Introduction
Malignant melanoma is the most dangerous type of
skin cancer, accounting for more than 75% of deaths related to skin
cancer. Patients with advanced melanoma with dissemination to
distant sites and visceral organs have a very poor prognosis, with
a median survival time of 6 months and a 5-year survival rate of
less than 5% (1). Currently, very
limited treatment options are available for metastatic melanoma.
Although multiple clinical trials have been initiated for novel
agents to treat melanoma, these have met with limited success, thus
highlighting the need for the development of new drugs and
identification of novel therapeutic targets for effective cancer
therapy.
The endoplasmic reticulum (ER) in eukaryotic cells
is required for several critical functions, including lipid and
cholesterol biosynthesis, maintenance of calcium homeostasis, and
transport of nascent proteins to subcellular organelles (2). Cellular stress conditions, such as
aberrant calcium signaling and accumulation of misfolded proteins
could lead to ER stress and activation of a counteractive reaction
called the ER stress response or unfolded protein response, which
serves to restore ER functionality and homeostasis (3,4).
However, prolonged induction of acute ER stress can lead to cell
death (5–9). Thus, while moderate ER stress
triggers cell survival signaling, severe stress may potentiate cell
death.
ER stress is regulated by two critical proteins,
glucose-regulated protein 78 (GRP78) and CCAAT/enhancer binding
protein homologous protein (CHOP) (10,11).
GRP78 is a key member of the HSP70 protein family that functions as
an ER chaperone involved in protein folding and assembly and
ER-mediated stress signaling (12). Overexpression of GRP78 has been
observed to cause aggressive tumor behavior and promote
angiogenesis in various solid tumors (13–17).
In melanoma, enhanced activation of GRP78 has been associated with
poor patient survival and increased disease progression through
downregulation of apoptosis and activation of unfolded protein
response mechanisms (18). De
Ridder et al found a link between humoral response to GRP78
and cancer progression in a murine model of melanoma (19). Studies have also demonstrated a
distinct role of GRP78 in drug resistance; GRP78 induced
doxorubicin resistance in dormant squamous carcinoma cells through
inhibition of BAX activation (20). Of note, GRP78 is expressed only on
the surface of cancer cells and not on the surface of normal cells,
making it an important target for therapeutic intervention
(17).
In contrast, prolonged expression of CHOP results in
cytotoxicity (21). Incremental
CHOP levels have been associated with increased apoptosis and
reduced tumor growth (22,23). Furthermore, numerous studies
indicate that knockdown of CHOP leads to significantly decreased
drug effects in cancer cells, confirming that CHOP plays a critical
role in mediating ER stress-induced cytotoxicity (24–26).
Thus, ER stress can be described as a double-edged sword: moderate
or chronic levels of ER stress can activate pro-survival cellular
signaling pathways through GRP78, whereas severe or acute levels of
ER stress can lead to cell death via activation of CHOP.
Autophagy is a self-digestive process that
facilitates lysosomal degradation of cytoplasmic proteins and
organelles as a means of maintaining cellular homeostasis and
adapting to different forms of stress (27,28).
Autophagy is primarily a mechanism of cell survival; however,
prolonged exposure of cells to deprivation conditions such as DNA
damage, oxidative stress, and starvation can lead to induction of
excessive autophagy, causing depletion of cellular organelles and
self-destruction (29,30). Thus, similarly to ER stress,
autophagy also plays a dual role in cancer. For instance, tumors
with activating mutations in Ras have been shown to employ
autophagy for survival (31).
Noteworthy, although nuclear p53 transactivates autophagy inducers
such as DRAM1 and sestrin2, cytoplasmic p53 inhibits autophagy
(32,33). Gene knockout of the autophagy
regulatory protein, Beclin-1, was found to increase tumor incidence
in mice with lymphoma and lung cancer (34,35).
Similarly, death-associated protein kinase (DAPK-1), which has
cancer metastasis suppressive properties, is activated following an
accumulation of unfolded proteins in cells, leading to ER stress
and initiation of autophagy through phosphorylation of Beclin-1
(36–38). Unfolded protein response, which is
triggered as an ER stress response, potentially induces autophagy;
binding of GRP78 to misfolded proteins leads to the release of the
3 ER membrane-associated proteins, PKR-like eIF2α kinase (PERK),
activation transcription factor-6 (ATF-6), and inositol-requiring
enzyme-1 (IRE-1) (39,40). Of note, although both PERK and
ATF-6 promote autophagy, IRE-1 attenuates the autophagic response
in cells. Furthermore, multiple recent studies have indicated that
ER stress can magnify autophagy and vice versa (41–44).
Hence, both ER stress and autophagy constitute valid therapeutic
targets, and inhibition of either or both of these processes could
lead to improved therapeutic outcomes.
25-epi Ritterostatin
GN1N, an analogue of cephalostatin 1
(Fig. 1), is a potent anticancer
agent with 50% inhibitory concentrations in the subnanomolar range
(45). Testing of this compound in
the NCI-60 cell line panel indicated that the compound is highly
effective against leukemia, melanoma lung, breast, renal, colon,
and prostate cancer cells (46,47).
Recent work by Kanduluru et al outlined the synthesis of
25-epi Ritterostatin GN1N (45). However, very little is known about
the mechanism of action of this novel inhibitor in cancer cells. In
this study, we aimed to delineate the mechanism of antitumor
activity of 25-epi Ritterostatin GN1N
in melanoma cells. We found that 25-epi Ritterostatin
GN1N triggered ER stress and autophagic cell
death in melanoma cells and inhibited tumor growth in mouse
xenografts. Importantly, 25-epi Ritterostatin
GN1N was therapeutically selective toward
melanoma cells, whereas, normal melanocytes were resistant to it.
These findings indicate that further study is warranted of
25-epi Ritterostatin GN1N as a
potential therapeutic agent for melanoma.
Materials and methods
Cell lines and reagents
A375 melanoma cells were purchased from American
Type Culture Collection (Manassas, VA, USA). WM35 cells were
purchased from the characterized cell line core at The University
of Texas MD Anderson Cancer Center (Houston, TX, USA). WM35 PKB
cells, which stably overexpress Akt, were a kind gift from Dr Jack
Arbiser (Emory University School of Medicine, Atlanta, GA, USA).
Normal human epidermal melanocytes were purchased from PromoCell
(Heidelberg, Germany). A375 and WM35/WM35 PKB cells maintained in
Dulbecco's modified Eagle's medium and RPMI-1640 medium
supplemented with 10% heat-inactivated fetal bovine serum were
grown in a cell culture incubator at 37°C with 5% CO2 in
humidified air. Normal melanocytes were grown in M254CF medium
supplemented with human melanocyte growth supplement-2 (Life
Technologies, Grand Island, NY, USA) in a cell culture incubator at
37°C with 5% CO2. The compound 25-epi
Ritterostatin GN1N was synthesized and
provided by the research group of Dr Philip L. Fuchs, Purdue
University (West Lafayette, IN, USA). 25-epi Ritterostatin
GN1N was first dissolved in dimethyl
sulfoxide and then further diluted in media to a final
concentration of 0.1%. Rhodamine 123, a fluorescent dye for the
detection of mitochondrial membrane potential, was purchased from
Life Technologies.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT)
and propidium iodide were purchased from Sigma-Aldrich (St. Louis,
MO, USA), and an Annexin V-FITC kit was purchased from BD
Pharmingen (San Jose, CA, USA). Antibodies were obtained from the
following commercial sources: β-actin from Calbiochem (San Diego,
CA, USA), GRP78 (BiP) and p62 from Santa Cruz Biotechnology (Santa
Cruz, CA, USA), LC3B from Novus Biologicals (Littleton, CO, USA),
and CHOP from Thermo Scientific (Rockford, IL, USA).
Cytotoxicity assays
The antiproliferative effect of 25-epi
Ritterostatin GN1N was determined by
performing the MTT assay. A375 and WM35 cells were plated in
triplicate in a 96-well plate (2,000 cells per well). After
overnight incubation, the cells were treated with log-scale serial
diluted concentrations of 25-epi Ritterostatin
GN1N and incubated at 37°C for 72 h, followed
by treatment with 50 µl of MTT reagent for 4 h. The cell
media was aspirated and the formazan precipitates were dissolved in
dimethyl sulfoxide. Absorbance at 570 nm was measured in a
Multiskan MK3 microplate-reader (Thermo Labsystem, Franklin, MA,
USA). The 50% inhibitory concentration values were then computed
using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA,
USA).
Cell apoptosis and necrosis were measured
using flow cytometry
Briefly, cells were harvested, washed in ice-cold
phosphate-buffered saline (PBS), and suspended in Annexin V binding
buffer. Cells were then stained with Annexin V-FITC for 15 min,
washed, and stained with propidium iodide. Cell death was
quantified using a BD Biosciences FACSCalibur flow cytometer
(Mountain View, CA, USA) and the results were analyzed using FlowJo
(TreeStar, Inc., Ashland, OR, USA).
Colony formation assay
To assess the effect of 25-epi Ritterostatin
GN1N on the colony formation capacity of
cells, we treated A375 and WM35 cells (5,000 cells per well) with
serial diluted concentrations of 25-epi Ritterostatin
GN1N for 2 weeks. At the end of this period,
the cells were fixed in methanol/acetic acid (10:1) fixation
solution and stained with Giemsa (Sigma-Aldrich). The stained cells
were photographed and counted.
Mitochondrial membrane potential
assay
Melanoma and normal melanocyte cells were stained
with 0.5 µM Rhodamine 123 during the last hour of treatment
with 25-epi Ritterostatin GN1N. The
cells were then harvested, washed, and suspended in PBS. The
mitochondrial membrane potential of the cells was measured using a
FACSCalibur flow cytometer and subsequent data analysis was
performed using FlowJo software.
Immunoblotting
Cellular protein extracts were separated using
standard SDS-PAGE and transferred onto a nitro cellulose membrane.
The membrane was probed for the indicated primary antibodies,
followed by the appropriate horseradish peroxidase-conjugated
secondary antibodies. The signal was developed using a
supersignal-enhanced chemiluminescence kit (Thermo Scientific).
In vivo antitumor activity assay
All animal experiments were performed according to
Institutional Animal Care and Use protocol at MD Anderson Cancer
Center. Ten athymic nude mice were each subcutaneously inoculated
with 2×106 WM35 PKB cells. The mice were divided into 2
groups (5 mice each); mice in the treatment group were injected
intraperitoneally with 2 mg/ kg 25-epi Ritterostatin
GN1N, and mice in the control group were
injected with an equal volume of PBS. The mice were monitored
routinely for tumor growth and body weight. Moribund animals were
sacrificed according to Institutional Animal Care and Use protocol,
and the time of death was recorded.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism version 6 (GraphPad Software, Inc.). Data are
represented as means ± standard error of the mean. 95% confidence
intervals were used to determine statistical significance.
Results
25-epi Ritterostatin
GN1N inhibits growth and viability of
melanoma cells at nanomolar concentrations
The effect of 25-epi Ritterostatin
GN1N on the viability of A375 and other
melanoma cells was determined by performing the MTT assay. Exposure
to log-scale serial diluted concentrations (0–100 nM) of
25-epi Ritterostatin GN1N for 72 h
resulted in concentration- and time-dependent cell death in
melanoma cells, with an average 50% inhibitory concentration of
90.2 nM across four melanoma cell lines (Fig 2A and B).
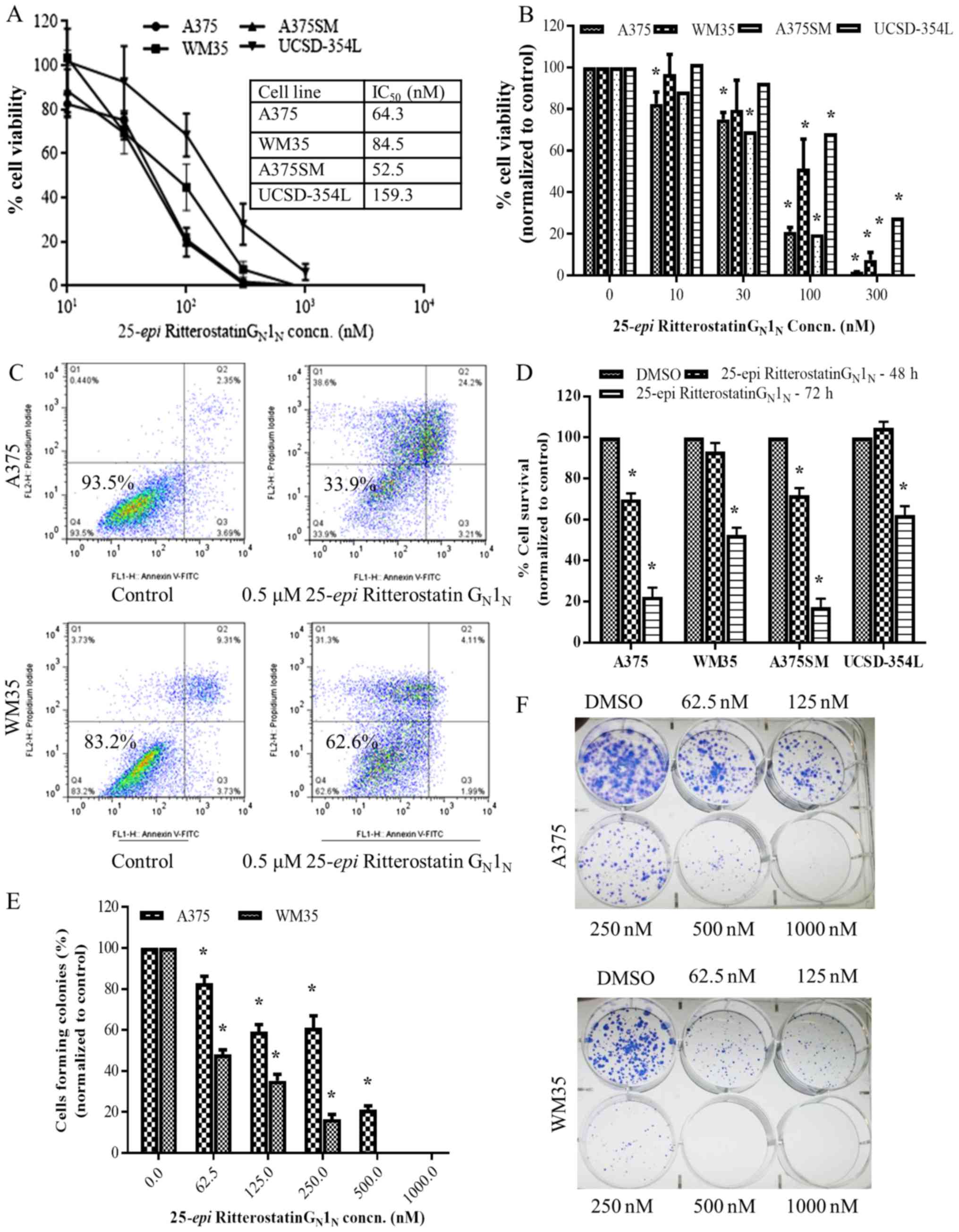 | Figure 225-epi Ritterostatin
GN1N is cytotoxic at nanomolar concentrations
in melanoma cells. (A and B) MTT assay results showing A375, WM35,
A375SM, and UCSD-354L melanoma cell viability after treatment with
various concentrations of 25-epi Ritterostatin
GN1N for 72 h. IC50, 50%
inhibitory concentration. (C) Percentage of viable WM35 and A375
cells after treatment with 0.5 µM 25-epi
Ritterostatin GN1N for 72 h, determined
according to Annexin V-FITC/propidium iodide staining and
subsequent flow cytometry analysis. (D) The bar graphs represent
the percentage of viable A375, WM35, A375SM, and UCSD-354L cells
(mean ± standard error) after treatment with 0.5 µM
25-epi Ritterostatin GN1N for 48 and
72 h, for 3 independent measurements, according to Annexin
V-FITC/propidium iodide staining analysis. (E and F) Giemsa
staining for colony formation assessment in A375 and WM35 cells
treated with varying concentrations of 25-epi Ritterostatin
GN1N [or dimethyl sulfoxide (DMSO) only] for
14 days. The bar graphs represent the mean (± standard error of the
mean) of three independent measurements. *p<0.05. |
To measure melanoma cell death after treatment with
25-epi Ritterostatin GN1N, we
performed Annexin V/propidium iodide staining in WM35, A375,
UCSD354L, and A375SM cells treated with 0.5 µM 25-epi
Ritterostatin GN1N (Fig. 2C and D). Results of a follow-up
colony-formation assay showed that 25-epi Ritterostatin
GN1N suppressed colony formation at
concentrations as low as 62.5 nM in melanoma cells (Fig. 2E and F). A375 cells, which were
relatively more sensitive than WM35 cells to 25-epi
Ritterostatin GN1N according to the MTT cell
viability assay, were comparatively resistant to 25-epi
Ritterostatin GN1N in the colony-formation
assay. A concentration dependent decrease in colony formation was
observed in melanoma cells at higher concentrations of
25-epi Ritterostatin GN1N. These
results demonstrate that 25-epi Ritterostatin
GN1N is cytotoxic and inhibits tumor growth
in melanoma cells at nanomolar concentrations.
Cell death caused by 25-epi Ritterostatin
GN1N is mediated through autophagy
Further investigation of the mechanisms of action of
25-epi Ritterostatin GN1N revealed
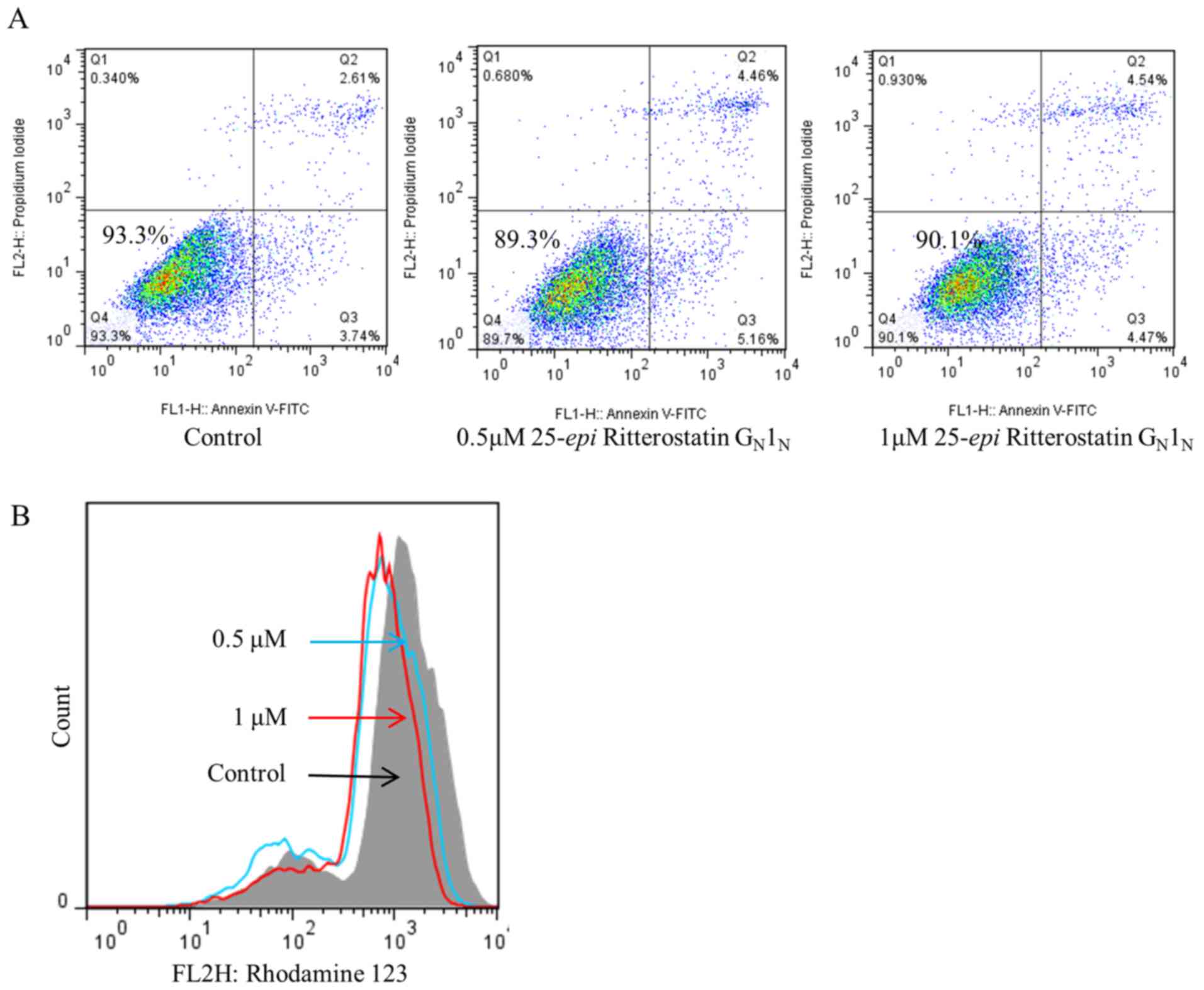
that cell death occurred as a result of autophagy. A massive
collapse of the mitochondrial membrane potential was observed in
melanoma cells treated with 0.5 and 1 µM 25-epi
Ritterostatin GN1N compared with untreated
cells (Fig. 3A and B). This
decrease in the mitochondrial membrane potential was
time-dependent.
To confirm the role of autophagy in mediating
cytotoxicity, we treated A375 cells with 25-epi
Ritterostatin GN1N in combination with
chloroquine. Chloroquine is a lysosomotrophic agent known to
promote rapid cell death in autophagic cells by inhibiting lysosome
acidification and degradation of autophagosomes, thus sensitizing
the cells to drug action (48,49).
For this experiment, 1 µM 25-epi Ritterostatin
GN1N was added to A375 cells pretreated with
10 µM chloroquine for 1 h. A significant increase in cell
death was observed in cells pretreated with chloroquine compared
with cells treated with 25-epi Ritterostatin
GN1N alone (Fig.
3C and D). Furthermore, cell death was rapid in the cells
pretreated with chloroquine; only 29.3% of cells were viable at 24
h after treatment (Fig. 3C).
However, the viability of A375 cells treated with 25-epi
Ritterostatin GN1N or chloroquine alone was
very similar to that of the control cells. Western blot analysis of
A375 cells treated with 0.5 µM 25-epi Ritterostatin
GN1N showed time-dependent lipidation of the
microtubule-associated autophagy marker protein, LC3B and increased
turnover of its phosphotidyl-ethanolamine-conjugated,
faster-migrating isoform, LC3B-II (Fig. 3E). The presence of LC3 protein in
autophagosomes and increased conversion of LC3-I to LC3-II has been
regarded as a key indicator of autophagy (50,51).
Taken together, the increased expression of LC3B-II,
collapse of mitochondrial membrane potential, and rapid apoptotic
cell death after pretreatment with chloroquine indicate that
25-epi Ritterostatin GN1N exerts its
antitumor effect in melanoma through induction of autophagy.
25-epi Ritterostatin
GN1N induces endoplasmic reticulum (ER)
stress in melanoma cells
Several studies have indicated that ER stress can
lead to activation of autophagy in cells (41,42).
Glucose-regulated protein 78 (GRP78) is thought to be a major
sensor of ER stress and is associated with melanoma progression and
increased drug resistance in melanoma and other cancers (18,52–54).
Since our findings indicated that autophagy was activated by
25-epi Ritterostatin GN1N in melanoma
cells, we investigated the effect of 25-epi Ritterostatin
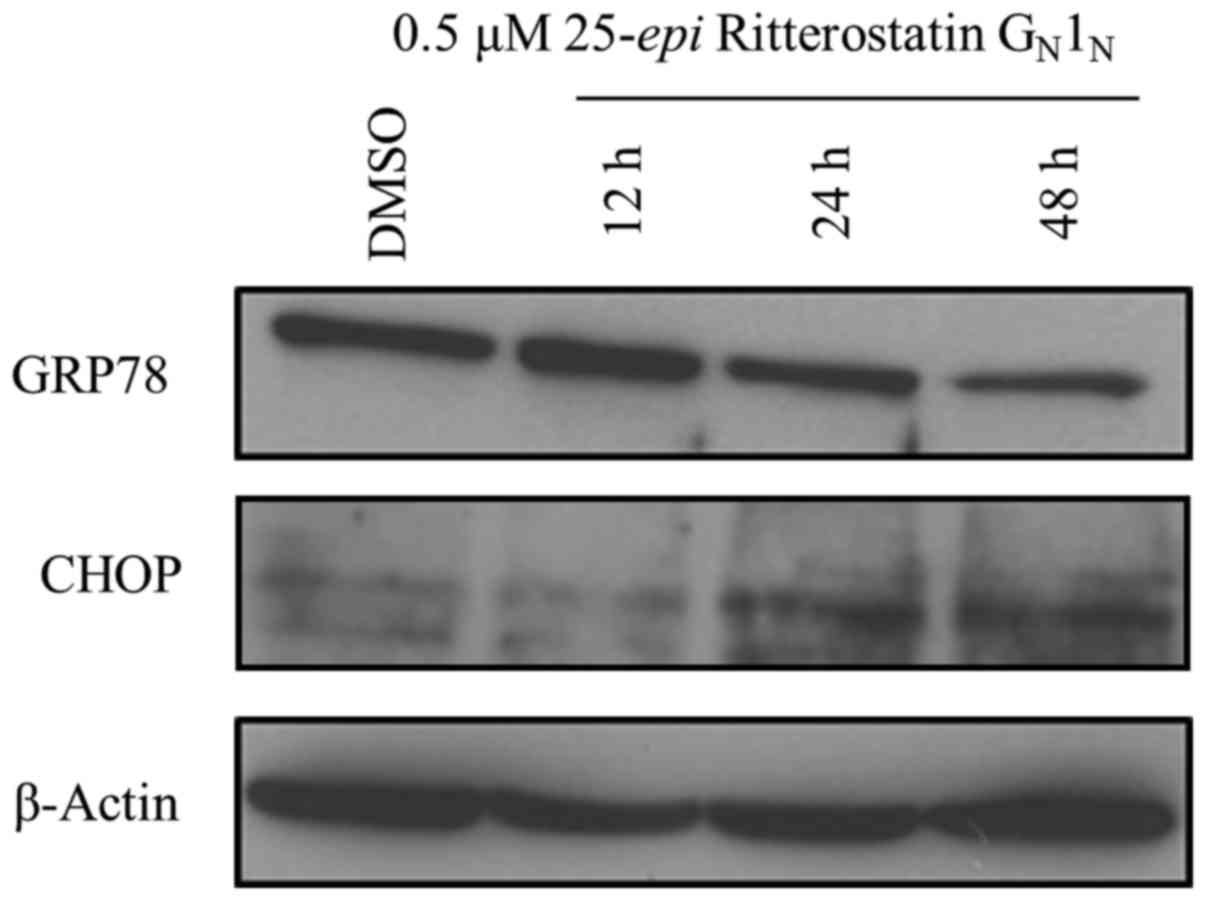
GN1N on GRP78 expression. 25-epi
Ritterostatin GN1N triggered a time-dependent
decrease in GRP78 expression in A375 cells. (Fig. 4). Noteworthy, an initial activation
of GRP78 expression was observed at 12 h after treatment with
25-epi Ritterostatin GN1N, before
decreasing at later time points. This early GRP78 activation might
be a result of its initial efforts to contain and neutralize ER
stress.
Simultaneously, we observed time-dependent
upregulated expression of CHOP, the ER stress marker protein known
to mediate cytotoxicity, in cells treated with 25-epi
Ritterostatin GN1N. Taken together, these
findings suggest that treatment with 25-epi Ritterostatin
GN1N triggered ER stress, leading to
autophagy and cell death.
25-epi Ritterostatin
GN1N exhibits minimal toxicity in normal
melanocytes
To determine whether the effects of 25-epi
Ritterostatin GN1N were specific to melanoma
cells, we also treated normal melanocytes with 0.5 and 1 µM
25-epi Ritterostatin GN1N and
performed Annexin V/propidium iodide staining to analyze cell
viability (Fig. 5A). We found that
96.6% of normal melanocytes (normalized to control) were viable
after treatment with 1µM 25-epi Ritterostatin
GN1N, compared with 36% of A375 cells treated
with 0.5µM 25-epi Ritterostatin
GN1N. Additionally, biochemical analysis for
changes in mitochondrial membrane potential indicated that the
normal melanocytes were resistant to the effect of 25-epi
Ritterostatin GN1N on mitochondrial membrane
integrity (Fig. 5B). Subsequent
immunoblotting analysis revealed no changes in the lipidation
expression profile of the autophagy marker protein LC3B (data not
shown). These findings indicate that the antitumor activity of
25-epi Ritterostatin GN1N is specific
to melanoma cells, with limited toxicity in normal melanocytes.
25-epi Ritterostatin
GN1N triggers inhibition of tumor growth in
vivo
Akt overexpression has been found to lead to rapid
tumor growth, increased angiogenesis, and a more pronounced
glycolytic mechanism in parental WM35 cells (55). Therefore, we used WM35 cells with
activated Akt expression (WM35 PKB cells) as a study model to
assess the antitumor activity of 25-epi Ritterostatin
GN1N. WM35 PKB cells (2×106) were
injected subcutaneously into female athymic nude mice, followed by
randomizing the mice into two groups of five each. The treatment
group received 2 mg/kg 25-epi Ritterostatin
GN1N 3 times per week via intraperitoneal
injection, and the control group received PBS. Tumor volume and
mouse body weight were recorded. Our results showed that
25-epi Ritterostatin GN1N
substantially suppressed melanoma tumor growth in the mouse
xenografts (Fig. 6A–C). In
addition, we found that body weight did not differ substantially
between the control (Fig. 6D) and
the treatment groups, suggesting that 25-epi Ritterostatin
GN1N does not cause any toxic effects in
mice.
Discussion
Malignant melanoma is one of the most aggressive
forms of cancer, with increasing incidence rates. Although multiple
clinical trials have been initiated with novel agents to treat
melanoma, very few have been successful, thus highlighting the need
to identify potential therapeutic agents through mechanistic
studies. Our results demonstrate that 25-epi Ritterostatin
GN1N exhibits antitumor activity in melanoma
cells by inducing autophagy and enhanced ER stress, with minimal
toxic effects in normal melanocytes, suggesting that further study
of 25-epi Ritterostatin GN1N as a
potential treatment for melanoma is necessary.
The chaperone protein GRP78 (also called BiP) acts
as the primary sensor of ER stress due to accumulation of unfolded
proteins in the ER lumen, which occurs as a result of oxidative
stress and cellular toxicity induced by calcium ionophores and
other agents (56). During ER
stress, GRP78 binds to these unfolded proteins, leading to the
release of resident ER transmembrane proteins such as PERK, ATF6,
and IRE1, which then activate downstream signaling cascades
promoting global repression of protein synthesis and subsequent
restoration of protein function (57–60).
GRP78 also supports cell survival by preventing caspase-7
activation and stabilization of mitochondrial function (61,62).
In melanoma, GRP78 has been linked to tumor progression and drug
resistance (18). Our results
showed that 25-epi Ritterostatin GN1N
inhibits GRP78 and activates CHOP expression in a time-dependent
fashion, leading to ER stress-mediated cell death.
Autophagy is a cellular self-digestive process that
facilitates degradation of long-lived proteins and damaged
organelles in cells to release energy and nutrients during stress
and starvation conditions. Numerous studies have indicated that
autophagy has a tumor-suppressive role in cancer (35). Work by Yue et al
demonstrated that activation of Beclin-1, the mammalian orthologue
for the yeast autophagy-related gene Atg6, inhibited in
vitro tumor cell proliferation and in vivo tumor growth
in mice (34), and also Sivridis
et al investigated the expression of two autophagy-related
proteins, Beclin-1 and LC3A, in 79 specimens of nodular cutaneous
melanoma (63). Their results
confirmed those from other researchers demonstrating that down
regulation of autophagic capacity is related to human
carcinogenesis (64,65). During autophagy, the light chain
protein LC3B is localized to autophagosomes and is therefore
considered a hallmark for autophagy. Our results demonstrate that
the cytotoxic effect of 25-epi Ritterostatin
GN1N is mediated by autophagy. In addition to
increased lipidation of LC3B, we observed a collapse of the
mitochondrial membrane potential. Importantly, the combination of
25-epi Ritterostatin GN1N with the
lysosomotrophic agent chloroquine resulted in rapid cell death.
Moreover, this induction of autophagy and loss of mitochondrial
membrane potential by 25-epi Ritterostatin
GN1N was not observed in normal melanocytes,
indicating that 25-epi Ritterostatin
GN1N has therapeutic selectivity to melanoma
cells.
To the best of our knowledge, the current study is
the first to characterize the in vitro and in vivo
antitumor effects of 25-epi Ritterostatin
GN1N in melanoma. We found that 25-epi
Ritterostatin GN1N inhibits melanoma cell
growth by activation of ER stress and autophagy. We also found that
intraperitoneal injection of 2 mg/kg 25-epi Ritterostatin
GN1N 3 times per week substantially reduced
the growth of melanoma in mice. Importantly, we observed no
significant change in body weight between mice treated with
25-epi Ritterostatin GN1N at this
dosage and control mice. These findings indicate that 25-epi
Ritterostatin GN1N could be a promising
treatment for melanoma. Nevertheless, additional in vivo and
clinical studies need to be conducted to ascertain the
effectiveness of this compound in inhibiting melanoma growth in
humans.
Abbreviations:
|
PI
|
propidium iodide
|
|
ER
|
endoplasmic reticulum
|
|
GRP78
|
glucose regulated protein 78
|
|
PBS
|
phosphate buffered saline
|
|
LC3
|
microtubule associated light chain
3
|
|
CHOP
|
CCAAT/enhancer binding protein
homologous protein
|
|
HSP70
|
heat shock protein 70
|
|
UPR
|
unfolded protein response
|
|
DAPK-1
|
death associated protein kinase
|
|
PERK
|
PKR-like EIF2α kinase
|
|
ATF-6
|
activation transcription factor-6
|
|
IRE-1
|
inositol requiring enzyme-1
|
|
BAX
|
Bcl-2 associated X
|
|
NHEM
|
normal human epidermal
melanocytes
|
|
DMSO
|
dimethyl sulfoxide
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide
|
|
FACS
|
fluorescence activated cell
sorting
|
|
Atg6
|
autophagy-related gene 6
|
References
|
1
|
Brown TJ and Nelson BR: Malignant
melanoma: A clinical review. Cutis. 63:275–278. 281–284.
1999.PubMed/NCBI
|
|
2
|
Alberts B, Lewis J, Raff M, Roberts K and
Walter P: Molecular Biology of the Cell. 5th edition. Garland
Science; New York, NY: pp. 13922008
|
|
3
|
Braakman I and Bulleid NJ: Protein folding
and modification in the mammalian endoplasmic reticulum. Annu Rev
Biochem. 80:71–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Malhotra JD and Kaufman RJ: The
endoplasmic reticulum and the unfolded protein response. Semin Cell
Dev Biol. 18:716–731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang CC, Chen LH, Gillespie S, Kiejda KA,
Mhaidat N, Wang YF, Thorne R, Zhang XD and Hersey P: Tunicamycin
sensitizes human melanoma cells to tumor necrosis factor-related
apoptosis-inducing ligand-induced apoptosis by up-regulation of
TRAIL-R2 via the unfolded protein response. Cancer Res.
67:5880–5888. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Noda I, Fujieda S, Seki M, Tanaka N,
Sunaga H, Ohtsubo T, Tsuzuki H, Fan GK and Saito H: Inhibition of
N-linked glycosylation by tunicamycin enhances sensitivity to
cisplatin in human head-and-neck carcinoma cells. Int J Cancer.
80:279–284. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Denmeade SR, Jakobsen CM, Janssen S, Khan
SR, Garrett ES, Lilja H, Christensen SB and Isaacs JT:
Prostate-specific antigen-activated thapsigargin prodrug as
targeted therapy for prostate cancer. J Natl Cancer Inst.
95:990–1000. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Treiman M, Caspersen C and Christensen SB:
A tool coming of age: Thapsigargin as an inhibitor of
sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol Sci.
19:131–135. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Johnson AJ, Hsu AL, Lin HP, Song X and
Chen CS: The cyclo-oxygenase-2 inhibitor celecoxib perturbs
intracellular calcium by inhibiting endoplasmic reticulum
Ca2+-ATPases: A plausible link with its anti-tumour
effect and cardiovascular risks. Biochem J. 366:831–837. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
11
|
Lee AS: The glucose-regulated proteins:
Stress induction and clinical applications. Trends Biochem Sci.
26:504–510. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J and Lee AS: Stress induction of
GRP78/BiP and its role in cancer. Curr Mol Med. 6:45–54. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xing X, Lai M, Wang Y, Xu E and Huang Q:
Overexpression of glucose-regulated protein 78 in colon cancer.
Clin Chim Acta. 364:308–315. 2006. View Article : Google Scholar
|
|
14
|
Fernandez PM, Tabbara SO, Jacobs LK,
Manning FC, Tsangaris TN, Schwartz AM, Kennedy KA and Patierno SR:
Overexpression of the glucose-regulated stress gene GRP78 in
malignant but not benign human breast lesions. Breast Cancer Res
Treat. 59:15–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shuda M, Kondoh N, Imazeki N, Tanaka K,
Okada T, Mori K, Hada A, Arai M, Wakatsuki T, Matsubara O, et al:
Activation of the ATF6, XBP1 and grp78 genes in human
hepatocellular carcinoma: A possible involvement of the ER stress
pathway in hepatocarcinogenesis. J Hepatol. 38:605–614. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luk JM, Lam CT, Siu AF, Lam BY, Ng IO, Hu
MY, Che CM and Fan ST: Proteomic profiling of hepatocellular
carcinoma in Chinese cohort reveals heat-shock proteins (Hsp27,
Hsp70, GRP78) up-regulation and their associated prognostic values.
Proteomics. 6:1049–1057. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arap MA, Lahdenranta J, Mintz PJ, Hajitou
A, Sarkis AS, Arap W and Pasqualini R: Cell surface expression of
the stress response chaperone GRP78 enables tumor targeting by
circulating ligands. Cancer Cell. 6:275–284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhuang L, Scolyer RA, Lee CS, McCarthy SW,
Cooper WA, Zhang XD, Thompson JF and Hersey P: Expression of
glucose-regulated stress protein GRP78 is related to progression of
melanoma. Histopathology. 54:462–470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
de Ridder GG, Ray R and Pizzo SV: A murine
monoclonal antibody directed against the carboxyl-terminal domain
of GRP78 suppresses melanoma growth in mice. Melanoma Res.
22:225–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ranganathan AC, Zhang L, Adam AP and
Aguirre-Ghiso JA: Functional coupling of 38-induced up-regulation
of BiP and activation of RNA-dependent protein kinase-like
endoplasmic reticulum kinase to drug resistance of dormant
carcinoma cells. Cancer Res. 66:1702–1711. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rutkowski DT, Arnold SM, Miller CN, Wu J,
Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D and Kaufman RJ:
Adaptation to ER stress is mediated by differential stabilities of
pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol.
4:e3742006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matsumoto M, Minami M, Takeda K, Sakao Y
and Akira S: Ectopic expression of CHOP (GADD153) induces apoptosis
in M1 myeloblastic leukemia cells. FEBS Lett. 395:143–147. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cho HY, Thomas S, Golden EB, Gaffney KJ,
Hofman FM, Chen TC, Louie SG, Petasis NA and Schönthal AH: Enhanced
killing of chemo-resistant breast cancer cells via controlled
aggravation of ER stress. Cancer Lett. 282:87–97. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rabik CA, Fishel ML, Holleran JL, Kasza K,
Kelley MR, Egorin MJ and Dolan ME: Enhancement of cisplatin
[cisdiammine dichloroplatinum (II)] cytotoxicity by
O6-benzylguanine involves endoplasmic reticulum stress. J Pharmacol
Exp Ther. 327:442–452. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sánchez AM, Martínez-Botas J,
Malagarie-Cazenave S, Olea N, Vara D, Lasunción MA and Díaz-Laviada
I: Induction of the endoplasmic reticulum stress protein
GADD153/CHOP by capsaicin in prostate PC-3 cells: A microarray
study. Biochem Biophys Res Commun. 372:785–791. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ravikumar B, Futter M, Jahreiss L,
Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM,
Narayanan U, Renna M, et al: Mammalian macroautophagy at a glance.
J Cell Sci. 122:1707–1711. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guo JY, Chen HY, Mathew R, Fan J,
Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM,
Karantza V, et al: Activated Ras requires autophagy to maintain
oxidative metabolism and tumorigenesis. Genes Dev. 25:460–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maiuri MC, Malik SA, Morselli E, Kepp O,
Criollo A, Mouchel PL, Carnuccio R and Kroemer G: Stimulation of
autophagy by the p53 target gene Sestrin2. Cell Cycle. 8:1571–1576.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tasdemir E, Maiuri MC, Galluzzi L, Vitale
I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C,
Harper F, et al: Regulation of autophagy by cytoplasmic p53. Nat
Cell Biol. 10:676–687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh
H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al:
Promotion of tumorigenesis by heterozygous disruption of the beclin
1 autophagy gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gozuacik D, Bialik S, Raveh T, Mitou G,
Shohat G, Sabanay H, Mizushima N, Yoshimori T and Kimchi A:
DAP-kinase is a mediator of endoplasmic reticulum stress-induced
caspase activation and autophagic cell death. Cell Death Differ.
15:1875–1886. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zalckvar E, Berissi H, Mizrachy L,
Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R
and Kimchi A: DAP-kinase-mediated phosphorylation on the BH3 domain
of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and
induction of autophagy. EMBO Rep. 10:285–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zalckvar E, Berissi H, Eisenstein M and
Kimchi A: Phosphorylation of Beclin 1 by DAP-kinase promotes
autophagy by weakening its interactions with Bcl-2 and Bcl-XL.
Autophagy. 5:720–722. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shen J, Chen X, Hendershot L and Prywes R:
ER stress regulation of ATF6 localization by dissociation of
BiP/GRP78 binding and unmasking of Golgi localization signals. Dev
Cell. 3:99–111. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Okada T, Yoshida H, Akazawa R, Negishi M
and Mori K: Distinct roles of activating transcription factor 6
(ATF6) and double-stranded RNA-activated protein kinase-like
endoplasmic reticulum kinase (PERK) in transcription during the
mammalian unfolded protein response. Biochem J. 366:585–594. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ding WX, Ni HM, Gao W, Hou YF, Melan MA,
Chen X, Stolz DB, Shao ZM and Yin XM: Differential effects of
endoplasmic reticulum stress-induced autophagy on cell survival. J
Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar
|
|
42
|
Yorimitsu T, Nair U, Yang Z and Klionsky
DJ: Endoplasmic reticulum stress triggers autophagy. J Biol Chem.
281:30299–30304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bernales S, McDonald KL and Walter P:
Autophagy counterbalances endoplasmic reticulum expansion during
the unfolded protein response. PLoS Biol. 4:e4232006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kanduluru AK, Banerjee P, Beutler JA and
Fuchs PL: A convergent total synthesis of the potent
cephalostatin/ritterazine hybrid -25-epi ritterostatin
GN1N. J Org Chem. 78:9085–9092. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shoemaker RH: The NCI60 human tumour cell
line anticancer drug screen. Nat Rev Cancer. 6:813–823. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
von Schwarzenberg K and Vollmar AM:
Targeting apoptosis pathways by natural compounds in cancer: Marine
compounds as lead structures and chemical tools for cancer therapy.
Cancer Lett. 332:295–303. 2013. View Article : Google Scholar
|
|
48
|
Fan C, Wang W, Zhao B, Zhang S and Miao J:
Chloroquine inhibits cell growth and induces cell death in A549
lung cancer cells. Bioorg Med Chem. 14:3218–3222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA
and Koh JY: Induction of lysosomal dilatation, arrested autophagy,
and cell death by chloroquine in cultured ARPE-19 cells. Invest
Ophthalmol Vis Sci. 51:6030–6037. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Papalas JA, Vollmer RT, Gonzalez-Gronow M,
Pizzo SV, Burchette J, Youens KE, Johnson KB and Selim MA: Patterns
of GRP78 and MTJ1 expression in primary cutaneous malignant
melanoma. Mod Pathol. 23:134–143. 2010. View Article : Google Scholar
|
|
53
|
Zheng HC, Takahashi H, Li XH, Hara T,
Masuda S, Guan YF and Takano Y: Overexpression of GRP78 and GRP94
are markers for aggressive behavior and poor prognosis in gastric
carcinomas. Hum Pathol. 39:1042–1049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Su R, Li Z, Li H, Song H, Bao C, Wei J and
Cheng L: Grp78 promotes the invasion of hepatocellular carcinoma.
BMC Cancer. 10:202010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Govindarajan B, Sligh JE, Vincent BJ, Li
M, Canter JA, Nickoloff BJ, Rodenburg RJ, Smeitink JA, Oberley L,
Zhang Y, et al: Overexpression of Akt converts radial growth
melanoma to vertical growth melanoma. J Clin Invest. 117:719–729.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: Coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Schindler AJ and Schekman R: In vitro
reconstitution of ER-stress induced ATF6 transport in COPII
vesicles. Proc Natl Acad Sci USA. 106:17775–17780. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shen J, Snapp EL, Lippincott-Schwartz J
and Prywes R: Stable binding of ATF6 to BiP in the endoplasmic
reticulum stress response. Mol Cell Biol. 25:921–932. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Haze K, Yoshida H, Yanagi H, Yura T and
Mori K: Mammalian transcription factor ATF6 is synthesized as a
transmembrane protein and activated by proteolysis in response to
endoplasmic reticulum stress. Mol Biol Cell. 10:3787–3799. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Reddy RK, Mao C, Baumeister P, Austin RC,
Kaufman RJ and Lee AS: Endoplasmic reticulum chaperone protein
GRP78 protects cells from apoptosis induced by topoisomerase
inhibitors: Role of ATP binding site in suppression of caspase-7
activation. J Biol Chem. 278:20915–20924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sun FC, Wei S, Li CW, Chang YS, Chao CC
and Lai YK: Localization of GRP78 to mitochondria under the
unfolded protein response. Biochem J. 396:31–39. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sivridis E, Koukourakis MI, Mendrinos SE,
Karpouzis A, Fiska A, Kouskoukis C and Giatromanolaki A: Beclin-1
and LC3A expression in cutaneous malignant melanomas: A biphasic
survival pattern for beclin-1. Melanoma Res. 21:188–195. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang J, Pan XL, Ding LJ, Liu DY, Da-Peng
Lei and Jin T: Aberrant expression of Beclin-1 and LC3 correlates
with poor prognosis of human hypopharyngeal squamous cell
carcinoma. PLoS One. 8:e690382013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Huang X, Bai HM, Chen L, Li B and Lu YC:
Reduced expression of LC3B-II and Beclin 1 in glioblastoma
multiforme indicates a downregulated autophagic capacity that
relates to the progression of astrocytic tumors. J Clin Neurosci.
17:1515–1519. 2010. View Article : Google Scholar : PubMed/NCBI
|