Introduction
Acute myeloid leukemia (AML) is a heterogenous
hematological malignancy involving the clonal expansion of myeloid
blasts in the bone marrow and peripheral blood with possible spread
to liver and spleen. An estimated 19,950 people were newly
diagnosed in 2016, 10,430 of whom will die from their disease
(1). The 5-year survival rate for
adult AML patients is only 26.6% based on 2006–2012 data, with a
median age of 67 years at diagnosis (1). Overexpression of anti-apoptotic Bcl-2
proteins such as Bcl-2, Bcl-xl, and Mcl-1 occurs frequently in AML
(2), which is widely associated
with tumor initiation, progression, and drug resistance. Most AML
patients become resistant to chemotherapy at some point in their
course and succumb to their disease. Therefore, there is an urgent
need to prevent chemoresistance or enhance chemo-sensitivity in a
selective fashion to lead to a higher cure rate and a lower toxic
burden.
Resisting cell death is a hallmark of cancer cells
that contributes to tumour progression and to chemoresistance
(3). Over the past three decades,
over 16 members of the Bcl-2 family protein were identified and
characterized (4). There are
proapoptosis BH3-only proteins (such as Bim and Bad), proapoptosis
multi-BH-domain proteins (such as Bak and Bax) and anti-apoptosis
proteins (including Bcl-2, Bcl-xl, Mcl-1, Bfl1 and Bclw). The
discovery of Bcl-2 started with a t(14;18) chromosomal
translocations in human follicular lymphoma (5,6).
This protein has since been shown to have a dominant role in the
survival of multiple lymphoid malignancies (7,8). The
pro-survival Bcl-xl protein, which was encoded by Bclx gene, was
associated with drug resistance and disease progression of
hematological malignancies (9).
The dependence of AML cells on the anti-apoptotic Bcl-2 protein can
be exploited for therapeutic effect using BH3 mimetics (10), a class of small molecules that
mimic the inhibitory features of BH3-only proteins (11). Cancer cells have greater
susceptibility to BH3 mimetic drugs than normal cells, partly
because they often have higher levels of anti-apoptosis proteins
and release more previously sequestered BH3-only proteins to
activate Bax and Bak (12,13).
ABT-737 (14) and
ABT-263 (15), both developed by
Abbvie Laboratories, displace pro-apoptotic proteins from Bcl-2 and
Bcl-xl and have synergistic toxicity with conventional
chemotherapeutics and radiation. They require Bax for cell killing
and causing MOMP in Bcl-2 dependent cancer cells (16,17),
thus confirming an on-target effect. However, on-target
thrombocytopenia caused by Bcl-xl inhibition limits the application
of ABT-263. For the treatment of cancers that depend on Bcl-2,
Bcl-2 selective inhibitor ABT-199 was created. ABT-199 does not
reduce platelet lifespan and is better tolerated than ABT-263
(18). These mimetics have shown
promising efficacy in various preclinical models and now in
advanced clinical trials for chronic lymphocytic leukemia(CLL) and
other malignancies (19–22).
Previously, our laboratory reported that small
molecule Bcl-2 inhibitors ApoG2 and BM-1197 have potent antitumor
effect on coloretal cancer cells (23,24).
APG-1252, a new BH3 mimetic that binds to Bcl-2 and Bcl-xl with
sub-nanomolar affinities (Ki <1 nM) (25), was demonstrated with better in
vivo antitumor activity than ABT-263 (26). APG-1252 achieved complete and
long-term tumor regression in both H146 and H1963 SCLC xenograft
models and avoid the commonly seen on-target toxicity when Bcl-xl
is inhibited. APG-1252 converts into a more active metabolite
APG-1252-12A (APG-1252-M1) in vivo. APG-1252-12A also binds
with high affinity to Bcl-2 and Bcl-xl (Ki <1 nM) and
is over ten times more active than APG-1252 (25). Regardless, the anti-tumor effect
and underlying mechanism of APG-1252-12A have not yet been
evaluated in leukemia. Herein, we report our detailed investigation
of APG-1252-12A in HL-60 cells. These data showed that APG-1252-12A
induced mitochondria-dependent apoptosis thus warranting further
investigation.
Materials and methods
Cells and reagents
The leukemia cell lines HL-60, MOLM-13, U937, THP-1
and MV4-11 were donated by State Key Laboratory of Oncology in
South China. Cells were cultured in RPMI-1640 (Gibco Life
Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (FBS-22A; Carpricorn Scientific Gmbh, Ebsdorfergrund,
Germany) and incubated at 37°C with 5% CO2. APG-1252-12A
was kindly provided by the Ascentage Pharma Group Corp Inc.
(Taizhou, China) and was dissolved in pure dimethyl sulfoxide
(DMSO; Sigma-Aldrich, St. Louis, MO, USA) with a stock
concentration of 40 mmol/l, stored at −20°C, and diluted in the
corresponding culture medium just before use.
Cell viability assay
Cell proliferation was determined by CellTiter
96AQueous MTS
(3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium,
inner salt) assay. AML cell lines were seeded 10,000–50,000 onto
96-well plates containing 200 µl of culture medium per well
and treated with APG-1252-12A of serial concentrations for 24, 48
and 72 h, respectively. After that, 40 µl of MTS (Promega,
Madison, WI, USA) was added to each well and reacted for another 4
h at 37°C. Then the absorbance value was measured with a
spectrophotometer at 490 nm. Cell viability was expressed as mean ±
SD of absorbance and analyzed with nonlinear regression on GraphPad
Prism version 6.0. The values were performed in triplicate as a
percentage relative to those obtained in untreated controls.
Apoptosis detection with nuclear
staining
The morphological assessment of apoptotic HL-60
cells was detected by Hoechst 33258 (Beyotime Institute of
Biotechnology, Jiangsu, China) staining. Cells (20,000) were plated
in each of 6-well plate and incubated with various concentrations
of APG-1252-12A and 0.1% DMSO for 24 h. The staining was performed
according to the manufacturer's protocol. The morphological
features of apoptosis were observed by fluorescence microscope
(Olympus, Tokyo, Japan).
Flow cytometry analysis of apoptosis and
cell cycle
Cell apoptosis was determined with an Annexin
V-propidium iodide (PI) apoptosis detection kit (KGA108; KeyGen
Biotech, Nanjing, China) by flow cytometry (Beckman Coulter,
Fullerton, CA, USA). Cells (200,000) were seeded into each well of
a 6-well plate and treated with indicated concentration of drug or
DMSO for 24 or 48 h. After treatment, cells were harvested and
washed twice with phosphate buffered saline (PBS), and resuspended
in 500 µl binding buffer containing 5 µl Annexin V
FITC and 5 µl propidium iodide (KeyGen Biotech). Experiments
were analyzed after incubating out of light in the staining
solution for 10 min.
Flow cytometry was performed to analyze cell cycle
position. After treatment, cells were collected, washed and fixed
in 70% cold ethanol at 4°C overnight. Next, the cells were
incubated with RNase for 30 min at 37°C, then stained with PI (Cell
cycle Detection kit, KeyGen) in the dark at 4°C for another 30 min.
Cells were analyzed with a FACS Calibur flow cytometer (Beckman
Coulter), and the data were analyzed using ModFit LT 3.2
software.
Western blot analysis
Cells were lysed with 1X Cell Lysis Buffer (#9803:
Cell Signaling Technology, Danvers, MA, USA), and protein
concentration was measured with the Pierce BCA protein assay kit.
Total cell lysates were extracted and separated by electrophoresis
in 8–15% SDS-polyacrylamide gel and transferred to PVDF membranes
(Roche, Basel, Switzerland). Following blockage in 5% non-fat milk,
PVDF membranes were incubated with anti-Mcl-1 (94296), Bcl-2
(4223), Bcl-xl (2764), anti-Bax (2772), anti-Bak (6947),
anti-cleaved caspase-3 (9661), anti-caspase-3 (9665),
anti-cytochrome c (4272), GAPDH (2118), anti-β-actin (4970, Cell
Signaling Technology) or anti-PARP-1 (sc-7150), anti-Bim antibody
(sc-374358; Santa Cruz Biotechnology Santa Cruz, CA, USA). The
secondary anti-mouse (sc-2005) and anti-rabbit (sc-2004) antibodies
were purchased from Santa Cruz Biotechnology. Antigen-antibody
complexes were detected using Bio-Rad Clarity™ western ECL
substrate and protein level were quantified by Image Lab (Bio-Rad
Laboratory, Hercules, CA, USA).
Mitochondrial cytochrome c release
assay
HL-60 cells were pretreated with 2.5 µmol/l
of APG-1252-12A for 6 h. Cytoplasmic fractionation was isolated
using the Cytosol/Mitochondria Fractionation kit (#QIA88: Merck
Millipore, Darmstadt, Germany). Following the kit recommendations,
cytosolic fractions were isolated from HL-60 cells. The amount of
cytochrome c in cytosol fraction was determined by western blot
analysis as described above.
Statistical analysis
IC50 values were calculated by non-liner
regression analysis with GraphPad Prism software v6.0 (GraphPad
Software, La Jolla, CA, USA). The results were expressed as the
mean ± standard error of mean (SEM) from at least three independent
experiments. One-way analysis of variance (ANOVA) was used to
compare the means between groups by SPSS 20.0 software. Differences
in P-value <0.05 were considered statistically significant.
Results
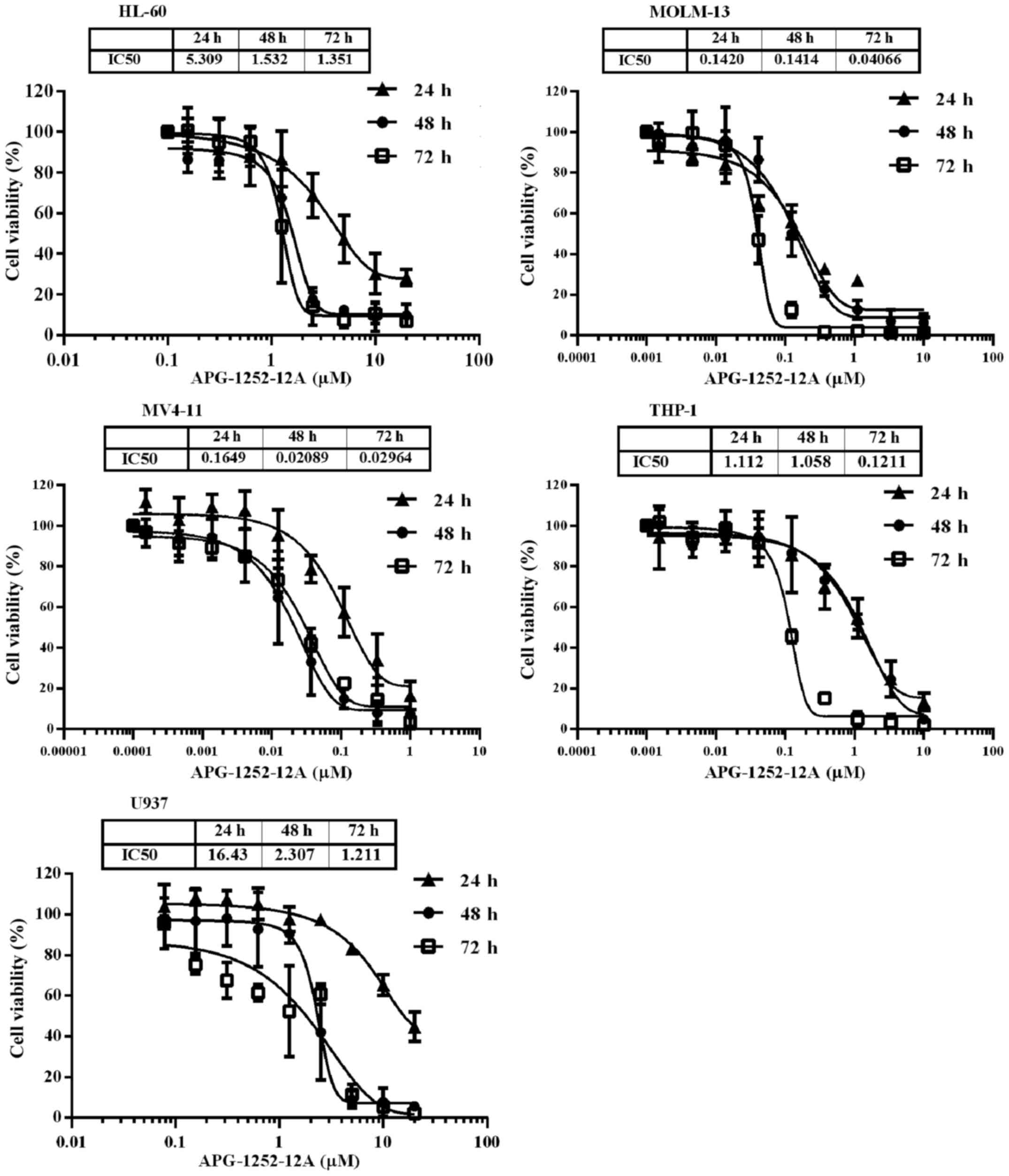
APG-1252-12A inhibits growth potently in
five leukemia cell lines
To test the potential utility of APG-1252-12A in
leukemia, we exposed five leukemia cell lines with increasing
concentrations of APG-1252-12A for 24, 48 and 72 h and then
determined the IC50 values. The viability of these cell
lines after treatment decreased significantly in a time- and
dose-dependent manner (Fig. 1).
The IC50 of APG-1252-12A ranged from <100 nM to
>1000 nM and MV4-11 was the most sensitive cell line (Table I).
 | Table IThe IC50 values
(µM) of APG-1252-12A in five AML cell lines. |
Table I
The IC50 values
(µM) of APG-1252-12A in five AML cell lines.
| Cell lines | 24 h | 48 h | 72 h |
|---|
| HL-60 | 5.35±1.04 | 1.52±0.30 | 1.40±0.43 |
| MOLM-13 | 0.15±0.06 | 0.14±0.03 | 0.04±0.01 |
| MV4-11 | 0.19±0.09 | 0.02±0.02 | 0.03±0.00 |
| THP-1 | 1.09±0.45 | 1.06±0.08 | 0.12±0.01 |
| U937 | 16.61±1.93 | 2.22±0.34 | 1.23±0.23 |
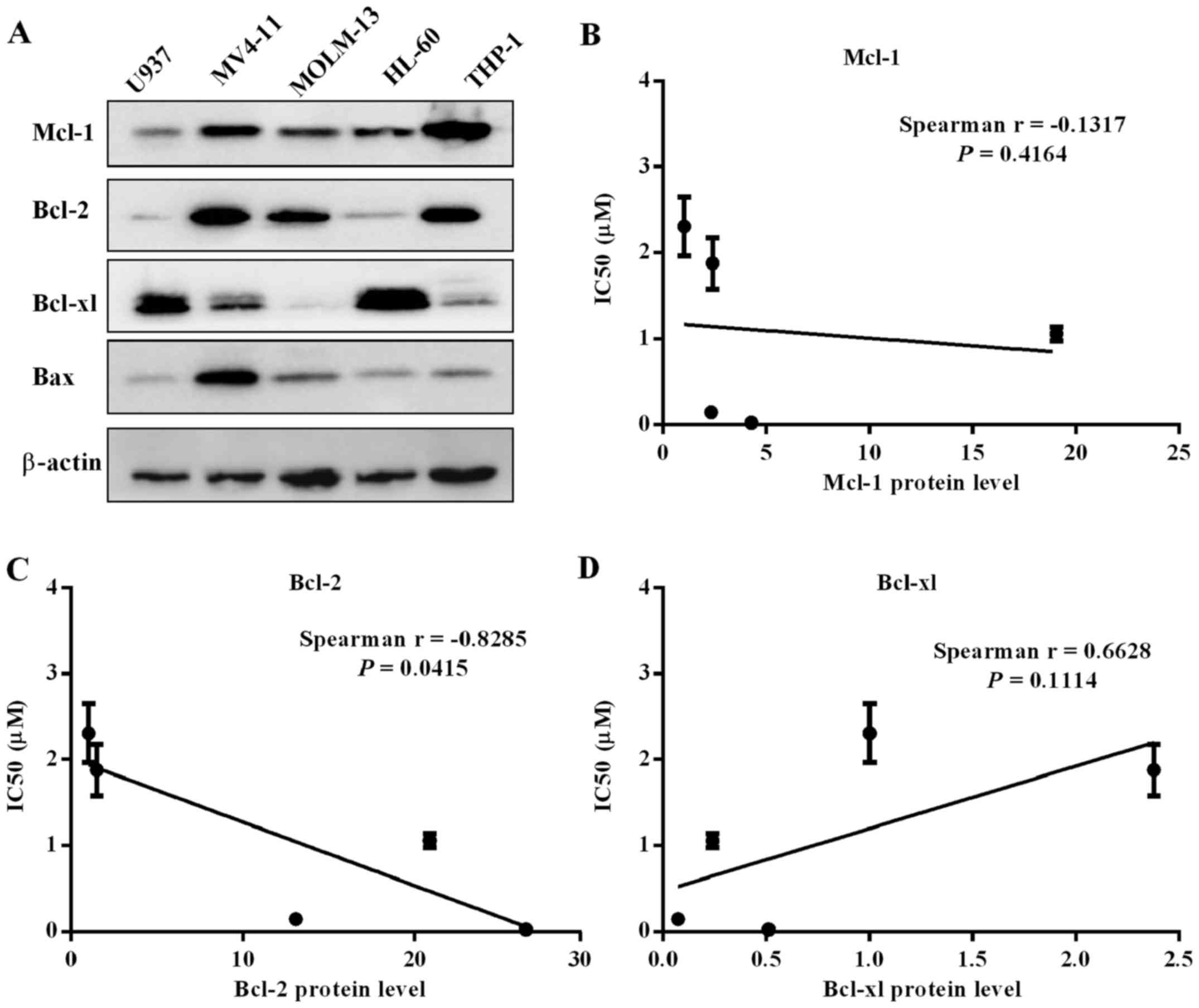
Bcl-2 family protein level in five
leukemia cell lines
To further clarify the on-target action of cell
killing via selectively binding with Bcl-1/Bcl-xl, we analyzed
whether there were correlates of cell line sensitivity to
APG-1252-12A. The expression of three Bcl-2 family proteins were
determined by western blot analysis (Fig. 2A). Spearman's analysis was
performed to assess correlation between IC50 values and
protein expression. The expression level of Bcl-2 correlated with
sensitivity to the drug, while levels of Bcl-xl and Mcl-1 had no
correlation with the drug sensitivity (Fig. 2B–D). The MV4-11 and MOLM-13 cells
with high levels of Bcl-2 protein and relatively low Bcl-xl
expression were more insensitive to APG-1252-12A. High expression
of Bcl-xl in HL-60 and U937 cells might explain the killing
mechanism of targeting Bcl-xl. THP-1 cell line had high level of
Bcl-2 and Mcl-1 as well as relatively low level of Bcl-xl which
supported that sensitivity to APG-1252-12A was correlated with
Bcl-2 protein level.
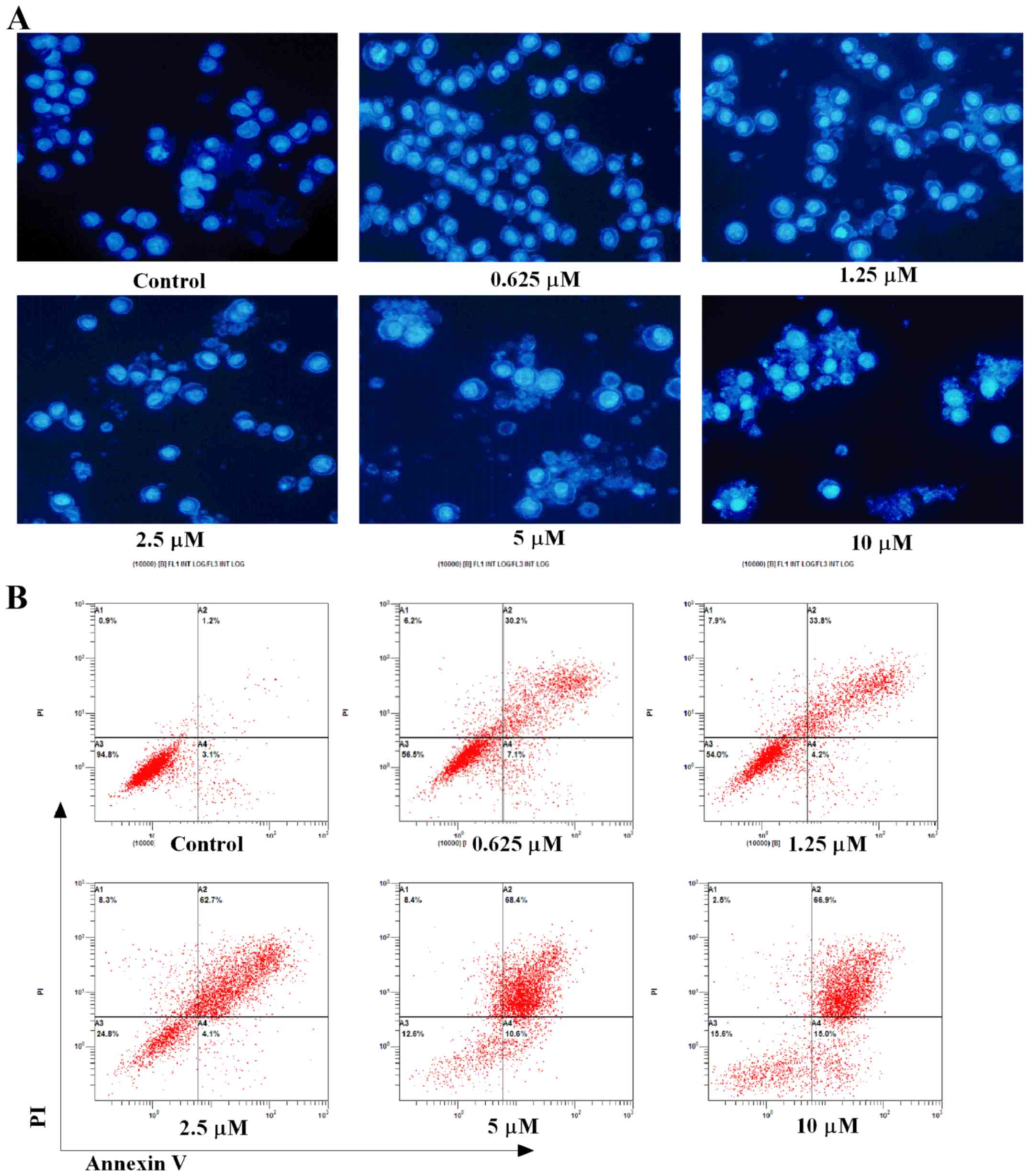
APG-1252-12A induces apoptosis in HL-60
cells
Hoechst 33258 staining and flow cytometry were used
to evaluate APG-1252-12A inducing apoptosis in HL-60 cells.
Increased apoptosis was shown by analysis of nuclei changes with
the electron microscopic analysis (Fig. 3A). The apoptotic bodies and nuclear
fragments were stained light blue, and the normal cells were
stained blue. The nuclei of the cells appeared normal, round and
large with regular contours in the control groups. Cells with
smaller nuclei and condensed chromatin were rare. By contrast, the
treated cells showed strong morphological alterations such as
nuclear shrinkage, intense fluorescence of nuclei and nuclear
fragmentation. Apoptosis detection by Annexin V and PI staining
showed that when treated with APG-1252-12A alone, dramatic increase
of Annexin V positive cells was seen in HL-60 cells (Fig. 3B). Flow cytometry also indicated
that treatment with increasing concentrations of the drug resulted
in a significant decrease of cell counts and induced apoptosis in a
dose-dependent manner. Time course analysis of cells exposed to
APG-1252-12A (10 µmol/l) revealed approximately 47% cell
death at 24 h, and substantially more pronounced lethality after 48
h (83%, Fig. 3C). An early and
late apoptotic cell distribution chart shows more late stage HL-60
apoptotic cells than early stage after 48 h treatment of
APG-1252-12A (Fig. 3D).
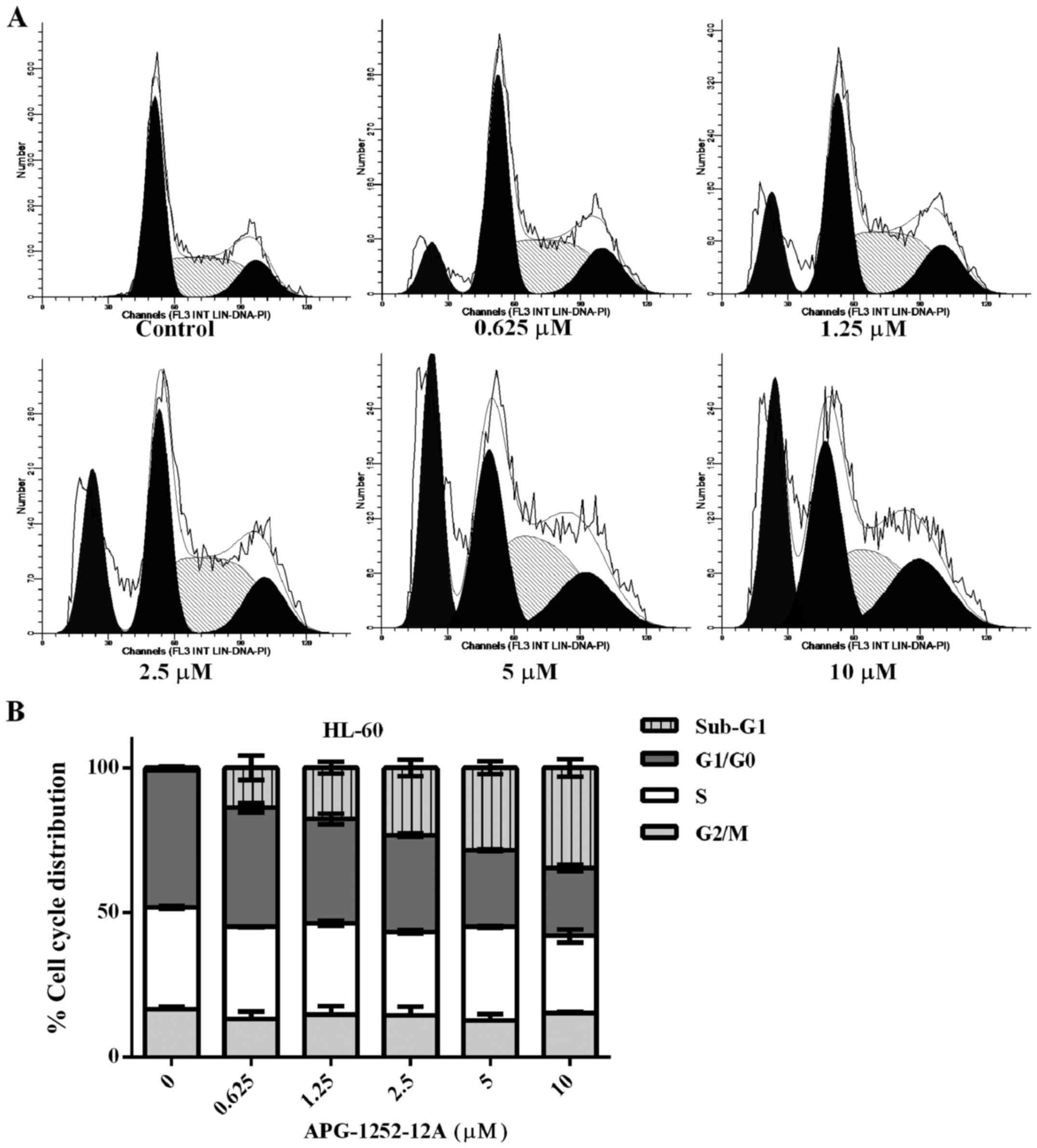
The sub-G1 phase increases after
APG-1252-12A treatment
Cell cycle analysis of the propidium iodine stained
DNA was performed in HL-60 cells. The percentage of cells in sub-G1
fraction increased significantly, pointing to APG-1252-12A- induced
cell death and DNA fragmentation (Fig.
4). Treatment with increasing concentrations of APG-1252-12A
resulted in a significant increase in the percentage of cells in
the sub-G1 phase. The remaining living cells showed no significant
increase in the percentage of cells in the S phase of the cell
cycle and the percentage of cells in the G1 and G2/M phase showed
similar results. No statistically significant correlation between
APG-1252-12A sensitivity and cell cycle was found.
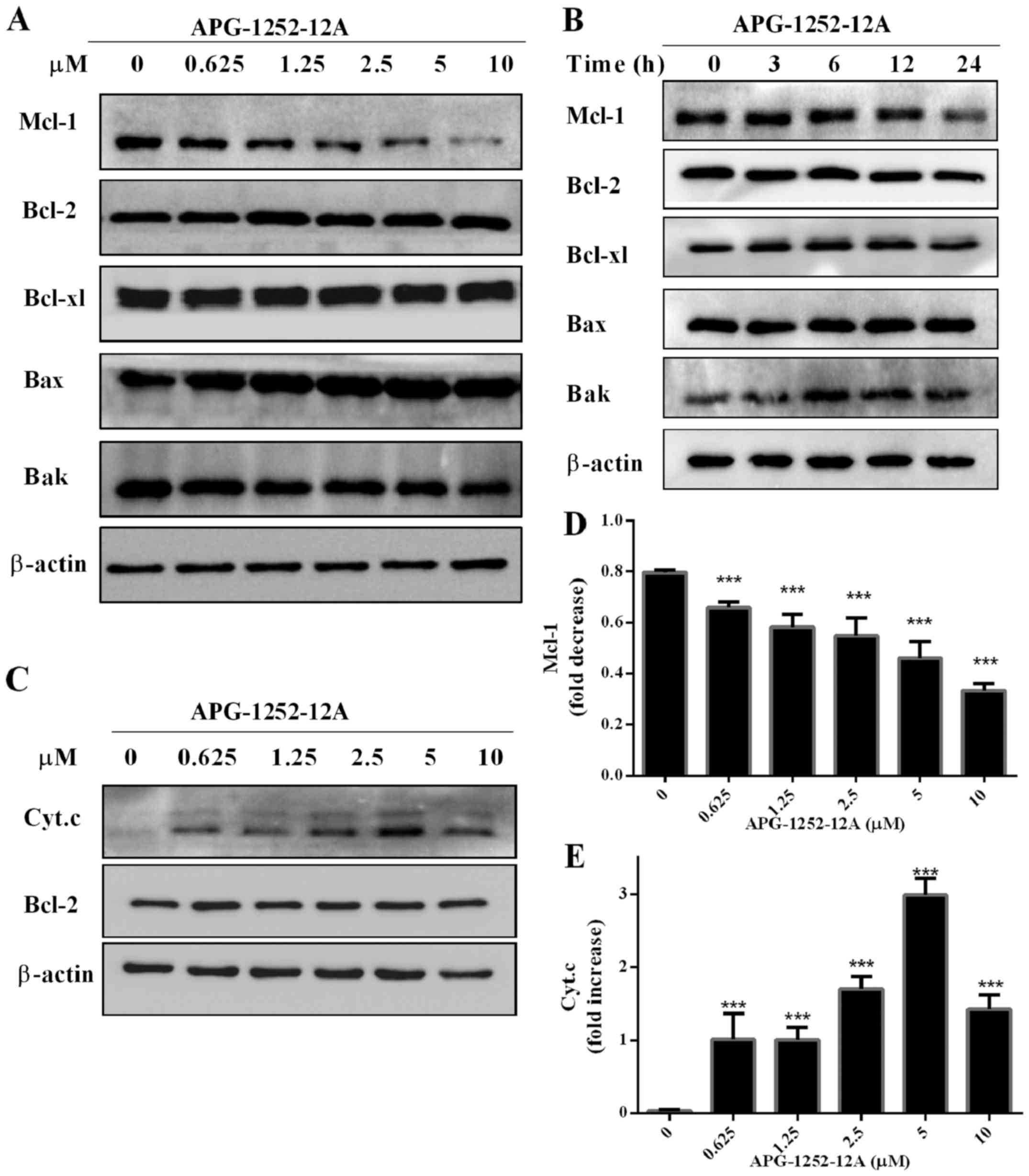
Effect of APG-1252-12A on Bcl-2 family
protein expression in HL-60 cells
To further investigate the effect of APG-1252-12A on
the protein expression level of Bcl-2 family members, we chose
HL-60 for analysis. After treating with serial concentrations of
APG-1252-12A for 24 h, there were no significant alternations in
the protein expression of Bcl-2 family except the suppression of
Mcl-1 in HL-60 cells (Fig. 5A and
D). A time course of Bcl-2, Bcl-xl, Bak and Bax protein levels
showed that the APG-1252-12A treatment in HL-60 cells did not
change their expression levels (Fig.
5B).
APG-1252-12A promotes cytochrome c
release in HL-60 cells
We also observed that cell death was induced by
APG-1252-12A that underwent cytochrome c release (Fig. 5C and E). Cytoplasmic cytochrome c
level was detected by western blot analysis. APG-1252-12A induced
cytochrome c release at a concentration of <0.625
µmol/l.
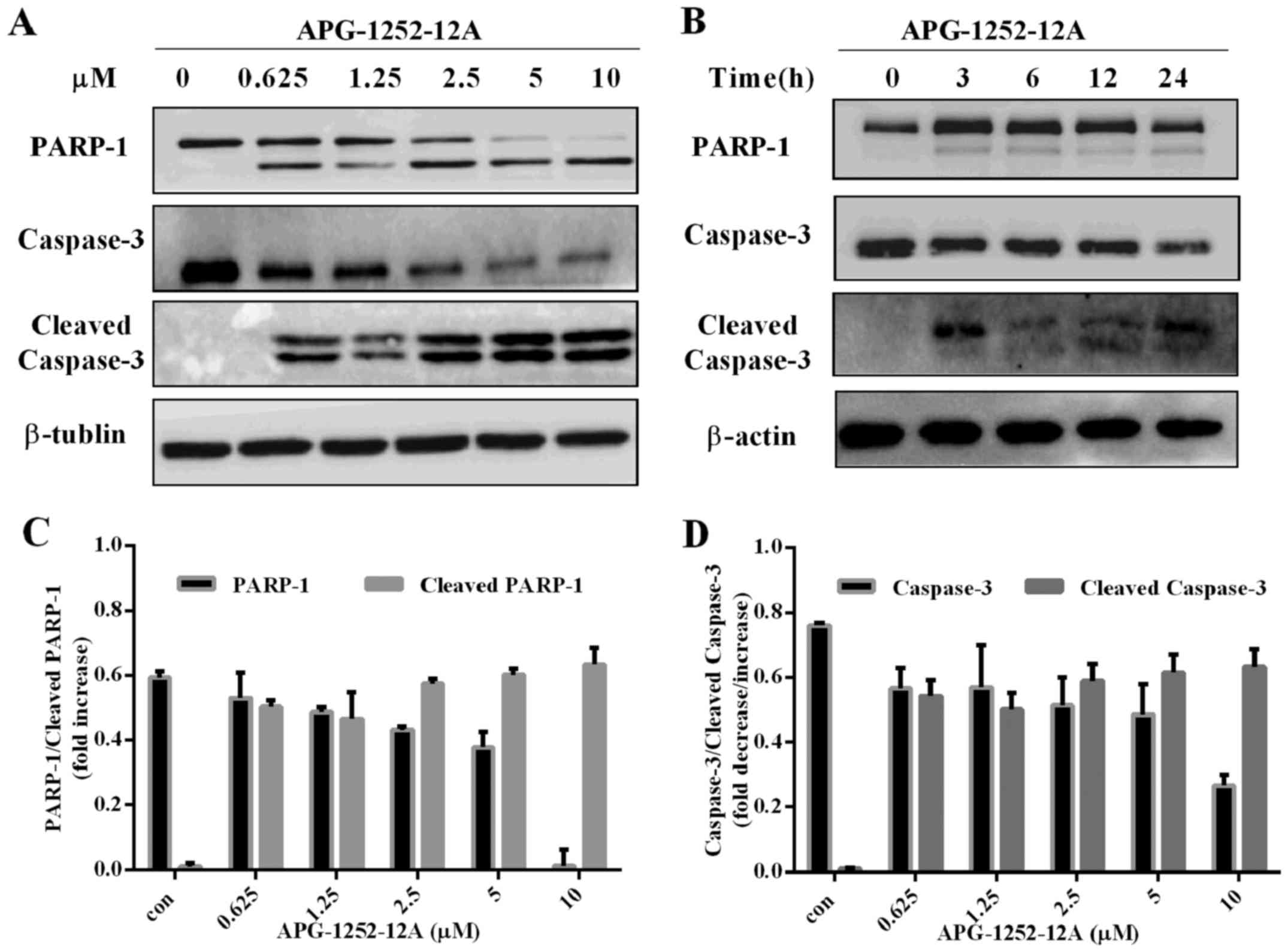
APG-1252-12A induces caspase-3 activation
in HL-60 cells
Western blot analysis was performed with antibodies
against PARP-1, caspase-3, and cleaved caspase-3. APG-1252-12A led
to increase of cleaved PARP, cleaved caspase-3 and decrease of
caspase-3 in HL-60 cells in a concentration-dependent manner
(Fig. 6A–D). The marked
cleavage/activation of caspases-3 and PARP-1 in HL-60 pronounced
the loss in mitochondrial membrane potential (MOMP). These findings
indicated that inhibition of anti-apoptotic Bcl-2 and Bcl-xl caused
MOMP, promoting cytochrome c release followed by caspase
activation.
Discussion
As multiple studies have implicated the role of
Bcl-2 family proteins in AML pathogenesis and prognosis (27–29),
small molecule BH3-mimetics that inhibit the anti-apoptotic
functions of Bcl-2 and Bcl-xL have been developed (15,18).
In clinical trials, ABT-263 significantly reduced tumour burden in
most patients with CLL as a single agent or in combination with
other conventional treatments (30,31).
Although thrombocytopenia limited the use of ABT-263 in patients,
the observed efficacy underscored the therapeutic potential of
selective Bcl-2 family inhibitors. ABT-199 was also investigated as
a single agent or in combination with other anti-cancer therapies
for CLL (20,32). Achievement of the primary end point
in the study led to the first successful US Food and Drug
Aministration (FDA) registration for ABT-199. Bai and colleagues
(25) identified APG-1252-12A as
an active metabolic product of APG-1252 in vivo, and bound
to Bcl-2 and Bcl-xl with sub-nanomalar affinities. In this study,
we tested the impact of Bcl-2/Bcl-xl dual inhibitor APG-1252-12A in
five leukemia cell lines. The results of MTS assay showed that
application of APG-1252-12A to leukemia cell lines significantly
inhibited cell proliferation. The IC50 was in low
nanomolar range, a range might be achievable in clinical trials. In
light of this observation, it is possible that the level of Bcl-2
family members might be related to sensitivity to APG-1252-12A.
We investigated whether the Bcl-2 protein level in
five leukemia cell lines was associated with the sensitivity to
APG-1252-12A. Leukemia cell lines express Bcl-2 with varied
expression of Bcl-xl. Though ABT-737 bound to Bcl-2 and Bcl-xl
proteins with similar affinities (14), it was surprising that the level of
Bcl-xl expression did not correlate with the sensitivity of AML
cells to APG-1252-12A. The levels of Bcl-xl and Mcl-1 had no
correlation with the drug sensitivity. It was found that increased
expression of Bcl-2 was associated with increased sensitivity to
Bcl-2 inhibitor which was similar to those previously reported
(10). The Bcl-2 protein level
correlated with cell line sensitivity to APG-1252-12A suggested an
on-target action of killing. The role of Bcl-2 in the survival of
tumor cells is well established, so the drugs that inhibited these
proteins might be useful therapeutically.
Herein, we demonstrated that HL-60 cells treated
with APG-1252-12A developed an accumulation of apoptotic cells. The
drug acted in a concentration- and time-dependent manner. Hoechst
staining and flow cytometry analysis of APG-1252-12A-treated HL-60
cells suggested the occurrence of apoptosis. Furthermore, the
effect of varying concentrations of inhibitors on cell cycle
distribution was determined by flow cytometric analysis. The
increase of the sub-G0/G1 phase of cells indicated typical late
stages of apoptosis.
Studies in cell lines and primary cells have
revealed that high expression of all anti-apoptotic Bcl-2 family
members, Bcl-2, Bcl-xl, Bcl-w, A1 and Mcl-1, were capable of
inhibiting the mitochondrial apoptotic pathway (33). We also investigated the mechanism
of antitumor activity of APG-1252-12A. Our findings are consistent
with the above studies that administration of APG-1252-12A to HL-60
cells rapidly induced hallmarks of apoptosis, including cytochrome
c release, caspase-3 and PARP-1 activation. Normally, releasing
cytochrome c from mitochondria to the cytoplasm is a critical
signal of caspase activation (34–36).
The occurrence of cytochrome c release suggested that APG-1252-12A
might induce in AML a form of apoptotic cell death that can include
caspase activation as an essential pathway. As MOMP and cytochrome
c release are usually viewed as characteristics of no return in
apoptosis, APG-1252-12A achieves a potent cell killing effect in
AML cell line. The Bcl-2 anti-apoptotic members are helical
proteins with an open groove that binds to the BH3 domain on the
proapoptotic partner. The anti-apoptotic Bcl-2-like proteins
provide a barrier against MOMP by binding proapoptotic BH3-only
protein (such as Bim and Bid) and keeping multidomain target (Bax
and Bak) in an inhibited state (37,38).
BH3 mimetic compounds, such as ABT-737 and ABT-263, binds to
anti-apoptotic Bcl-2 family proteins and liberates proapoptotic
BH3-only proteins. The proapoptotic BH3-only protein stimulate
apoptosis not only by binding anti-apoptotic Bcl-2-like proteins to
release Bax and Bak but also by directly activating Bax and Bak.
Previous discoveries revealed that Bax and Bak have important roles
in unleashing the effector phase of mitochondrial apoptosis and
must change shape to cause MOMP and apoptosis (39–41).
The ability of APG-1252-12A to induce HL-60
mitochondrial apoptosis was confirmed in vitro. These
findings have some implications for the investigation of
APG-1252-12A, suggesting that inhibiting Bcl-2 and Bcl-xl protein
could activate the intrinsic apoptotic pathway. Our work might
provide a foundation for studies in APG-1252-12A as a single agent
in vivo which can be exploited as a potential therapeutic
drug in AML.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (NSFC: 81101671) and Natural Science
Foundation of Guangdong Province (2016A030313280).
References
|
1
|
National Cancer Institute: SEER Stat Fact
Sheets: Acute Myeloid Leukemia (AML). NIH. http://seer.cancer.gov/statfacts/html/amyl.html.
Accessed May 19. 2016
|
|
2
|
Irish JM, Anensen N, Hovland R, Skavland
J, Borresen-Dale A-L, Bruserud O, Nolan GP and Gjertsen BT: Flt3
Y591 duplication and Bcl-2 overexpression are detected in acute
myeloid leukemia cells with high levels of phosphorylated wild-type
p53. Blood. 109:2589–2596. 2007. View Article : Google Scholar
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Delbridge AR, Grabow S, Strasser A and
Vaux DL: Thirty years of BCL-2: Translating cell death discoveries
into novel cancer therapies. Nat Rev Cancer. 16:99–109. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsujimoto Y, Cossman J, Jaffe E and Croce
CM: Involvement of the bcl-2 gene in human follicular lymphoma.
Science. 228:1440–1443. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cleary ML, Smith SD and Sklar J: Cloning
and structural analysis of cDNAs for bcl-2 and a hybrid
bcl-2/immunoglobulin transcript resulting from the t(14;18)
translocation. Cell. 47:19–28. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vaux DL, Cory S and Adams JM: Bcl-2 gene
promotes haemopoietic cell survival and cooperates with c-myc to
immortalize pre-B cells. Nature. 335:440–442. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang JZ, Sanger WG, Greiner TC, Staudt
LM, Weisenburger DD, Pickering DL, Lynch JC, Armitage JO, Warnke
RA, Alizadeh AA, et al: The t(14;18) defines a unique subset of
diffuse large B-cell lymphoma with a germinal center B-cell gene
expression profile. Blood. 99:2285–2290. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Minn AJ, Rudin CM, Boise LH and Thompson
CB: Expression of bcl-xL can confer a multidrug resistance
phenotype. Blood. 86:1903–1910. 1995.PubMed/NCBI
|
|
10
|
Pan R, Hogdal LJ, Benito JM, Bucci D, Han
L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, et al:
Selective BCL-2 inhibition by ABT-199 causes on-target cell death
in acute myeloid leukemia. Cancer Discov. 4:362–375. 2014.
View Article : Google Scholar
|
|
11
|
Ni Chonghaile T and Letai A: Mimicking the
BH3 domain to kill cancer cells. Oncogene. 27(Suppl 1): S149–S157.
2008. View Article : Google Scholar
|
|
12
|
Merino D, Khaw SL, Glaser SP, Anderson DJ,
Belmont LD, Wong C, Yue P, Robati M, Phipson B, Fairlie WD, et al:
Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737
and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood.
119:5807–5816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Del Gaizo Moore V, Schlis KD, Sallan SE,
Armstrong SA and Letai A: BCL-2 dependence and ABT-737 sensitivity
in acute lymphoblastic leukemia. Blood. 111:2300–2309. 2008.
View Article : Google Scholar
|
|
14
|
Oltersdorf T, Elmore SW, Shoemaker AR,
Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges
J, Hajduk PJ, et al: An inhibitor of Bcl-2 family proteins induces
regression of solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tse C, Shoemaker AR, Adickes J, Anderson
MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et
al: ABT-263: A potent and orally bioavailable Bcl-2 family
inhibitor. Cancer Res. 68:3421–3428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gavathiotis E, Suzuki M, Davis ML, Pitter
K, Bird GH, Katz SG, Tu H-C, Kim H, Cheng EH-Y, Tjandra N, et al:
BAX activation is initiated at a novel interaction site. Nature.
455:1076–1081. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Konopleva M, Contractor R, Tsao T, Samudio
I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi Y-X, Sneed T, et al:
Mechanisms of apoptosis sensitivity and resistance to the BH3
mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 10:375–388.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Souers AJ, Leverson JD, Boghaert ER,
Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH,
Fairbrother WJ, et al: ABT-199, a potent and selective BCL-2
inhibitor, achieves antitumor activity while sparing platelets. Nat
Med. 19:202–208. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vandenberg CJ and Cory S: ABT-199, a new
Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive
Myc-driven mouse lymphomas without provoking thrombocytopenia.
Blood. 121:2285–2288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roberts AW, Davids MS, Pagel JM, Kahl BS,
Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR,
Gressick L, et al: Targeting BCL2 with venetoclax in relapsed
chronic lymphocytic leukemia. N Engl J Med. 374:311–322. 2016.
View Article : Google Scholar
|
|
21
|
San-Miguel JF: Consolidation therapy in
myeloma: A consolidated approach? Blood. 120:2–3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roberts AW, Advani RH, Kahl BS, Persky D,
Sweetenham JW, Carney DA, Yang J, Busman TB, Enschede SH,
Humerickhouse RA, et al: Phase 1 study of the safety,
pharmacokinetics, and anti-tumour activity of the BCL2 inhibitor
navitoclax in combination with rituximab in patients with relapsed
or refractory CD20 lymphoid malignancies. Br J Haematol.
170:669–678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li T, Yuan G, Zhang L, Ye L, Li S, Fan Y
and Sun J: ApoG2 inhibits the antiapoptotic protein, Mcl1, and
induces mitochon-driadependent apoptosis in human colorectal cancer
cells. Mol Med Rep. 12:6976–6984. 2015.PubMed/NCBI
|
|
24
|
Ye L, Yuan G, Xu F, Sun Y, Chen Z, Chen M,
Li T, Sun P, Li S and Sun J: The small-molecule compound BM-1197
inhibits the antiapoptotic regulators Bcl-2/Bcl-xL and triggers
apoptotic cell death in human colorectal cancer cells. Tumour Biol.
36:3447–3455. 2015. View Article : Google Scholar
|
|
25
|
Bai L, Chen J, Liu L, McEachern D, Aguilar
A, Zhou H, Yang CY, Wang H, Wen J, Wang G, et al: BM-1252
(APG-1252): A potent dual specific Bcl-2/Bcl-xL inhibitor that
achieves complete tumor regression with minimal platelet toxicity.
Eur J Cancer. 50:109–110. 2014. View Article : Google Scholar
|
|
26
|
Wang H, Wang G, Du Z, Wu M, McEachern D,
Aguilar A, Lin Y, Lin X, Wen J, Gu L, et al: Preclinical studies of
a dual Bcl-2/Bcl-xL inhibitor APG-1252 with strong anti-tumor
efficacy and significantly reduced platelet toxicity. Eur J Cancer.
50:176–177. 2014. View Article : Google Scholar
|
|
27
|
Glaser SP, Lee EF, Trounson E, Bouillet P,
Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, et
al: Anti-apoptotic Mcl-1 is essential for the development and
sustained growth of acute myeloid leukemia. Genes Dev. 26:120–125.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Högstrand K, Hejll E, Sander B, Rozell B,
Larsson LG and Grandien A: Inhibition of the intrinsic but not the
extrinsic apoptosis pathway accelerates and drives MYC-driven
tumorigenesis towards acute myeloid leukemia. PLoS One.
7:e313662012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Del Poeta G, Venditti A, Del Principe MI,
Maurillo L, Buccisano F, Tamburini A, Cox MC, Franchi A, Bruno A,
Mazzone C, et al: Amount of spontaneous apoptosis detected by
Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML).
Blood. 101:2125–2131. 2003. View Article : Google Scholar
|
|
30
|
Roberts AW, Seymour JF, Brown JR, Wierda
WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DCS, Xiong H, et al:
Substantial susceptibility of chronic lymphocytic leukemia to BCL2
inhibition: Results of a phase I study of navitoclax in patients
with relapsed or refractory disease. J Clin Oncol. 30:488–496.
2012. View Article : Google Scholar
|
|
31
|
Ackler S, Mitten MJ, Foster K, Oleksijew
A, Refici M, Tahir SK, Xiao Y, Tse C, Frost DJ, Fesik SW, et al:
The Bcl-2 inhibitor ABT-263 enhances the response of multiple
chemotherapeutic regimens in hematologic tumors in vivo. Cancer
Chemother Pharmacol. 66:869–880. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stilgenbauer S, Eichhorst B, Schetelig J,
Coutre S, Seymour JF, Munir T, Puvvada SD, Wendtner C-M, Roberts
AW, Jurczak W, et al: Venetoclax in relapsed or refractory chronic
lymphocytic leukaemia with 17p deletion: A multicentre, open-label,
phase 2 study. Lancet Oncol. 17:768–778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar
|
|
34
|
Liu X, Kim CN, Yang J, Jemmerson R and
Wang X: Induction of apoptotic program in cell-free extracts:
Requirement for dATP and cytochrome c. Cell. 86:147–157. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: A
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Willis SN and Adams JM: Life in the
balance: How BH3-only proteins induce apoptosis. Curr Opin Cell
Biol. 17:617–625. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and
Korsmeyer SJ: Proapoptotic BAX and BAK: A requisite gateway to
mitochondrial dysfunction and death. Science. 292:727–730. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Czabotar PE, Westphal D, Dewson G, Ma S,
Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al:
Bax crystal structures reveal how BH3 domains activate Bax and
nucleate its oligomerization to induce apoptosis. Cell.
152:519–531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dewson G and Kluck RM: Mechanisms by which
Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci.
122:2801–2808. 2009. View Article : Google Scholar : PubMed/NCBI
|