Introduction
Distal metastasis of tumors is the main cause of
mortality in patients with cancer. Invasion and metastasis are
regarded as important factors in the assessment of malignant
behavior of tumors and have been extensively examined in the field
of cancer research (1–3). Prostate cancer, the most common type
of cancer in the male urinary and reproductive system, is difficult
to treat after the occurrence of local invasion and distal
metastasis. Moreover, few targets associated with metastasis and
malignancy have been identified. Therefore, elucidation of the
novel mechanisms of prostate cancer metastasis and identification
of potential targets for the diagnosis and treatment of highly
malignant prostate cancers are needed.
Proteomics-based technologies are currently used for
the large-scale analysis of proteins expressed in cells and tissues
(4). These methods represent
effective tools for the identification of tumor-associated
antigens, particularly for identifying cellular membrane antigens
(5,6). For example, in our previous study, we
analyzed the differential protein expression in PC-3M cells and
PC-3 cells using two-dimensional liquid phase chromatographic
fractionation followed by matrix-assisted laser
desorption/time-of-flight mass spectrometry (MALDI-TOF/MS)
(unpublished data). ATP synthase and caveolin were found to be
upregulated in highly metastatic PC-3M cells compared with low less
metastatic PC-3 cells using additional bioinformatics analysis.
F1Fo-ATPsynthase, which is exclusively localized in
the inner membrane of the mitochondria, catalyzes the rate-
limiting step of ATP formation in eukaryotic cells. However, more
evidence has shown that F1Fo-ATP synthase is ectopically expressed
on the plasma membrane of tumor cells and some types of normal
cells, such as endothelial cells and adipocytes (7). Proteomic analysis of membrane
fractions has indicated that some highly malignant cell types show
higher ectopic expression of ATP synthase than less malignant cells
(8,9). Ectopic ATP synthase, including ATP
synthase beta subunit (ATP5B), is localized on lipid rafts or
caveolae of the cytoplasmic membrane (10–12).
However, the mechanisms responsible for the transport of this
protein have not been established, and it is unclear how ectopic
ATP5B promotes the invasion and metastasis of prostate cancer.
Accordingly, in the present study, we analyzed the
localization and expression of ATP5B in PC-3M cells and explored
the effects of this protein on the biological behavior of PC-3M
cells in order to determine its role in the metastasis of prostate
cancer.
Materials and methods
Cell lines and cell culture
PC-3 human prostate cancer, PC-3M highly metastatic
human prostate cancer, and normal prostate cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in F12,
RPMI-1640, or H-DMEM, respectively, supplemented with 10% fetal
bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin
(Amresco, Solon, OH, USA). The cell lines were grown as adherent
monolayer cultures at 37°C with 5% CO2 in a humidified
incubator. Human paraffin-embedded prostate cancer tissue specimens
(T4, N0, M0 and G1) were obtained from the Pathology Department of
the Jilin University.
Subtractive bio-panning in vitro
PC-3M and PC-3 cells were used as positive target
cells, whereas normal prostate cells were used as negative absorber
cells. Normal prostate cells were grown to log phase, washed three
times with phosphate- buffered saline (PBS), and blocked with 1%
bovine serum albumin (BSA) for 30 min at 37°C. Approximately,
2×1011 pfu phages were incubated with normal prostate
cells at 37°C for 30 min with gentle agitation. After incubation,
the supernatant containing unbound phages was incubated with
blocked PC-3 cells at 37°C for 30 min with gentle agitation. Then,
the supernatant containing unbound phages was incubated with
blocked PC-3M cells at 37°C for 30 min with gentle agitation. The
PC-3M cells were washed three times with 0.1% PBST (PBS plus 0.1%
Tween-20, v/v) to remove the unbound phages. Phages bound to the
cell membrane were then eluted with 1 ml of 0.2 M glycine (pH 2.2)
for 15 min on ice and immediately neutralized with 200 μl of
1 M Tris-HCl (pH 9.1). The eluted phages were amplified, purified
and titrated according to the manufacturer's instructions.
Subsequently, 2×1011 pfu phages were subjected to the
next round of bio-panning.
Enzyme-linked immunosorbent assay
(ELISA)
PC-3M and PC-3 cells were seeded into 96-well plates
(1×104 cells/well). The cells were washed twice and
fixed with 4% paraformaldehyde for 20 min at room temperature. A
solution of H2O2 (3%, 100 μl/well) was
added, and the plates were placed at room temperature for 30 min to
inhibit endogenous peroxidase activity. The cells were then blocked
with 5% BSA at 4°C overnight. The phages were added to PC-3M and
PC-3 cells (3×1011 pfu, 100 μl/well) and
incubated at 37°C for 2 h. After washing with 0.5% TBST three
times, the cells were incubated with 200 μl of mouse
anti-M13 monoclonal antibodies (1:5,000; Santa Cruz Biotechnology,
Santa Cruz, CA, USA) at 37°C for 1.5 h. Next, cells were washed
three times with 0.1% TBST and then incubated with 200 μl
horseradish peroxidase (HRP)-labeled goat anti-mouse antibody IgG
(1:5,000; Santa Cruz Biotechnology) at 37°C for 1 h. After washing
three times with 0.1% TBST, the cells were incubated with 100
μl TMB at 37°C for 10 min, and the reaction was then
terminated by adding 200 μl of 2M
H2SO4. The absorbance was then measured at
450 nm using an ELISA plate reader. Cell-free wells were used as
controls.
DNA sequencing of the positive phage
clones
The selected positive phage clones were used to
extract DNA for sequencing analysis. First, 65 μl
polyethylene glycol (PEG)/NaCl was added to 200 μl phages at
room temperature for 10 min. The mixture was then centrifuged at
12,000 rpm for 10 min at 4°C. The precipitate was suspended in
iodide buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA and 4 M NaI],
followed by ethanol precipitation at room temperature for 10 min.
Single-stranded DNA (ssDNA) was recovered and dissolved in TE
buffer [10 mM Tris-HCl (pH 8.0) and 1 mM EDTA]. DNA sequencing of
the selected phages was carried out by Sangon Biotech Co., Ltd.
(Shanghai, China). The primer used for sequencing was -96 gIII
5′-CCCTCATAGTTAGCGTAACG-3′.
Cell immunofluorescence assay
PC-3M and PC-3 cells were cultured on coverslips
overnight. One group of cells was then incubated with
2×1011 pfu phages at 37°C for 8 h, washed three times
with TBS, and fixed with 4% paraformaldehyde for 20 min at room
temperature. Cells were then blocked in 5% BSA for 1 h at 37°C and
incubated with mouse anti-M13 monoclonal antibodies (1:250),
anti-ATP5B antibodies (1:200; Santa Cruz Biotechnology) and
anti-E-cadherin antibodies (1:200; Santa Cruz Biotechnology) at 4°C
overnight. After washing three times with PBS, the cells were then
incubated with anti-mouse Alexa Fluor 488 (1:600; Cell Signaling
Technology, Danvers, MA, USA) or anti-rabbit Alexa Fluor 555
(1:600; Cell Signaling Technology) at 37°C for 30 min. Cells were
then washed three times with PBS, nuclei were stained with Hoechst
33342 and the slides were observed using a laser scanning confocal
microscope (LSCM).
Affinity chromatography and liquid
chromatography tandem mass spectrometry (LC-MS/MS) analyses
MS and LC-MS/MS analyses were employed to detect and
identify the corresponding target proteins of the identified short
peptide binding to ATP5B (B04). Sephrose-4B was pre-incubated with
B04, and the Sephrose-4B coupled with B04 was incubated with
membrane proteins of PC-3M cells for 1 h to obtain the
corresponding B04-binding proteins. The eluted proteins were
identified by LC-MS/MS analyses. LTQ Velos was used with the search
database ipi.HUMAN.v3.53 protein database. The filter parameters
for SEQUEST results were as follows: charge, +1 and Xcorr, ≥1.9;
charge, +2 and Xcorr, ≥ 2.2; charge, +3 and Xcorr, ≥3.75; DelCN,
0.1. The MS/MS data, including the mass values, intensity and
charge of the precursor ions, were analyzed with the Mascot 2.0
against the Swiss-Prot protein database.
Immunoelectromicroscopy
The cells were fixed with of 4% paraformaldehyde at
4°C for 20 min and washed in PBS three times. Cells were then
incubated with 5% BSA for 1 h, and the primary anti-ATP5B antibody
(diluted 1:100) was applied at 4°C overnight. After three 5-min
washes with PBS, and cells were incubated with the secondary
antibody (goat antimouse) labeled with gold colloid (Wuhan Boster
Biological Technology, Ltd., Wuhan, China) at room temperature for
1 h. Cells were washed in PBS three times, and immune complexes
were pelleted by centrifugation (3,000 rpm for 5 min). The
suspensions were mixed with an equal volume of 3% phosphotungstic
acid (pH 7.0) placed on formvar-coated, carbonized copper grids and
observed under a transmission electron microscope (TEM; Phillips
201; Norelco, Eindhoven, The Netherlands).
Flow cytometric analysis
PC-3M cells were fixed with 4% paraformaldehyde for
15 min and then washed twice with staining buffer. Cells were then
blocked with 5% BSA in PBS and incubated with antibodies against
ATP5B for 45 min at 4°C after washing twice. Secondary antibodies
(Alexa Fluor 488 rabbit anti-mouse IgG [H+L]) were used at a 1:500
dilution for 30 min at 4°C. The stained cells were then analyzed
using a flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA).
The percentage of cells was documented using ModFit software
(Becton-Dickinson).
Western blotting
Cells were lysed with RIPA lysis buffer containing 1
mM phenylmethylsulfonyl fluoride (PMSF; Beyotime Institute of
Biotechnology, Shanghai, China) on ice for 30 min. Total cellular
proteins were assayed using a BCA kit (Beyotime Institute of
Biotechnology). Equal amounts of protein were subjected to sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and
then electrophoretically transferred to polyvinylidene fluoride
(PVDF) membranes. Membranes were blocked with 5% non-fat dry milk
in PBS with 0.1% Tween-20 for 1 h at 37°C and the incubated with
anti-ATP5B, anti-B04, anti-β-actin and anti-GAPDH antibodies at 4°C
overnight. Finally, membranes were incubated with HRP-conjugated
secondary antibodies for 1 h. The signals of detected proteins were
visualized on an enhanced chemiluminescence (ECL; Beyotime
Institute of Biotechnology) western blotting detection system
(Amersham Biosciences, Inc., Piscataway, NJ, USA).
Cell Counting kit-8 (CCK-8) assays
PC-3M cells were seeded in 96-well plates at
1×104 cells/well in triplicate and incubated with B04 or
08K (as a control) for 6, 12, 24 or 48 h. After incubation, cells
were detected using CCK-8 assays. Twenty microliters of CCK-8
solution was added to each well. The absorbance was measured at 450
nm to determine the number of viable cells in each well. All
procedures were performed in triplicate.
Transwell assays
Cell invasion were analyzed using Transwell assays.
The upper chambers were coated with 60 μl Matrigel (dilution
1:8), and 3×104 cells were then seeded in the upper
chamber of each well in serum-free medium containing B04 or 08K.
Additionally, 600 μl of RPMI-1640 media supplemented with
10% fetal calf serum (FCS) was added to the lower chamber. The
cells were incubated for 24 h, after which the cells on the upper
side of the filter were removed by wiping with a cotton swab. The
cells that had invaded to the lower surface were fixed with 4%
paraformaldehyde and stained with Hoechst 33342. The average number
of invaded cells was determined by counting the cells in 10 random
fields (magnification, ×100). Three independent experiments were
performed with triplicate wells.
Tumor metastasis assay
Chicken embryo chorioallantoic membranes (CAMs) were
used to explore the effects of B04 on the migration of PC-3M. PC-3M
cells were cultured with B04 or 08K for 24 h as previously
described. Cells were detached from the culture dish with 2 mM EDTA
in PBS, counted, resuspended in 50 ml of PBS with Ca2+
and Mg2+ and inoculated (105–106
cells) onto the CAM of a 9-day-old chick embryo in which an
artificial air sac was created (13). After 50 h of incubation, the lower
half of the CAM was removed, placed in a sterile 14-ml
polypropylene tube, snap-frozen in liquid nitrogen and stored
frozen at −80°C. The frozen tissue was crushed in lysis buffer with
sterile 5-ml pipettes. Genomic DNA was isolated from the lower CAMs
using a DNA Isolation kit (Gentra Systems, Inc., Big Lake, MN, USA)
according to the manufacturer's specifications. The specific
primers for human Alu sequences were as follows: Alu sense,
5′-ACGCCTGTAATCCCAGCACTT-3′ and Alu antisense,
5′-TCGCCCAGGCTGGAGTGCA-3′, which produced a band of 220 bp. The
primers were positioned in the most conserved areas of the Alu
sequence (14), with the first
monomer at nucleotides 21–40 and the second monomer at nucleotides
263–245. Polymerase chain reaction (PCR) was performed according to
the following conditions: 95°C for 2 min, followed by 30 cycles of
95°C for 30 sec, 63°C for 30 sec and 72°C for 30 sec. The reaction
mixture contained 1 μg of genomic DNA as a template, 1X PCR
buffer, 1.5 mM MgCl2, 50 μM dNTPs, 1.0 μM
each of Alu sense and antisense primers, 1±2 U of AmpliTaq Gold,
and 0.1 μCi fresh 32P-α-dCTP. The PCR products
were electrophoresed on a 7% polyacrylamide gel at 100 V for 1 h,
dried and exposed to film at −80°C. The bands were quantified by
densitometric scanning using a Gel Scan XL (Pharmacia Biotech,
Uppsala, Sweden).
Statistical analysis
All data were summarized as the mean ± standard
deviations (SDs) from at least three independent experiments.
Statistical analysis was performed using the SPSS Statistics 21.
Differences between the means were analyzed using t-tests and
one-way analysis of variance (ANOVA). A two-tailed P<0.05 was
considered statistically significant.
Results
Selection and identification of the phage
clone B04 specifically bound to the membrane proteins expressed on
PC-3M cells
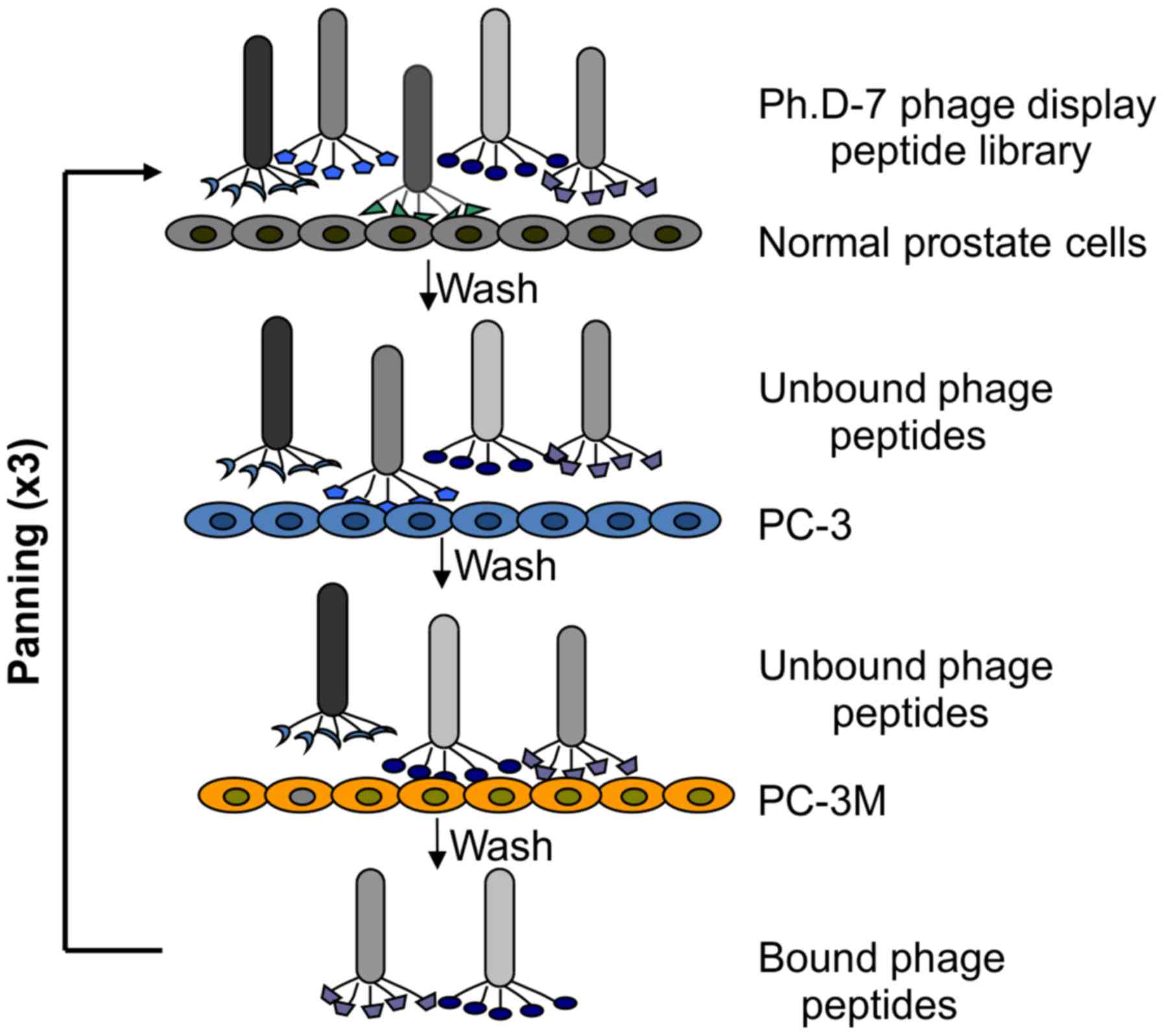
Phages that specifically bound to PC-3M cells were
identified through three rounds of in vitro subtractive
bio-panning with PC-3 cells and normal prostate cells. In each
round, a series of phages binding to the PC-3M cells instead of
control cells was obtained and amplified for subsequent panning.
The unbound phages were removed by washing with PBST. Fourteen
positive phages were significantly selected and enriched (B01-14).
The phage subtractive panning procedure is shown in Fig. 1. The 14 positive phage clones were
selected for DNA sequencing. The peptide sequences were then
deduced from the DNA sequences. The 14 peptides were then
designated B01-14 (as shown in Table
I). The amino acid sequences of the 14 peptides were further
analyzed and compared with the known proteins. B04 and B08 shared a
common amino acid sequence: GWTPVI. Similar amino acid sequences,
i.e., TP and PVI, had higher occurrence rates. Similar sequences
were identified by Clustal W and National Center of Biotechnology
Information BLAST analysis between the peptides displayed by the
phages and known proteins. The results showed that the peptides had
the same sequences as 32 species of vascular endothelial cell
surface proteins (data not shown), suggesting that highly
metastatic prostate cancer may exhibit unique protein expression
and molecular mechanisms to facilitate interactions with vascular
endothelial cells.
 | Table IPeptide sequences of positive phage
clones. |
Table I
Peptide sequences of positive phage
clones.
| Phage clone | DNA (21 bp) | Peptides |
|---|
| B01 |
GCTCCTCATTTGCTTCGTCCT | APHLLRP |
| B02 |
TTTCGGGGGCTGGAGAGTGGT | FRGLESG |
| B03 |
ACGCGTATGGATCTGAAGTTG | TRMDLKL |
| B04 |
TGTGGTTGGACTCCGGTTATT | CGWTPVI |
| B05 |
GGGACTGCGACTCATCCTACG | GTATHPT |
| B06 |
ACTCCGAAGTATGATAATCAT | TPKYDNH |
| B07 |
TCGCCGACGTGGAGTTTTTTG | SPTWSFL |
| B08 |
TCTGGTTGGACTCCGGTTATT | SGWTPVI |
| B09 |
GGTACTAAGCTTACTTCTTTT | GTKLTSF |
| B10 |
AATGCTCCGGTTATTTTGCTT | NAPVILL |
| B11 |
AATCCGCCTTTGCTTACGCAT | NPPLLTH |
| B12 |
ACGCCGCCTCCTTTTAGGATT | TPPPFRI |
| B13 |
ACTTTTCGTTTGATGACTGAT | TFRLMTD |
| B14 |
GCGGTTAGTCGTGATATGCGT | AVSRDMR |
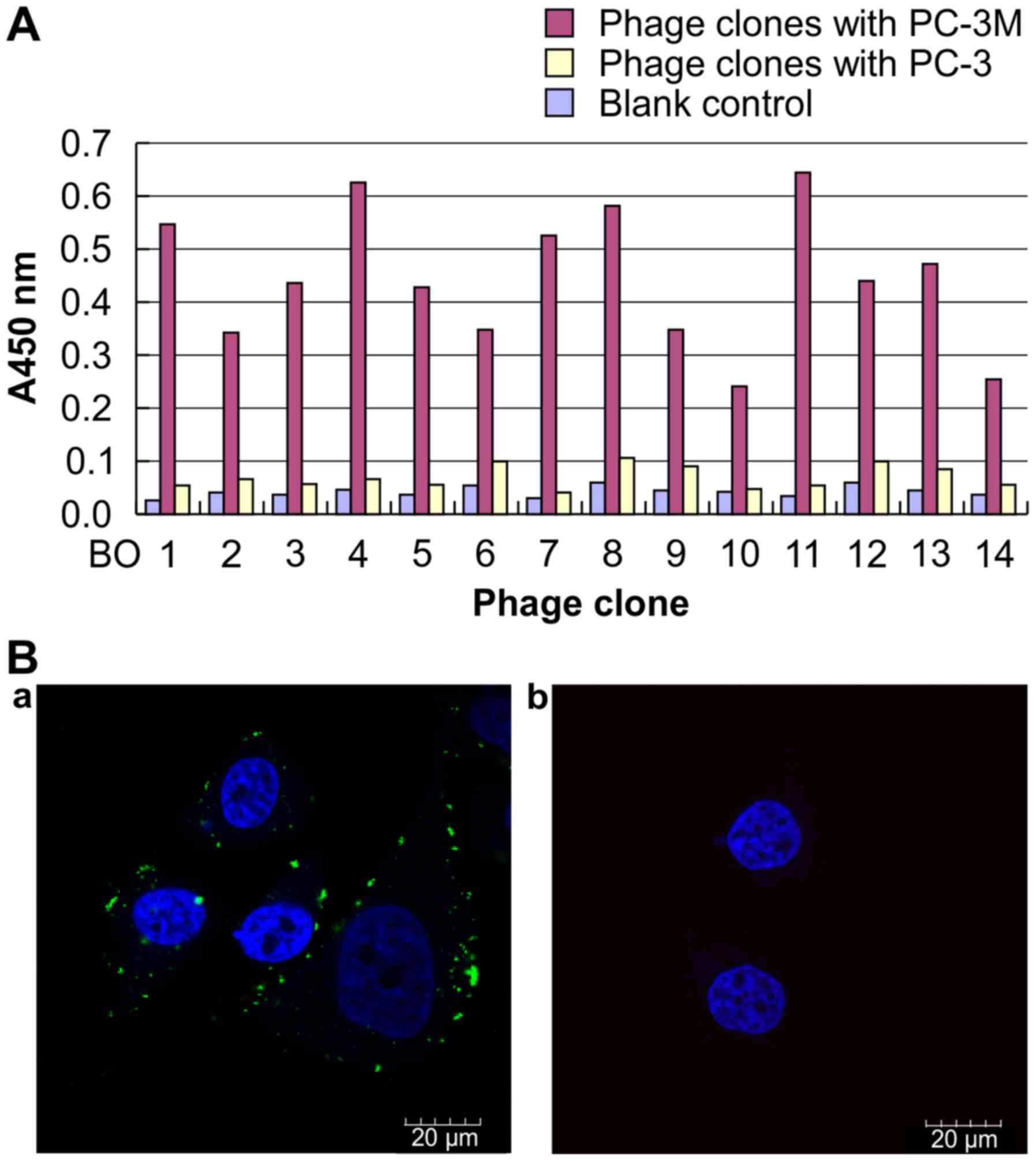
Cellular ELISA and immunofluorescence assays were
performed to determine the affinities of the 14 phage clones to
PC-3M cells. The results of ELISA demonstrated that the 14 peptides
specifically bound to PC-3M cells but not to PC-3 cells. B04, B08
and B11 peptides showed the highest binding ability to PC-3M cells
(Fig. 2A). Immunofluorescence
assays confirmed these findings (Fig.
2B). We chose the corresponding phage clones B04 and B08 (with
the most consensus sequences) for further identification. Here, we
focus on the results of B04. B04 was further confirmed to bind
specifically with the cell surface proteins of PC-3M cells by
immunofluorescence analysis (Fig.
2B-a), however, specific binding of B04 were not observed on
the cellular surface of PC-3 cells (Fig. 2B-b). Therefore, the short peptide
B04 was used as a hypothetical ligand for screening of
metastasis-related proteins on the surface of PC-3M cells.
B04 binds to ATP5B on PC-3M cells
First, we extracted the plasma proteins and cellular
membrane proteins from PC-3M cells and used B04 as the primary
antibody to identify B04-binding proteins by western blotting. The
results indicated that a specific 60-kDa protein from the cellular
membrane fraction of PC-3M cells bound to B04; no bands were
observed from the plasma fraction of PC-3M cells (Fig. 3A). These data were consistent with
the results of cell immunofluorescence assays (Fig. 2B).
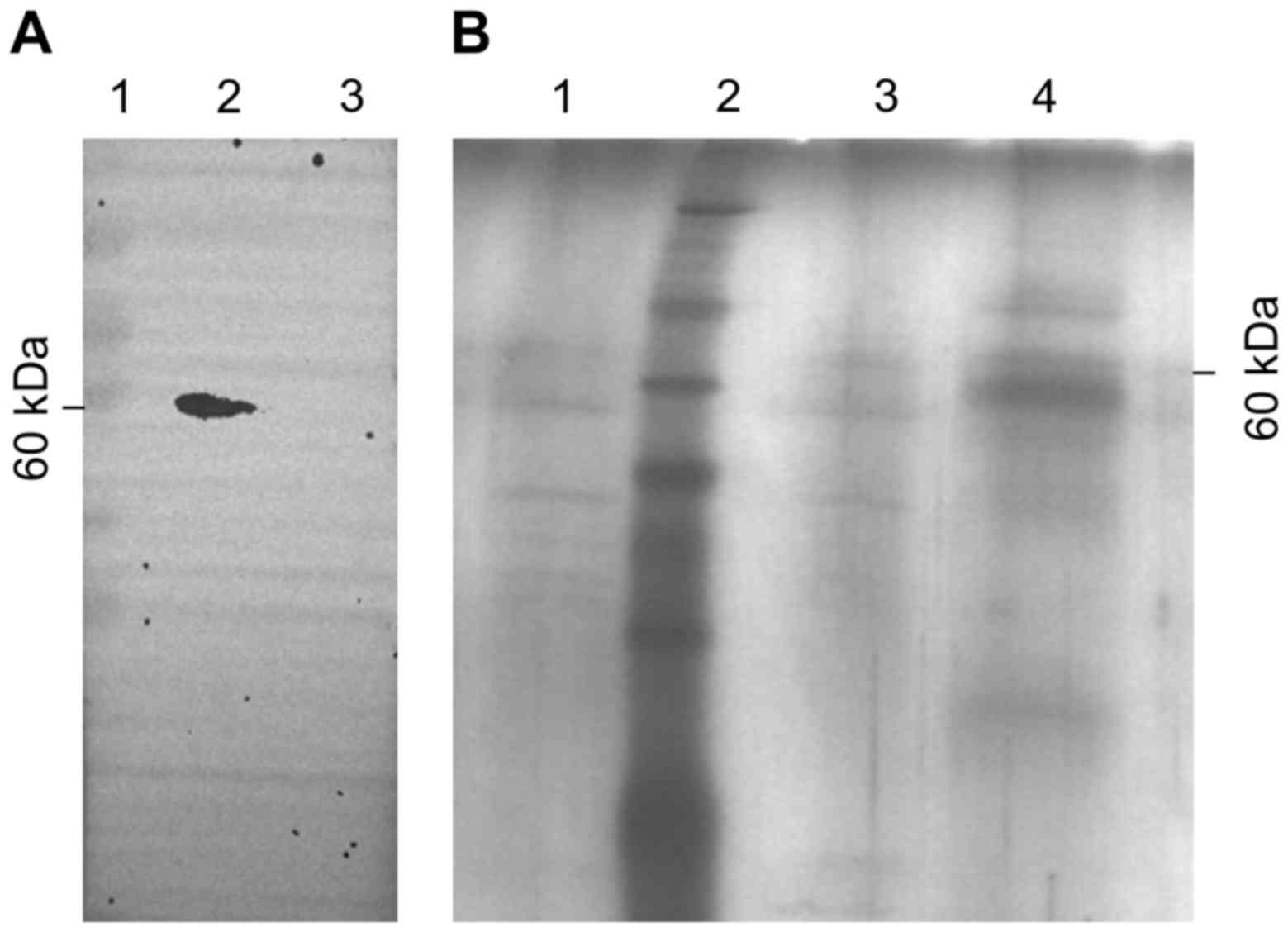 | Figure 3Binding of PC-3M membrane proteins to
B04. (A) Western blot analysis of the target binding protein of
B04. B04 was used as a primary antibody, and the black arrow p60
indicates the target protein of B04. Lane 1, marker; lane 2,
membrane proteins of PC-3M cells; lane 3, plasma proteins of PC-3M
cells. (B) PC-3M membrane p60 proteins binding to B04 were detected
by silver staining. Sephrose-4B was pre-incubated with B04, and the
Sephrose-4B coupled with B04 was then incubated with membrane
proteins of PC-3M cells to obtain the corresponding B04-binding
proteins. The flow-through fraction, wash fractions, and elution
fractions were separated by SDS-PAGE and gels were visualized by
silver staining. Lane 1, flow-through fraction; lane 2, marker;
lane 3, wash fractions; lane 4, elution fractions. |
Next, we confirmed these findings using affinity
chromatography after pre-incubation of Sephrose-4B with B04. The
Sephrose-4B used for coupling with B04 was incubated with cellular
membrane proteins of PC-3M cells. The flow-through fraction, wash
fractions and elution fractions were separated by SDS-PAGE and
visualized by silver staining. A specific band of 60 kDa was found
in the eluate, whereas no significant bands were found in the
flow-through fraction or the wash fractions (Fig. 3B). The results above indicate that
B04 bound to a protein with a molecular weight of 60 kDa on the
PC-3M cell membrane.
Therefore, the p60 band was excised from the gel and
further analyzed by MALDI-MS. Ten proteins were consistently
identified in each of three independent experiments (Table II).
| Table IIMALDI-TOF-MS/MS analysis of 60-kDa
peptides. |
Table II
MALDI-TOF-MS/MS analysis of 60-kDa
peptides.
| Protein | No. of amino
acids | Molecular weight
(kDa) | Transmembrane
domain |
|---|
| ALB | 609 | 66 | Transmembrane
protein |
| HSPA5 | 654 | 71 | |
| SLC25A5 | 298 | 32.7 | |
| ACTB | | | |
| TUBA1C | 451 | 49.6 | |
| GAPDH | | | |
| EEF1G | 487 | 53.5 | |
| EEF1A2 | | | |
| ATP5B | 529 | 58.1 | Transmembrane
protein |
| TRAP1 | 704 | 77.5 | |
Taken together, these findings of immunofluorescence
analysis, western blotting and MS confirmed that there was a
protein expressed on the cell membrane of PC-3M cells that bound to
B04. After excluding housekeeping proteins and plasma proteins,
ATP5B was identified as the final B04-target protein.
ATP5B is expressed on the extracellular
surface of PC-3M cells
We used immunofluorescence analysis to examine the
expression and localization of ATP5B on PC-3M cells. As shown in
Fig. 4A, ATP5B was found to be
localized both on the cellular membrane and in the cytoplasm of
PC-3M cells. However, positive staining was only found in the
cytoplasm of PC-3 cells. Thus, we next performed
immunoelectromicroscopy to further define the localization of ATP5B
on PC-3M cells using gold particles (Fig. 4B). From the results of transmission
electron microscopy (TEM), gold particles were detected both on the
cellular membrane and in the cytoplasm of PC-3M cells, indicating
expression on organelle membranes. The distribution of gold
particles on the cellular membrane of PC-3M cells was detected on
microvilli (Fig. 4B-a) and on the
smooth surface of the cells (Fig.
4B-b). The gold particles were distributed on microvilli at a
much higher frequency than that on the smooth surface of the cells;
this phenomenon may be associated with the increased energy demand
in highly metastatic tumor cells. However, no gold particles were
observed on the cellular membrane of PC-3 cells (Fig. 4B-c and -d). We further investigated
the surface localization of ATP5B in prostate cancer tissues using
double immunofluorescence staining of ATP5B and E-Ca with
paraffin-embedded prostate cancer tissue specimens. The expression
of the ATP5B was obviously positive on the cellular membrane of
prostate carcinoma specimens (Fig.
4C).
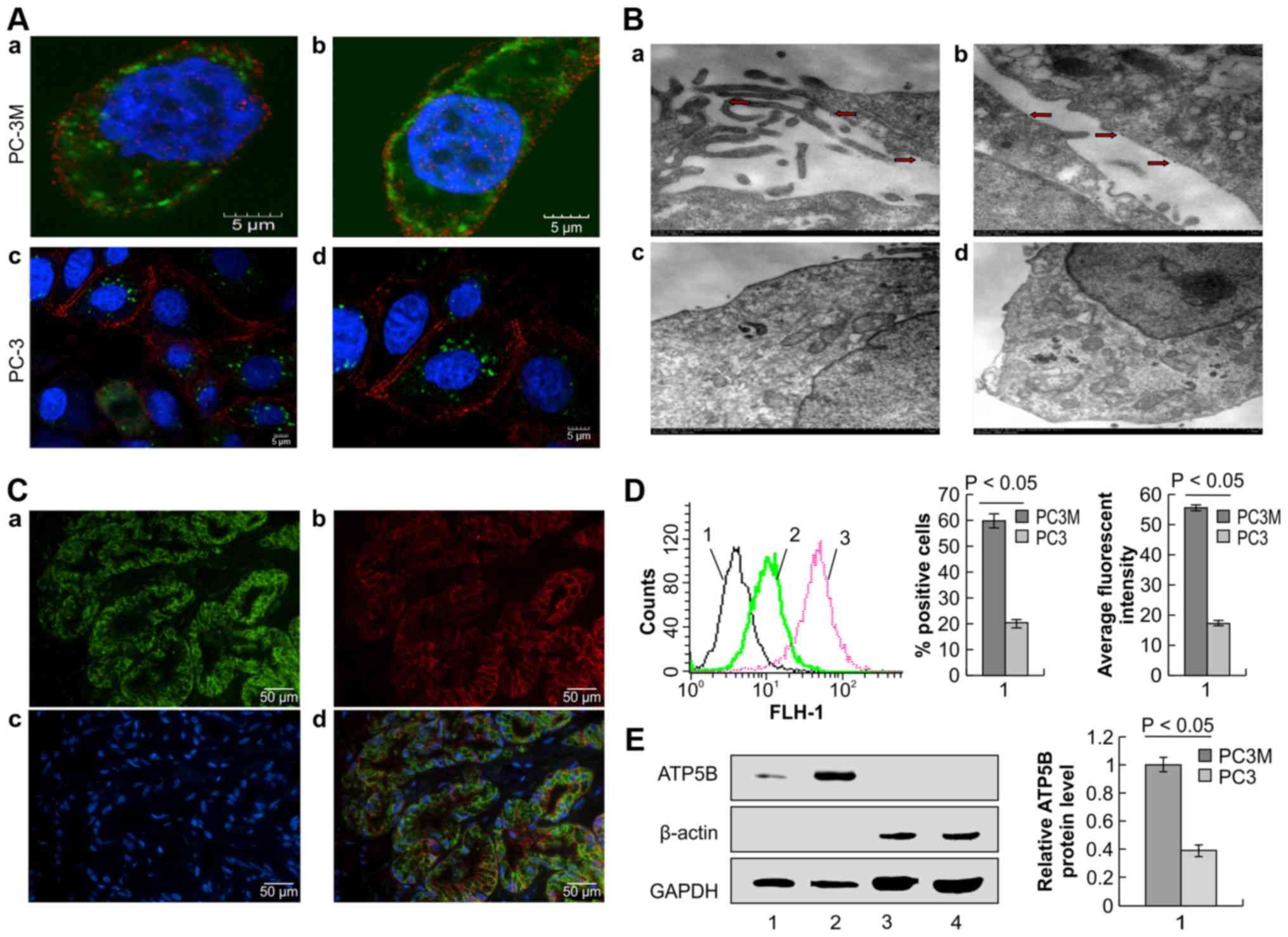 | Figure 4Subcellular localization of ATP5B.
(A) Immunofluorescence localization of the ATP5B on the surface of
PC-3M cells by confocal microscopy. Cells were stained with
anti-ATP5B and anti-E-Ca antibodies, followed by secondary antibody
staining. Non-permeabilized PC-3M cells are shown in a and b
(magnification, ×3000), and non-permeabilized PC-3 cells are shown
in c (magnification, ×1500) and d (magnification, ×2000). ATP5B
(green), E-Ca (red), and nuclei (blue) are shown. (B)
Immunostaining of PC-3M cells with anti-ATP5B antibodies by
immunoelectromicroscopy. Gold particles on the cellular membrane
are indicated by red arrows. (C) Immunocytochemistry double
staining of ATP5B and E-Ca using paraffin-embedded tissue
specimens. a, ATP5B (green); b, E-Ca (red); c, nuclei (blue); d,
merged image of a-c. Magnification, ×200. (D) Flow cytometric
analysis of the localization of ATP5B. 1, PC-3M and PC-3 cells
incubated with Alexa Fluor 488 IgG; 2, PC-3 cells incubated with
antibodies against ATP5B; 3, PC-3M cells incubated with antibodies
against ATP5B. (E) Western blotting was used to detect ATP5B
expression. Lane 1, membrane proteins expressed in PC-3 cells; lane
2, membrane proteins expressed in PC-3M cells; lane 3, plasma
proteins expressed in PC-3 cells; lane 4, plasma proteins expressed
in PC-3M cells. GAPDH and β-actin were used as references for
cellular membrane proteins and plasma proteins, respectively. |
Next, we used flow cytometry to further confirm the
presence of extracellular ATP5B on PC-3M cells with anti-ATP5B
antibodies. ATP5B was present on the surface of PC-3M cells, and
the percentage of ATP5B-positive cells and the fluorescence
intensity of ATP5B were significantly higher in PC-3M cells than in
PC-3 cells (61.68±3.94 vs. 20.23±1.81%, respectively, for the
percentage of ATP5B-positive cells; 53.87±5.14 vs. 17.65±0.19,
respectively, for the fluorescence intensity; Fig. 4D, P<0.05). To further confirm
the localization of ATP5B on the cellular membrane of PC-3M cells,
plasma proteins and membrane proteins were separated from PC-3M and
PC-3 cells, and anti-ATP5B antibodies were used to detect the
expression of ATP5B in these fractions. The results indicated that
ATP5B was expressed on the cellular membrane of PC-3M cells and
that the expression ratio of ATP5B on PC-3M cells was significantly
higher than that on PC-3 cells (Fig.
4E; P<0.05). From the above results, we concluded that ATP5B
was ectopically expressed on the cellular membrane of PC-3M
cells.
Reduction of membrane-bound ATP5B levels
inhibit proliferation, invasion and metastasis of PC-3M cells
ATP5B is generally localized to the inner
mitochondrial membrane where it functions to catalyze the
rate-limiting step of ATP formation in eukaryotic cells (15). Our above-described results suggest
that ATP5B partially translocated to the cellular membrane of PC-3M
cells. Next, we investigated the potential functions of ectopically
expressed ATP5B on the cellular membrane of PC-3M cells. First, we
examined the spreading ability of PC-3M cells pre-incubated with
B04 at different concentrations for 48 h. As shown in Fig. 5A, the impact of B04 on PC-3M cell
spreading was concentration-dependent; higher concentrations of B04
resulted in greater inhibition of cell spreading. Moreover, CCK-8
assays indicated that B04 inhibited the proliferation of PC-3M
cells in vitro (Fig. 5B).
Cell growth curves showed that cell proliferation was significantly
inhibited by B04 as compared with that in the control group.
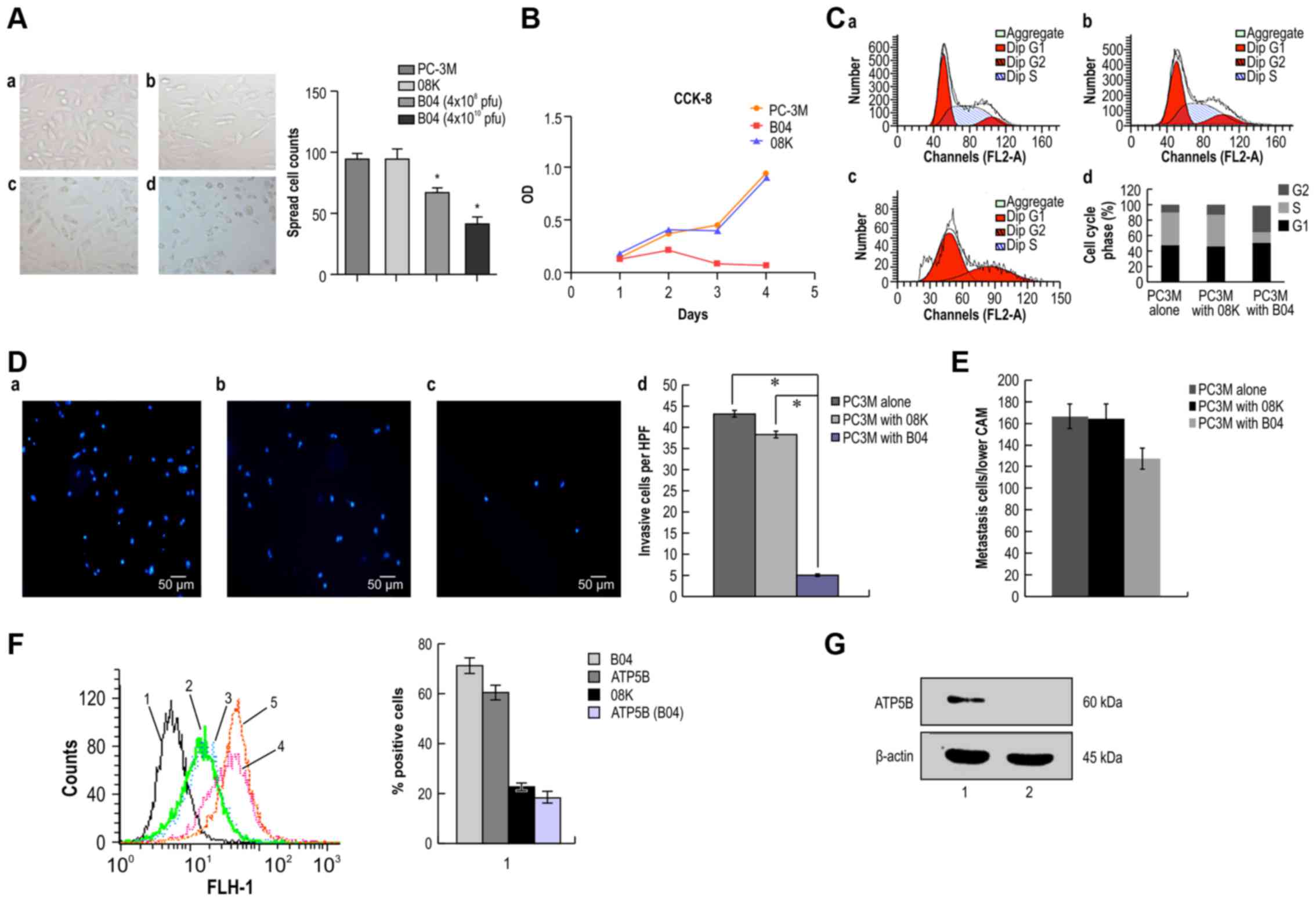 | Figure 5B04 inhibits the proliferation,
invasion and metastasis of PC-3M cells by targeting the cell
surface protein ATP5B. (A) Effects of B04 on the spreading of PC-3M
cells (magnification, ×100). (a) PC-3M cells alone; (b) PC-3M cells
with 08K (M13 without a seven-peptide gene sequence); (c) PC-3M
cells with a lower concentration of B04; (d) PC-3M cells with a
higher concentration of B04. A bar graph showing the spread cell
counts of each group is given in the right panel. (B) The
inhibition rates were determined by CCK-8 assays. Values are the
average of triplicate experiments and are represented as the mean ±
SD. (C) Cell cycle distribution of PC-3M cells in vitro.
PC-3M cells were treated with B04, 08K, or nothing (PC-3M cells
alone) for 24 h, and the fraction of cells in each phase of the
cell cycle was evaluated by flow cytometry. (a) PC-3M cells alone
as a control; (b) PC-3M cells with 08K as a control; (c) PC-3M
cells with B04; (d) cell cycle distribution in each group. (D)
Effects of B04 on PC-3M invasion in Transwell assays.
Representative photographs (a-c) and quantification (d) are shown.
Cell nuclei were stained with Hoechst 33342 (blue). (a) PC-3M cells
alone; (b) PC-3M cells with 08K; (c) PC-3M cells with B04
(4×1010 pfu). Magnification, ×100. (E) Effects of B04 on
PC-3M cell metastasis in the chicken embryo chorioallantoic
membrane (CAM). PC-3M cells were cultured with B04 or 08K, and
105–106 cells were inoculated onto the CAM.
After 50 h of incubation, genomic DNA was isolated, and human Alu
sequence PCR amplification was performed to determine the number of
metastatic cells in each group. *P<0.05 for comparisons between
the two groups. (F) Flow cytometry was used to determine the
effects of B04 on ATP5B expression. 1, PC-3M cells incubated with
Alexa Fluor 488 IgG (control); 2, PC-3M cells incubated with
anti-08K antibodies after pre-incubation with 08K; 3, PC-3M cells
were incubated with anti-ATP5B antibodies after pre-incubation with
B04 for 24 h; 4, PC-3M cells were incubated with anti-B04
antibodies after pre-incubation with B04; 5, PC-3M cells were
incubated with anti-ATP5B antibodies. (G) ATP5B bound to B04. Total
proteins from PC-3M and PC-3 cells were separated by SDS-PAGE,
transblotted and incubated with excessive amounts of B04.
Anti-ATP5B antibodies were then added. β-actin was used as a
control. Lane 1, total proteinsfrom PC-3 cells; lane 2, total
proteins from PC-3M cells. |
Based on these results, we examined whether the
anti-proliferative effects of B04 were caused by induction of cell
cycle arrest through analysis of the cell cycle distribution. The
results showed that B04 significantly affected the cell cycle
distribution, leading to cell cycle arrest at the G2/M phase; the
percentage of cells in the G2/M phase increased from 9.91% in
control cells to 34.28% in B04-treated cells. A corresponding
decrease in the percentage of cells in the S phase was also
observed (Fig. 5C).
In Transwell assays (Fig. 5D), we found that the invasive
ability of PC-3M cells incubated with B04 was significantly
decreased compared with that of PC-3M cells alone and PC-3M cells
incubated with the control phage 08K (P<0.01). In addition, the
results of analysis with the CAM model showed that B04
significantly inhibited tumor metastasis, with an inhibition ratio
of 23.41% (Fig. 5E), compared with
that in the 08K group.
The above results demonstrated that B04 could
effectively inhibit the aggressive properties of PC-3M cells,
including proliferation, spreading, migration and invasion, i.e.,
the characteristic malignant behavior of highly metastatic tumors.
To further confirm the role of specific binding of B04 with
ectopically expression ATP5B on the cellular membrane of PC-3M
cells in the inhibition of the malignant behavior, we used
competitive inhibition experiments with B04 and anti- ATP5B
antibodies. The results showed that ectopic expression of ATP5B was
significantly decreased in B04 pretreated cells compared with that
in the control group, incubated with blank phage. Specifically,
ATP5B expression decreased from 61.68±3.94% in the PC-3M cells
alone group and 65.77±5.75% in the 08K pre-incubated group to
17.62%±2.5% in the B04 pre-incubated group (P<0.05; Fig. 5F). Moreover, the binding rate of
B04 on PC-3M cells (71.31±2.58%) was significantly higher than that
of 08K (22.53±0.94%) on PC-3M cells (P<0.05; Fig. 5F).
Finally, we confirmed the binding of B04 and ATP5B
using competitive binding assays after SDS-PAGE and transblotting
of proteins onto PVDF membranes. The results showed that ATP5B on
PC-3M cells were specifically blocked by B04 (Fig. 5G).
Discussion
Treatment failure in patients with prostate cancer
can frequently be explained by osseous metastasis and invasion to
neighboring organs. Advances in predicting the metastasis of
prostate cancer will improve survival rates in patients with
prostate cancer. Therefore, in the present study, we aimed to
identify novel metastasis-related proteins using PC-3M and PC-3
cells, which exhibit differential levels of malignant behavior,
coupled with a 7-mer phage library screening. using this strategy,
we identified B04 as a phage peptide that bound to ATP5B
specifically on PC-3M cells. Furthermore, blockade of ATP5B with
B04 inhibited malignant behavior such as proliferation and invasion
of PC-3M cells. These findings provide important insights into the
use of phage technology for identification of metastasis-associated
proteins and shed light on the role of ATP5B in metastatic prostate
cancer.
Several strategies have been used for the
identification of tumor-associated proteins, including serological
analysis of recombinant cDNA expression libraries, ribosome
display, tumor-specific antibody cloning and phage antibody
libraries (8). In the present
study, we used a phage antibody library for screening specific
short peptides that could target unique protein expression
patterns. Peptides have the advantages of high clonal diversity,
small size, rapid binding kinetics, and low immunogenicity as
detection probes (16,17). Here, we also used bio-panning with
normal prostate cells and PC-3 cells to decrease non-specific
binding to PC-3M cells in each round. This analysis identified B04
as a ligand for a metastatic protein on PC-3M cells, and subsequent
proteomic analysis with MS/MS identified ATP5B as a B04-target
protein. These results highlight the applicability of this
methodological strategy in the identification of metastasis-related
proteins.
ATP5B is part of the catalytic core of the enzyme
complex that catalyzes the rate-limiting step of ATP formation in
eukaryotic cells. The F1 portion is extramembranous and constitutes
the catalytic core, whereas the F0 portion is largely embedded in
the membrane and functions as a proton channel involved in proton
translocation (18,19). ATP synthases (including ATP5B) are
thought to be localized exclusively on the inner mitochondrial
membrane. However, in this study, ATP5B was identified from the
membranous fraction of PC-3M cells, which indicated the potential
translocation of ATP5B from the inner mitochondrial membrane to the
outer cellular membrane. These data were further confirmed using
double immunofluorescence staining and immunoelectromicroscopy,
consistent with previous reports by Chi and Pizzo (7) and Ma et al (20). Additionally, we found that ATP5B
was localized to the surface of cells in a specimen from a patient
with highly malignant prostate cancer. Our results also suggested
that ectopic ATP synthase localized to the caveolae of the cell
membrane, with the F1 portion directed into the extracellular
space. Some studies have also reported that other subunits of the
ATP synthase, such as the α and β subunits of the F1 domain and the
β- and δ-subunits of F0, can be found on the outside of the
membranes of tumor cells and some types of normal cells, including
hepatocytes, keratinocytes, adipocytes, endothelial cells and
cardiomyocytes (21–24), suggesting that translocation is
possible. The enzyme may therefore be called cell surface ATP
synthase or ecto-ATP synthase (ecto- ATPase, ecto-F1-ATPase), and
ectopically localized ATP5B may be called ecto-ATP5B.
Ectopic ATP synthase has been reported as a
structural element and asan enzyme involved in ATP synthesis and
proton transport, particularly in some normal cells. Moreover, ATP
synthase on the cell surface has been shown to bind a variety of
factors, and the β subunit has been shown to be a target of the
hormone enterostatin. F1Fo-ATP synthase on the hepatocyte cell
surface is localized in caveolae/lipid rafts and functions as a
receptor for HDL apolipoprotein A-I, mediating HDL endocytosis. The
entire F1Fo-ATP synthase has activity in the endothelial cell
surface, with ability to synthesize ATP and transport protons.
Moser and colleagues (25,26) identified endothelial cell surface
ATPase as a receptor for angiostatin, which inhibits
vascularization by suppression of endothelial surface ATP
metabolism. Following ATP synthesis, intracellular protons are
pumped out of the cell to prevent acidosis. When the activity of
ectopic ATP synthase is blocked, the proliferation and migration of
cells could be inhibited, particularly when the cells were cultured
with a low extracellular pH. Notari et al (27) reported that pigment
epithelium-derived factor (PEDF) functions as a potent blocker of
angiogenesis in vivo, inhibits endothelial cell migration
and tubule formation, reduces the amount of extracellular ATP
produced by endothelial cells, and binds with high affinity to
ATP5B on the surfaces of endothelial cells. PEDF also blocks the
binding between ATP5B and angiostatin. ATP synthesis products may
excite the purinoceptor, which has been reported to induce many
biological effects, including endothelial cell proliferation and
angiogenesis (28). However, the
ectopic expression of ATP5B and its potential roles in malignant
cells have not been described in detail. Chi and Pizzo (29) reported that ATP5B is expressed on
the surface of tumor cells and that treatment with angiostatin or
antibodies against ATP5B inhibits the synthase enzyme, suggesting
that the activity of ATP5B is independent of the malignant status
of cells. For ectopic ATP5B, its specificity to tumors relies on
its increased catalytic activity in acidic and hypoxic
microenvironments when compared with normal tissues. Consistent
with these above findings, our current results showed that ATP5B
translocated from the inner mitochondrial membrane to the outer
surface of PC-3M cells and that incubation of PC-3M cells with the
short peptide B04 reduced the ectopic expression of ATP5B on the
PC-3M cellular membrane. Moreover, the proliferation, invasion and
metastasis of PC-3M cells were significantly inhibited by
specifically blocking the cell surface protein ATP5B. These results
indicated that the translocation and ectopic expression of ATP5B on
PC-3M cells may be involved in the invasion and metastasis of
prostate cancer.
The signaling pathways through which ectopic ATP5B
regulates the biological behavior of PC-3M cells are unclear. In
our previous MALDI-TOF/MS studies, we found that ATP synthase and
caveolin were upregulated in PC-3M cells. In accordance with
studies by Ma and colleagues (20)
and Chi and Pizzo (29), the
purinergic signaling pathway through P2X and P2Y receptors may
function to regulate the cell surface ATP synthase. Thus, the
following pathway may be involved in this process. First, with the
synthesis of ATP, protons are pumped out of the cells. As autocrine
agents in the caveolae, ATP synthesis products may excite the
purinoceptor (P2Y) to promote cell proliferation, leading to tumor
growth and angiogenesis. Cell surface F1Fo-ATP synthase generates
ATP and ADP, which may participate in purinergic signaling through
P2X and P2Y receptors. Purinergic signaling is implicated in cell
growth, proliferation, chemotaxis, inflammatory responses, hypoxic
injury and apoptosis (30–34). P2X receptors are ATP-gated,
Ca2+-permeable channels that mediate rapid,
non-selective passage of cations (Na+, K+ and
Ca2+), resulting in an increase in intracellular
Ca2+ (35). P2Y
receptors are G-protein-coupled receptors that regulate cell
proliferation through various pathways (36). However, the mechanistic basis of
the potential roles of ectopic ATP5B pathways in cancer metastasis
is still unclear.
In summary, using a short peptide, B04, identified
from screening of a phage peptide library, we identified a
metastasis-related protein, ATP5B, located on the cellular surface
of PC-3M cells. Ectopic ATP5B was critical for the proliferation,
invasion and metastasis of PC-3M cells, and may be a potential
biomarker or therapeutic target in highly metastatic tumors.
However, the mechanisms through which ATP5B translocates to the
cellular membrane of highly metastatic prostate cancer cells and
the potential effects of this protein on the metastatic potential
of prostate cancer need to be examined more fully in future
studies.
Acknowledgments
We thank Dr F. William Orr for his critical
suggestions on the organization of the manuscript. The present
study was supported by the Jilin Provincial Science and Technology
Projects (grant nos. 20150101126JC and 20130102084JC).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kauffman EC, Robinson VL, Stadler WM,
Sokoloff MH and Rinker-Schaeffer CW: Metastasis suppression: The
evolving role of metastasis suppressor genes for regulating cancer
cell growth at the secondary site. J Urol. 169:1122–1133. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hanash SM, Madoz-Gurpide J and Misek DE:
Identification of novel targets for cancer therapy using expression
proteomics. Leukemia. 16:478–485. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao J, Gao Y, Ju Y, Yang J, Wu Q, Zhang J,
Du X, Wang Z, Song Y, Li H, et al: Proteomics-based generation and
characterization of monoclonal antibodies against human liver
mitochondrial proteins. Proteomics. 6:427–437. 2006. View Article : Google Scholar
|
|
6
|
Liu B, Huang L, Sihlbom C, Burlingame A
and Marks JD: Towards proteome-wide production of monoclonal
antibody by phage display. J Mol Biol. 315:1063–1073. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chi SL and Pizzo SV: Cell surface F1Fo ATP
synthase: A new paradigm? Ann Med. 38:429–438. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu ZJ, Song QF, Jiang SS, Song Q, Wang W,
Zhang GH, Kan B, Chen LJ, Yang JL, Luo F, et al: Identification of
ATP synthase beta subunit (ATPB) on the cellsurface as a non-small
cell lung cancer (NSCLC) associated antigen. BMC Cancer. 9:162009.
View Article : Google Scholar
|
|
9
|
Dowling P, Meleady P, Dowd A, Henry M,
Glynn S and Clynes M: Proteomic analysis of isolated membrane
fractions from superin- vasivecancer cells. Biochim Biophys Acta.
1774:93–101. 2007. View Article : Google Scholar
|
|
10
|
Kim BW, Choo HJ, Lee JW, Kim JH and Ko YG:
Extracellular ATP isgenerated by ATP synthase complex in adipocyte
lipid rafts. Exp Mol Med. 36:476–485. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hong D, Jaron D, Buerk DG and Barbee KA:
Transport- dependent calcium signaling in spatially segregated
cellular caveolar domains. Am J Physiol Cell Physiol.
294:C856–C866. 2008. View Article : Google Scholar
|
|
12
|
Bae TJ, Kim MS, Kim JW, Kim BW, Choo HJ,
Lee JW, Kim KB, Lee CS, Kim JH, Chang SY, et al: Lipid raft
proteome reveals ATP synthase complex in the cell surface.
Proteomics. 4:3536–3548. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ribatti D: Chicken chorioallantoic
membrane angiogenesis model. Methods Mol Biol. 843:47–57. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kariya Y, Kato K, Hayashizaki Y, Himeno S,
Tarui S and Matsubura K: Revision of consensus sequence of human
Alu repeats a review. Gene. 53:1–10. 1987. View Article : Google Scholar
|
|
15
|
Boyer PD: The ATP synthase - a splendid
molecular machine. Annu Rev Biochem. 66:717–749. 1997. View Article : Google Scholar
|
|
16
|
Larimer BM, Thomas WD, Smith GP and
Deutscher SL: Affinity maturation of an ERBB2-targeted SPECT
imaging peptide by in vivo phage display. Mol Imaging Biol.
16:449e582014. View Article : Google Scholar
|
|
17
|
Goetz M and Wang TD: Molecular imaging in
gastrointestinal endoscopy. Gastroenterology. 138:828e33.e8212010.
View Article : Google Scholar
|
|
18
|
Senior AE: ATP synthesis by oxidative
phosphorylation. Physiol Rev. 68:177–231. 1988.PubMed/NCBI
|
|
19
|
Stock D, Leslie AG and Walker JE:
Molecular architecture of the rotary motor in ATP synthase.
Science. 286:1700–1705. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma Z, Cao M, Liu Y, He Y, Wang Y, Yang C,
Wang W, Du Y, Zhou M and Gao F: Mitochondrial F1Fo-ATP synthase
translocates to cell surface in hepatocytes and has high activity
in tumor-like acidic and hypoxic environment. Acta Biochim Biophys
Sin (Shanghai). 42:530–537. 2010. View Article : Google Scholar
|
|
21
|
Champagne E, Martinez LO and Collet Xand
Barbaras R: Ecto-F1Fo ATP synthase/F1 ATPase: metabolic and
immunological functions. Curr Opin Lipidol. 17:279–284. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jung KH, Song SH, Paik JY, Koh BH, Choe
YS, Lee EJ, Kim BT and Lee KH: Direct targeting of tumor cell
F1F0 ATP-synthase by radioiodine angiostatin
in vitro and in vivo. Cancer Biother Radiopharm. 22:704–712. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Das B, Mondragon MO, Sadeghian M, Hatcher
VB and Norin AJ: A novel ligand in lymphocyte-mediated
cytotoxicity: Expression of the beta subunit of H+
transporting ATP synthase on the surface of tumor cell lines. J Exp
Med. 180:273–281. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arakaki N, Nagao T, Niki R, Toyofuku A,
Tanaka H, Kuramoto Y, Emoto Y, Shibata H, Magota K and Higuti T:
Possible role of cell surface H -ATP synthase in the extracellular
ATP synthesis and proliferation of human umbilical vein endothelial
cells. Mol Cancer Res. 1:931–939. 2003.PubMed/NCBI
|
|
25
|
Moser TL, Stack MS, Asplin I, Enghild JJ,
Højrup P, Everitt L, Hubchak S, Schnaper HW and Pizzo SV:
Angiostatin binds ATP synthase on the surface of human endothelial
cells. Proc Natl Acad Sci USA. 96:2811–2816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moser TL, Kenan DJ, Ashley TA, Roy JA,
Goodman MD, Misra UK, Cheek DJ and Pizzo SV: Endothelial cell
surface F1-F0 ATP synthase is active in ATP synthesis and is
inhibited by angiostatin. Proc Natl Acad Sci USA. 98:6656–6661.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Notari L, Arakaki N, Mueller D, Meier S,
Amaral J and Becerra SP: Pigment epithelium-derived factor binds to
cell- surface F1-ATP synthase. FEBS J. 277:2192–2205.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rumjahn SM, Yokdang N, Baldwin KA, Thai J
and Buxton IL: Purinergic regulation of vascular endothelial growth
factor signaling in angiogenesis. Br J Cancer. 100:1465–1470. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chi SL and Pizzo SV: Angiostatin is
directly cytotoxic to tumor cells at low extracellular pH: A
mechanism dependent on cell surface-associated ATP synthase. Cancer
Res. 66:875–882. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schwiebert EM and Zsembery A:
Extracellular ATP as a signaling molecule for epithelial cells.
Biochim Biophys Acta. 1615:7–32. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bodin P and Burnstock G: Increased release
of ATP from endothelial cells during acute inflammation. Inflamm
Res. 47:351–354. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Di Virgilio F: Dr. Jekyll/Mr. Hyde: The
dual role of extracellular ATP. J Auton Nerv Syst. 81:59–63. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ralevic V and Burnstock G: Receptors for
purines and pyrimidines. Pharmacol Rev. 50:413–492. 1998.PubMed/NCBI
|
|
34
|
Wen LT and Knowles AF: Extracellular ATP
and adenosine induce cell apoptosis of human hepatoma Li-7A cells
via the A3 adenosine receptor. Br J Pharmacol. 140:1009–1018. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schwiebert LM, Rice WC, Kudlow BA, Taylor
AL and Schwiebert EM: Extracellular ATP signaling and P2X
nucleotide receptors in monolayers of primary human vascular
endothelial cells. Am J Physiol Cell Physiol. 282:C289–C301. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Burnstock G: Purinergic signaling and
vascular cell proliferation and death. Arterioscler Thromb Vasc
Biol. 22:364–373. 2002. View Article : Google Scholar : PubMed/NCBI
|