüringen, Germany).
Western blotting and antibodies
Western blotting was performed as previously
described (26,27). Anti-PGRMC1 rabbit polyclonal
antibody, and anti-LC3B mouse monoclonal antibody (mAb) were
purchased from Abcam Inc. (Abcam, Cambridge, UK). Anti-GAPDH mouse
mAb, and horseradish peroxidase (HRP)-conjugated goat anti-mouse
and anti-rabbit IgG were purchased from Santa Cruz Biotechnologies
(Santa Cruz, CA, USA). Anti-Bcl-2 mouse mAb, anti-Bax rabbit mAb,
anti-phospho-AKT (Ser473) mouse mAb, and anti-AKT mouse mAb, were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Statistical analysis
Numerical data were expressed as mean ± SD. Two
group comparisons were analyzed by two-sided Student's t-test.
p-values were calculated, and p<0.05 was considered significant.
All experiments were performed at least in triplicate.
Availability of data and materials
Literature collection was performed by using
electronic databases PubMed, Cochrane Library, and Web of Science.
All statistical analyses were executed by using SPSS 20.0 software
(IBM, Chicago, IL, USA). Raw and processed data are stored with the
corresponding author of this report and are available upon
request.
Results
Effect of hyperoside on viability of
ovarian cancer cells
To determine the effect of hyperoside on viability
of human ovarian cancer cells, we used SKOV-3 and HO-8910 cells,
all of which were exposed to increasing concentrations of
hyperoside (0, 50 or 100 µM) for 24, 48 and 72 h. As shown
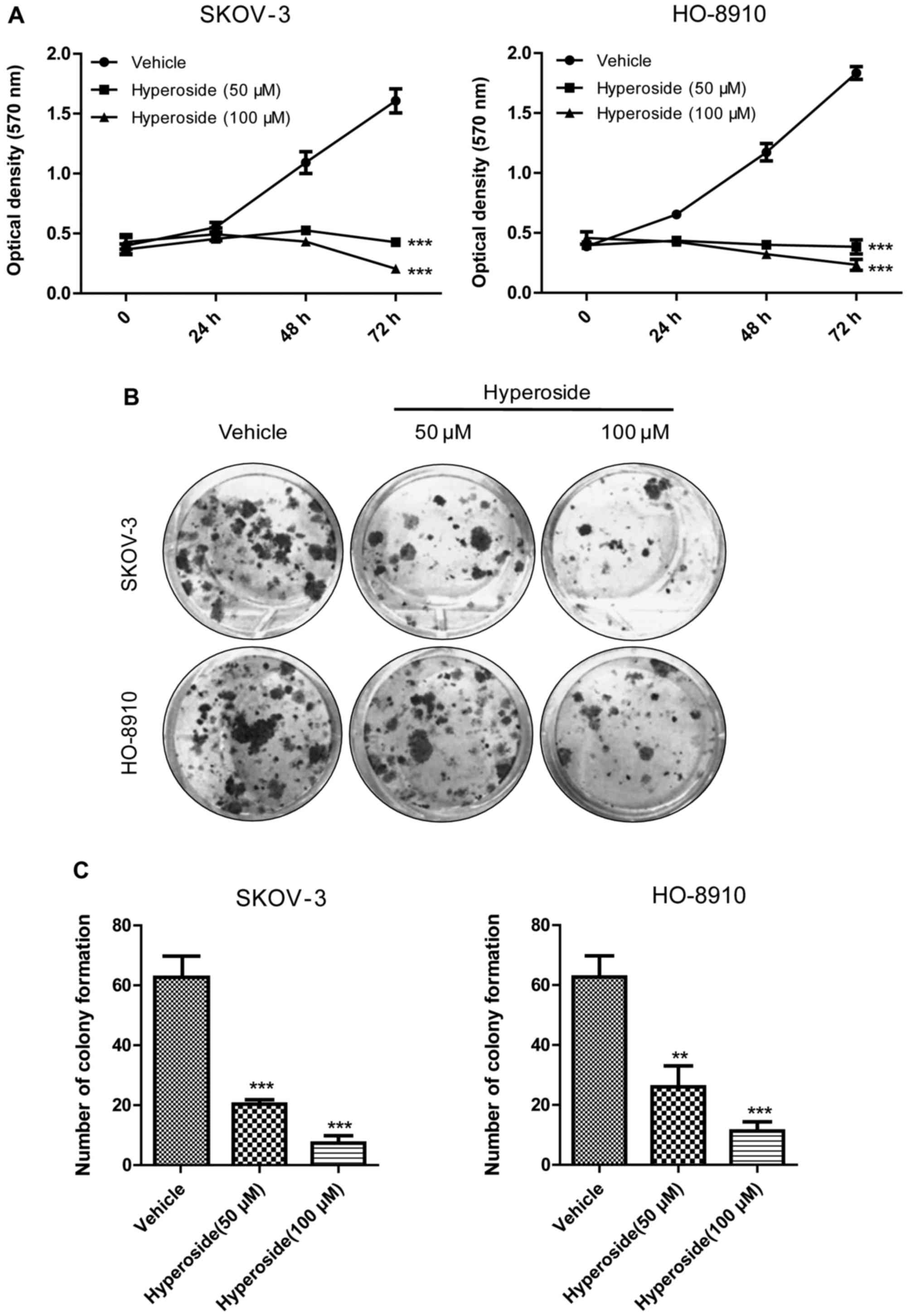
in Fig. 1A hyperoside inhibited
the proliferation of both ovarian cancer cell lines in a dose- and
time-dependent manner. Concordantly, in the colony formation assay
(Fig. 1B and C), hyperoside
suppressed the colony formation of both cancer cells in a
dose-dependent manner. Briefly, in SKOV-3 cells, the number of
colony formation of 50 or 100 µM group was 20.33±0.88 or
7.33±1.15 respectively, compared with the control group
(62.67±6.76). Similarly, in HO-8910 cells, the number of colony
formation of 50 or 100 µM group was 26.00±4.04 or
11.33±1.76, compared with the control group (64.88±5.71). Together,
these data indicate that hyperoside can effectively inhibit the
proliferation of ovarian cancer cells.
Hyperoside induces apoptosis and
autophagy in ovarian cancer cells
As hyperoside has a potent anti-proliferative
potential in ovarian cancer cells, we suspected that hyperoside
induces apoptosis in these tumor cells. Thus, SKOV-3 or HO-8910
cells were treated with various concentrations of hyperoside (0, 50
or 100 µM) for 48 h, stained with Annexin V/PI, subjected to
flow cytometry to determine the apoptosis rate. As we can see in
Fig. 2A, the treatment of ovarian
cancer cells with various concentrations of hyperoside led to an
obvious dose-dependent improvement in both early and late stages of
apoptosis. The apoptotic indices were 0.97±0.29, 29.70±1.65, and
47.53±1.59% in SKOV-3 and 3.47±1.31, 30.53±1.03, and 56.50±2.30% in
HO-8910 at 0, 50 and 100 µM concentrations of hyperoside,
respectively. Additionally, we observed characteristics of vacuolar
structure in cancer cells which were exposed to hyperoside,
comparing to that of vehicle- or oroxylin A-treated cells (Fig. 2C). Subsequently, as shown in
Fig. 2D, the characteristic was
validated as autophagic vacuoles by the MDC staining. Considering
the linkage between apoptosis and autophagy, we inferred that
autophagy may be involved in the hyperoside-induced apoptosis. Then
flow cytometry and western blotting were carried out to determine
the expression of LC3B, which is a specific marker of autophagy. As
shown in Fig. 2E and F, hyperoside
dose-dependently increased the level of LC3B in both SKOV-3 (1.82-
to 2.97-fold of activation normalized to the control) and HO-8910
(2.36- to 3.77-fold of activation normalized to the control) cells,
which indicated that autophagy plays a role at least in part in the
hyperoside-induced apoptosis. Concordantly, the expression of
Bax/Bcl-2 protein were also in line with the case, where hyperoside
treatment downregulated the level of bcl-2 (0.43–0.17 in SKOV-3 or
0.52–0.36 in HO-8910) but increased the expression of bax
(1.67–2.56 or 2.56–3.49) in a dose-dependent manner. Together,
these results suggest an autophagy-associated cell death by
hyperoside in ovarian cancer cells.
 | Figure 2Hyperoside induces apoptosis and
autophagy in ovarian cancer cells. (A) Effect of hyperoside on
apoptosis of SKOV-3 and HO-8910 cells. SKOV-3 and HO-8910 cells
were incubated with hyperoside at 0, 50 and 100 µM. Cells
were collected, and apoptotic cells were examined at 48 h post
incubtion. x- and y-axes indicated Annexin V and propidium iodide
staining intensities, respectively. (B) Summary of percentages of
total apoptotic cells in (A), ***p<0.001, vs.
vehicle-treated cells. (C) Morphological alteration of SKOV-3 cells
upon hyperoside (100 µM) treatment, compared with vehicle-
(negative control) or oroxylin A- (disparity control) treated
cells. Photographs of hyperoside depict vacuolar structure in
SKOV-3 cells (left, original magnification, ×100; right, ×400). (D)
Monodansycadaverine (MDC) staining. SKOV-3 and HO-8910 cells
treated with hyperoside (100 µM) for 48 h were incubated
with MDC (0.05 mM) for 20 min and observed under a fluorescence
microscope (left, original magnification, ×100). Inserts are high
magnification micrographs of the boxed regions (right, ×400). (E)
Representative flow cytometry histograms for LC3B expression in
SKOV-3 cells treated as in (A). Cells were stained with anti-LC3B
MAb and fluorescein isothiocyanate-labeled IgG was used as
secondary antibody. (F) Western blot analysis of LC3B, Bcl-2 and
Bax in SKOV-3 and HO-8910 cells incubated with hyperoside at 0, 50
and 100 µM for 48 h. The relative level of LC3B-II, Bcl-2
and Bax were determined by quantitative densitometry compared to
GAPDH. The relative value of LC3B-II, Bcl-2 and Bax in vehicle
group was considered to be one for comparison, respectively. |
Inhibition of autophagy blocks
hyperoside-induced apoptotic cell death
Autophagy and apoptosis may act independently in
parallel pathways or may influence one another (29). Autophagy may cooperate with
apoptosis to promote cell death (30). To test whether hyperoside-induced
apoptosis is dependent on autophagy, we investigated the apoptotic
effect following autophagy inhibition by 3-MA after hyperoside
exposure. SKOV-3 or HO-8910 cells were treated with hyperoside (100
µM) in the presence or absence of 3-MA (2 mM) for 48 h. As
shown in Fig. 3A, 3-MA pretreatment significantly decreased
the LC3B-II/LC3B-I ratio of hyperoside-treated SKOV-3 cells from
1.69- to 0.87-fold, compared to the cells treated with hyperoside
alone. Similar result was also determined by flow cytometry assay
(Fig. 3B). Notably, 3-MA
pretreatment markedly attenuated the hyperoside-induced
upregulation of Bax (4.59- to 0.53-fold activation) and restored
the hyperoside-induced downregulation of Bcl-2 (0.66- to 0.92-fold
activation) (Fig. 3A). Finally,
assays were carried out to access the effect of 3-MA on cell
apoptosis. As expected, SKOV-3 cells with 3-MA pretreatment were
restored from hyperoside-induced apoptosis with an apoptotic ratio
of 11.21±0.87%, compared to the cells without 3-MA pretreatment of
31.24±0.90%. These results suggest that hyperoside-induced
apoptosis of ovarian cancer cells is at least partly dependent on
autophagy.
Hyperoside-induced autophagy is dependent
on the PGRMC1/AKT pathway
Classical AKT signaling has been shown to be engaged
in autophagy (31). Thus, we
attempted to find out whether AKT signaling also plays a role in
hyperoside-induced autophagy. SKOV-3 cells pretreated with
hyperoside (100 µM) for 48 h, were subjected to western
blotting. As shown in Fig. 4D,
hyperoside inhibited the expression of p-AKT (0.59-fold activation
normalized to the control), which suggested that AKT signaling
inactivation may be utilized by hyperoside to induce autophagy.
PGRMC1 has taken a great part in tumorigenesis by promoting cell
invasion and by withstanding drug stress (32). As is the case with autophagy,
PGRMC1 promotes recycling substrate to help tumor cells survive
(25). However, in the condition
of hyperoside, where autophagy is pointing to apoptosis, the effect
of PGRMC1 on cell viability and apoptosis is still unclear. Thus,
an overexpression construct of PGRMC1 (pPGRMC1) as well as a
knockdown shRNAs were transfected into SKOV-3 cells for hyperoside
treatment, respectively. Interestingly, PGRMC1 overexpression
significantly promoted hyperoside-induced autophagy and cell
apoptosis (Fig. 4A–C). Apoptotic
ratio of SKOV-3 cells treated with hyperoside plus pPGRMC1 was
increased to 32.31±3.30 from 2.50±1.50% of hyperoside alone treated
cells. In contrast, when expression of PGRMC1 was knocked down by
shRNA, hyperoside-induced autophagy and apoptosis were also
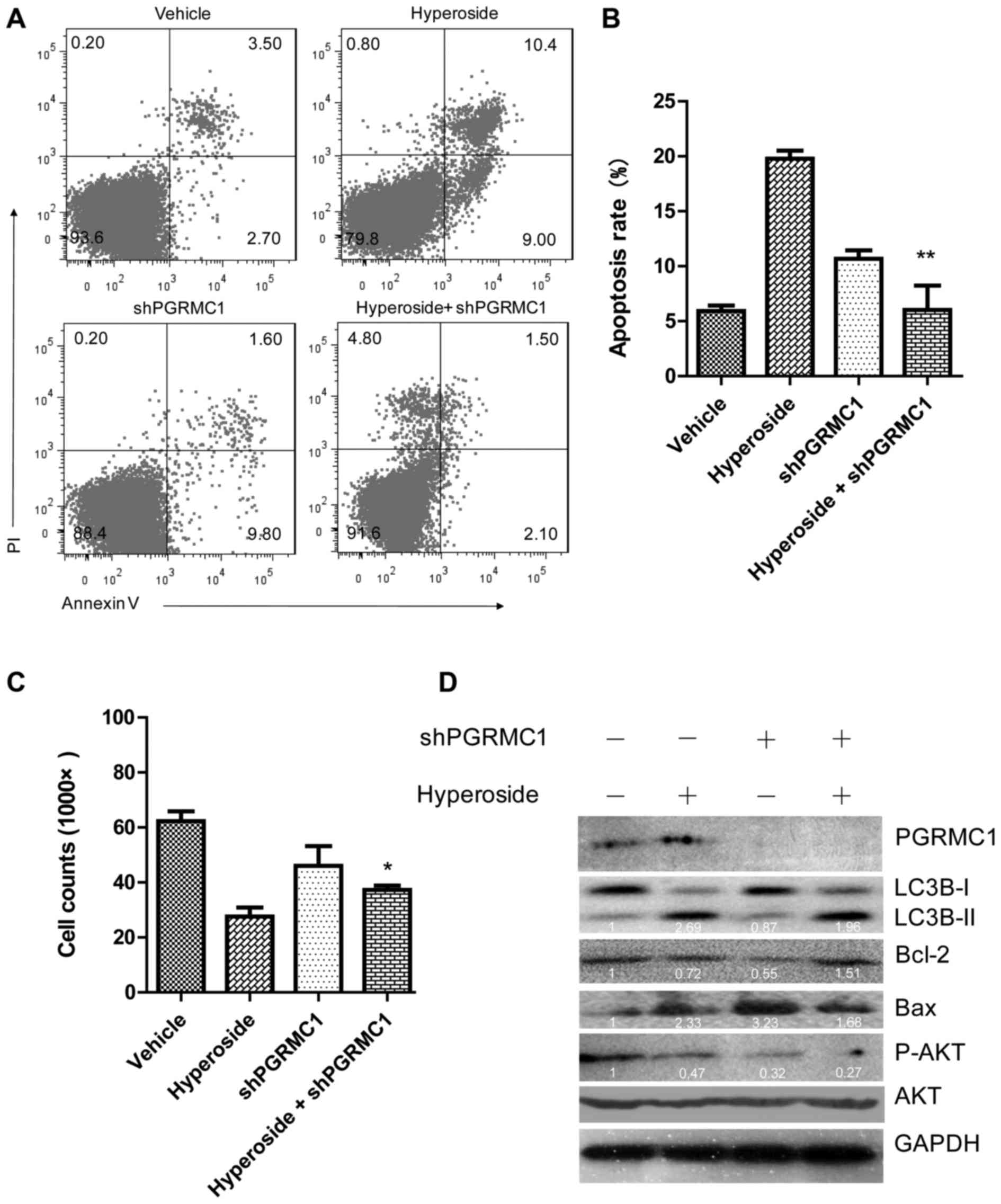
abrogated (Fig. 5A–C). Apoptotic
ratio of hyperoside plus shPGRMC1-treated SKOV-3 cells was
decreased to 6.02±1.28%, compared to that of hyperoside alone
treated cells of 19.78±0.72%. Concordantly, at the protein level,
PGRMC1 overexpression enhanced LC3B expression in
hyperoside-treated SKOV-3 cells (2.77- to 3.21-fold activation) as
well as the Bax expression (1.59–1.93), while Bcl-2 expression
(0.62–0.34) was decreased (Fig.
4D). Conversely, PGRMC1 knockdown inhibited LC3B (2.69–1.96)
and Bax expression (2.33–1.68), while Bcl-2 expression (0.72–1.51)
was elevated (Fig. 5D). Also of
note is that PGRMC1 alone enhanced phosphorylation of AKT in SKOV-3
cells without hyperoside-treatment (1.71-fold activation), but in
the presence of hyperoside, phosphorylation of AKT was reversely
exhausted (Fig. 4D). Although the
knockdown of PGRMC1 failed to give a significant increase of p-AKT
in hyperoside-treated cells (Fig.
5D), PGRMC1/AKT axis at least play a partial role in
hyperoside-induced autophagy.
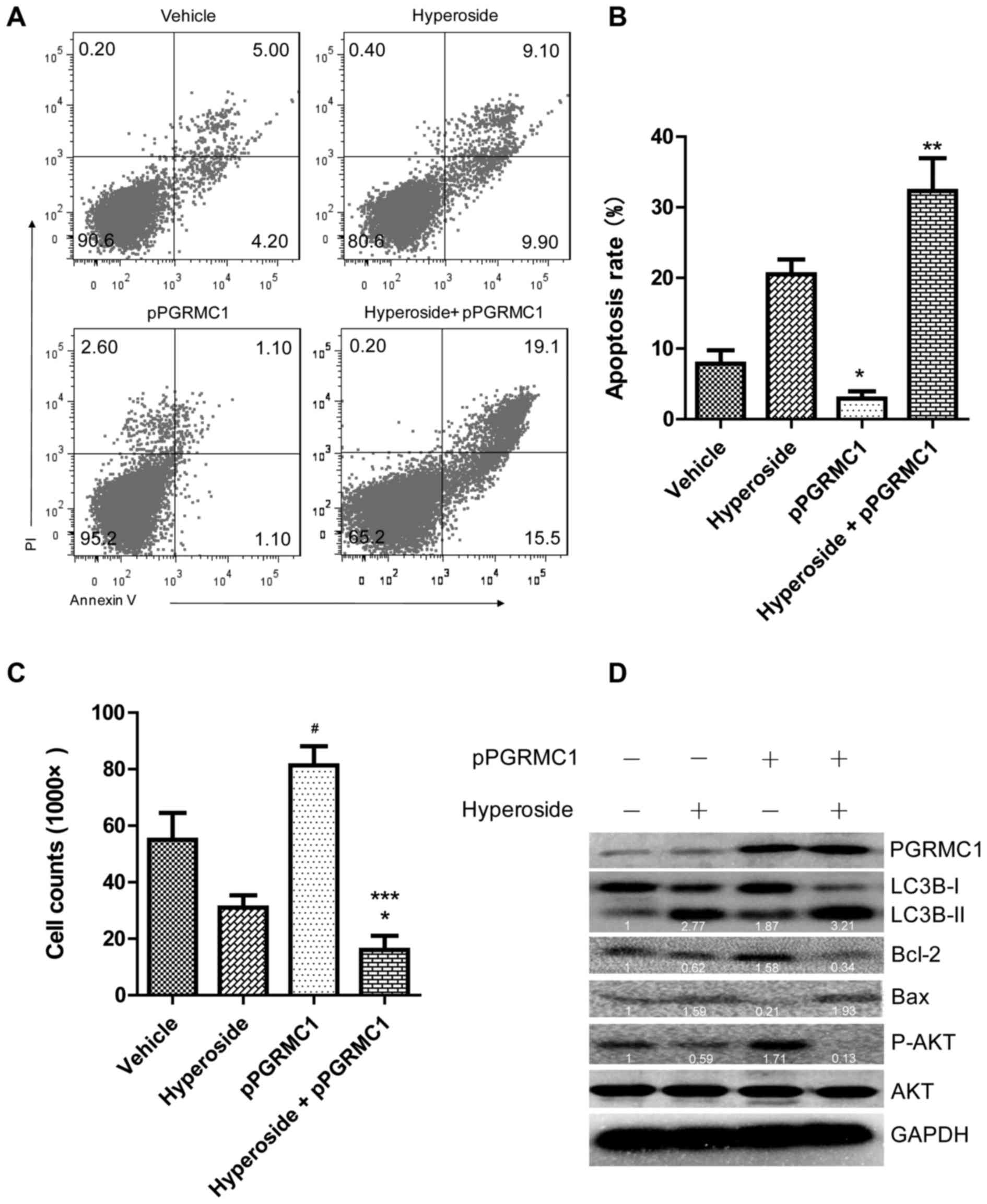 | Figure 4PGRMC1 overexpression induces
autophagy and apoptosis in SKOV-3 cells. SKOV-3 cells transfected
with or without pPGRMC1 (4 µg) for 24 h were subjected to
hyperoside (100 µM) and inculcated for another 24 h. (A)
Cells were collected, and incubated with Annexin V and propidium
iodide for apoptosis analysis. (B) Summary of percentages of total
apoptotic cells in (A), *p<0.05, vs. vehicle-treated
cells, ***p<0.001, vs. hyperoside-treated cells. (C)
Cells of the samples were trypsin digested, resuspended in a total
volume of 200 µl medium, and cell number was counted by
double-blind method. The results represent the mean ± SD; a
representative experiment repeated three times is shown.
*p<0.05, vs. vehicle-treated cells; and
***p<0.001, vs. hyperoside-treated cells. (D)
Cellular proteins were lysed and subjected to western blot analysis
for levels of LC3, Bcl-2, Bax and AKT. |
PGRMC1 colocalizes with LC3B to promote
hyperoside-induced autophagic cell death and sensitizes ovarian
cancer cells to cisplatin treatment
As described in our previous study (26), cisplatin-treatment of ovarian
cancer would lead to an overexpression of PGRMC1, which then will
induce cancer chemoresistance and promote cell survival. However,
in this study, we also found a potential of PGRMC1 in promoting
hyperoside-induced autophagic cell death. Thus, we inferred that
hyperoside may synergistically with cisplatin kill ovarian cancer
cells, especially when PGRMC1 is overexpressed. Cell viability and
apoptosis of ovarian cancer cells subjected to hyperoside,
cisplatin or a combination of both were assessed. As shown in
Fig. 6C, cisplatin, at a level of
20 µM, did not significantly inhibit SKOV-3 cell
proliferation. However, when in the presence of hyperoside,
cisplatin at the same concentration significantly blocked the
proliferation of the cells. Moreover, plenty of autophagic vacuoles
emerged in cisplatin plus hyperoside treated SKOV-3 cells by the
MDC staining, compared to that of cells treated with cisplatin
alone (Fig. 6D). In agreement with
these findings, cisplatin alone could not induce apparent
apoptosis, whereas cisplatin plus hyperoside induced a large body
of apoptosis (Fig. 6A). Notably,
co-localization of PGRMC1 with LC-3B in SKOV-3 cells was also
determined by immunofluorescence staining as shown in Fig. 6E. Cisplatin alone induced massive
expression of PGRMC1, but had no significant effect on LC-3B
expression. However, in the presence of hyperoside, abundant PGRMC1
induced by cisplatin co-localized with LC-3B to promote autophagic
cell death, thus confirming a striking role of PGRMC1 in
hyperoside-induced autophagic cell death. Taken together, in
ovarian cancer cells especially the drug-resistant ones where
PGRMC1 is overexpressed, hyperoside may utilize this 'Achilles'
heel' of PGRMC1 to promote autophagic cell death.
 | Figure 6Hyperoside synergizes with cisplatin
to promote cell death of SKOV-3 in which LC3B colocalized with
PGRMC1. (A) SKOV-3 cells treated with hyperoside (100 µM),
cisplatin (20 µM) or hyperoside plus cisplatin were
subjected to apoptosis assay. (B) A histogram of percentages of
total apoptotic cells is presented with the means ± SE from three
independent experiments. ***p<0.001, vs. hyperoside-
or cisplatin-treated cells. (C) MTT assay for viability of SKOV-3
cells exposed to hyperoside, cisplatin or hyperoside plus
cisplatin, respectively. ***p<0.001 and
**p<0.01, vs. cisplatin-treated cells. (D) MDC
staining of SKOV-3 cells treated with hyperoside or hyperoside plus
cisplatin. Green fluorescence indicates autophagic vacuoles
(original magnification, ×100). (E) Fluorescence microscopy of
SKOV-3 cells incubated for 24 h with hyperoside, cisplatin or
hyperoside plus cisplatin, then stained for PGRMC1 (green) and LC3B
(red) with rabbit-anti-PGRMC1 and mouse-anti-LC3B antibodies,
respectively. FITC-tagged anti-rabbit IgG and CY3-tagged anti-mouse
IgG were used as second antibodies. Arrows indicate colocalization
of PGRMC1 and LC3B. 4′,6′-diamidino-2-phenylindole (DAPI) (blue)
stained nuclei. |
Discussion
Ovarian cancer causes the most mortality in
gynecological malignancies (33).
However, to date, limited therapeutic measures such as surgical
resection and platinum-based chemotherapy have shown weak efficacy
(33–36). Progesterone receptor membrane
component (PGRMC) 1 plays a vital role in the chemoresistance of
the cancer. Accordingly, as shown in our previous study, PGRMC1 is
elevated in cisplatin-treated HO-8910 cells (26), while also elevated in SKOV-3 cells
and ovarian cancer tissues as described by Peluso (37,38).
The mechanism by which PGRMC1 mediated chemoresistance is: i)
enhancing expression of cytochrome P450 to accelerate drug
metabolism (39,40); ii) promoting cancer cell migration
and invasion to evade cytotoxicity (38,40,41);
and, iii) activating signaling pathways to avoid apoptosis
(42). For this reason, ligands
targeting PGRMC1 have shown a promising potential in ovarian cancer
therapy (43). However, in this
study, we first report a feeble effect of the tumor-enhancing
protein PGRMC1, which has been utilized by hyperoside to promote
autophagy and induce apoptosis in ovarian cancer cells.
In this study, we demonstrated that hyperoside
dose-dependently inhibits the proliferation and colony formation of
both SKOV-3 and HO-8910 cells. Furthermore, Annexin V/PI double
staining showed a dose-dependent apoptotic effect of hyperoside on
the cells. This indicated that hyperoside inhibits cell viability
by inducing apoptosis. Notably, dose-dependent apoptosis by
hyperoside constantly corresponded to a dose-dependent expression
of LC3B-II, which suggesting an involvement of autophagy in the
hyperoside-mediated apoptosis. Autophagy is an evolutionarily
conserved intracellular catabolic process that is used by all cells
to degrade dysfunctional or unnecessary cytoplasmic components
through delivery to the lysosome (44). However, the role of autophagy in
tumor formation is crucial but ambiguous. On one hand, through
intracellular recycling, autophagy provides substrates that enable
tumor cells to survive the metabolic stress in the tumor
microenvironment and promotes tumor progression (44). On the other hand, imbalanced
autophagy functions in tumor suppression through directly
restricting cell proliferation by inducing cell death (45). To verify the role of autophagy in
this case, an autophagy inhibitor, 3-MA was used to block the
autophagy by hyperoside. As expected, 3-MA exposure not only gave a
reduction in the amount of LC3B protein, but also restored the
conversion of LC3B-I to LC3B-II in hyperoside-treated ovarian
cancer cells. Autophagy and apoptosis function in parallel tracks
but sometimes they also engage in a complex interplay in both
physiological and pathological settings (46). Herein, the interplay gave a causal
relationship with the fact that autophagy evoked apoptosis in
hyperoside-treated ovarian cancer cells. As a result of the
autophagy inhibition by 3-MA, the level of Bcl-2 was elevated while
the level of Bax was decreased, suggesting a subsequent reversion
of apoptosis by 3-MA in hyperoside-treated cancer cells.
Despite these findings, however, the molecular basis
of crosstalk is still poorly understood. AKT signaling is now
acknowledged to be the crucial pathway which manipulates the
autophagy processing (31,47). Given a linkage between AKT
signaling and PGRMC1/2 family by our previous study (26), we inferred that PGRMC1/2 family may
also take part in the hyperoside-mediated autophagy and apoptosis.
In fact, Mir and colleagues identified an association between
PGRMC1 and LC3B in A549 cells where PGRMC1 shows cytoprotective
effects (25). The role of PGRMC1
in hyperoside-induced autophagy and apoptosis was explored.
Regarding an elevation of PGRMC1 by cisplatin in ovarian cancer
cells, we suspected hyperoside induced-autophagy was the result of
an alteration of the PGRMC1 expression. However, treatment of
hyperoside from 0 to 100 µM did not alter the expression
profile of PGRMC1 (data not shown). We attempted to clarify the
role of PGRMC1 in hyperoside-mediated autophagy and apoptosis. A
recombinant PGRMC1 overexpressing plasmid was transfected into the
SKOV-3 cells. Our results are in line with the findings by Mir
et al (25) that PGRMC1
overexpression enhanced autophagic flux while it was inhibited upon
PGRMC1 knockdown. However, beyond our expectations and as
contradiction to the notions of PGRMC1 having tumor-promoting
capacity, in the presence of hyperoside, overexpression of PGRMC1
led to cell death of ovarian cancer. Simultaneously, PGRMC1
overexpression in SKOV-3 cells with hyperoside exposure elevated
the level of LC3B-II, a conversion from LC3B-I constantly indicates
a formation of autophagosomes. Concordantly, PGRMC1 knockdown by
specific shRNA significantly abrogated hyperoside-induced
autophagic cell death and decreased LC3B-II values. All these
results indicate a tumor-inhibiting instead of tumor-promoting
effect of PGRMC1 in hyperoside-treated ovarian cancer cells.
Crosstalk between the apoptotic and autophagic
machineries is emerging as a recurring theme with particular
importance for Bcl-2 family proteins in bridging the two pathways
(48). In the present study, Bcl-2
family had also taken part in the PGRMC1-dependent autophagy and
apoptosis by hyperoside. Indeed, ectopic PGRMC1 elevated Bcl-2
expression and dropped Bax expression, yet, in the presence of
hyperoside, ectopic PGRMC1 reversed the expression profile of the
proteins by dropping Bcl-2 and increasing Bax. The role of Bcl-2
family in relation to autophagy and apoptosis is complicated and is
still unclear, but in this study, we showed-experimental evidence
that Bcl-2 family plays a role at least in part in the
hyperoside-induced autophagy and apoptosis.
Autophagy shows either cytoprotective or cytotoxic
effect on cell fate depending on the context. In this study, in the
presence of hyperoside, extremely imbalanced autophagic flux led to
cytotoxic effect on ovarian cancer cells. In particular, PGRMC1
colocalized with LC3B to promote autophagy and apoptosis. LC3 (or
MAP1LC3) is an ubiquitin like ortholog of yeast Atg8 (49), which is required for autophagy
(50). Accordingly, PGRMC1 binds
to LC3B-II and is essential for the degradative activity of
autophagy (25). PGRMC1 engages in
the fusion of autophagosomes with lysosomes (51), which in turn activate mitochondrial
apoptosis (52,53). Thus, the tumor-inhibiting effect of
PGRMC1 by hyperoside given in this study may ascribe to the
combination of these two molecules that collaboratively triggered
imbalanced autophagic flux and led to subsequent apoptotic cell
death.
Pathophysiologically and coincidentally, in
vitro and in vivo cisplatin treatment lead to PGRMC1
overexpression in ovarian cancer cells. Thus, utilization of the
'Achilles' heel' of PGRMC1 which functions as autophagy-enhancing
and tumor-inhibiting effect by hyperoside may benefit and highlight
a possible cure of the ovarian cancer patients.
In conclusion, in this study, we explored the
effectiveness of hyperoside on treatment of ovarian cancer cells.
Mechanistically, a central role of PGRMC1 played in the action was
determined. We found that PGRMC1 colocalized with LC3B to
participate in the autophagy and apoptosis by hyperoside. PGRMC1
overexpression significantly promoted hyperoside-induced autophagy
and apoptosis, while PGRMC1 knockdown abrogated the action.
Additionally, in ovarian cancer cells where PGRMC1 is
overexpressed, hyperoside enhanced sensitivity of cells to
cisplatin treatment. Although we found the involvement of Bcl-2
family in the hyperoside-mediated autophagy and apoptosis, the
exact role of the family particularly in bridging autophagy and
apoptosis processes should be further investigated.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (81503368 and 81603358), and
Natural Science Foundation of Ministry of Science and Technology of
Jiangsu Province (BK 20151003). The authors are indebted to all the
colleagues whose names were not included in the author list, but
who participated in our study.
References
|
1
|
Salzberg M, Thurlimann B, Bonnefois H,
Fink D, Rochlitz C, von Moos R and Senn H: Current concepts of
treatment strategies in advanced or recurrent ovarian cancer.
Oncology. 68:293–298. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Middleton E Jr, Kandaswami C and
Theoharides TC: The effects of plant flavonoids on mammalian cells:
Implications for inflammation, heart disease, and cancer. Pharmacol
Rev. 52:673–751. 2000.PubMed/NCBI
|
|
3
|
Zou Y, Lu Y and Wei D: Antioxidant
activity of a flavonoid-rich extract of Hypericum perforatum L. in
vitro. J Agric Food Chem. 52:5032–5039. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Piao MJ, Kang KA, Zhang R, Ko DO, Wang ZH,
You HJ, Kim HS, Kim JS, Kang SS and Hyun JW: Hyperoside prevents
oxidative damage induced by hydrogen peroxide in lung fibroblast
cells via an antioxidant effect. Biochim Biophys Acta.
1780:1448–1457. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ku SK, Zhou W, Lee W, Han MS, Na M and Bae
JS: Anti-inflammatory effects of hyperoside in human endothelial
cells and in mice. Inflammation. 38:784–799. 2015. View Article : Google Scholar
|
|
6
|
Ku SK, Kwak S, Kwon OJ and Bae JS:
Hyperoside inhibits high-glucose-induced vascular inflammation in
vitro and in vivo. Inflammation. 37:1389–1400. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lü P: Inhibitory effects of hyperoside on
lung cancer by inducing apoptosis and suppressing inflammatory
response via caspase-3 and NF-κB signaling pathway. Biomed
Pharmacother. 82:216–225. 2016. View Article : Google Scholar
|
|
8
|
Boukes GJ and van de Venter M: The
apoptotic and autophagic properties of two natural occurring
prodrugs, hyperoside and hypoxoside, against pancreatic cancer cell
lines. Biomed Pharmacother. 83:617–626. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan J and Horvitz HR: A first insight
into the molecular mechanisms of apoptosis. Cell. 116:S53–S56.
51following S59. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yaacoub K, Pedeux R, Tarte K and
Guillaudeux T: Role of the tumor microenvironment in regulating
apoptosis and cancer progression. Cancer Lett. 378:150–159. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ivanov VN, Bhoumik A and Ronai Z: Death
receptors and melanoma resistance to apoptosis. Oncogene.
22:3152–3161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ahmed IS, Rohe HJ, Twist KE and Craven RJ:
PGRMC1 (progesterone receptor membrane component 1) associates with
epidermal growth factor receptor and regulates erlotinib
sensitivity. J Biol Chem. 285:24775–24782. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kabe Y, Nakane T, Koike I, Yamamoto T,
Sugiura Y, Harada E, Sugase K, Shimamura T, Ohmura M, Muraoka K, et
al: Haem-dependent dimerization of PGRMC1/Sigma-2 receptor
facilitates cancer proliferation and chemoresistance. Nat Commun.
7:110302016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Szczesna-Skorupa E and Kemper B:
Progesterone receptor membrane component 1 inhibits the activity of
drug-metabolizing cytochromes P450 and binds to cytochrome P450
reductase. Mol Pharmacol. 79:340–350. 2011. View Article : Google Scholar :
|
|
15
|
Zheng K, Li Y, Wang S, Wang X, Liao C, Hu
X, Fan L, Kang Q, Zeng Y, Wu X, et al: Inhibition of
autophagosome-lysosome fusion by ginsenoside Ro via the
ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to
5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage
checkpoint. Autophagy. 12:1593–1613. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kohli L, Kaza N, Lavalley NJ, Turner KL,
Byer S, Carroll SL and Roth KA: The pan erbB inhibitor PD168393
enhances lysosomal dysfunction-induced apoptotic death in malignant
peripheral nerve sheath tumor cells. Neurooncol. 14:266–277.
2012.
|
|
17
|
Ghadimi MP, Lopez G, Torres KE, Belousov
R, Young ED, Liu J, Brewer KJ, Hoffman A, Lusby K, Lazar AJ, et al:
Targeting the PI3K/mTOR axis, alone and in combination with
autophagy blockade, for the treatment of malignant peripheral nerve
sheath tumors. Mol Cancer Ther. 11:1758–1769. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stanton MJ, Dutta S, Zhang H, Polavaram
NS, Leontovich AA, Hönscheid P, Sinicrope FA, Tindall DJ, Muders MH
and Datta K: Autophagy control by the VEGF-C/NRP-2 axis in cancer
and its implication for treatment resistance. Cancer Res.
73:160–171. 2013. View Article : Google Scholar :
|
|
20
|
de la Cruz-Morcillo MA, Valero ML,
Callejas-Valera JL, Arias-González L, Melgar-Rojas P, Galán-Moya
EM, García-Gil E, García-Cano J and Sánchez-Prieto R: P38MAPK is a
major determinant of the balance between apoptosis and autophagy
triggered by 5-fluorouracil: Implication in resistance. Oncogene.
31:1073–1085. 2012. View Article : Google Scholar
|
|
21
|
Weidhaas JB, Babar I, Nallur SM, Trang P,
Roush S, Boehm M, Gillespie E and Slack FJ: MicroRNAs as potential
agents to alter resistance to cytotoxic anticancer therapy. Cancer
Res. 67:11111–11116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu Y, Cao L, Yang L, Kang R, Lotze M and
Tang D: microRNA 30A promotes autophagy in response to cancer
therapy. Autophagy. 8:853–855. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu H, He Z, von Rütte T, Yousefi S,
Hunger RE and Simon HU: Down-regulation of autophagy-related
protein 5 (ATG5) contributes to the pathogenesis of early-stage
cutaneous melanoma. Sci Transl Med. 5:202ra1232013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leng S, Hao Y, Du D, Xie S, Hong L, Gu H,
Zhu X, Zhang J, Fan D and Kung HF: Ursolic acid promotes cancer
cell death by inducing Atg5-dependent autophagy. Int J Cancer.
133:2781–2790. 2013.PubMed/NCBI
|
|
25
|
Mir SU, Schwarze SR, Jin L, Zhang J,
Friend W, Miriyala S, St Clair D and Craven RJ: Progesterone
receptor membrane component 1/Sigma-2 receptor associates with
MAP1LC3B and promotes autophagy. Autophagy. 9:1566–1578. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu X, Han Y, Fang Z, Wu W, Ji M, Teng F,
Zhu W, Yang X, Jia X and Zhang C: Progesterone protects ovarian
cancer cells from cisplatin-induced inhibitory effects through
progesterone receptor membrane component 1/2 as well as AKT
signaling. Oncol Rep. 30:2488–2494. 2013.PubMed/NCBI
|
|
27
|
Zhu X, Guo Y, Yao S, Yan Q, Xue M, Hao T,
Zhou F, Zhu J, Qin D and Lu C: Synergy between Kaposi's
sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein
in promotion of angiogenesis and oncogenesis: Role of the AKT
signaling pathway. Oncogene. 33:1986–1996. 2014. View Article : Google Scholar
|
|
28
|
Xue M, Yao S, Hu M, Li W, Hao T, Zhou F,
Zhu X, Lu H, Qin D, Yan Q, et al: HIV-1 Nef and KSHV oncogene K1
synergistically promote angiogenesis by inducing cellular miR-718
to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res.
42:9862–9879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu H and Zhang Y: Life and death partners
in post-PCI restenosis: Apoptosis, autophagy, and the cross-talk
between them. Curr Drug Targets. Jun 24–2016.Epub ahead of print.
PubMed/NCBI
|
|
30
|
Dalby KN, Tekedereli I, Lopez-Berestein G
and Ozpolat B: Targeting the prodeath and prosurvival functions of
autophagy as novel therapeutic strategies in cancer. Autophagy.
6:322–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang RC, Wei Y, An Z, Zou Z, Xiao G,
Bhagat G, White M, Reichelt J and Levine B: Akt-mediated regulation
of autophagy and tumorigenesis through Beclin 1 phosphorylation.
Science. 338:956–959. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cahill MA, Jazayeri JA, Catalano SM,
Toyokuni S, Kovacevic Z and Richardson DR: The emerging role of
progesterone receptor membrane component 1 (PGRMC1) in cancer
biology. Biochim Biophys Acta. 1866:339–349. 2016.PubMed/NCBI
|
|
33
|
Grabowski JP and Sehouli J: Current
management of ovarian cancer. Minerva Med. 106:151–156.
2015.PubMed/NCBI
|
|
34
|
Herzog TJ: The current treatment of
recurrent ovarian cancer. Curr Oncol Rep. 8:448–454. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Absolom K, Eiser C, Turner L, Ledger W,
Ross R, Davies H, Coleman R, Hancock B, Snowden J and Greenfield D;
Late Effects Group Sheffield: Ovarian failure following cancer
treatment: Current management and quality of life. Hum Reprod.
23:2506–2512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rodriguez-Freixinos V, Mackay HJ,
Karakasis K and Oza AM: Current and emerging treatment options in
the management of advanced ovarian cancer. Expert Opin
Pharmacother. 17:1063–1076. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Peluso JJ: Non-genomic actions of
progesterone in the normal and neoplastic mammalian ovary. Semin
Reprod Med. 25:198–207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Peluso JJ: Progesterone signaling mediated
through progesterone receptor membrane component-1 in ovarian cells
with special emphasis on ovarian cancer. Steroids. 76:903–909.
2011.PubMed/NCBI
|
|
39
|
Oda S, Nakajima M, Toyoda Y, Fukami T and
Yokoi T: Progesterone receptor membrane component 1 modulates human
cytochrome p450 activities in an isoform-dependent manner. Drug
Metab Dispos. 39:2057–2065. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Albrecht C, Huck V, Wehling M and Wendler
A: In vitro inhibition of SKOV-3 cell migration as a distinctive
feature of progesterone receptor membrane component type 2 versus
type 1. Steroids. 77:1543–1550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ahmed IS, Rohe HJ, Twist KE, Mattingly MN
and Craven RJ: Progesterone receptor membrane component 1 (Pgrmc1):
A heme-1 domain protein that promotes tumorigenesis and is
inhibited by a small molecule. J Pharmacol Exp Ther. 333:564–573.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Peluso JJ, Liu X, Gawkowska A, Lodde V and
Wu CA: Progesterone inhibits apoptosis in part by PGRMC1-regulated
gene expression. Mol Cell Endocrinol. 320:153–161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
van Waarde A, Rybczynska AA, Ramakrishnan
NK, Ishiwata K, Elsinga PH and Dierckx RA: Potential applications
for sigma receptor ligands in cancer diagnosis and therapy. Biochim
Biophys Acta. 1848B:2703–2714. 2015. View Article : Google Scholar
|
|
44
|
Lin L and Baehrecke EH: Autophagy, cell
death, and cancer. Mol Cell Oncol. 2:e9859132015. View Article : Google Scholar
|
|
45
|
Gewirtz DA: Cytoprotective and
nonprotective autophagy in cancer therapy. Autophagy. 9:1263–1265.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rubinstein AD, Eisenstein M, Ber Y, Bialik
S and Kimchi A: The autophagy protein Atg12 associates with
antiapoptotic Bcl-2 family members to promote mitochondrial
apoptosis. Mol Cell. 44:698–709. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mann SS and Hammarback JA: Molecular
characterization of light chain 3. A microtubule binding subunit of
MAP1A and MAP1B. J Biol Chem. 269:11492–11497. 1994.PubMed/NCBI
|
|
50
|
Tsukada M and Ohsumi Y: Isolation and
characterization of autophagy-defective mutants of Saccharomyces
cerevisiae. FEBS Lett. 333:169–174. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ostenfeld MS, Høyer-Hansen M, Bastholm L,
Fehrenbacher N, Olsen OD, Groth-Pedersen L, Puustinen P,
Kirkegaard-Sørensen T, Nylandsted J, Farkas T, et al: Anti-cancer
agent siramesine is a lysosomotropic detergent that induces
cytoprotective autophagosome accumulation. Autophagy. 4:487–499.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ishisaka R, Kanno T, Akiyama J, Yoshioka
T, Utsumi K and Utsumi T: Activation of caspase-3 by lysosomal
cysteine proteases and its role in
2,2′-azobis-(2-amidinopropane)dihydrochloride (AAPH)-induced
apoptosis in HL-60 cells. J Biochem. 129:35–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stoka V, Turk B, Schendel SL, Kim TH,
Cirman T, Snipas SJ, Ellerby LM, Bredesen D, Freeze H, Abrahamson
M, et al: Lysosomal protease pathways to apoptosis. Cleavage of
bid, not pro-caspases, is the most likely route. J Biol Chem.
276:3149–3157. 2001. View Article : Google Scholar
|