Introduction
Colorectal cancer is one of the most common cancers
in the world. Patients in advanced stages with metastatic lesions
appear to have a poor prognosis (1). Clinical studies have found that
colorectal cancer cells prefer to metastasize to the liver over
other organs, which results in poor prognosis. Therefore, it is
important to identify and understand the factors involved in the
progression of colorectal cancer metastasis to the liver.
PRL-3 belongs to the family of protein tyrosine
phosphatases (PTPs), which has been demonstrated to play an
important role in colorectal cancer metastasis in the liver
(2). PTPs regulate phosphorylation
of many important signaling molecules that are involved in cell
proliferation, migration and prognosis (3). PRL-3 is normally expressed in heart
and skeletal muscle. However, studies have found that PRL-3 is
significantly overexpressed in metastatic cells and is moderately
expressed in primary lesions of colorectal cancer (4). Moreover, the expression of PRL-3 in
primary colorectal cancer lesions indicates poor prognosis and
shortened survival (5). Therefore,
PRL-3 appears to be a biomarker for colorectal cancer, and, in
particular, a biomarker for colorectal cancer liver metastasis.
However, the mechanisms of regulating liver metastasis are still
uncertain. Our previous research found that PRL-3 promoted
colorectal cancer cell proliferation through TNF-α secretion, which
also induced the activation of a Ca2+-activated
K+ channel (KCNN4) (6).
We demonstrated that PRL-3 facilitated epithelial mesenchymal
transition (EMT) in colorectal cancer cells (7), indicating that PRL-3 strongly
influenced the biological characteristics of tumor cells. Moreover,
we studied the tumor microenvironment and found that PRL-3
integrated with tumor associated macrophages (TAMs), induced TAM
secreting inflammatory cytokines such as IL-6 and IL-8, which then
enhanced colorectal cancer cell invasion (8).
Tumor cell growth and proliferation require large
quantities of bioenergy and biomaterials. In recent years,
increasing number of studies have found that an important hallmark
of cancer cells is metabolism reprogramming, which was first
proposed by Warburg (termed the Warburg effect) in the 1920s
(9,10). Unlike normal cells, most cancer
cells exhibit a high rate of glycolysis rather than oxidative
phosphorylation and therefore produce large amounts of lactate,
leading to a decrease in extracellular pH, which facilitates cancer
cell removal (11). The activation
of glycolytic pathways promotes tumor cells to adapt to fast
proliferation. Furthermore, glycolysis-associated enzymes and
molecules are highly expressed in tumor cells, which also play an
important role in cancer (12).
Inflammation is an important risk factor for
colorectal cancer, but the mechanisms underlying this effect of
inflammation on colorectal cancer cells are still not fully
understood. It has been indicated that tumor associated
inflammation may affect the proliferation, metastasis and
angiogenesis of tumor cells (13).
Notably, our previous study also found that inflammatory cytokines
IL-6 and IL-8, which were secreted by TAMs, enhanced colorectal
cancer cells invasion (8).
However, the association between inflammatory cytokines and
glycolysis metabolism is still uncertain.
In the present study, we aimed to determine whether
PRL-3 is involved in the metabolism reprogramming of colorectal
cancer cells. This investigation revealed that PRL-3 promotes
glycolysis through secretion of IL-8 in colorectal cancer cells,
leading to an increase of glucose consumption and lactate
production, reduced intercellular reactive oxygen species (ROS)
levels and induced overexpression of glycolysis enzymes and
molecules, contributing to enhanced tumor cell proliferation and
invasion.
Materials and methods
Samples and patients
Colorectal cancer cell samples were collected from
47 patients admitted to the Department of Gastroenteropancreatic
Surgery of Sun Yat-sen Memorial Hospital, Sun Yat-sen University,
between 2013 and 2016. Specimens were collected immediately after
tumor removal. All samples were collected with informed consent
according to the Internal Review and the Ethics Boards of the Sun
Yat-sen Memorial Hospital of Sun Yat-sen University. The protocol
was approved by the Ethics Committee of Sun Yat-sen Memorial
Hospital.
Cell cultures and treatments
LoVo colorectal cancer cells were purchased from the
Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai,
China). Cells were transfected with PAcGFP-PRL-3 (LoVo-P) or PAcGFP
(LoVo-C) using Lipofectamine 3000. Cells were stored at Sun Yat-sen
Memorial Hospital (6). Cells were
cultured in RPMI-1640 medium and 10% fetal bovine serum (FBS), with
100 mg/ml penicillin. The cells were incubated at 37°C, 5%
CO2 in a humidified atmosphere.
Reagents and antibodies
Lipofectamine 3000 was purchased from Sigma-Aldrich
(St. Louis, MO, USA). Fetal bovine serum (FBS) was purchased from
Biological Industries (Kibbutz Beit Haemek, Israel). RPMI was
purchased from Invitrogen (Carlsbad, CA, USA). TRIzol and Prime
Script RT were purchased from Takara Bio (Dalian, China). The siRNA
was purchased from Shanghai GenePharma, Co., Ltd. (Shanghai,
China). Antibodies against GAPDH (cat. no. ab8245), IL-8 (cat. no.
ab18672), Glut1 (cat. no. ab115730), PKM2 (cat. no. ab38237), HK2
(cat. no. ab104836), LDHA (cat. no. ab125683) were purchased from
Abcam (Cambridge, MA, USA).
Western blot assay
Cells were washed with phosphate-buffered saline
(PBS) and then lysed on ice with RIPA buffer containing 1% PMSF.
The Bradford assay was used to detect protein concentration.
Denatured proteins were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis, transferred to PVDF
membranes and then blocked in 5% non-fat milk. Membranes were
washed 3 times with Tris-buffered saline + 0.1% Tween-20 (TBST),
incubated overnight at 4°C with relevant primary antibodies, and
then washed and incubated with horseradish peroxidase-conjugated
secondary antibodies for 1 h at room temperature. Labeled proteins
and relative band intensities were visualized and measured with
Quantity One software (Bio-Rad Laboratories, Hrercules, CA,
USA).
RNA extraction and real-time quantitative
RT-PCR
Total RNA was isolated using TRIzol reagent, and 500
ng RNA was reverse transcribed using PrimeScript RT according to
the manufacturer's protocol. Quantitative real-time RT-PCR was
performed using the LightCycler 480 (Roche, Basel, Switzerland) and
SYBR assays (Takara Bio). Primers were designed to detect GAPDH,
PRL-3, Glut1, PKM2, HK2, LDHA and IL-8. The primers used for
qRT-PCR are shown in Table I. Each
sample contained 1X SYBR Premix Ex Taq™, 0.2 μM of each
forward and reverse primer and 500 ng template cDNA in a final
volume of 20 μl. Cycling parameters were set as follows:
denaturation at 95°C for 30 sec, followed by 40 amplification
cycles (95°C for 5 sec and 60°C for 20 sec). For relative
quantification, 2−ΔCt was used to calculate the fold
change in gene expression. All experiments were performed in
triplicate.
 | Table IOligonucleotide sequence of qRT-PCR
primers. |
Table I
Oligonucleotide sequence of qRT-PCR
primers.
| Gene | Forward primer | Reverse primer | Amplicon |
|---|
| GAPDH |
AATGGGCAGCCGTTAGGAAA |
GCGCCCAATACGACCAAATC | 168 |
| PRL-3 |
ACACATGCGCTTCCTCATCA |
GTCACTTCACACACACGCAC | 111 |
| Glut1 |
GGCTTCTCCAACTGGACCTC |
CCGGAAGCGATCTCATCGAA | 176 |
| HK2 |
CAAGAAGCTCCCACTGGGTT |
CAACGTCTCTGCCTTCCACT | 122 |
| PKM2 |
GTCTGGGAGGAAAGTCGCTC |
GGCGGAAGGACACAGATTCA | 104 |
| LDHA |
CATGGCCTGTGCCATCAGTA |
AGATATCCACTTTGCCAGAGACA | 158 |
| IL-8 |
CCACCGGAAGGAACCATCTC |
TTCCTTGGGGTCCAGACAGA | 279 |
Glycolysis consumption and lactate
production
Glucose and lactate assay kits were purchased from
Sigma-Aldrich to determine the concentrations of glucose and
lactate in the culture medium, respectively. Cells were seeded on
6-well plates at a density of 1×105 cells/well and the
medium was changed to DMEM after incubation overnight. The
concentrations of glucose and lactate were measured according to
the manufacturer's protocol.
Measurement of intracellular reactive
oxygen species (ROS)
Intracellular ROS levels were detected by
H2DCF-DA (Invitrogen). The cells were cultured in a
96-well plate. Cells were washed with PBS before incubation with
H2DCF-DA for 30 min, and ROS levels were examined at
excitation and emission wavelength of 485 and 520 nm. Cell numbers
were normalized before the measurement.
Inflammatory cytokine array analysis
The RayBio Cytokine Analysis array (RayBiotech,
Norcross, GA, USA), consisting of 40 different inflammatory
cytokine antibodies spotted onto a membrane, was used in the
present study. Cytokine array membranes were blocked for 30 min and
then incubated with samples at 37°C for 1 h. Then membranes were
washed and incubated with diluted biotin-conjugated antibodies at
37°C for 2 h. After the membranes were washed, 1,000-fold diluted
horseradish peroxidase-conjugated streptavidin was added and
incubation was continued for 2 h. Membranes were then washed
thoroughly and exposed to detection buffer in the dark. By
comparing the signal intensities, relative expression levels of
cytokines were made. The intensities of signals were quantified by
densitometry.
Immunohistochemistry
Using primary antibodies against PRL-3, Glut1, PKM2,
HK2, LDHA and IL-8, the tissue slides were incubated overnight at
room temperature. Secondary staining with Alexa Fluor 555
conjugated donkey anti-rabbit and Alexa Fluor 488 conjugated goat
anti-mouse secondary antibodies was performed at room temperature
for 60 min. Images were taken with a Zeiss LSM 700 laser scanning
microscope (Carl Zeiss, Oberkochen, Germany) with a core data
acquisition system (Applied Precision, Bratislava, Slovakia). For
control experiments, primary antibody was substituted with normal
rabbit serum.
Wound scratch test and invasion
assays
Cell migration was measured by the movement of cells
into a scraped area created by the tip of a 200 μl pipette.
The degree of 'wound closure' was examined after 24 and 48 h. After
cell adherence, the remaining gap was then measured using light
microscopy and quantified. Invasion assays were performed using
105 cells/well added to a Matrigel invasion chamber. FBS
was added to the lower chamber, and the number of cells invaded
from the top chamber after 24 and 48 h was measured with a
spectrophotometer.
Statistical analysis
Statistical analysis was performed using the SPSS.
Data from three separate experiments were reported as the means ±
SD. Statistical significance between the samples was assessed by
the Student's-t test where P<0.05 was considered to be
statistically significant.
Results
PRL-3 promotes glycolysis in colorectal
cancer cells
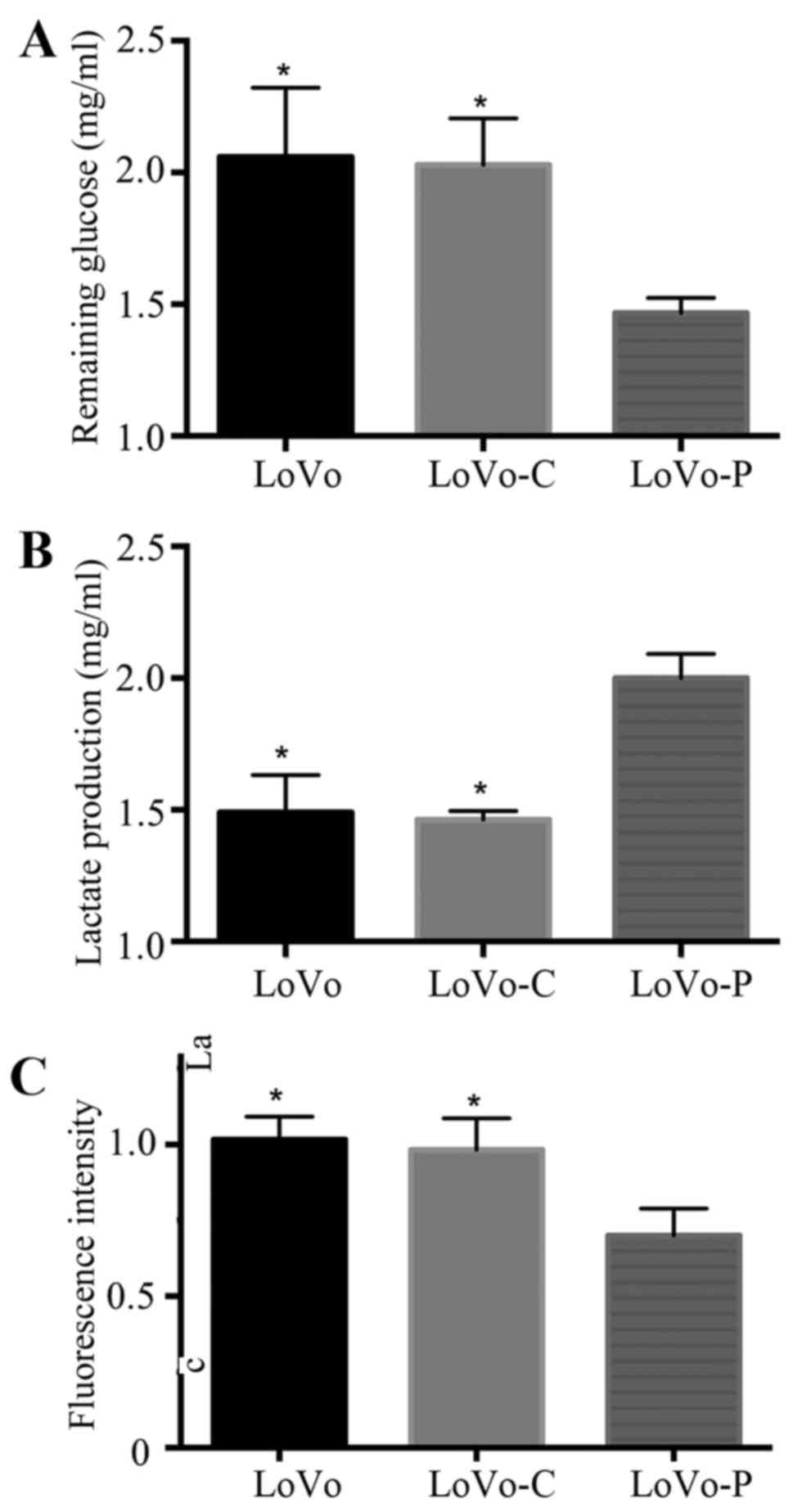
Tumor cells reprogram their metabolism from
oxidative phosphorylation to glycolysis to meet huge energy demands
and for large amounts of biomass (14). A glycolytic feature is often
characterized by glucose assumption, lactate production and reduced
intracellular ROS levels. To examine the role of PRL-3 in
glycolysis of colorectal cancer cells, we used glucose consumption
assays, lactate production assays and intracellular ROS measurement
assays to analyze glycolysis in PRL-3 overexpressed colorectal
cancer cells (LoVo-P), control cells (LoVo-C) and wild-type cells
(LoVo). We found that overexpression of PRL-3 promoted glucose
consumption, lactate production and reduced the intracellular ROS
levels. However, LoVo-C and LoVo cells did not exhibit the same
trends (Fig. 1).
PRL-3 promotes Glut1, HK2, PKM2 and LDHA
expression in colorectal cancer cells
The progression of glycolysis contains more than ten
metabolic reactions that are catalyzed by a number of enzymes or
molecules. For example, Glut1 transports glucose across the plasma
membrane, which plays an important role in rate-limiting glucose
metabolism. Additionally, HK2, PKM2 and LDHA are important
rate-limiting enzymes in glycolysis. To examine whether
glycolysis-associated molecules and enzymes may be regulated by
PRL-3 in colorectal cancer cells, we analyzed mRNA expressions of
Glut1, HK2, PKM2 and LDHA. Compared to LoVo-C and LoVo cells, the
data showed that expression of Glut1, HK2, PKM2 and LDHA were all
increased in LoVo-P cells (Fig.
2A). Consistent with these results, western blot analysis also
showed the same trends (Fig. 2B and
C).
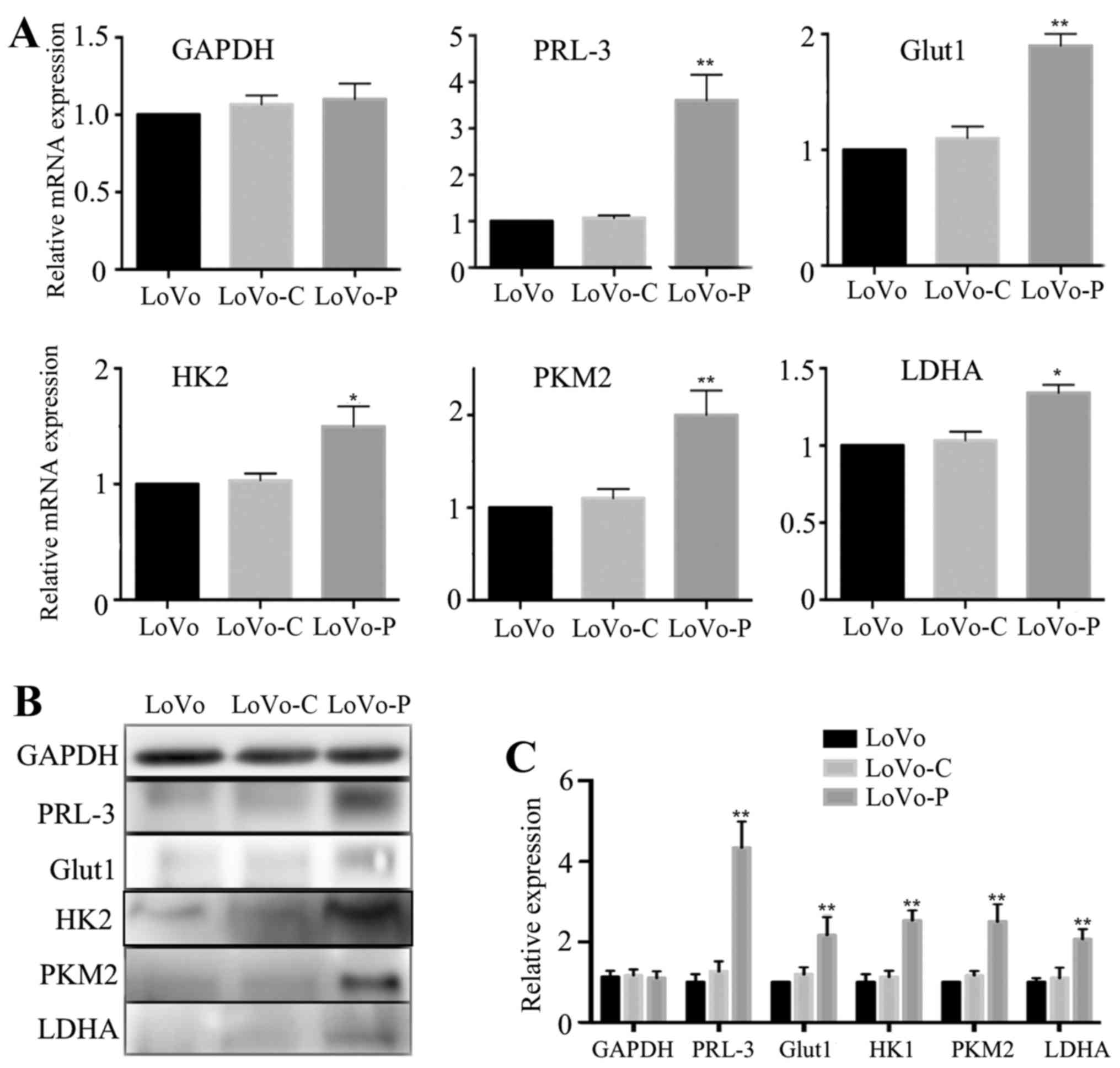 | Figure 2PRL-3 promotes Glut1, HK2, PKM2 and
LDHA expression in colorectal cancer cells. (A) GAPDH, PRL-3,
Glut1, HK2, PKM2 and LDHA mRNA expression levels in LoVo, LoVo-C
and LoVo-P cells. (B and C) GAPDH, PRL-3, Glut1, HK2, PKM2 and LDHA
protein expression levels in LoVo, LoVo-C and LoVo-P cells.
*P<0.05, **P<0.01. |
PRL-3 improves IL-8 expression in
colorectal cancer cells
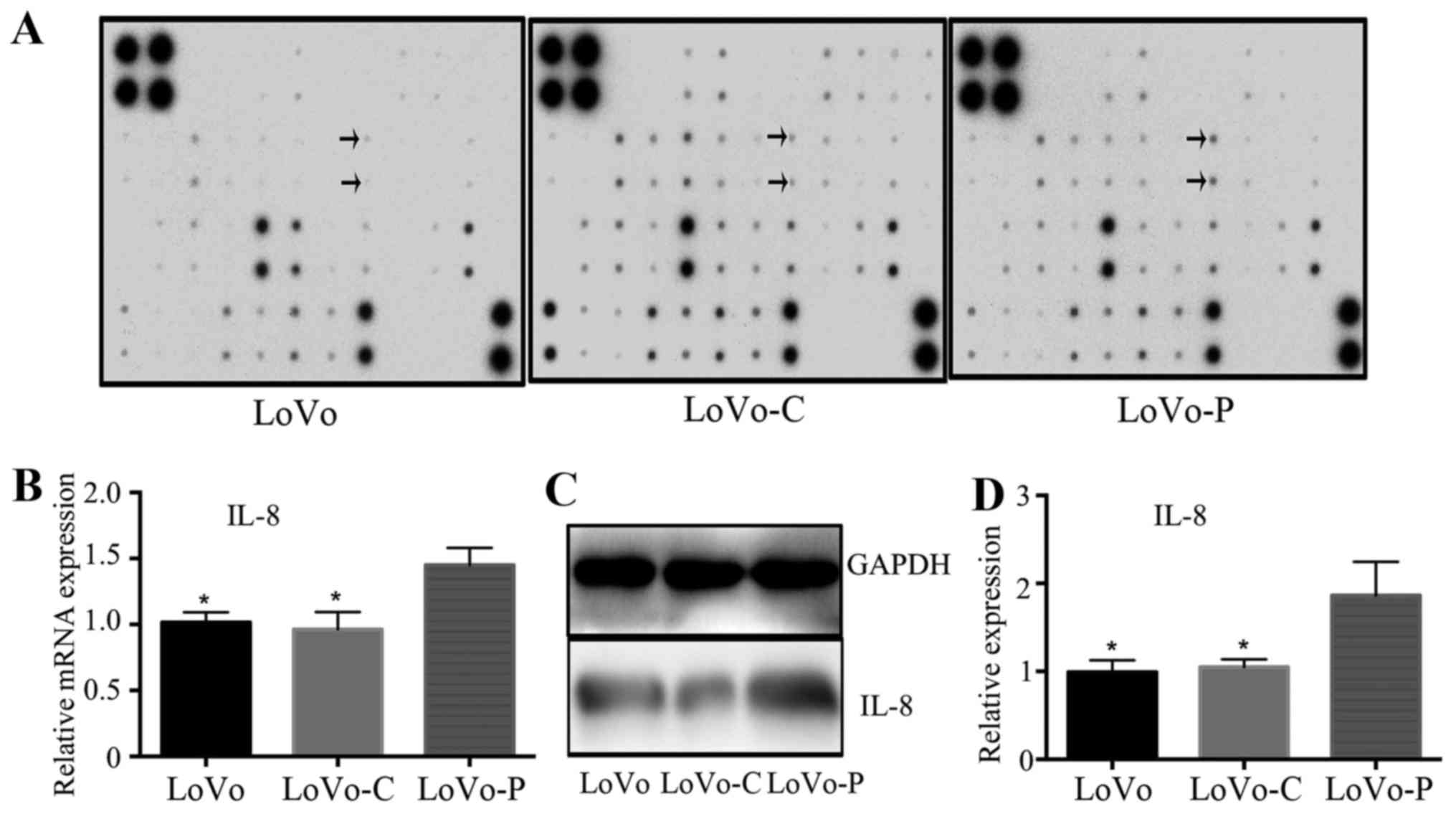
We next explored the inflammatory cytokine
expression in supernatants by using inflammatory cytokine antibody
array to find the expression differences between LoVo-P, LoVo-C and
LoVo cells. Each membrane contained 40 inflammatory cytokines
(Table II), relative inflammatory
cytokine expression was compared between between LoVo-P, LoVo-C and
LoVo cells separately. Results showed significant upregulation of
IL-8 expression in LoVo-P cells (Fig.
3A). RT-PCR showed that IL-8 gene was upregulated in LoVo-P
cells (Fig. 3B). In line with the
mRNA level, the protein level of IL-8 showed the same trends
(Fig. 3C and D). These data
suggested that PRL-3 improved the expression of IL-8 in colorectal
cancer cells.
| Table IIInflammatory cytokines on the
membrane. |
Table II
Inflammatory cytokines on the
membrane.
| A | B | C | D | E | F | G | H | I | J | K | L |
|---|
| 1 | POS | POS | NEG | NEG | Eotaxin | Eotaxin-2 | G-CSF | GM-CSF | ICAM1 | IFN-γ | I-309 | IL-1α |
| 2 | POS | POS | NEG | NEG | Eotaxin | Eotaxin-2 | G-CSF | GM-CSF | ICAM1 | IFN-γ | I-309 | IL-1α |
| 3 | IL-1β | IL-2 | IL-3 | IL-4 | IL-6 | IL-6sR | IL-7 | IL-8 | IL-10 | IL-11 | IL-12p40 | IL-12p70 |
| 4 | IL-1β | IL-2 | IL-3 | IL-4 | IL-6 | IL-6sR | IL-7 | IL-8 | IL-10 | IL-11 | IL-12p40 | IL-12p70 |
| 5 | IL-13 | IL-15 | IL-16 | IL-17 | IP-10 | MCP-1 | MCP-2 | M-CSF | MIG | MIP-1α | MIP-1β | MIP-1δ |
| 6 | IL-13 | IL-15 | IL-16 | IL-17 | IP-10 | MCP-1 | MCP-2 | M-CSF | MIG | MIP-1α | MIP-1β | MIP-1δ |
| 7 | RANTES | TGF-β | TNF-α | TNF-β | sTNF-RI | sTNF-RII | PDGF-BB | TIMP-2 | BLANK | BLANK | NEG | POS |
| 8 | RANTES | TGF-β | TNF-α | TNF-β | sTNF-RI | sTNF-RII | PDGF-BB | TIMP-2 | BLANK | BLANK | NEG | POS |
IL-8 mediates the promotion of glycolysis
in colorectal cancer cells
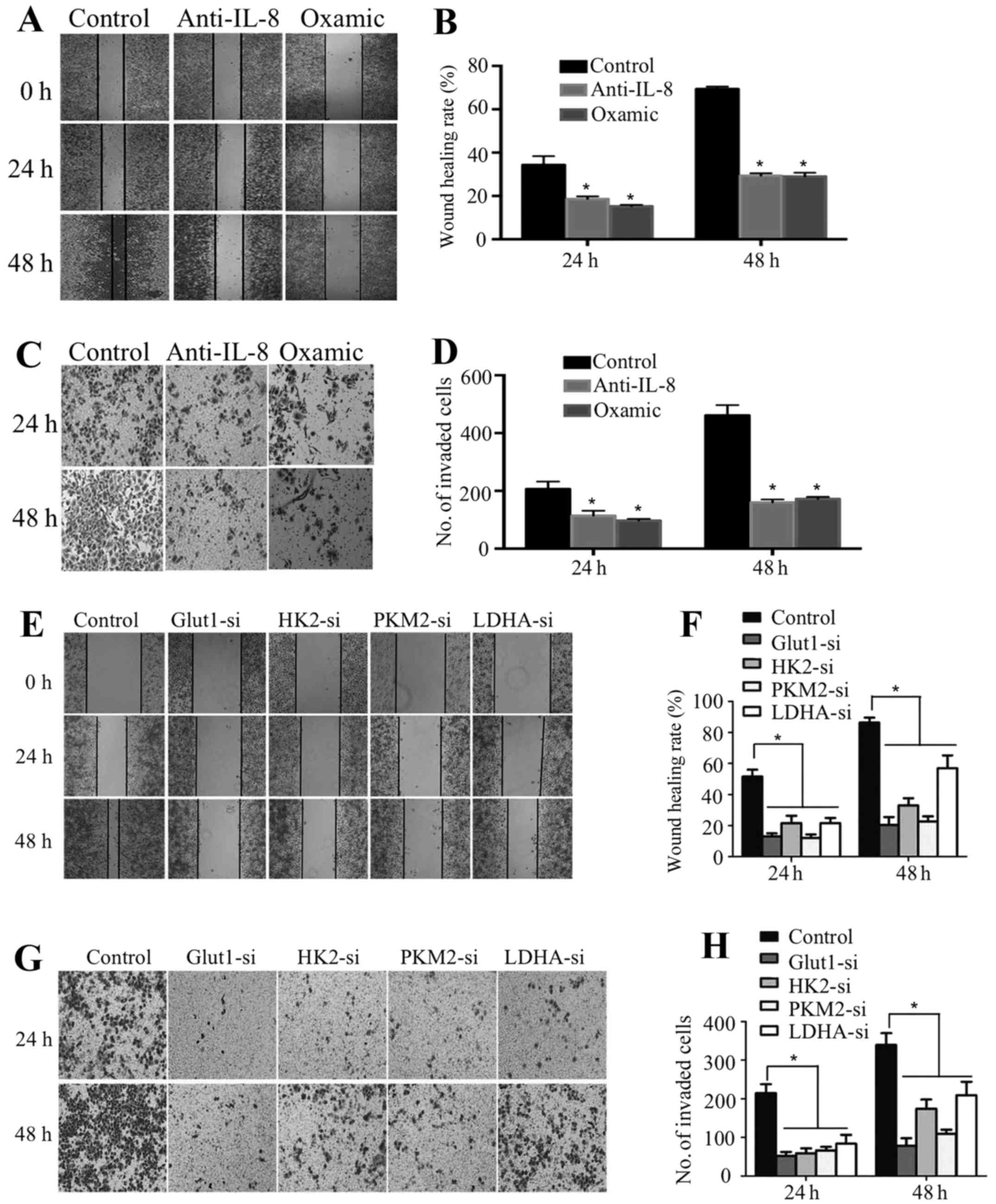
To explore whether IL-8 mediates the promotion of
glycolysis, we used anti-IL-8 antibody to neutralize IL-8 function.
The addition of anti-IL-8 antibody to the culture medium increased
the remaining glucose, reduced the lactate production and increased
intracellular ROS levels of cancer cells in a dose-dependent
manner, whereas an isotype-matched IgG (10 μg/ml) did not
have same effects (Fig. 4A–C),
indicating the role of IL-8 in colorectal cancer cells glycolysis.
In order to examine whether PRL-3 improves glycolysis related
molecules and enzymes through IL-8, we examined the effect of IL-8
on the expression of Glut1, HK2, PKM2 and LDHA in LoVo-P cells.
RT-PCR and western blot analysis showed these molecules and enzymes
were significantly reduced after anti-IL-8 antibody was added into
the culture medium (Fig. 4D and
F).
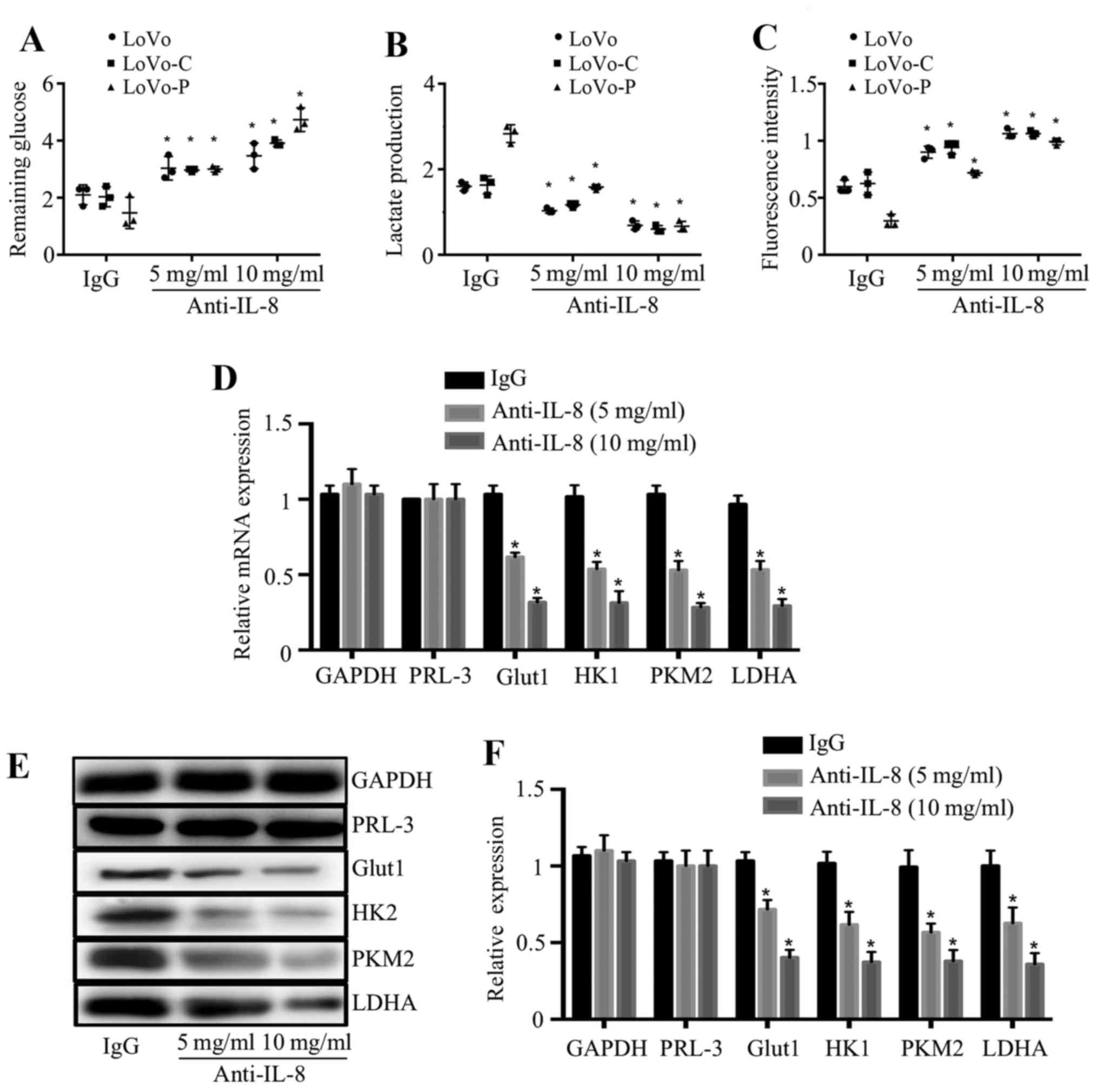 | Figure 4IL-8 mediates the promotion of
glycolysis in colorectal cancer cells. (A–C). Remaining glucose,
lactate production and intracellular ROS levels were detected in
LoVo, LoVo-C, LoVo-P cells which were pretreated with an
isotype-matched IgG control (IgG, 10 mg/ml) or anti-IL-8 antibody
at 5 or 10 mg/ml. (D) mRNA expression of GAPDH, PRL-3, Glut1, HK2,
PKM2 and LDHA in LoVo-P cells which were pretreated with anti-IL-8
antibody at 5 or 10 mg/ml. (E and F) Protein expression of GAPDH,
PRL-3, Glut1, HK2, PKM2 and LDHA in LoVo-P cells which were
pretreated with anti-IL-8 antibody at 5 or 10 mg/ml.
*P<0.05. |
PRL-3 improves growth and invasion via
glycolysis in colorectal cancer cells
Lactate and glycolysis-associated enzymes and
molecules have been found to play important roles in improving
cancer cell metastasis (15–19).
To identify the role of PRL-3 induced glycolysis through IL-8 on
colorectal cancer cell growth and invasion, we inhibited lactate by
pretreating colorectal cancer cells with oxamic acid, or we
inhibited Glut1, HK2, PKM2 or LDHA expression by siRNA, or we
inhibited IL-8 by pretreating anti-IL-8 antibody. We found that
LoVo-P cells exhibited decreased motility and invasion when oxamic
acid or anti-IL-8 antibody was added (Fig. 5A–D). Moreover, our data also showed
decreased motility and invasion of LoVo-P cells when the expression
of Glut1, HK2, PKM2 or LDHA was inhibited (Fig. 5E–H).
Correlation between PRL-3 and Glut1, HK2,
PKM2, LDHA and IL-8 in CRC patients
To explore the association between PRL-3, IL-8,
Glut1, HK2, PKM2 and LDHA in clinical patient tissues, we performed
IHC and scored the results of 47 patients with colorectal cancer.
We first analyzed the expression of PRL-3 in tissues from clinical
colorectal carcinoma samples. Consistent with our previous
research, PRL-3 was rarely expressed in adjacent normal colorectal
lesions but was overexpressed in colorectal carcinomas lesions.
Furthermore, we analyzed the protein expression of Glut1, HK2,
PKM2, LDHA and IL-8 and found that they were overexpressed in tumor
tissues and positively correlated with PRL-3 expression (Fig. 6A). IHC scores of PRL-3, Glut1, HK2,
PKM2, LDHA and IL-8 were remarkably higher in tumor tissues than
those in normal adjacent tissues (Fig.
6B and Table III).
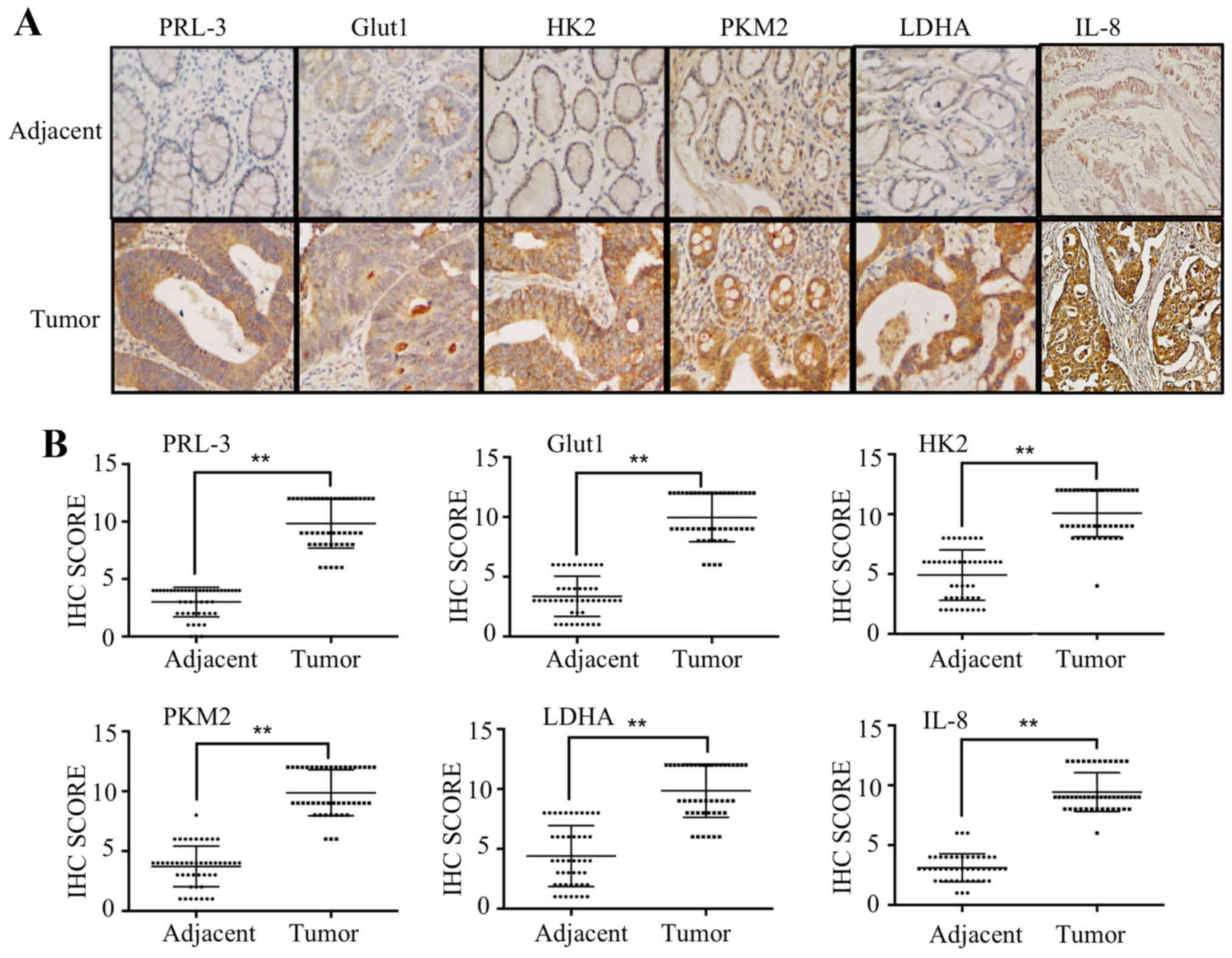 | Figure 6Correlation between PRL-3 and Glut1,
HK2, PKM2, LDHA, IL-8 in CRC patients. (A) The expression of PRL-3,
Glut1, HK2, PKM2, LDHA and IL-8 in human adjacent normal and tumor
tissues was evaluated by immunohistochemistry. (B) IHC scores of
PRL-3, Glut1, HK2, PKM2, LDHA and IL-8 in 47 tumor and
corresponding adjacent normal tissues. *P<0.05. |
| Table IIIAssociation of PRL-3 and Glut1, HK2,
PKM2, LDHA, IL-8 expression in 47 colorectal cancer patients. |
Table III
Association of PRL-3 and Glut1, HK2,
PKM2, LDHA, IL-8 expression in 47 colorectal cancer patients.
| Adjacent
tissues | Tumor tissues |
|---|
| Number | 47 | 47 |
| PRL-3
expression | | |
| No staining | 3 | 0 |
| Weak staining | 44 | 0 |
| Intermediate
staining | 0 | 14 |
| Strong
staining | 0 | 43 |
| IHC score, mean ±
SE | 3.0±1.3 | 9.8±2.1 |
| P-value | <0.05 | |
| Glut1
expression | | |
| No staining | 0 | 0 |
| Weak staining | 37 | 4 |
| Intermediate
staining | 10 | 6 |
| Strong
staining | 0 | 37 |
| IHC score, mean ±
SE | 3.4±1.7 | 9.9±2.0 |
| P-value | <0.05 | |
| HK2 expression | | |
| No staining | 0 | 0 |
| Weak staining | 20 | 1 |
| Intermediate
staining | 27 | 10 |
| Strong
staining | 0 | 36 |
| IHC score, mean ±
SE | 4.9±2.1 | 10.1±2.0 |
| P-value | <0.05 | |
| PKM2
expression | | |
| No staining | 0 | 0 |
| Weak staining | 34 | 0 |
| Intermediate
staining | 13 | 9 |
| Strong
staining | 0 | 38 |
| IHC score, mean ±
SE | 3.7±1.7 | 9.9±2.0 |
| P-value | <0.05 | |
| LDHA
expression | | |
| No staining | 0 | 0 |
| Weak staining | 25 | 0 |
| Intermediate
staining | 22 | 14 |
| Strong
staining | 0 | 33 |
| IHC score, mean ±
SE | 4.4±2.5 | 9.9±2.2 |
| P-value | <0.05 | |
| IL-8
expression | | |
| No staining | 0 | 0 |
| Weak staining | 44 | 0 |
| Intermediate
staining | 3 | 10 |
| Strong
staining | 0 | 37 |
| IHC score, mean ±
SE | 3.1±1.2 | 9.4±1.6 |
| P-value | <0.05 | |
Discussion
Reprogrammed metabolism, which fuels tumor cells
replication, growth and invasion, was added to the hallmarks of
cancer (20). Many studies have
explored the mechanisms of tumor cell unlimited growth and altered
metabolism. In the present study, we found that PRL-3 improves
glycolysis of colorectal cancer cells, which contributes to cancer
cells proliferation and invasion in vitro. Our previous
research revealed that inflammatory cytokine IL-8, which was
secreted by tumor associated macrophage, promoted colorectal cancer
cell invasion (8). Our current
experiments showed that PRL-3 improved IL-8 expression in
colorectal cancer cells, and IL-8 participates in the promotion of
glycolysis by PRL-3. To the best of our knowledge, this is the
first report indicating the association between PRL-3 and tumor
metabolism reprogram, furthermore, our research uncovered the role
of inflammatory cytokine IL-8 in glycolysis.
Various research has been made into the key steps of
metastatic process influenced by PRL-3. For example, PRL-3
repressed various target genes which participate in cell cycle
arrest to give cell unlimited proliferative advantage (21). PRL-3 promotes PI3K-AKT activity,
which is an important driver of cell proliferation and survival
(22). PRL-3 has been associated
in regulation of focal adhesion components, such as Src, integrin
and paxillin, which induced cell motility (23). PRL-3 promoted cell invasion by
increasing MMP2 activity (23),
induced EMT by acting upstream of PI3K/AKT signaling (24). PRL-3 increased the expression of
VEGF and promoted tumor cell angiogenesis (25). However, little attention has been
given to the relationship between PRL-3 and tumor metabolism
reprogram. In the present study, remaining glucose in the culture
medium of LoVo-P cells is less than LoVo and LoVo-C cells, which
means LoVo-P cells consumed more glucose. The culture medium did
not contain FBS to eliminate possible interference caused by cell
growth rate. Besides, lactate production level was significantly
higher and intracellular ROS level was lower in LoVo-P cells,
indicating the function of PRL-3 in improving colorectal cancer
cell glycolysis. Moreover, we also found the expression of
glycolysis related molecules and enzymes Glut1, HK2, PKM2 and LDHA
were increased in LoVo-P cells, these findings provide evidence
that PRL-3 promotes colorectal cancer cell glycolysis.
Our previous research showed the association between
PRL-3 and inflammation in tumor microenvironment. Tumor associated
macrophages secreted IL-6 and IL-8 enhanced colorectal cancer cells
invasion, however, the mechanism remained unclear. Previous
research on the association between inflammation and cancer mainly
focused on tumor growth, angiogenesis, EMT, invasion, colonization
and recruitment (26–31). Besides, activation of several
signal pathways was found to be involved in chronic inflammation
such as NF-κB (32). Notably,
recent studies revealed that activation of NF-κB increased
glycolysis in the inflammatory environment (33). In the present study, we integrated
the association between IL-8 and glycolysis. Inflammatory cytokine
antibody array showed upregulation of IL-8 in LoVo-P cells,
suggesting correlation between PRL-3 and IL-8. Furthermore,
glycolysis of colorectal cancer cells was inhibited when IL-8 was
neutralized, and inhibition was more significant in LoVo-P cells,
indicating the important role of IL-8 in improving colorectal
cancer cell glycolysis by PRL-3. Tumor metabolism reprogram
available cell proliferation and even invasion since glycolysis
produces more biological materials than oxidative phosphorylation
(34). This study confirmed the
function of glycolysis in colorectal cancer cell proliferation and
invasion. Various research has explored the correlation between
glycolysis and metastasis. For example, decreased pH may facilitate
the invasion of tumor cells by promoting adjacent non-tumor cell
apoptosis (35), and it has been
found that by TGF-β dependent regulation of MMP2, lactate promotes
tumor migration (36). Moreover,
Glut1 was found to be correlated with MMP-2, which is important in
degrading the basement membrane and improving cancer cell invasion
(37); Hexokinase 2 was found to
be a potent factor which is associated with cancer cell migration
(17); PKM2 was found to promote
cancer cell migration via activation of STAT signal pathway
(18). In the present study, when
lactate was neutralized or glycolysis related molecules and enzymes
were inhibited, colorectal cancer cell invasion was repressed
significantly, which is consistent with other research.
Furthermore, we showed high expression of PRL-3, Glut1, HK2, PKM2,
LDHA and IL-8 in tumor legion of colorectal cancer, and the
positive correlation between PRL-3 and other molecules, indicating
the possible clinical therapeutic strategies for colorectal cancer
patients.
In summary, this study demonstrated that PRL-3
improved glycolysis of colorectal cancer cells via the secretion of
IL-8. However, the detailed mechanism is still unknown and will be
investigated in our following research.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of Guangdong Province (no.
2016A030313353), the National Natural Science Foundation of China
(no. 81602539), the International Science and Technology
Cooperation Program of Guangdong Province (no. 2013B051000025) and
the Science and Technology Project of Guangdong Province (no.
2015A050502021).
References
|
1
|
Ferlay J1, Soerjomataram I, Dikshit R,
Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in globocan 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Saha S, Bardelli A, Buckhaults P,
Velculescu VE, Rago C, St Croix B, Romans KE, Choti MA, Lengauer C,
Kinzler KW, et al: A phosphatase associated with metastasis of
colorectal cancer. Science. 294:1343–1346. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Aidaroos AQ and Zeng Q: PRL-3
phosphatase and cancer metastasis. J Cell Biochem. 111:1087–1098.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang Y, Liu XQ, Rajput A, Geng L, Ongchin
M, Zeng Q, Taylor GS and Wang J: Phosphatase PRL-3 is a direct
regulatory target of TGFbeta in colon cancer metastasis. Cancer
Res. 71:234–244. 2011. View Article : Google Scholar
|
|
5
|
Molleví DG, Aytes A, Padullés L,
Martínez-Iniesta M, Baixeras N, Salazar R, Ramos E, Figueras J,
Capella G and Villanueva A: PRL-3 is essentially overexpressed in
primary colorectal tumours and associates with tumour
aggressiveness. Br J Cancer. 99:1718–1725. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lai W, Chen S, Wu H, Guan Y, Liu L, Zeng
Y, Zhao H, Jiang J and Chu Z: PRL-3 promotes the proliferation of
LoVo cells via the upregulation of KCNN4 channels. Oncol Rep.
26:909–917. 2011.PubMed/NCBI
|
|
7
|
Lai W, Liu L, Zeng Y, Wu H, Xu H, Chen S
and Chu Z: KCNN4 channels participate in the EMT induced by PRL-3
in colorectal cancer. Med Oncol. 30:5662013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu H, Lai W, Zhang Y, Liu L, Luo X, Zeng
Y, Wu H, Lan Q and Chu Z: Tumor-associated macrophage-derived IL-6
and IL-8 enhance invasive activity of lovo cells induced by Prl-3
in a kcnn4 channel-dependent manner. BMC Cancer. 14:3302014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brahimi-Horn MC, Chiche J and Pouysségur
J: Hypoxia signalling controls metabolic demand. Curr Opin Cell
Biol. 19:223–229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martins SF, Amorim R, Viana-Pereira M,
Pinheiro C, Costa RF, Silva P, Couto C, Alves S, Fernandes S,
Vilaça S, et al: Significance of glycolytic metabolism-related
protein expression in colorectal cancer, lymph node and hepatic
metastasis. BMC Cancer. 16:5352016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen KY, Liu X, Bu P, Lin CS, Rakhilin N,
Locasale JW and Shen X: A metabolic signature of colon cancer
initiating cells. Conf Proc IEEE Eng Med Biol Soc. 2014:4759–4762.
2014.
|
|
13
|
Erreni M, Mantovani A and Allavena P:
Tumor-associated macrophages (TAM) and inflammation in colorectal
cancer. Cancer Microenviron. 4:141–154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu X, Li J, Sun X, Guo Y, Chu D, Wei L, Li
X, Yang G, Liu X, Yao L, et al: Tumor suppressor NDRG2 inhibits
glycolysis and glutaminolysis in colorectal cancer cells by
repressing c-Myc expression. Oncotarget. 6:26161–26176. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Estrella V, Chen T, Lloyd M, Wojtkowiak J,
Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg
JM, Sloane BF, et al: Acidity generated by the tumor
microenvironment drives local invasion. Cancer Res. 73:1524–1535.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wellberg EA, Johnson S, Finlay-Schultz J,
Lewis AS, Terrell KL, Sartorius CA, Abel ED, Muller WJ and Anderson
SM: The glucose transporter GLUT1 is required for ErbB2-induced
mammary tumorigenesis. Breast Cancer Res. 18:1312016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Katagiri M, Karasawa H, Takagi K, Nakayama
S, Yabuuchi S, Fujishima F, Naitoh T, Watanabe M, Suzuki T, Unno M,
et al: Hexokinase 2 in colorectal cancer: A potent prognostic
factor associated with glycolysis, proliferation and migration.
Histol Histopathol. 1:117992016.
|
|
18
|
Yang P and Li Z, Fu R, Wu H and Li Z:
Pyruvate kinase M2 facilitates colon cancer cell migration via the
modulation of STAT3 signalling. Cell Signal. 26:1853–1862. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xian ZY, Liu JM, Chen QK, Chen HZ, Ye CJ,
Xue J, Yang HQ, Li JL, Liu XF and Kuang SJ: Inhibition of LDHA
suppresses tumor progression in prostate cancer. Tumour Biol.
36:8093–8100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Min SH, Kim DM, Heo YS, Kim HM, Kim IC and
Yoo OJ: Downregulation of p53 by phosphatase of regenerating liver
3 is mediated by MDM2 and PIRH2. Life Sci. 86:66–72. 2010.
View Article : Google Scholar
|
|
22
|
Basak S, Jacobs SBR, Krieg AJ, Pathak N,
Zeng Q, Kaldis P, Giaccia AJ and Attardi LD: The
metastasis-associated gene Prl-3 is a p53 target involved in
cell-cycle regulation. Mol Cell. 30:303–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peng L, Xing X, Li W, Qu L, Meng L, Lian
S, Jiang B, Wu J and Shou C: PRL-3 promotes the motility, invasion,
and metastasis of LoVo colon cancer cells through PRL-3-integrin
beta1-ERK1/2 and-MMP2 signaling. Mol Cancer. 8:1102009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ming J, Liu N, Gu Y, Qiu X and Wang EH:
PRL-3 facilitates angiogenesis and metastasis by increasing ERK
phosphorylation and up-regulating the levels and activities of
Rho-A/C in lung cancer. Pathology. 41:118–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grivennikov SI and Karin M: Dangerous
liaisons: STAT3 and NF-kappaB collaboration and crosstalk in
cancer. Cytokine Growth Factor Rev. 21:11–19. 2010. View Article : Google Scholar
|
|
27
|
Zumsteg A and Christofori G: Corrupt
policemen: Inflammatory cells promote tumor angiogenesis. Curr Opin
Oncol. 21:60–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM
and Zhou BP: Stabilization of snail by NF-kappaB is required for
inflammation-induced cell migration and invasion. Cancer Cell.
15:416–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu Y and Zhou BP: Inflammation: A driving
force speeds cancer metastasis. Cell Cycle. 8:3267–3273. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nguyen DX, Bos PD and Massagué J:
Metastasis: From dissemination to organ-specific colonization. Nat
Rev Cancer. 9:274–284. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo JL, Maeda S, Hsu LC, Yagita H and
Karin M: Inhibition of NF-kappaB in cancer cells converts
inflammation- induced tumor growth mediated by TNFalpha to
TRAIL-mediated tumor regression. Cancer Cell. 6:297–305. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Greten FR, Eckmann L, Greten TF, Park JM,
Li ZW, Egan LJ, Kagnoff MF and Karin M: IKKbeta links inflammation
and tumorigenesis in a mouse model of colitis-associated cancer.
Cell. 118:285–296. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dáňová K, Klapetková A, Kayserová J,
Šedivá A, Špíšek R and Jelínková LP: NF-κB, p38 MAPK, ERK1/2, mTOR,
STAT3 and increased glycolysis regulate stability of
paricalcitol/dexamethasone-generated tolerogenic dendritic cells in
the inflammatory environment. Oncotarget. 16:14123–14138. 2015.
View Article : Google Scholar
|
|
34
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Williams AC, Collard TJ and Paraskeva C:
An acidic environment leads to p53 dependent induction of apoptosis
in human adenoma and carcinoma cell lines: Implications for clonal
selection during colorectal carcinogenesis. Oncogene. 18:3199–3204.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baumann F, Leukel P, Doerfelt A, Beier CP,
Dettmer K, Oefner PJ, Kastenberger M, Kreutz M, Nickl-Jockschat T,
Bogdahn U, et al: Lactate promotes glioma migration by
TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro
Oncol. 11:368–380. 2009. View Article : Google Scholar :
|
|
37
|
Ito S, Fukusato T, Nemoto T, Sekihara H,
Seyama Y and Kubota S: Coexpression of glucose transporter 1 and
matrix metalloproteinase-2 in human cancers. J Natl Cancer Inst.
94:1080–1091. 2002. View Article : Google Scholar : PubMed/NCBI
|