Introduction
Chronic myeloid leukemia (CML) is a hematological
malignancy arising from hematopoietic stem cell transformation
(1,2). Targeting BCR-ABL protein with
tyrosine kinase inhibitors (TKIs) has profoundly improved the
survival of CML patients. However, there are still challenges:
firstly, approximately 40% CML patients relapse with BCR-ABL
mutation and these patients are resistant to most TKIs; secondly,
CML patients in blast crisis (BC) are insensitive to TKIs (3,4);
thirdly, CML stem cells are intrinsically insensitive to TKIs. It
is urgent to explore novel strategies for CML treatment.
Combination therapy and immunotherapy are potential way to
circumvent these problems. Combination treatments with
hydroxychloroquine (HCQ) and traditional agents have been
investigated recently (5–12). HCQ enhances the sensitivity of CML
cells to TKIs, even in primary CML stem cells (13,14).
The combination of HCQ with imatinib almost completely eliminates
CML stem cells in vitro. Based on this finding, a randomized
phase II clinical trial (NCT01227135) is now underway (2,15).
HCQ can also potentiate the anti-CML efficiency of other agents
including suberoylanilide hydroxamic acid, perifosine, asparaginase
and diosgenin (6–12). In addition, HCQ can strengthen the
efficacy of immunotherapy (16–19).
By inhibiting hypoxia-induced autophagy in breast cancer cells, HCQ
facilitates natural killer cell (NK)-mediated elimination of tumor
cells (18). HCQ also enhances
cytotoxic T lymphocyte (CTL)-mediated lysis of melanoma cells
(19).
Vγ9Vδ2 T cell is important immune cell in peripheral
blood and are attractive candidate for the elimination of leukemia
cells (20–23). Consistent with an earlier study, we
have previously shown that Vγ9Vδ2 T cells can effectively recognize
and kill CML cells (23–25). Both HCQ and Vγ9Vδ2 T cells are
promising in CML treatment, but the effects of HCQ on the
elimination of CML cells by Vγ9Vδ2 T cells are unknown.
The anti-CML effects of HCQ mainly rely on autophagy
inhibition (26). However, some
studies have revealed that HCQ also can exert anticancer effects
independent of autophagy (27,28).
In the present study, we uncovered a previously unknown
autophagy-independent mechanism by which HCQ enhanced the
sensitivity of CML cells to Vγ9Vδ2 T cell-mediated lysis. HCQ
enhanced the sensitivity by promoting the translocation of ULBP4
from the cytoplasm of CML cells to the membrane, which is important
for the recognition of cancer cells by Vγ9Vδ2 T cells (24,29,30).
Our results revealed an unknown mechanism of HCQ in treating CML,
and provide the first evidence that combining HCQ with Vγ9Vδ2T
immunotherapy represents a promising treatment for CML.
Materials and methods
Reagents
Hydroxychloroquine sulfate (HCQ) was purchased from
Selleck Chemicals (Houston, TX, USA). Zoledronate was from Novartis
(Basel, Switzerland). IL2 was purchased from PeproTech (Rocky Hill,
NJ, USA).
Vγ9Vδ2 T cell preparation
Vγ9Vδ2 T cells were prepared from peripheral blood
samples. Peripheral blood mononuclear cells (PBMCs) were isolated
from fresh blood samples of healthy volunteers and cultured in
RPMI-1640 medium (Corning Costar, Corning, NY, USA) containing 10%
fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) for 10–14 days.
PBMCs were stimulated with 400 nM zoledronate and 300 IU/ml
recombinant human IL2 for 72 h. IL2 was renewed every 3 days at the
same concentration and lymphocytes were kept at 1.5×106
cells/ml. Ten to fourteen days later, the purity of Vγ9Vδ2 T cells
was determined by flow cytometry using mAbs for TCRVδ2 and
indicated a purity ≥90%.
Leukemia cell culture
Human CML cell lines K562 and K562/GO1 were
purchased from the Institute of Hematology at the Chinese Academy
of Medical Sciences (Tianjin, China). K562/GO1 cell is an imatibin
resistant CML cell line, which showed increased level of BCR-ABL
(31). The cell lines were
cultured (0.5 cells ×106/ml) in complete RPMI-1640
medium (Corning Costar) containing 10% FBS (Gibco).
Bone marrow samples were taken from two CML patients
and bone marrow mononuclear cells were separated by Ficoll-Hypaque
gradient centrifugation (Haoyang Biological Manufacture, Co., Ltd.,
Tianjin, China). CML stem cells were labelled with anti-CD34 FITC
antibody and selected using anti-FITC magnetic beads (Miltenyi
Biotec, Bergisch Gladbach, Germany). The percentage of
CD34-positive cells was >85%. Informed consent was obtained in
accordance with the Declaration of Helsinki from all patients and
volunteers. Approval for the study was obtained from the Ethics
Committee of the First Affiliated Hospital of Zhejiang
University.
Vγ9Vδ2 T cell cytotoxicity assay
To test the cytotoxicity of Vγ9Vδ2 T cells, a flow
cytometric cytotoxicity assay was performed using CFSE (Life
Technologies, Grand Island, NY, USA) and propidium iodide (PI;
Multi Science, Hangzhou, China) (32). K562 and K562/GO1 cells were
pretreated with 30 µM HCQ for 8 h and control cells were
pretreated with an equal volume of phosphate-buffered saline (PBS).
Then cells were harvested and washed twice with PBS. After CFSE
staining, the cells were resuspended in serum-free medium and
plated into 96-well plates, and Vγ9Vδ2 T cells were added at
effector target ratios (E:T) of 20:1, 10:1, or 5:1. In addition,
150 IU/ml IL-2 was added to the wells. Wells containing only
labelled target cells were also prepared to evaluate background
levels of cell death. Cells were incubated for 4 h at 37°C and 5%
CO2, then stained with PI and analyzed by flow
cytometry. Cytolytic activity was calculated based on the
percentage of dead target cells (CFSE+ PI+).
In some experiments, effector cells were pretreated with 10
µg/ml anti-NKG2D neutralizing mAb (eBioscience, Inc., San
Diego, CA, USA) or isotype control mAb (eBioscience) for 1 h at
room temperature. Fluorescence was analysed using a Flow Cytometry
FC500 system (Beckman Coulter, Inc., Miami, FL, USA) and data were
analyzed using the CXP flow cytometry software.
Vγ9Vδ2 T cell degranulation assay
Degranulation of Vγ9Vδ2 T cells was evaluated using
the lysosomal marker CD107a as previously described (33). CML cell lines and primary CML cells
were pretreated with HCQ or PBS. Cells were incubated with Vγ9Vδ2 T
cells at an E:T ratio of 1:1 in a 96-well plate, and 5 µl
Alexa647-CD107 (BioLegend, San Diego, CA, USA) was added to all
wells. As positive control wells, 0.05 µg/ml phorbol ester
(PMA) and 1 µg/ml ionomycin (Multi Science) were added.
Wells containing Vγ9Vδ2 T cells only were prepared as negative
controls to establish background levels of degranulation. Cells
were incubated for 1 h at 37°C, then 3 µl GolgiStop (BD
Biosciences, Franklin Lakes, NJ, USA) was added. Cells were further
incubated for 3 h, and then washed and incubated with an anti-Vδ2
TCR antibody (eBioscience) to label Vγ9Vδ2 T cells for analyzing by
flow cytometry. The level of Vγ9Vδ2 T cell degranulation was
determined by the percentage of CD107-positive Vγ9Vδ2 T cells.
Toxicity assay of HCQ to CML cell
lines
CML cell lines were cultured at 2×105
cells/ml in the presence of 30 µM HCQ for 8 h at 37°C and 5%
CO2. Control CML cells were treated with equal volumes
of PBS. Cells were washed twice with PBS, and then stained with 7.5
µM PI for 0.5 h. Cell viability was tested by flow cytometry
and PI-positive cells were considered dead.
Flow cytometric analysis
Cells were washed and resuspended in 100 µl
PBS, then incubated with 5 µl fluorophore-conjugated mAb at
4°C in the dark for 30 min. Mouse anti-human PE-ULBP1,
PE-ULBP2/5/6, PE-ULBP3, APC-ULBP4 and PE-MICA/B (R&D Systems,
Minneapolis, MN, USA), FITC-CD34 and PE-Vδ2TCR (eBioscience)
antibodies were used. Isotype antibodies were used to assess
non-specific staining. For intracellular staining, cells were fixed
and permeabilized using a fixation/permeabilization kit (BD
Biosciences) according to the manufacturer's instructions before
mAb labelling.
Protein extraction and western blot
analyses
Equal numbers of cells were lysed using RIPA lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China)
containing phenyl-methane sulfonyl fluoride. Cell lysates were
separated using sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and proteins were transferred onto
nitrocellulose membranes. The membranes were blocked in 5% BSA (BBI
Life Science, Shanghai, China), then incubated with primary
antibodies and secondary antibodies (LI-COR Biosciences, Lincoln,
NE, USA). The following primary antibodies were used:
Anti-P62/SQSTM1 (Abcam, Cambridge, MA, USA), anti-ULBP4 (R&D
Systems), anti-LC3, anti-ATG7 and anti-β-actin (Cell Signaling
Technology, Danvers, MA, USA). Immunoreactive bands were visualized
using an Odyssey infrared imaging system (LI-COR Biosciences).
Short hairpin RNA (shRNA) preparation and
transfection
Lentiviral vectors containing shRNA against ATG7 or
the corresponding control shRNA were synthesized by Shanghai
GenePharma, Co., Ltd. (Shanghai, China). Lentiviruses were produced
in 293T cells by transfecting the lentiviral expression vector and
packaging vectors (psPAX2 and pMD2.G; Addgene, Cambridge, MA, USA)
using Attractenen transfection reagent (Qiagen, Valencia, CA, USA).
After enriching, the lentiviruses were transfected into K562 and
K562/GO1 cells. Transfection efficiency was estimated by evaluating
GFP expression. The effects of autophagy inhibition were tested by
western blot analysis.
Confocal immunofluorescence
microscopy
HCQ- or PBS-treated K562 cells were placed onto
adhesion microscope slides (Citoglas, Haimen, China) and fixed in
4% methanol (Biotech Well, Shanghai, China). After fixation, cells
were permeabilized with 0.1% Triton X-100 and incubated in a
primary anti-ULBP4 (Abcam) antibody followed by a CY3-tagged
secondary antibody (Wuhan Boster Biological Technology Ltd., Wuhan,
China) to stain ULBP4 protein. The green fluorescent dye Dio
(Invitrogen, Carlsbad, CA, USA) was used to stain the cell
membrane. The nucleus was counter-stained with DAPI. Images were
captured using a two-photon confocal microscope (Olympus Corp.,
Tokyo, Japan).
Transmission electron microscopy
K562 and K562/GO1 cells were treated with 30
µM HCQ or PBS for 8 h. Harvested cells were fixed with 2.5%
glutaraldehyde overnight at 4°C and then post-fixed in a solution
containing 1% osmium tetroxide and dehydrated through an alcohol
series. Fixed samples were sectioned and stained with 3% uranyl
acetate and Reynolds lead citrate. Samples were imaged using a
transmission electron microscope (Philips TECAN 10; Philips
Electronic N.V, Amsterdam, The Netherlands).
Statistical analyses
Data were from three independent experiments and
expressed as mean ± SD. All data were analyzed using SPSS 7.0
software with ANOVA or two-tailed Student's t-test. P<0.05 was
considered statistically significant.
Results
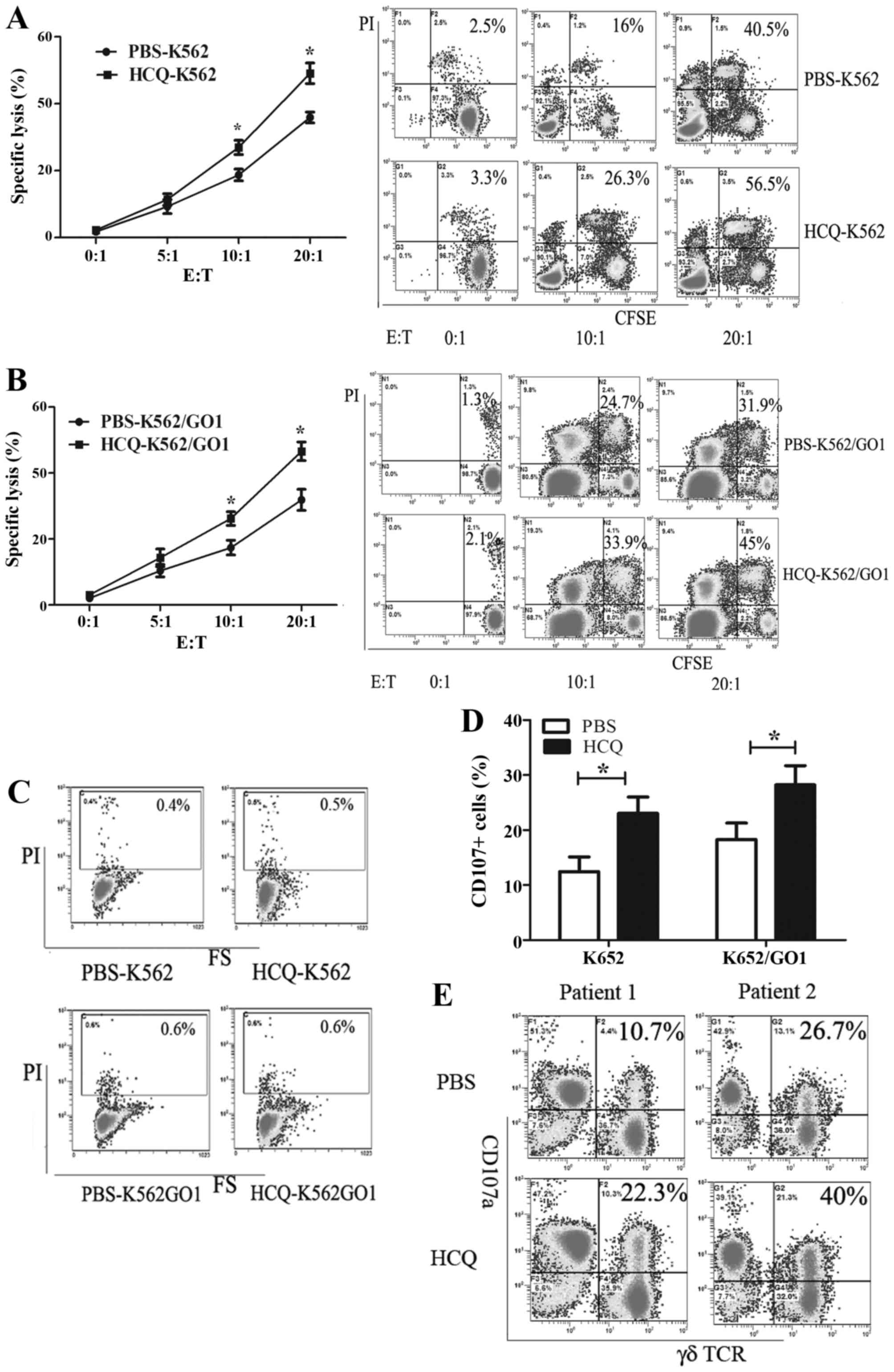
HCQ sensitizes CML cells to Vγ9Vδ2T
cell-mediated lysis
To investigate whether HCQ can affect the
susceptibility of CML cells to Vγ9Vδ2 T cell-mediated lysis, the
human CML cell lines K562 and K562/GO1 pretreated with HCQ or PBS
were used as target cells and Vγ9Vδ2 T cells as effector cells. CML
cells were co-cultured with Vγ9Vδ2 T cells for 4 h at different
effector target ratios (E:T) and cell death was quantified by PI
staining. Background cell death was similar in HCQ-pretreated cells
and control cells, but HCQ-pretreated cells showed higher
sensitivity to Vγ9Vδ2 T cell-mediated lysis than control cells. The
specific lysis of K562 by Vγ9Vδ2 T cell increased from 35.9±1.64 to
47.1±5.97% (P<0.05) and from 18.72±3.73 to 24.95±2.345%
(P<0.05) at E:T ratios of 20:1 and 10:1 (Fig. 1A). Similarly, the lysis of K562/GO1
increased from 31.7±3.9 to 46.6±1.85% (P<0.05) and from 17.4±2.2
to 26.2±3.06% (P<0.05) at E:T ratios of 20:1 and 10:1 (Fig. 1B). To confirm that enhanced CML
cell death was not caused by toxicity of HCQ to CML cells, we
performed a toxicity assay. HCQ treatments (30 µM) for 8 h
did not reduce cell viability (Fig.
1C).
To confirm the afore-mentioned results, we examined
degranulation of Vγ9Vδ2 T cells using CD107a as a marker. CD107a
expression was evaluated by flow cytometry after Vγ9Vδ2 T cell
interaction with HCQ or PBS pretreated K562 and K562/GO1 cells. The
expression of CD107a increased in Vγ9Vδ2 T cells co-cultured with
HCQ-pretreated CML cell lines compared with Vγ9Vδ2 T cells
co-cultured with control CML cells. The percentage of CD107a
positive Vγ9Vδ2 T cells co-cultured with K562 cells increased from
12.45±4.66 to 23.05±4.98% (P<0.05), and from 18.3±3 to 28.2±3.5%
(P<0.05) in Vγ9Vδ2 T cells co-cultured with K562/GO1 (Fig. 1D). We validated this result in
primary CML stem cells isolated from two CML patients. HCQ
pretreated primary CML cells accelerated Vγ9Vδ2 T cell
degranulation (Fig. 1E).
Collectively, our results demonstrate that HCQ can enhance the
susceptibility of CML cell lines and CML primary stem cells to
Vγ9Vδ2 T cell-mediated lysis.
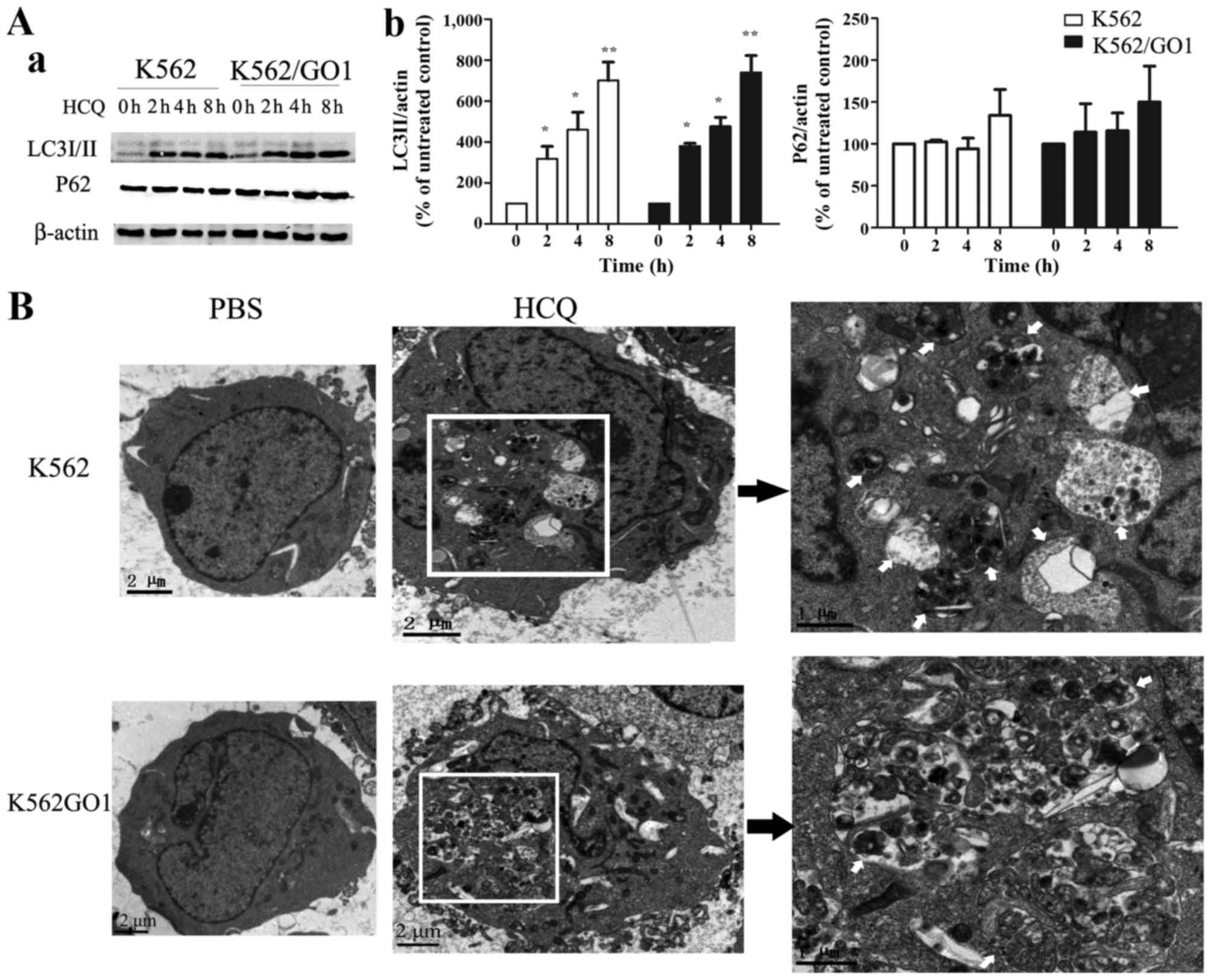
HCQ inhibits autophagy in CML cells
HCQ is an important autophagy inhibitor and some
studies have proven that inhibiting autophagy promotes the
elimination of cancer cells by NK and CTL cells (18,19).
To explore the mechanisms involved in the sensitizing effects of
HCQ, we first tested whether HCQ can inhibit autophagy in CML
cells. During autophagy, microtubule-associated protein light
chain-3II (LC3II) is selectively expressed on the autophagosomal
membrane and can be easily detected as autophagosome biomarkers
(34,35). We evaluated LC3II expression in
K562 and K562/GO1 cells after HCQ treatment. As shown in Fig. 2A, LC3II accumulation was dependent
upon the exposure time to HCQ. After 2 h of exposure LC3II begins
to accumulate, and with increasing exposure time autophagy
inhibition increased. HCQ treatment also increased P62 expression
(Fig. 2A), which correlated with
autophagy inhibition (36).
Furthermore, transmission electron microscopy showed that
autophagic vacuoles were not present in control CML cells. HCQ
treatment induced the accumulation of autophagic vacuoles in CML
cells containing some organelles and electron-dense inclusions
(Fig. 2B). In summary,
immunoblotting and transmission electron microscopy showed that HCQ
significantly inhibits autophagy in CML cells.
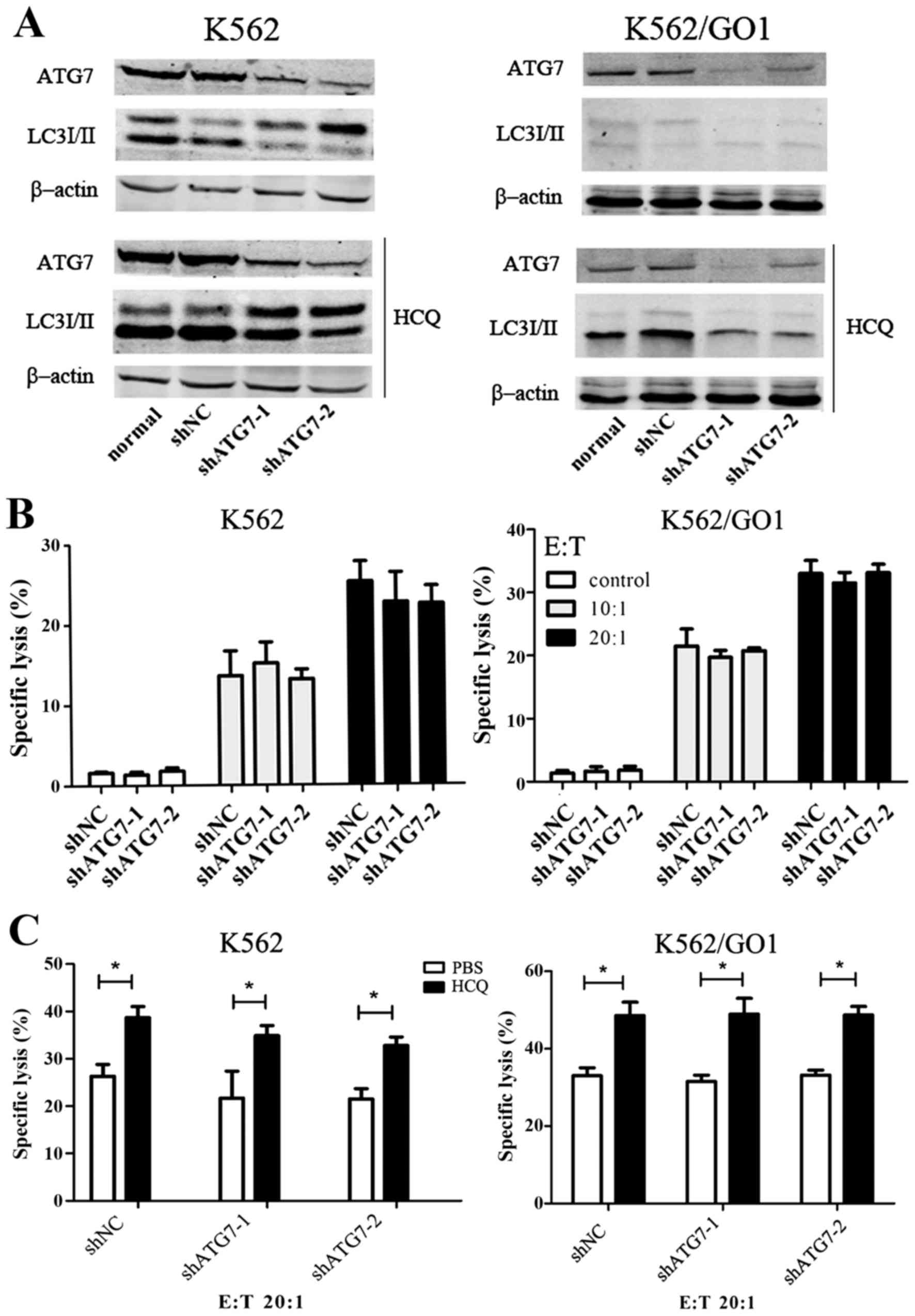
HCQ sensitizes CML cells to Vγ9Vδ2T
cell-mediated lysis independent of autophagy
HCQ can inhibit autophagy in CML cells, but whether
HCQ sensitizes CML cells to Vγ9Vδ2 T cell-mediated lysis by
inhibiting autophagy is unknown. We targeted the autophagy protein
ATG7 with shRNA to inhibit autophagy. Contrast to control shRNA,
shATG7 significantly silenced ATG7 protein expression, and
inhibited autophagy as shown by the disappearance of LC3II even in
the presence of HCQ (Fig. 3A).
Autophagy inhibition with shATG7 did not increase the sensitivity
of K562 and K562/GO1 cells to Vγ9Vδ2 T cytotoxicity (Fig. 3B). This suggests that HCQ
sensitizing CML cells to Vγ9Vδ2 T cytotoxicity is not through
autophagy inhibition. ATG7 knockdown prevents autophagy in early
stages while HCQ blocks later stages of autophagy. To further
confirm that the sensitizing effects of HCQ are
autophagy-independent, we tested whether HCQ enhances the
sensitivity of autophagy-defective CML cells to Vγ9Vδ2 T cells.
After knocking down ATG7 in CML cells, we pretreated these cells
with PBS or HCQ and co-cultured with Vγ9Vδ2 T cells. HCQ sensitized
autophagy-defective K562 and K562/GO1 cells to Vγ9Vδ2 T cell
cytotoxicity (Fig. 3C). We
conclude that HCQ sensitizes K562 and K562/GO1 cells to Vγ9Vδ2 T
cell-mediated lysis independent of autophagy.
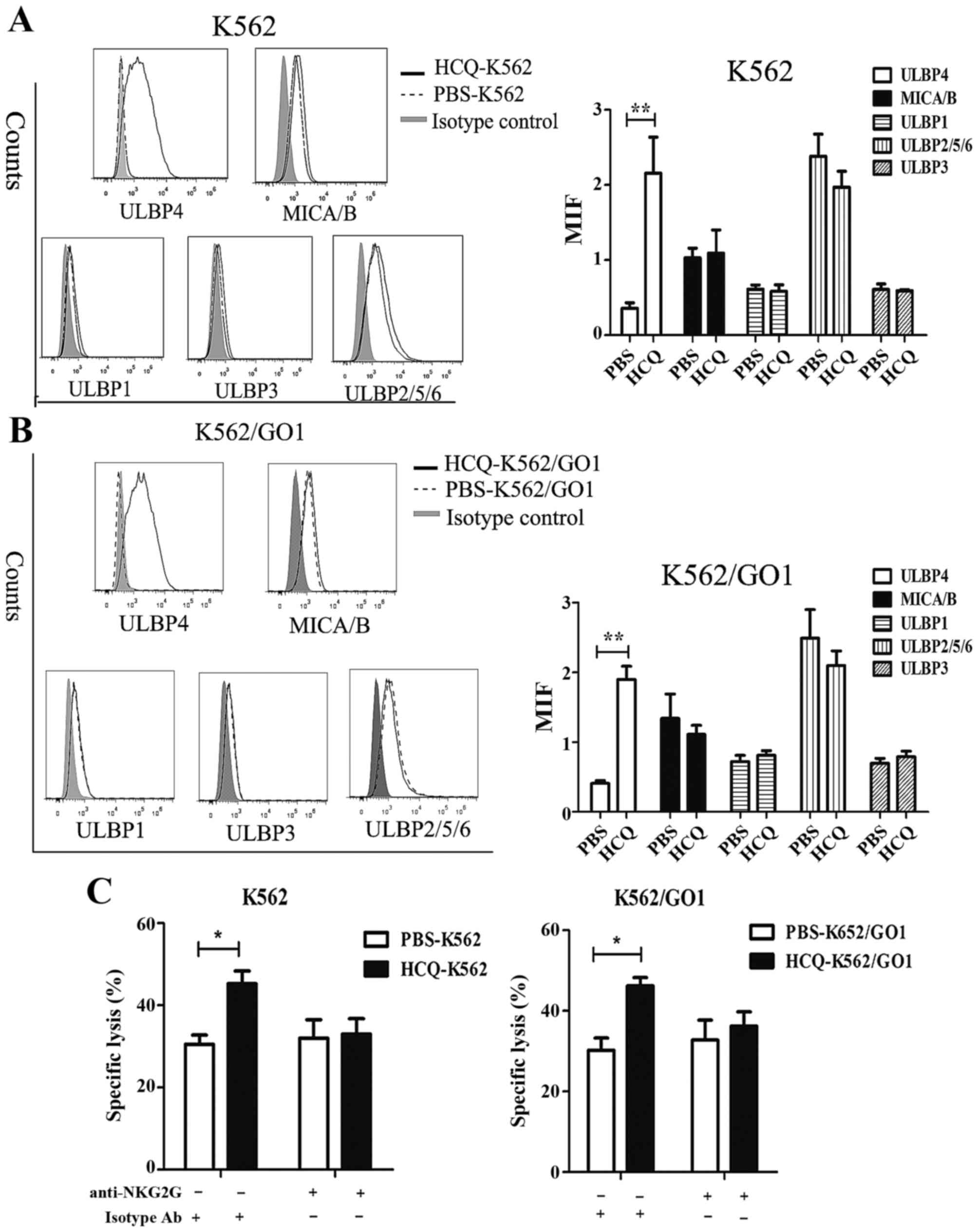
HCQ sensitizes CML cells to Vγ9Vδ2 T
cell-mediated lysis by inducing ULBP4 expression on CML
membrane
Although the recognition of target cell is mainly
TCR mediated, Vγ9Vδ2T cells also can utilize activating receptor
NKG2D. NKG2D is a NK cell-activating receptor, which recognizes
stress-inducing ligands including MICA, MICB and UL16-binding
proteins (ULBP1-6) in cancer cells (37–39).
We and others have previously shown that the interaction of NKG2D
with its ligands is important for recognition of leukemia cells by
Vγ9Vδ2 T cell and activation of Vγ9Vδ2 T cells. We speculated that
HCQ may affect the expression of NKG2D ligands in CML cells.
Consistent with our hypothesis, flow cytometric analysis showed
that HCQ induced ULBP4 expression on the cell membrane, and the
expression of other NKG2D ligands was not affected (Fig. 4A and B). ULBP4 was not expressed on
the membrane of K562 and K562/GO1 cells, but after HCQ treatment
for 8 h the mean fluorescence intensity (MFI) of ULBP4 increased
from 0.359±0.072 to 2.157±0.78 in K562 cells and from 0.41±0.04 to
1.9±0.19 in K562/GO1 cells (Fig. 4A
and B). Next, we investigated whether HCQ-induced expression of
ULBP4 is involved in sensitizing CML cells to Vγ9Vδ2 T
cell-mediated lysis. We incubated effector cells with anti-NKG2D
antibody to block the interaction of NKG2D with its ligands and
then performed cytotoxicity assays. HCQ increased the sensitivity
of CML cells to Vγ9Vδ2 T cell-mediated lysis, and blocking the
interaction of NKG2D with its ligands almost completely abrogated
this sensitizing effect (Fig. 4C).
Notably, control CML cell did not express ULBP4 on the membrane,
and blocking interaction of NKG2D with its ligands did not affect
the sensitivity of control CML cells to Vγ9Vδ2 T cell-mediated
lysis (Fig. 4C). This implies that
ULBP4 is much more important than other NKG2D ligands in Vγ9Vδ2 T
cell activation and CML cell elimination, consistent with previous
findings (39). Taken together,
these results indicate that HCQ sensitizes CML cells to Vγ9Vδ2 T
cell-mediated cytotoxicity by inducing ULBP4 expression on the CML
cell membrane.
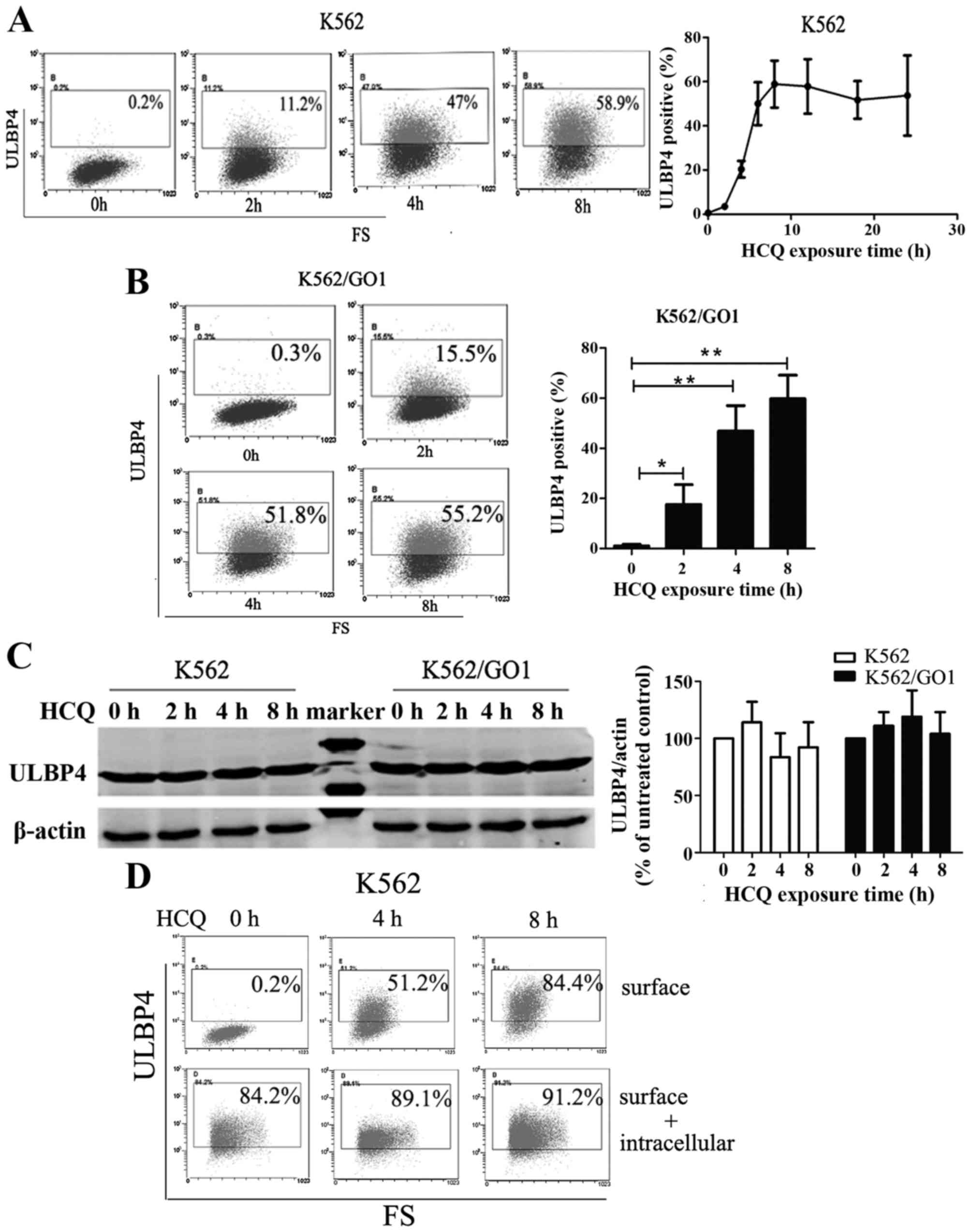
HCQ induces ULBP4 translocation from the
cytoplasm to the cell membrane
To explore the mechanisms behind HCQ-induced
expression of ULBP4 on the CML cell membrane, we monitored ULBP4
expression over time after HCQ treatment. HCQ induced the
expression of ULBP4 in a time-dependent manner (Fig. 5A and B). ULBP4 was not expressed on
K562 and K562/GO1 cells before HCQ treatment but the percentage of
ULBP4-positive cells began to increase after HCQ exposure for 2 h.
After 8 h of HCQ exposure, the percentage of ULBP4-positive cells
rose to 52.6±9.7% in K562 cells and 61.4±8.3% in K562/GO1 cells and
remained relatively stable for 24 h (Fig. 5A and B). Next, we tested the
regulation of ULBP4 synthesis or degradation by HCQ. Western
blotting showed that HCQ does not affect the total amount of ULBP4
protein in CML cells (Fig. 5C). To
verify this result, we examined ULBP4 expression by flow cytometry
after cells were fixed and permeabilized. HCQ exposure increased
the expression of ULBP4 on the cell surface but did not affect the
total expression of ULBP4 on the cell surface plus that within the
cell (Fig. 5D). Furthermore, we
found that ULBP4 was not expressed on the cell membrane of
HCQ-untreated K562 and K562/GO1 cells, but it accumulated in the
cytoplasm (Fig. 5E). These results
implied that HCQ may induce ULBP4 translocation from the cytoplasm
to the membrane. To confirm this hypothesis, we evaluated the
expression of ULBP4 in K562 cells by confocal microscopy. HCQ
treatment redistributed ULBP4 from the cytoplasm to the cell
membrane. Without HCQ treatment, ULBP4 was distributed throughout
the cytoplasm and HCQ treatment relocalized ULBP4 to the membrane
(Fig. 5F). Taken together, these
findings show that HCQ does not affect ULBP4 synthesis and
degradation, but relocates ULBP4 from the cytoplasm to the
membrane.
Discussion
HCQ is known to be an effective autophagy inhibitor.
However, HCQ has multiple functions (28,40).
HCQ suppresses antigen processing and presentation, inhibits
prostaglandin and cytokine synthesis, modulates toll-like
receptors, and affects matrix metalloproteinase levels in serum
(40–42). In the present study, we revealed
that HCQ enhances the susceptibility of CML cells to Vγ9Vδ2 T
cell-mediated cytotoxicity. HCQ sensitized imatinib-resistant and
imatinib-sensitive CML cells to Vγ9Vδ2 T cell-mediated lysis, and
HCQ pretreated CML cells accelerated the degranulation of Vγ9Vδ2 T
cells. These results indicate that HCQ combination with
immunotherapy may be an effective and safe choice for CML patients,
regardless of responsiveness to imatinib.
Autophagy in carcinoma is involved in immunotherapy
resistance. Hypoxia-induced autophagy compromised the
susceptibility of cancer cells to CTL cytotoxicity (19,43).
In breast cancer cells and renal carcinoma cells, hypoxia-induced
autophagy selectively degraded NK cell-derived granzyme B and
impaired the susceptibility of carcinomas to NK-mediated lysis
(18,44). Inhibiting autophagy can enhance the
sensitivity of carcinoma cells to NK- and CTL-mediated lysis under
hypoxic conditions. We have shown that HCQ can inhibit autophagy in
CML cells. However, HCQ sensitizes CML cells to Vγ9Vδ2 T
cell-mediated lysis in an autophagy-independent manner. Knockdown
of ATG7 effectively blocked autophagy, but did not enhance the
sensitivity of CML cells to Vγ9Vδ2 T cells. Furthermore, HCQ
increased the sensitivity of autophagy-incompetent CML cell to
Vγ9Vδ2 T cell-mediated cytotoxicity. These findings indicated that
autophagy is not involved in resistance to Vγ9Vδ2 T cell-mediated
lysis in CML cells. We speculate that the level of basal autophagy
in CML cells is too low to have an effect. Consistent with this
hypothesis, previous studies have demonstrated that BCR-ABL
protein, which is a hallmark of CML, can downregulate autophagy in
PI3K/AKT/mTORC1-dependent or -independent manner (45,46),
and BCR-ABL-expressing cells exhibit low basal autophagy (47).
In addition to TCR-dependent activation, Vγ9Vδ2 T
cell activity is tightly regulated by the NK-like active receptor
NKG2D (20,48,49).
The NKG2D ligands MICA/B and ULBP1-6 are specifically expressed on
microorganism-infected cells and various tumor cells. They mark
these cells as targets for Vγ9Vδ2 T and other immune cells
(50–52). Previous studies have shown that
ULBP4, one of NKG2D ligands, plays a key role in γδT cell
activation (39). Consistent with
this, we have demonstrated that specific lysis of leukemia cells by
Vγ9Vδ2 T cells correlates positively with ULBP4 expression on the
leukemia cell surface (unpublished data). We tested the expression
of NKG2D ligands in CML cells after HCQ treatment. Expression of
NKG2D ligands was absent (ULBP4, ULBP1 and ULBP3) or only weakly
detected (MICA/B and ULBP2/5/6) in CML cells. HCQ treatment
specifically induced the expression of ULBU4 on the membrane of CML
cells. Blocking the interaction of NKG2D with its ligands
completely deleted the sensitizing effects of HCQ. This result
suggests that HCQ sensitizes CML cells to Vγ9Vδ2 T cytotoxicity by
inducing ULBP4 expression on the cell membrane. Vγ9Vδ2 T cells
recognize tumors via three mechanisms: preferential involvement of
the TCR, preferential involvement of NKG2D, or a combination of
both (24,50). We showed that blocking interaction
of NKG2D with its ligands decreased Vγ9Vδ2 T cell-mediated lysis to
HCQ-pretreated CML cells expressing ULBP4 on the membrane, but not
in control cells that did not show ULBP4 expression at the membrane
(Fig. 4B). This indicates that
ULBP4 is major NKG2D ligand in CML cells recognized by NKG2D during
Vγ9Vδ2 T cell activation, and Vγ9Vδ2 T cells recognize CML cells
pretreated with or without HCQ in different ways. Vγ9Vδ2 T cells
may recognize PBS-treated CML cells through preferential
involvement of the TCR and recognize HCQ-treated CML cells through
a combination of NKG2D and TCR.
NKG2D ligands mark cancer cells for recognition by
immune cells, but tumor cells can detain NKG2D ligands
intracellularly to evade immune surveillance (53–55).
Human melanomas prevent NK cell-mediated cytotoxity through
sequestration of MICA in the endoplasmic reticulum (53). ULBP4 was not detected at the
membrane of HCQ-untreated CML cells, but it accumulated in the
cytoplasm. This may indicate an immune escape mechanism of CML
cells to avoid elimination by Vγ9Vδ2 T and NK cells. Fortunately,
HCQ can overcome this obstacle and induces ULBP4 expression at the
cell surface. Chemotherapy strategies using HCQ promote
NKG2D-dependent elimination of CML cells by Vγ9Vδ2 T cells.
Recently published studies have emphasized the importance of
post-translational regulation in NKG2D ligand synthesis (54–56).
The present study reveals that HCQ does not affect the synthesis or
degradation of ULBP4 but promotes ULBP4 translocation from the
cytoplasm to the cell membrane. However, the mechanisms behind
ULBP4 translocation remain elusive. Some studies have demonstrated
that N-linked glycosylation regulated the translocation of MICA/B
from the cytoplasm to the membrane (57,58).
However, we have shown that tunicamycin, a selective inhibitor of
N-linked glycosylation, did not abolish HCQ-induced ULBP4
expression (data not shown). This suggests that N-linked
glycosylation may not be involved in regulating ULBP4
translocation. Transportation of MICB and ULBP2 from the cytoplasm
to the membrane is regulated through endosomal and lysosomal
pathways (59,60). Whether these pathways are involved
in ULBP4 transportation remains to be elucidated.
Our results uncovered a novel autophagy-independent
mechanism of HCQ in CML treatment, and imply that combination
treatments with HCQ and Vγ9Vδ2 T cells represent a potential
strategy for CML, especially for the relapse and TKI-resistant
patients.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81470307, 81570146,
81500114, 81370644 and 81670148).
References
|
1
|
Daley GQ, Van Etten RA and Baltimore D:
Induction of chronic myelogenous leukemia in mice by the
P210bcr/abl gene of the Philadelphia chromosome. Science.
247:824–830. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Helgason GV, Karvela M and Holyoake TL:
Kill one bird with two stones: Potential efficacy of BCR-ABL and
autophagy inhibition in CML. Blood. 118:2035–2043. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Branford S, Rudzki Z, Walsh S, Parkinson
I, Grigg A, Szer J, Taylor K, Herrmann R, Seymour JF, Arthur C, et
al: Detection of BCR-ABL mutations in patients with CML treated
with imatinib is virtually always accompanied by clinical
resistance, and mutations in the ATP phosphate-binding loop
(P-loop) are associated with a poor prognosis. Blood. 102:276–283.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sawyers CL, Hochhaus A, Feldman E, Goldman
JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F,
Deininger MW, et al: Imatinib induces hematologic and cytogenetic
responses in patients with chronic myelogenous leukemia in myeloid
blast crisis: Results of a phase II study. Blood. 99:3530–3539.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zeng X, Zhao H, Li Y, Fan J, Sun Y, Wang
S, Wang Z, Song P and Ju D: Targeting Hedgehog signaling pathway
and autophagy overcomes drug resistance of BCR-ABL-positive chronic
myeloid leukemia. Autophagy. 11:355–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carew JS, Nawrocki ST, Kahue CN, Zhang H,
Yang C, Chung L, Houghton JA, Huang P, Giles FJ and Cleveland JL:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tong Y, Liu YY, You LS and Qian WB:
Perifosine induces protective autophagy and upregulation of ATG5 in
human chronic myelogenous leukemia cells in vitro. Acta Pharmacol
Sin. 33:542–550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song P, Ye L, Fan J, Li Y, Zeng X, Wang Z,
Wang S, Zhang G, Yang P, Cao Z, et al: Asparaginase induces
apoptosis and cytoprotective autophagy in chronic myeloid leukemia
cells. Oncotarget. 6:3861–3873. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang S, Fan J, Wang Q, Ju D, Feng M, Li
J, Guan ZB, An D, Wang X and Ye L: Diosgenin induces ROS-dependent
autophagy and cytotoxicity via mTOR signaling pathway in chronic
myeloid leukemia cells. Phytomedicine. 23:243–252. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen JJ, Long ZJ, Xu DF, Xiao RZ, Liu LL,
Xu ZF, Qiu SX, Lin DJ and Liu Q: Inhibition of autophagy augments
the anticancer activity of α-mangostin in chronic myeloid leukemia
cells. Leuk Lymphoma. 55:628–638. 2014. View Article : Google Scholar
|
|
11
|
Kamitsuji Y, Kuroda J, Kimura S, Toyokuni
S, Watanabe K, Ashihara E, Tanaka H, Yui Y, Watanabe M, Matsubara
H, et al: The Bcr-Abl kinase inhibitor INNO-406 induces autophagy
and different modes of cell death execution in Bcr-Abl-positive
leukemias. Cell Death Differ. 15:1712–1722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yogalingam G and Pendergast AM: Abl
kinases regulate autophagy by promoting the trafficking and
function of lysosomal components. J Biol Chem. 283:35941–35953.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calabretta B and Salomoni P: Inhibition of
autophagy: A new strategy to enhance sensitivity of chronic myeloid
leukemia stem cells to tyrosine kinase inhibitors. Leuk Lymphoma.
52(Suppl 1): 54–59. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bellodi C, Lidonnici MR, Hamilton A,
Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi
T, Yacobi R, et al: Targeting autophagy potentiates tyrosine kinase
inhibitor-induced cell death in Philadelphia chromosome-positive
cells, including primary CML stem cells. J Clin Invest.
119:1109–1123. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Townsend KN, Hughson LR, Schlie K, Poon
VI, Westerback A and Lum JJ: Autophagy inhibition in cancer
therapy: Metabolic considerations for antitumor immunity. Immunol
Rev. 249:176–194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang X, De Vera ME, Buchser WJ, Romo de
Vivar Chavez A, Loughran P, Beer Stolz D, Basse P, Wang T, Van
Houten B, Zeh HJ III, et al: Inhibiting systemic autophagy during
interleukin 2 immunotherapy promotes long-term tumor regression.
Cancer Res. 72:2791–2801. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu S, Cao L, Yu Y, Yang L, Yang M, Liu K,
Huang J, Kang R, Livesey KM and Tang D: Inhibiting autophagy
potentiates the anticancer activity of IFN1α/IFNα in chronic
myeloid leukemia cells. Autophagy. 9:317–327. 2013. View Article : Google Scholar :
|
|
18
|
Baginska J, Viry E, Berchem G, Poli A,
Noman MZ, van Moer K, Medves S, Zimmer J, Oudin A, Niclou SP, et
al: Granzyme B degradation by autophagy decreases tumor cell
susceptibility to natural killer-mediated lysis under hypoxia. Proc
Natl Acad Sci USA. 110:17450–17455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Noman MZ, Janji B, Kaminska B, Van Moer K,
Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib
F, et al: Blocking hypoxia-induced autophagy in tumors restores
cytotoxic T-cell activity and promotes regression. Cancer Res.
71:5976–5986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bonneville M, O'Brien RL and Born WK:
Gammadelta T cell effector functions: A blend of innate programming
and acquired plasticity. Nat Rev Immunol. 10:467–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gertner-Dardenne J, Castellano R,
Mamessier E, Garbit S, Kochbati E, Etienne A, Charbonnier A,
Collette Y, Vey N and Olive D: Human Vγ9Vδ2 T cells specifically
recognize and kill acute myeloid leukemic blasts. J Immunol.
188:4701–4708. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meeh PF, King M, O'Brien RL, Muga S,
Buckhalts P, Neuberg R and Lamb LS Jr: Characterization of the
gammadelta T cell response to acute leukemia. Cancer Immunol
Immunother. 55:1072–1080. 2006. View Article : Google Scholar
|
|
23
|
Siegers GM, Felizardo TC, Mathieson AM,
Kosaka Y, Wang XH, Medin JA and Keating A: Anti-leukemia activity
of in vitro-expanded human gamma delta T cells in a xenogeneic Ph
leukemia model. PLoS One. 6:e167002011. View Article : Google Scholar
|
|
24
|
D'Asaro M, La Mendola C, Di Liberto D,
Orlando V, Todaro M, Spina M, Guggino G, Meraviglia S, Caccamo N,
Messina A, et al: Vγ9Vδ2 T lymphocytes efficiently recognize and
kill zoledronate-sensitized, imatinib-sensitive, and
imatinib-resistant chronic myelogenous leukemia cells. J Immunol.
184:3260–3268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu KN, Wang YJ, He Y, Hu YX, Fu HR, Sheng
LX, Wang BS, Fu S and Huang H: Dasatinib promotes the potential of
proliferation and antitumor responses of human γδT cells in a
long-term induction ex vivo environment. Leukemia. 28:206–210.
2014. View Article : Google Scholar
|
|
26
|
Mishima Y, Terui Y, Mishima Y, Taniyama A,
Kuniyoshi R, Takizawa T, Kimura S, Ozawa K and Hatake K: Autophagy
and autophagic cell death are next targets for elimination of the
resistance to tyrosine kinase inhibitors. Cancer Sci. 99:2200–2208.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maes H, Kuchnio A, Peric A, Moens S, Nys
K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, et
al: Tumor vessel normalization by chloroquine independent of
autophagy. Cancer Cell. 26:190–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maycotte P, Aryal S, Cummings CT, Thorburn
J, Morgan MJ and Thorburn A: Chloroquine sensitizes breast cancer
cells to chemotherapy independent of autophagy. Autophagy.
8:200–212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chalupny NJ, Sutherland CL, Lawrence WA,
Rein-Weston A and Cosman D: ULBP4 is a novel ligand for human
NKG2D. Biochem Biophys Res Commun. 305:129–135. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bacon L, Eagle RA, Meyer M, Easom N, Young
NT and Trowsdale J: Two human ULBP/RAET1 molecules with
transmembrane regions are ligands for NKG2D. J Immunol.
173:1078–1084. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qi J, Peng H, Gu ZL, Liang ZQ and Yang CZ:
Establishment of an imatinib resistant cell line K562/G01 and its
characterization. Zhonghua Xue Ye Xue Za Zhi. 25:337–341. 2004.In
Chinese. PubMed/NCBI
|
|
32
|
Godoy-Ramirez K, Mäkitalo B, Thorstensson
R, Sandström E, Biberfeld G and Gaines H: A novel assay for
assessment of HIV-specific cytotoxicity by multiparameter flow
cytometry. Cytometry A. 68:71–80. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gertner J, Wiedemann A, Poupot M and
Fournié JJ: Human gammadelta T lymphocytes strip and kill tumor
cells simultaneously. Immunol Lett. 110:42–53. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Klionsky DJ1, Baehrecke EH, Brumell JH,
Chu CT, Codogno P, Cuervo AM, Debnath J, Deretic V, Elazar Z,
Eskelinen EL, et al: Comprehensive glossary of autophagy-related
molecules and processes (2nd edition). Autophagy. 7:1273–1294.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rincon-Orozco B, Kunzmann V, Wrobel P,
Kabelitz D, Steinle A and Herrmann T: Activation of Vγ9Vδ2T cells
by NKG2D. J Immunol. 175:2144–2151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vantourout P and Hayday A:
Six-of-the-best: Unique contributions of γδT cells to immunology.
Nat Rev Immunol. 13:88–100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kong Y, Cao W, Xi X, Ma C, Cui L and He W:
The NKG2D ligand ULBP4 binds to TCRgamma9/δ2 and induces
cytotoxicity to tumor cells through both TCRgammadelta and NKG2D.
Blood. 114:310–317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Solomon VR and Lee H: Chloroquine and its
analogs: A new promise of an old drug for effective and safe cancer
therapies. Eur J Pharmacol. 625:220–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lesiak A, Narbutt J, Sysa-Jedrzejowska A,
Lukamowicz J, McCauliffe DP and Wózniacka A: Effect of chloroquine
phosphate treatment on serum MMP-9 and TIMP-1 levels in patients
with systemic lupus erythematosus. Lupus. 19:683–688. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Savarino A, Di Trani L, Donatelli I, Cauda
R and Cassone A: New insights into the antiviral effects of
chloroquine. Lancet Infect Dis. 6:67–69. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Akalay I, Janji B, Hasmim M, Noman MZ,
André F, De Cremoux P, Bertheau P, Badoual C, Vielh P, Larsen AK,
et al: Epithelial-to-mesenchymal transition and autophagy induction
in breast carcinoma promote escape from T-cell-mediated lysis.
Cancer Res. 73:2418–2427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Messai Y, Noman MZ, Janji B, Hasmim M,
Escudier B and Chouaib S: The autophagy sensor ITPR1 protects renal
carcinoma cells from NK-mediated killing. Autophagy. Feb
25–2015.Epub ahead of print. Update please. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sheng Z, Ma L, Sun JE, Zhu LJ and Green
MR: BCR-ABL suppresses autophagy through ATF5-mediated regulation
of mTOR transcription. Blood. 118:2840–2848. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Calabretta B and Salomoni P: Suppression
of autophagy by BCR/ABL. Front Biosci (Schol Ed). 4:453–460. 2012.
View Article : Google Scholar
|
|
47
|
Altman BJ, Jacobs SR, Mason EF, Michalek
RD, MacIntyre AN, Coloff JL, Ilkayeva O, Jia W, He YW and Rathmell
JC: Autophagy is essential to suppress cell stress and to allow
BCR-Abl-mediated leukemogenesis. Oncogene. 30:1855–1867. 2011.
View Article : Google Scholar :
|
|
48
|
Bauer S, Groh V, Wu J, Steinle A, Phillips
JH, Lanier LL and Spies T: Activation of NK cells and T cells by
NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Raulet DH and Guerra N: Oncogenic stress
sensed by the immune system: Role of natural killer cell receptors.
Nat Rev Immunol. 9:568–580. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wrobel P, Shojaei H, Schittek B, Gieseler
F, Wollenberg B, Kalthoff H, Kabelitz D and Wesch D: Lysis of a
broad range of epithelial tumour cells by human γδ T cells:
Involvement of NKG2D ligands and T-cell receptor-versus
NKG2D-dependent recognition. Scand J Immunol. 66:320–328. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gomes AQ, Correia DV and Silva-Santos B:
Non-classical major histocompatibility complex proteins as
determinants of tumour immunosurveillance. EMBO Rep. 8:1024–1030.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Das H, Groh V, Kuijl C, Sugita M, Morita
CT, Spies T and Bukowski JF: MICA engagement by human
Vgamma2Vdelta2 T cells enhances their antigen-dependent effector
function. Immunity. 15:83–93. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fuertes MB, Girart MV, Molinero LL,
Domaica CI, Rossi LE, Barrio MM, Mordoh J, Rabinovich GA and
Zwirner NW: Intracellular retention of the NKG2D ligand MHC class I
chain-related gene A in human melanomas confers immune privilege
and prevents NK cell-mediated cytotoxicity. J Immunol.
180:4606–4614. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nice TJ, Deng W, Coscoy L and Raulet DH:
Stress-regulated targeting of the NKG2D ligand Mult1 by a
membrane-associated RING-CH family E3 ligase. J Immunol.
185:5369–5376. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja
H, Sampaio KL, Eknigk U, Rammensee HG and Steinle A: Selective
intracellular retention of virally induced NKG2D ligands by the
human cytomegalovirus UL16 glycoprotein. Eur J Immunol. 33:194–203.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Raulet DH, Gasser S, Gowen BG, Deng W and
Jung H: Regulation of ligands for the NKG2D activating receptor.
Annu Rev Immunol. 31:413–441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Andresen L, Skovbakke SL, Persson G,
Hagemann-Jensen M, Hansen KA, Jensen H and Skov S: 2-deoxy
D-glucose prevents cell surface expression of NKG2D ligands through
inhibition of N-linked glycosylation. J Immunol. 188:1847–1855.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mellergaard M, Skovbakke SL, Schneider CL,
Lauridsen F, Andresen L, Jensen H and Skov S: N-glycosylation of
aspara-gine 8 regulates surface expression of major
histocompatibility complex class I chain-related protein A (MICA)
alleles dependent on threonine 24. J Biol Chem. 289:20078–20091.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Agüera-González S, Boutet P, Reyburn HT
and Valés-Gómez M: Brief residence at the plasma membrane of the
MHC class I-related chain B is due to clathrin-mediated
cholesterol-dependent endocytosis and shedding. J Immunol.
182:4800–4808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Uhlenbrock F, Hagemann-Jensen M, Kehlet S,
Andresen L, Pastorekova S and Skov S: The NKG2D ligand ULBP2 is
specifi-cally regulated through an invariant chain-dependent
endosomal pathway. J Immunol. 193:1654–1665. 2014. View Article : Google Scholar : PubMed/NCBI
|