Introduction
Approximately 1.4 million new cases of colon cancer
are diagnosed each year worldwide. Colon cancer is the third most
common cancer in Korea, and its incidence is increasing in Asian
countries, including Korea (1,2).
Intensive efforts have been made to identify a causative factor for
this major cancer, and epidemiologic studies indicate that a
western style diet is associated with a high incidence of colon
cancer (3–5).
Reactive oxygen species (ROS) modulate growth
signals and activate gene expression, leading to sustained
proliferation of cancer cells (6–8). In
cancer cells, ROS-induced phosphorylation of c-Jun N-terminal
kinase (JNK) translates oncogenic signals, supporting cellular
proliferation through the activation of activating protein-1, in
addition to the proliferation signals mediated by extracellular
regulated kinase (ERK) (9).
Therefore, ROS likely play an important role in promoting tumor
development.
Flavonoids are biologically active polyphenolic
compounds widely distributed in plants, and luteolin,
3′,4′,5,7-tetrahydroxyflavone, is rich in vegetables and flowers.
Flavonoids including luteolin have been focused on anticancer
properties via various anticancer mechanisms (10–13).
Luteolin have anti-inflammatory, antiallergic, anticancer,
antioxidant and ROS scavenging activities (14–16).
Luteolin acts as an antimetastatic agent via decrease of MMP-2 and
MMP-9, iNOS and COX-2 (17,18).
Luteolin enhanced the expression of Nrf2 and activates
glutathione-S-transferase (GST)-α and GST-μ (19). Luteolin induces growth arrest by
inhibiting Wnt/β-catenin/GSK-3β signaling pathway and induces
apoptosis by caspase-3 in human colon cancer cells (20–23).
Thus, the objective was to determine whether
luteolin induces HT-29 cell death through an antioxidant
effect.
Materials and methods
Materials
Luteolin was obtained from Professor Sam Sik Kang of
Seoul National University (Seoul, Republic of Korea). The xanthine,
xanthine oxidase, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO),
propidium iodide,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
2′,7′-dichlorofluorescein diacetate (DCF-DA), dihydrorhodamine
(DHR) 123, N-acetyl-L-cysteine (NAC), reduced glutathione (GSH),
buthionine sulfoximine (BSO), diethyldithiocarbamate (DEDTC),
3-amino-1,2,4-triazole (ATZ), epinephrine, and L-ascorbic acid
(vitamin C) were purchased from Sigma-Aldrich (St. Louis, MO, USA);
7-amino-4-chloromethylcoumarin (CMAC) was obtained from Invitrogen
(Poole, Dorset, UK).
5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine
chloride (JC-1) and Rhod2-AM were purchased from Molecular Probes
(Eugene, OR, USA). Primary antibodies against manganese superoxide
dismutase (MnSOD), catalase (CAT), cytochrome c, and β-actin
were purchased from Santa Cruz Biotechnology (Dallas, TX, USA), and
primary antibodies against Bcl-2, Bax, caspase-3, caspase-9,
phospho-ERK, ERK, phospho-JNK, JNK, phospho-p38, and p38 were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Cell culture
Human colorectal cancer (HT-29) and normal human
colon (FHC) cells were obtained from the American Type Culture
Collection (Rockville, MD, USA) and maintained at 37°C in an
incubator with a humidified atmosphere of 5% CO2. HT-29
cells were cultured in Dulbecco's modified Eagle's medium (DMEM)
containing 10% heat-inactivated fetal calf serum (FCS),
streptomycin (100 μg/ml), and penicillin (100 U/ml). Normal
human colon FHC cells were cultured in a 1:1 mixture of Ham's F12
and DMEM containing HEPES (25 mM), cholera toxin (10 ng/ml,
Calbiochem-Novabiochem Corp., La Jolla, CA, USA), insulin (5
μg/ml), transferrin (5 μg/ml), hydrocortisone (100
ng/ml), and 10% FCS.
Cell viability
The effect of luteolin on the viability of cells was
determined using the MTT assay, which is based on the reduction of
a tetrazolium salt by mitochondrial dehydrogenase in viable cells
(24). Cells were treated with
luteolin at various concentrations. After 48 h, 50 μl MTT
stock solution (2 mg/ml) was added to each well to obtain a total
reaction volume of 200 μl. After incubation for 4 h, the
plate was centrifuged at 800 × g for 5 min followed by aspiration
of the supernatants. Formazan crystals present in each well were
dissolved in 150 μl DMSO and the absorbance at 540 nm was
measured on a scanning multi-well spectrophotometer.
Detection of superoxide radical
Superoxide radical was produced by the reaction of
the xanthine/xanthine oxidase system and reacted with spin trap
DMPO. The DMPO-·OOH adduct was detected using electron spin
resonance (ESR) spectroscopy (25). The ESR spectrum was recorded at 2.5
min after mixing in a phosphate buffered solution (pH 7.4) with 20
μl of 6 M DMPO, 20 μl xanthine oxidase (0.25 U/ml),
20 μl xanthine (5 mM), and 20 μl luteolin (final 50
μg/ml) using the JES-FA ESR spectrometer (JEOL, Tokyo,
Japan). The parameters of the ESR spectrometer were set at the
following conditions: magnetic field, 336.5 mT; power, 1.00 mW;
frequency, 9.4380 GHz; modulation amplitude, 0.2 mT; gain, 500;
scan time, 0.5 min; scan width, 10 mT; time constant, 0.03 sec; and
temperature, 25°C.
Detection of hydroxyl radical
Hydroxyl radical was generated by the Fenton
reaction. Hydroxyl radical reacted with DMPO and the resultant
DMPO-·OH adduct was detected using an ESR spectrometer (25). The ESR spectrum was recorded 2.5
min after mixing in a phosphate buffered solution (pH 7.4) with 0.2
ml of 0.3 M DMPO, 0.2 ml of 10 mM FeSO4, 0.2 ml of 10 mM
H2O2, and luteolin using an ESR spectrometer.
The parameters of the ESR spectrometer were set at the following
conditions: magnetic field, 336.5 mT; power, 1.00 mW; frequency,
9.4380 GHz; modulation amplitude, 0.2 mT; gain, 200; scan time, 0.5
min; scan width, 10 mT; time constant, 0.03 sec; and temperature,
25°C.
Intracellular ROS measurement
Image analysis for the generation of intracellular
ROS was achieved by seeding cells on a coverslip-loaded 6-well
plate at a density of 2×105 cells/well. At 16 h after
plating, cells were treated with luteolin at a concentration of 50
μg/ml. After 24 h, 100 μM DCF-DA was added to each
well and cells were incubated for an additional 30 min at 37°C.
After washing with phosphate buffered saline (PBS), the stained
cells were mounted onto a microscope slide in mounting medium
(Dako, Carpinteria, CA, USA). Microscopic images were collected
using the Laser Scanning Microscope 5 PASCAL program (Carl Zeiss,
Jena, Germany) on a confocal microscope. In addition, cells were
treated with luteolin at 50 μg/ml and incubated for an
additional 48 h at 37°C. After addition of 25 μM DCF-DA
solution, the fluorescence signal of 2′, 7′-dichlorofluorescein was
detected using a Perkin Elmer LS-5B spectrofluorometer (26).
Mitochondrial ROS measurement
The cells were seeded in a 96-well plate at a
density of 2×104 cells/well. At 16 h after plating, the
cells were treated with luteolin at 50 μg/ml and incubated
for 24 h. After addition of 20 μM DHR 123 solution for 10
min, fluorescence was detected using a Perkin Elmer LS-5B
spectrofluorometer. For image analysis of the generation of
mitochondrial ROS, cells were seeded on a coverslip-loaded six-well
plate at a density of 2×105 cells/well. At 16 h after
plating, cells were treated with luteolin at a concentration of 50
μg/ml. After 24 h, the medium was changed, 20 μM DHR
123 was added to each well, and the plate was incubated for an
additional 30 min at 37°C. After washing with PBS, the stained
cells were mounted onto a microscope slide in mounting medium.
Images were acquired using the Laser Scanning Microscope 5 PASCAL
program (Carl Zeiss) on a confocal microscope.
Measurement of superoxide dismutase (SOD)
activity
Cells were seeded in a culture dish at a density of
1×105 cells/ml, and at 16 h after plating they were
treated with luteolin at 50 μg/ml and incubated for
additional times as indicated. The cells were then washed with cold
PBS and scraped. The harvested cells were suspended in 10 mM
phosphate buffer (pH 7.5) and lysed on ice by sonicating twice for
15 sec. Triton X-100 (1%) was then added to the lysates and
incubated for 10 min on ice. The lysates were clarified by
centrifugation at 5000 × g for 10 min at 4°C to remove cellular
debris, and the protein content of the supernatant was determined.
SOD activity was assessed by detecting the inhibition of
auto-oxidation of epinephrine (27) as follows: 50 μg protein was
added to 50 mM phosphate buffer (pH 10.2) containing 0.1 mM EDTA
and 0.4 mM epinephrine. Epinephrine rapidly undergoes
auto-oxidation at pH 10 to produce adrenochrome, a pink colored
product that can be measured at 480 nm using a UV/VIS
spectrophotometer in kinetic mode. SOD inhibits the auto-oxidation
of epinephrine. The rate of inhibition was monitored at 480 nm. SOD
activity was expressed as units/mg protein, and one unit of enzyme
activity was defined as the amount of enzyme required for 50%
inhibition of auto-oxidation of epinephrine.
Western blot analysis
Harvested cells were lysed on ice for 30 min in 100
μl lysis buffer [120 mM NaCl, 40 mM Tris (pH 8.0), and 0.1%
NP 40] and centrifuged at 13,000 × g for 15 min. Supernatants were
collected from the lysates and protein concentrations were
determined. Aliquots of the lysates (40 μg of protein) were
boiled for 5 min, electrophoresed on a 10% SDS-polyacrylamide gel,
and transferred onto nitrocellulose membranes, which were
subsequently incubated with primary antibodies. The membranes were
further incubated with secondary anti-immunoglobulin-G-horseradish
peroxidase conjugates (Pierce, Rockford, IL, USA). Protein bands
were detected using an enhanced chemiluminescence western blotting
detection kit (Amersham, Little Chalfont, Buckinghamshire, UK).
Measurement of catalase activity
Aliquots containing 50 μg protein were added
to 50 mM phosphate buffer (pH 7.0) containing 100 mM
H2O2. The reaction mixture was incubated for
2 min at 37°C and the absorbance was monitored at 240 nm for 5 min.
The change in absorbance with time was proportional to the
breakdown of H2O2 (27). Catalase activity was expressed as
units/mg protein and one unit of enzyme activity was defined as the
amount of enzyme required to break down 1 μM
H2O2.
Detection of GSH level
Intracellular GSH content was measured with a
commercial colorimetric assay kit (GSH-400) from Oxis International
(Portland, OR, USA). After treatment with luteolin for 24 h, cells
were harvested and homogenized in a metaphosphoric working
solution. After centrifugation, 50 μl R1 solution (a
solution of a chromogenic reagent in HCl) was added to 900
μl supernatant, followed by gentle vortex mixing. Following
the addition of 50 μl R2 solution (30% NaOH), the mixtures
were incubated at 25±3°C for 10 min. After centrifugation, the
absorbance of the clear supernatant was measured at 400 nm. The
intracellular GSH level was also determined using CMAC, a
GSH-sensitive fluorescence dye. Cells were incubated with 5
μM CMAC for 30 min in the dark and CMAC fluorescence images
were analyzed with a Zeiss Axiovert 200 inverted microscope at an
excitation wavelength of 351 nm and an emission wavelength of 380
nm (28).
Nuclear staining with Hoechst 33342
Cells were treated with luteolin at 50 μg/ml
and the mixture was incubated for 48 h. Hoechst 33342 (1.5
μl of a 10 mg/ml stock solution), a DNA-specific fluorescent
dye, was added to each well and incubated for 10 min at 37°C. The
stained cells were then observed under a fluorescent microscope
equipped with a CoolSNAP-Pro color digital camera to examine the
degree of nuclear condensation (29).
DNA fragmentation
Cells were treated with luteolin at 50 μg/ml
and the mixture was incubated for 48 h. Cellular DNA fragmentation
was assessed using a cytoplasmic histone-associated DNA
fragmentation kit from Roche Diagnostics (Mannheim, Germany)
according to the manufacturer's instructions.
Detection of sub-G1
populations
Cells were treated with luteolin at 50 μg/ml
and the mixture was incubated for 48 h. Flow cytometry was
performed to determine the content of apoptotic sub-G1
hypodiploid cells (30). The cells
were harvested, fixed in 1 ml of 70% ethanol for 30 min at 4°C,
washed twice with PBS, and then incubated in 1 ml PBS containing
100 μg propidium iodide and 100 μg RNase A for 30 min
in the dark at 37°C. Flow cytometric analysis was performed and the
proportion of sub-G1 hypodiploid cells was assessed by
the histograms generated using the computer programs Cell Quest and
Mod-Fit (Becton-Dickinson, Mountain View, USA).
Detection of mitochondrial membrane
potential
Mitochondrial membrane potential (Δψ) was analyzed
using JC-1, a lipophilic cationic fluorescence dye. Cells were
harvested, and after changing the media, JC-1 (10 μg/ml) was
added to each well and incubated for an additional 30 min at 37°C.
After washing with PBS, the stained cells were assayed using flow
cytometer.
Determination of mitochondrial
Ca2+ level
Mitochondrial Ca2+ levels were determined
by Rhod2-AM (250 nM), a mitochondrial Ca2+ sensitive
fluorescence dye. Cells were incubated with 250 nM Rhod2-AM in PBS
including Ca2+ for 30 min in the dark. Rhod2-AM
fluorescence images were analyzed with a Zeiss Axiovert 200
inverted microscope at an excitation wavelength of 552 nm and an
emission wavelength of 581 nm (31). In addition, cells incubated with
250 nM Rhod2-AM in PBS including Ca2+ for 30 min in the
dark were detected using a flow cytometer (Becton-Dickinson).
Transient transfection and small
interfering RNA (siRNA) treatment
Overnight-seeded cells at 1.0×105
cells/ml were transfected for 4 h with 10 nM specific siRNAs or
equal molar amounts of mismatched siRNA controls. Control siRNA
(sc-37007, Santa Cruz Biotechnology). ERK-1, JNK, and p38 knockdown
was performed using siRNAs against ERK-1 (sc-29308, Santa Cruz
Biotechnology), JNK (5′-AAAAAGAA TGTCCTACCTTCT-3′), and p38 (5′-AGC
CCA GCA ACC TAG CTG T-3′), respectively, which were transfected
into cells using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA,
USA) following the manufacturer's instructions. At 24 h after
transfection, the cells were treated with 50 μg/ml luteolin
for 24 h and examined by immunoblotting.
Statistical analysis
All measurements were performed in triplicate and
all values represent the mean ± standard error of the mean (SEM).
The results were subjected to analysis of variance (ANOVA) using
Tukey's test to analyze differences. P<0.05 was considered
significant.
Results
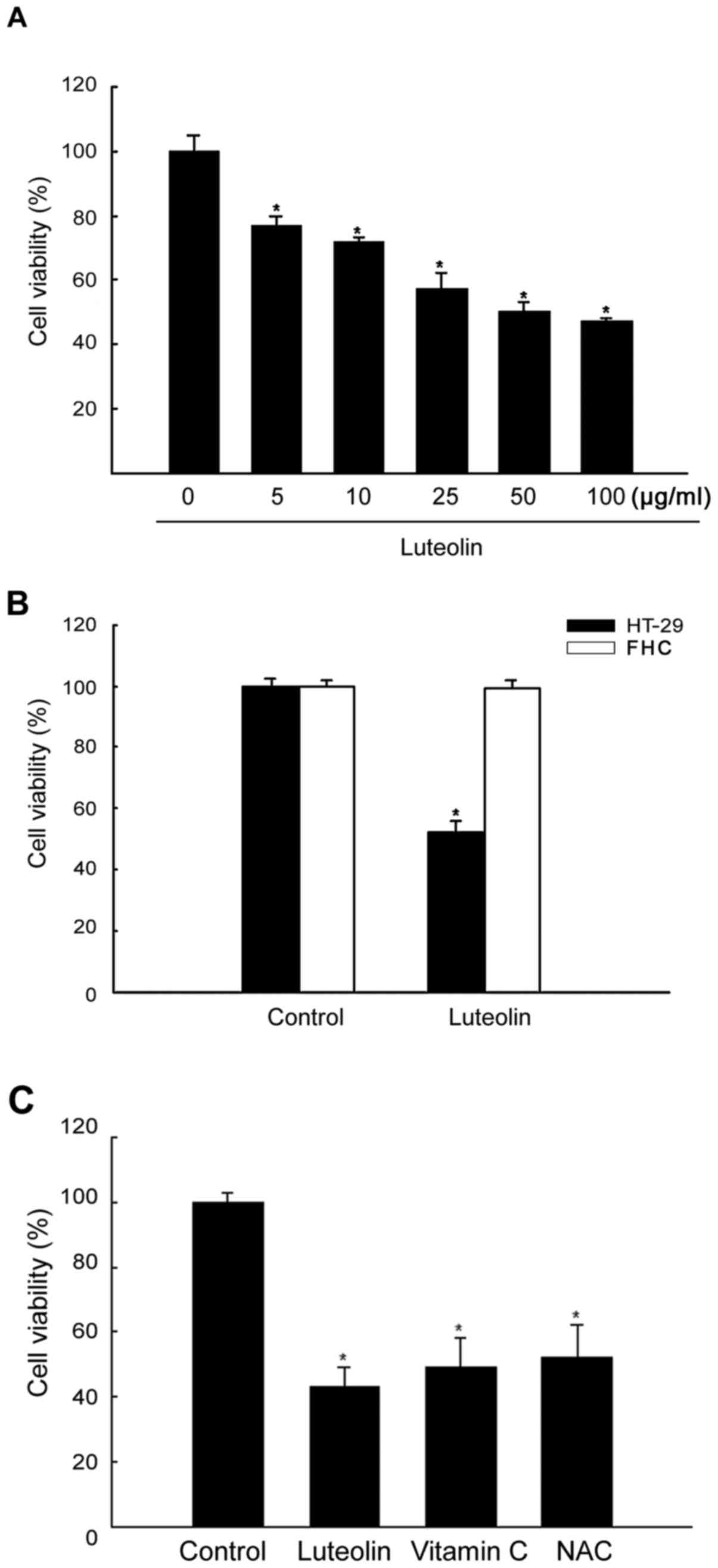
Luteolin inhibits the growth of human
colon cancer cells
The effects of luteolin on the proliferation of
HT-29 cells were assessed at 48 h using the MTT assay, and
IC50 (50% growth inhibitory concentration) value was
determined. At 48 h, luteolin had cytotoxic effects at different
concentrations. As shown in Fig.
1A, cell viability was inhibited by luteolin in a
dose-dependent manner as follows: 79% at 5 μg/ml, 70% at 10
μg/ml, 60% at 25 μg/ml, 50% at 50 μg/ml, and
42% at 100 μg/ml. Among the tested concentrations, 50
μg/ml luteolin was selected for further investigations. The
cytotoxicity of luteolin in normal colon cells (FHC) was
investigated using the MTT assay. As shown in Fig. 1B, the viability of luteolin-treated
FHC cells was higher than that of luteolin-treated HT-29 cells.
Since luteolin is an antioxidant compound, the effect of known
antioxidant agents on HT-29 cell viability was determined. As shown
in Fig. 1C, vitamin C and NAC,
which are antioxidant agents, decreased cell viability compared
with that in untreated cells.
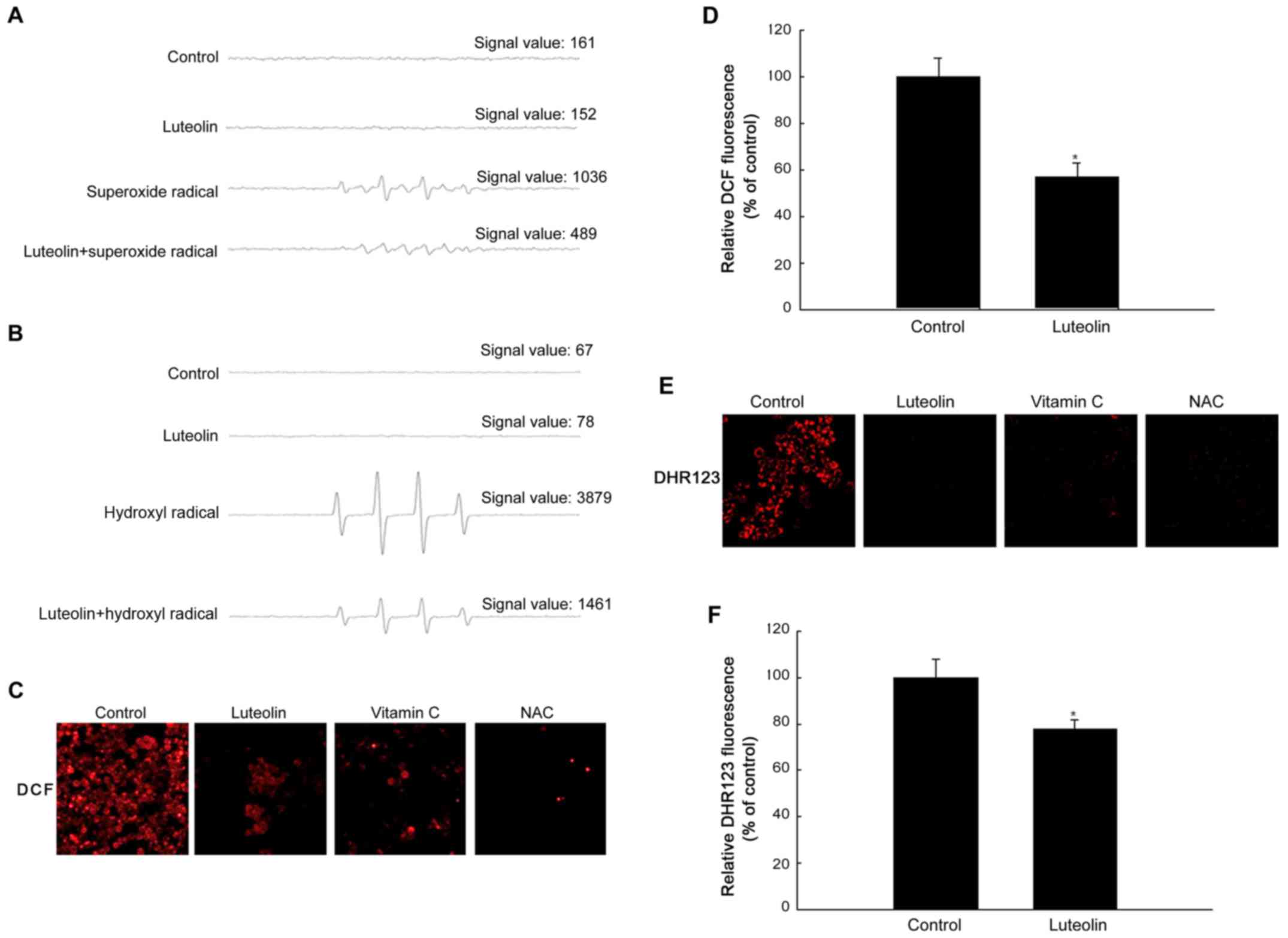
Luteolin shows ROS scavenging
activity
To determine the ROS scavenging effects of luteolin,
the production of super-oxide and hydroxyl radical was assessed.
The superoxide radical produced via reaction of the xanthin/xanthin
oxidase system and the hydroxyl radical generated via the Fenton
reaction (FeSO4+H2O2) in a
cell-free system were detected by ESR spectrometry. The ESR results
revealed that a specific signal was not clearly detected in the
control and in the 50 μg/ml luteolin-treated group; however,
the superoxide radical signal increased to up to 1036 in the
xanthin/xanthin oxidase system, and luteolin treatment decreased
the superoxide radical signal to 489 (Fig. 2A). The hydroxyl radical signal
increased to up to 3879 in the
FeSO4+H2O2 system, and luteolin
treatment decreased the hydroxyl radical signal to 1461 (Fig. 2B). The scavenging effect of
luteolin on intra cellular ROS in HT-29 cells was measured. As
shown in Fig. 2C, 50 μg/ml
luteolin significantly decreased the intensity of the DCF signal
compared with that in the control. Consistently, antioxidant agents
significantly decreased the intensity of the DCF signal. The red
fluorescence intensity of ROS measured using a confocal microscope
was decreased in luteolin-treated cells at 50 μg/ml. In
addition, ROS levels detected with a spectrofluorometer were
significantly lower in luteolin-treated than in untreated cells
(Fig. 2D). To examine the
scavenging effect of luteolin on mitochondrial ROS in HT-29 cells,
the DHR 123 fluorescence dye was used to detect mitochondrial ROS
in cells treated with or without luteolin. Confocal microscopy
revealed that luteolin reduced the red fluorescence intensity of
mitochondrial ROS in untreated cells (Fig. 2E). Fluorescence spectrometric data
revealed that luteolin treatment decreased the level of
mitochondrial ROS compared with that in untreated cells (Fig. 2F).
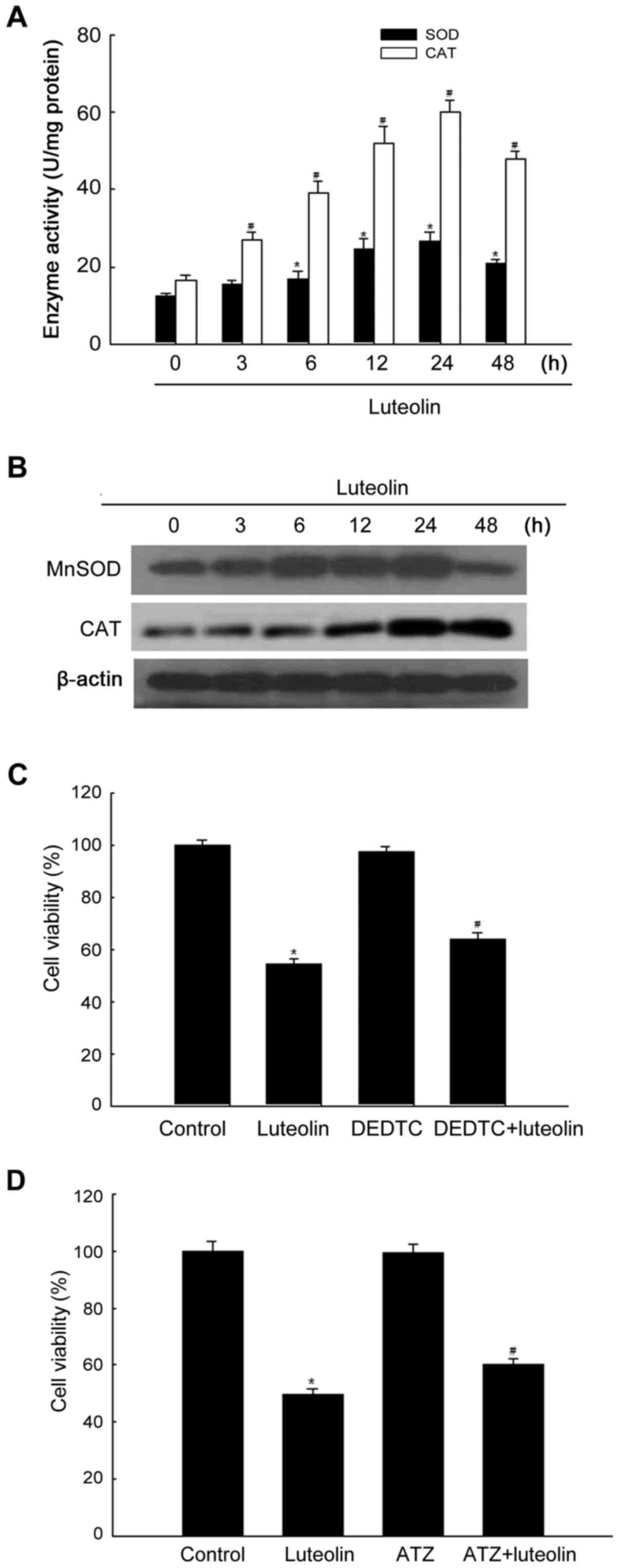
Effects of luteolin on antioxidant
enzymes in HT-29 cells
Assessment of the effect of luteolin on the activity
of antioxidant enzymes showed that luteolin increased SOD and CAT
activity in a time-dependent manner until 24 h (Fig. 3A). The protein levels of MnSOD and
CAT increased in response to treatment with 50 μg/ml
luteolin for 24 h (Fig. 3B). To
determine whether MnSOD activity was related to luteolin-induced
cytotoxicity, cells were treated with the MnSOD inhibitor DEDTC and
subjected to the MTT assay. As shown in Fig. 3C, DEDTC treatment attenuated the
cytotoxic effect of luteolin. To examine the effect of CAT on
luteolin-induced cytotoxicity, cells were pretreated with the CAT
inhibitor ATZ (32). As shown in
Fig. 3D, ATZ treatment attenuated
the cytotoxic effect of luteolin.
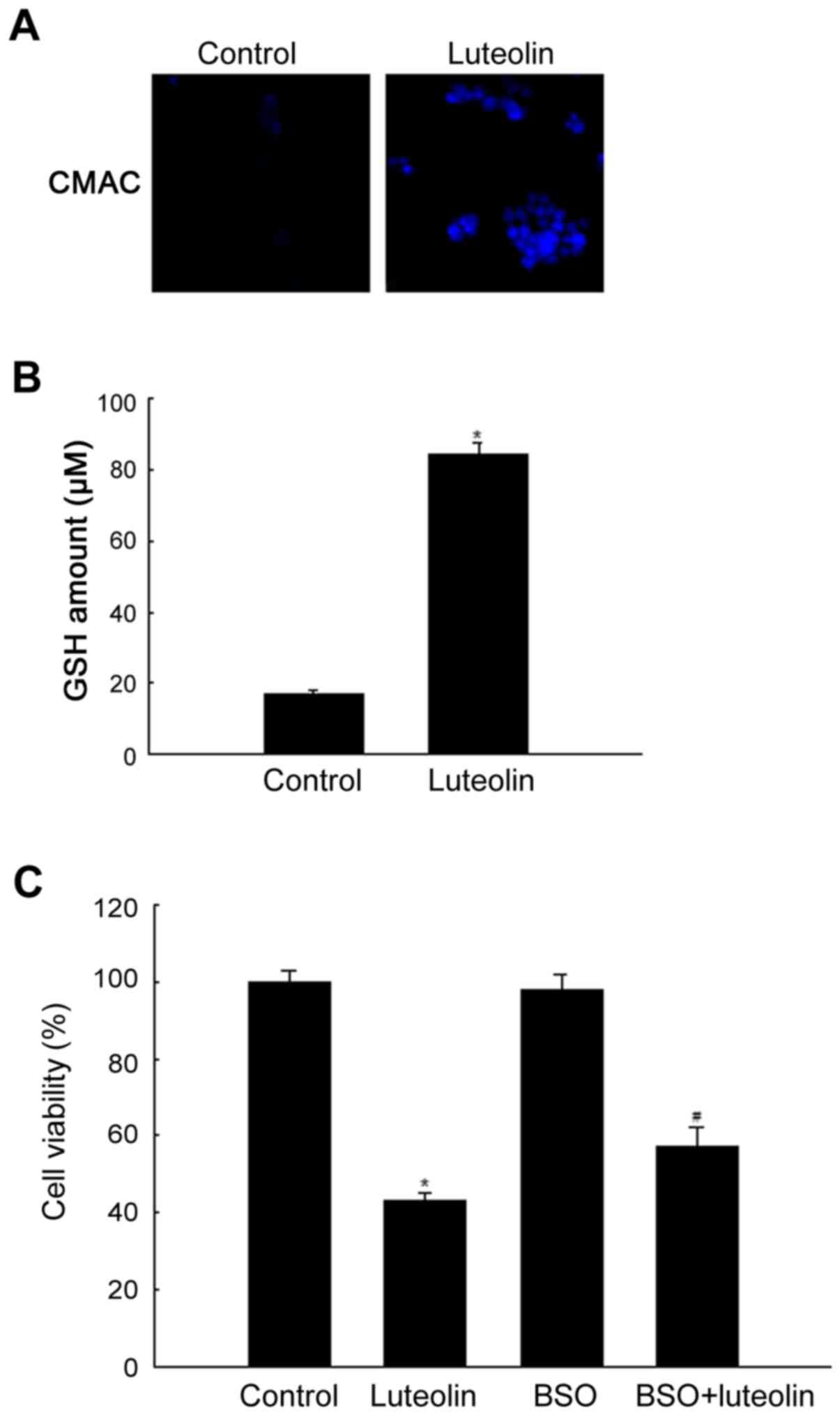
Effect of luteolin on glutathione
GSH is one of the most abundant intracellular
antioxidants, and determination of changes in the level of GSH
provides a method to monitor oxidative stress within cells. The
GSH-sensitive fluorescent dye, CMAC, can be used as a probe to
evaluate the level of intracellular GSH (28). As shown in Fig. 4A, cells treated with luteolin at 50
μg/ml and stained with CMAC showed higher blue fluorescence
intensity of cellular GSH measured using a confocal microscope than
untreated cells. This pattern was confirmed by measurement of the
cellular GSH level. Cellular GSH level was significantly higher in
luteolin-treated than in control cells (Fig. 4B). To determine whether GSH was
related to luteolin-induced cytotoxicity, cells were treated with
the glutathione synthesis inhibitor BSO and subjected to the MTT
assay. As shown in Fig. 4C, BSO
treatment attenuated the cytotoxic effect of luteolin.
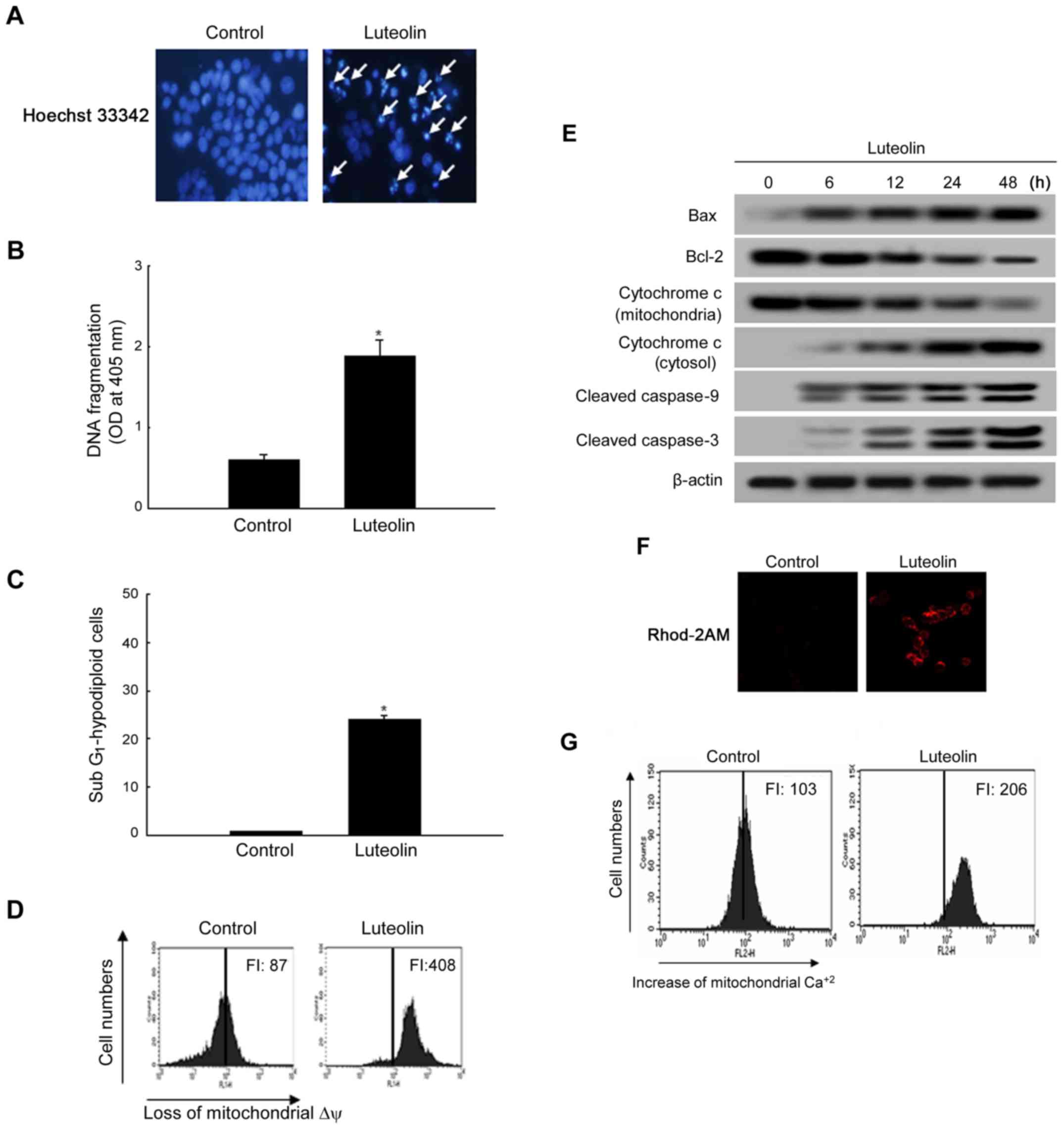
Luteolin induces apoptosis by the
mitochondrial pathway
To study the cytotoxic effect of luteolin via
apoptotic cell death, cell nuclei were stained with Hoechst 33342
and assessed by microscopy. The microscopic images in Fig. 5A show that control cells had intact
nuclei, whereas luteolin-treated cells had significant nuclear
fragmentation, which is characteristic of apoptosis. In addition,
the levels of DNA fragmentation were higher in the luteolin-treated
group than in the control group (Fig.
5B). In addition to the morphological evaluation, the effect of
luteolin on apoptosis in HT-29 cells was confirmed by apoptotic
sub-G1 DNA analysis. As shown in Fig. 5C, luteolin-treated cells were 25%
in the apoptotic sub-G1 DNA content compared with 4% of
the apoptotic sub-G1 DNA content in control group.
During the apoptotic process, the mitochondrial membrane pore
opening induces the loss of mitochondrial ∆ψ, which induces the
release of cytochrome c from mitochondria. Luteolin
treatment resulted in the loss of ∆ψ, as measured with the JC-1 dye
(Fig. 5D). The effect of luteolin
on apoptosis-related protein expression was determined by western
blot analysis of the antiapoptotic and proapoptotic proteins Bcl-2
and Bax. As shown in Fig. 5E,
luteolin upregulated Bax but downregulated Bcl-2 expression. The
loss of ∆ψ induces the release of cytochrome c from
mitochondria. As shown in Fig. 5E,
luteolin induced the release of cytochrome c from
mitochondria to the cytosol. The level of active (cleaved)
caspase-9 was examined by western blotting because this enzyme is
activated in response to mitochondrial membrane disruption
(33). As shown in Fig. 5E, luteolin upregulated the active
form of caspase-9 and caspase-3. Excessive Ca2+ within
mitochondria can induce apoptosis by opening the mitochondrial
permeability transition pore (34); therefore, we investigated whether
mitochondrial Ca2+ changes occurred in response to
luteolin. Our data demonstrated that luteolin at 50 μg/ml
caused a sustained elevation of mitochondrial Ca2+
compared with that in the control. These data were determined by
confocal (Fig. 5F) imaging and
FACS (Fig. 5G) analysis using
Rhod2-AM.
Effect of luteolin on the MAPK signaling
pathway
The MAPK signaling pathway is activated in response
to certain cellular stress conditions, and it is implicated in
cellular death or survival signaling (35,36).
To investigate whether the MAPK pathway is involved in
luteolin-induced apoptosis, the activity of MAPK was assessed in
luteolin-treated cells. Control cells showed low or undetectable
levels of phosphorylated MAPK, whereas luteolin at 50 μg/ml
increased the phosphorylation of all three MAPK in a time-dependent
manner (Fig. 6A). Next, the
effects of the specific ERK, JNK, and p38 MAP kinase inhibitors
U0126, SP600125, and SB203580 on luteolin-induced apoptosis were
examined by the MTT assay. As shown in Fig. 6B, luteolin significantly reduced
the cell viability, whereas pretreatment with SP600125 and SB203580
except U0126 attenuated the cytotoxic effect of luteolin. Similar
results were obtained in response to transfection with
MAPK-specific siRNAs (Fig.
6C).
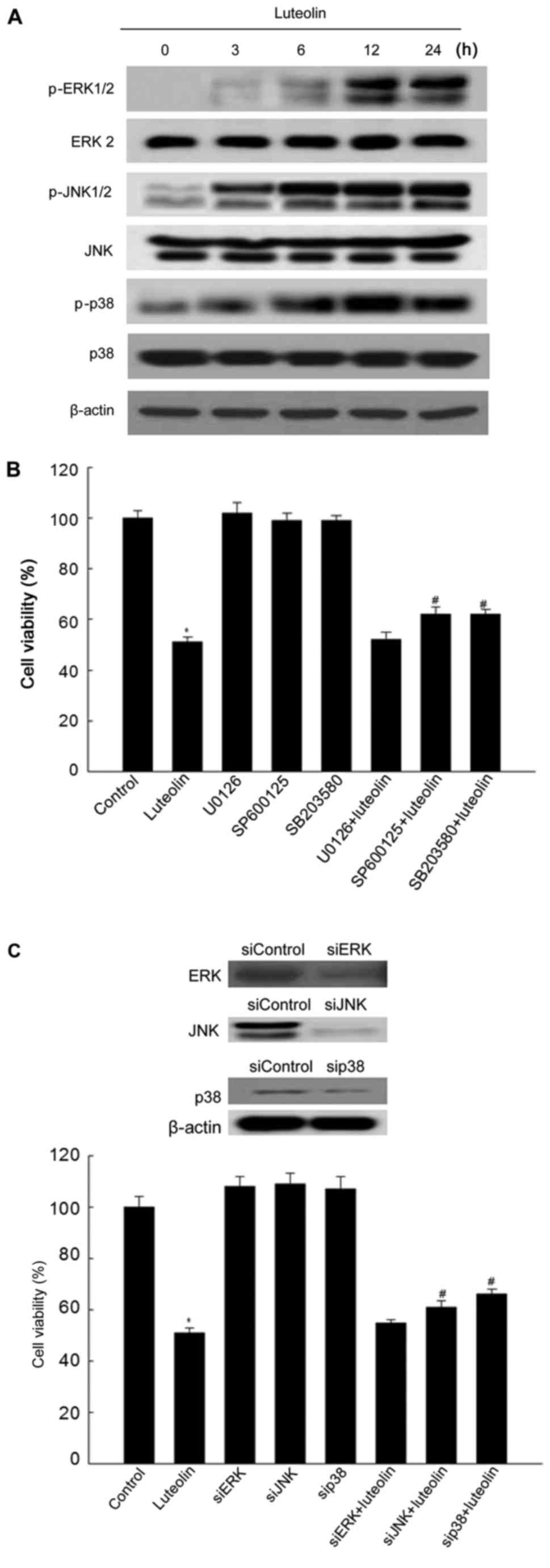 | Figure 6Effect of luteolin on the MAPK
signaling pathway. Cells were treated with luteolin at 50
μg/ml for 24 h to detect the expression of MAPK signaling
proteins. (A) Cell lysates were electrophoresed and immunoblotted
using anti-ERK2, anti-phospho-ERK1/2, anti-JNK, anti-phospho-JNK,
anti-p38, and anti-phospho-p38 antibodies. (B) The cell viability
after pretreatment with the ERK, JNK, and p38 MAPK inhibitors
U0126, SP600125, and SB203580 and treatment with luteolin was
determined by the MTT assay. *Significantly different
from control cells (P<0.05); #significantly different
from luteolin-treated cells (P<0.05). (C) The viability of HT-29
cells after transfection with siRNAs against ERK, JNK, and p38 and
treatment with luteolin was determined by the MTT assay.
*Significantly different from control cells (P<0.05);
#significantly different from luteolin-treated cells
(P<0.05). |
Discussion
Cancer cells need an increased energy supply to
support high rates of metabolism. Normally, ATP is produced with
high efficiency through oxidative phosphorylation in mitochondria.
An alternative metabolic pathway is adopted (enhanced glycolysis)
when mitochondrial ATP production is compromised. The enhanced
oxidative stress observed in cancer cells can result not only from
ROS overproduction but also from low levels or inactivation of
antioxidant mechanisms (37). The
enhanced constitutive oxidative stress renders tumor cells highly
dependent on endogenous antioxidants to protect them from
continuous intracellular ROS injury (38).
Luteolin has anticancer activity mediated by its
role as a DNA topoisomerase II poison, its inhibition of invasive
activity, and the induction of cell cycle arrest and apoptosis
(39–41).
The present data indicated that luteolin induced
apoptosis in HT-29 cells by a caspase-dependent pathway with
mitochondrial involvement. In the present study, we used a direct
approach to examine the scavenging activity of luteolin with
hydroxyl and superoxide radical, and the results suggested that
luteolin has strong antioxidant activity. To examine the role of
ROS in luteolin-induced apoptosis, ROS production was examined
using an intracellular oxidant-sensitive fluorescent probe, DCF-DA,
and a mitochondrial oxidant-sensitive fluorescent probe, DHR123.
The results showed that treatment with luteolin had ROS scavenging
effect compared with that in the control, indicating that
antioxidant effect of luteolin induced cell death in HT-29
cells.
Polyphenolic antioxidants are scavengers of free
radicals and modifiers of various enzymatic functions. Luteolin is
a flavonoid that contains two phenolic structures. To determine
whether luteolin-induced HT-29 cell apoptosis was associated with
the antioxidant properties of luteolin, we examined the antioxidant
activity of luteolin. Decreased activity and expression of
mitochondrial MnSOD was reported in certain colorectal carcinomas,
probably accounting for increased superoxide radical production
(42). Accumulation of superoxide
radical stimulates cell growth by altering the redox status of
transcriptional factors and cell cycle regulatory proteins
(43). Moreover, induced
overexpression of MnSOD suppresses malignant phenotypes in
experimental in vitro model. Therefore, MnSOD is considered
to be a tumor suppressor that acts indirectly via ROS. In the
present study, we examined the expression of the MnSOD and CAT
proteins during luteolin-induced apoptosis. Our results showed that
the MnSOD and CAT inhibitors DEDTC and ATZ attenuated
luteolin-induced apoptosis. MnSOD and CAT act as tumor suppressor
genes in several human cancer cells (44). The mechanism by which MnSOD
suppresses cancer development remains unknown. However, the
expression of this antioxidant enzyme may play a significant role
in maintaining cellular redox status. The tumor suppressor effects
of MnSOD overexpression are mediated in part by the modulation of
specific oncogenes. The relationship between MnSOD expression, the
modulation of DNA-binding activity, and the transcriptional
activation of redox-sensitive oncoproteins and tumor suppressor
proteins was reported recently (45–48).
The present data indicated that luteolin increased GSH levels in
HT-29 cells. This was confirmed by experiments using the
glutathione synthesis inhibitor BSO, which indicated that GSH was
related to luteolin-induced apoptosis. In addition, the present
study demonstrated that JNK and p38 MAPK pathway was involved in
apoptosis induced by luteolin. Taken together, the results of the
present study suggest that luteolin-induced apoptosis is associated
with the antioxidant properties of luteolin.
Acknowledgments
This work was supported by grant from the Basic
Science Research Program (NRF-2016R1A6A3A11932235) through the
National Research Foundation of Korea (NRF) funded by the Ministry
of Education, Science and Technology, and by grant from the Basic
Research Laboratory Program (NRF-2017R1A4A1014512) by NRF grant
funded by the Korea government (MSIP).
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Lee HH, Kim SK, Choi HH, Kim HK, Kim SS,
Chae HS, Cho H and Cho YS: Post-colonoscopy colorectal cancers in
average-risk Korean subjects with a normal initial colonoscopy.
Turk J Gastroenterol. 27:17–22. 2016. View Article : Google Scholar
|
|
3
|
Vargas AJ and Thompson PA: Diet and
nutrient factors in colorectal cancer risk. Nutr Clin Pract.
27:613–623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang SY, Kim YS, Lee JE, Seol J, Song JH,
Chung GE, Yim JY, Lim SH and Kim JS: Dietary protein and fat intake
in relation to risk of colorectal adenoma in Korean. Medicine
(Baltimore). 95:e54532016. View Article : Google Scholar
|
|
5
|
Mehta RS, Nishihara R, Cao Y, Song M, Mima
K, Qian ZR, Nowak JA, Kosumi K, Hamada T, Masugi Y, et al:
Association of dietary patterns with risk of colorectal cancer
subtypes classified by fusobacterium nucleatum in tumor tissue.
JAMA Oncol. 6374:20162017.
|
|
6
|
Nogueira V and Hay N: Molecular pathways:
Reactive oxygen species homeostasis in cancer cells and
implications for cancer therapy. Clin Cancer Res. 19:4309–4314.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harris IS, Treloar AE, Inoue S, Sasaki M,
Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA,
et al: Glutathione and thioredoxin antioxidant pathways synergize
to drive cancer initiation and progression. Cancer Cell.
27:211–222. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schumacker PT: Reactive oxygen species in
cancer: A dance with the devil. Cancer Cell. 27:156–157. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ho BY, Wu YM, Chang KJ and Pan TM:
Dimerumic acid inhibits SW620 cell invasion by attenuating
H2O2-mediated MMP-7 expression via JNK/C-Jun
and ERK/C-Fos activation in an AP-1-dependent manner. Int J Biol
Sci. 7:869–880. 2011. View Article : Google Scholar :
|
|
10
|
Impei S, Gismondi A, Canuti L and Canini
A: Metabolic and biological profile of autochthonous Vitis vinifera
L. ecotypes. Food Funct. 6:1526–1538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Potze L, di Franco S, Kessler JH, Stassi G
and Medema JP: Betulinic acid kills colon cancer stem cells. Curr
Stem Cell Res Ther. 11:427–433. 2016. View Article : Google Scholar
|
|
12
|
Pandurangan AK and Esa NM: Luteolin, a
bioflavonoid inhibits colorectal cancer through modulation of
multiple signaling pathways: A review. Asian Pac J Cancer Prev.
15:5501–5508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gismondi A, Di Marco G, Canuti L and
Canini A: Antiradical activity of phenolic metabolites extracted
from grapes of white and red Vitis vinifera L. cultivars. Vitis.
56:19–26. 2017.
|
|
14
|
Kumar S and Pandey AK: Chemistry and
biological activities of flavonoids: An overview. Scientific World
Journal. 2013:1627502013. View Article : Google Scholar
|
|
15
|
Zhang YC, Gan FF, Shelar SB, Ng KY and
Chew EH: Antioxidant and Nrf2 inducing activities of luteolin, a
flavonoid constituent in Ixeris sonchifolia Hance, provide
neuroprotective effects against ischemia-induced cellular injury.
Food Chem Toxicol. 59:272–280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kasala ER, Bodduluru LN, Barua CC and
Gogoi R: Antioxidant and antitumor efficacy of Luteolin, a dietary
flavone on benzo(a) pyrene-induced experimental lung
carcinogenesis. Biomed Pharmacother. 82:568–577. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pandurangan AK, Dharmalingam P, Sadagopan
SK and Ganapasam S: Luteolin inhibits matrix metalloproteinase 9
and 2 in azoxymethane-induced colon carcinogenesis. Hum Exp
Toxicol. 33:1176–1185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pandurangan AK, Ananda Sadagopan SK,
Dharmalingam P and Ganapasam S: Inhibitory effect of luteolin on
azoxymethane induced colon carcinogenesis: Involvement of iNOS and
COX-2. Pharmacogn Mag. 10:306–310. 2014. View Article : Google Scholar
|
|
19
|
Pandurangan AK, Ananda Sadagopan SK,
Dharmalingam P and Ganapasam S: Luteolin, a bioflavonoid inhibits
Azoxymethane-induced colorectal cancer through activation of Nrf2
signaling. Toxicol Mech Methods. 24:13–20. 2014. View Article : Google Scholar
|
|
20
|
Pandurangan AK, Dharmalingam P, Sadagopan
SK, Ramar M, Munusamy A and Ganapasam S: Luteolin induces growth
arrest in colon cancer cells through involvement of
Wnt/β-catenin/GSK-3β signaling. J Environ Pathol Toxicol Oncol.
32:131–139. 2013. View Article : Google Scholar
|
|
21
|
Ashokkumar P and Sudhandiran G: Luteolin
inhibits cell proliferation during Azoxymethane-induced
experimental colon carcinogenesis via Wnt/β-catenin pathway. Invest
New Drugs. 29:273–284. 2011. View Article : Google Scholar
|
|
22
|
Attoub S, Hassan AH, Vanhoecke B, Iratni
R, Takahashi T, Gaben AM, Bracke M, Awad S, John A, Kamalboor HA,
et al: Inhibition of cell survival, invasion, tumor growth and
histone deacetylase activity by the dietary flavonoid luteolin in
human epithelioid cancer cells. Eur J Pharmacol. 651:18–25. 2011.
View Article : Google Scholar
|
|
23
|
Pandurangan AK and Ganapasam S: Cytotoxic
effect of luteolin on human colorectal cancer cell line (HCT-15):
Crucial involvement of reactive oxygen species. Middle East J
Cancer. 4:177–182. 2013.
|
|
24
|
Safi W, Kuehnl A, Nüssler A, Eckstein HH
and Pelisek J: Differentiation of human CD14+ monocytes:
An experimental investigation of the optimal culture medium and
evidence of a lack of differentiation along the endothelial line.
Exp Mol Med. 48:e2272016. View Article : Google Scholar
|
|
25
|
Kimura S, Inoguchi T, Yamasaki T, Yamato
M, Ide M, Sonoda N, Yamada K and Takayanagi R: A novel DPP-4
inhibitor teneligliptin scavenges hydroxyl radicals: In vitro study
evaluated by electron spin resonance spectroscopy and in vivo study
using DPP-4 deficient rats. Metabolism. 65:138–145. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim HB and Yoo BS: Propolis inhibits
UVA-induced apoptosis of human keratinocyte HaCaT cells by
scavenging ROS. Toxicol Res. 32:345–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carrillo MC, Kanai S, Nokubo M and Kitani
K: (−) deprenyl induces activities of both superoxide dismutase and
catalase but not of glutathione peroxidase in the striatum of young
male rats. Life Sci. 48:517–521. 1991. View Article : Google Scholar
|
|
28
|
Tauskela JS, Hewitt K, Kang LP, Comas T,
Gendron T, Hakim A, Hogan M, Durkin J and Morley P: Evaluation of
glutathione-sensitive fluorescent dyes in cortical culture. Glia.
30:329–341. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fernando PM, Piao MJ, Kang KA, Ryu YS,
Hewage SR, Chae SW and Hyun JW: Rosmarinic acid attenuates cell
damage against UVB radiation-induced oxidative stress via enhancing
antioxidant effects in human HaCaT cells. Biomol Ther (Seoul).
24:75–84. 2016. View Article : Google Scholar
|
|
30
|
Nicoletti I, Migliorati G, Pagliacci MC,
Grignani F and Riccardi C: A rapid and simple method for measuring
thymocyte apoptosis by propidium iodide staining and flow
cytometry. J Immunol Methods. 139:271–279. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang D and Armstrong JS: Bax and the
mitochondrial permeability transition cooperate in the release of
cytochrome c during endoplasmic reticulum-stress-induced apoptosis.
Cell Death Differ. 14:703–715. 2007. View Article : Google Scholar
|
|
32
|
Gold-Smith F, Fernandez A and Bishop K:
Mangiferin and cancer: Mechanisms of action. Nutrients. 8:3962016.
View Article : Google Scholar :
|
|
33
|
Tait SW and Green DR: Mitochondrial
regulation of cell death. Cold Spring Harb Perspect Biol.
5:0087062013. View Article : Google Scholar
|
|
34
|
Cao XH, Zhao SS, Liu DY, Wang Z, Niu LL,
Hou LH and Wang CL: ROS-Ca(2+) is associated with mitochondria
permeability transition pore involved in surfactin-induced MCF-7
cells apoptosis. Chem Biol Interact. 190:16–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cheng YY, Yang JS, Tsai SC, Liaw CC, Chung
JG, Huang LJ, Lee KH, Lu CC, Chien HC, Tsuzuki M, et al: The newly
synthesized
2-(3-hydroxy-5-methoxyphenyl)-6,7-methylenedioxyquinolin-4-one
triggers cell apoptosis through induction of oxidative stress and
upregulation of the p38 MAPK signaling pathway in HL-60 human
leukemia cells. Oncol Rep. 28:1482–1490. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hsieh CJ, Kuo PL, Hsu YC, Huang YF, Tsai
EM and Hsu YL: Arctigenin, a dietary phytoestrogen, induces
apoptosis of estrogen receptor-negative breast cancer cells through
the ROS/p38 MAPK pathway and epigenetic regulation. Free Radic Biol
Med. 67:159–170. 2014. View Article : Google Scholar
|
|
37
|
Schulze A and Harris AL: How cancer
metabolism is tuned for proliferation and vulnerable to disruption.
Nature. 491:364–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang P, Feng L, Oldham EA, Keating MJ and
Plunkett W: Superoxide dismutase as a target for the selective
killing of cancer cells. Nature. 407:390–395. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee LT, Huang YT, Hwang JJ, Lee AY, Ke FC,
Huang CJ, Kandaswami C, Lee PP and Lee MT: Transinactivation of the
epidermal growth factor receptor tyrosine kinase and focal adhesion
kinase phosphorylation by dietary flavonoids: Effect on invasive
potential of human carcinoma cells. Biochem Pharmacol.
67:2103–2114. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sonoda M, Nishiyama T, Matsukawa Y and
Moriyasu M: Cytotoxic activities of flavonoids from two Scutellaria
plants in Chinese medicine. J Ethnopharmacol. 91:65–68. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lim DY, Jeong Y, Tyner AL and Park JH:
Induction of cell cycle arrest and apoptosis in HT-29 human colon
cancer cells by the dietary compound luteolin. Am J Physiol
Gastrointest Liver Physiol. 292:G66–G75. 2007. View Article : Google Scholar
|
|
42
|
Miar A, Hevia D, Muñoz-Cimadevilla H,
Astudillo A, Velasco J, Sainz RM and Mayo JC: Manganese superoxide
dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers
for tumor progression and metastasis in prostate, colon, and lung
cancer. Free Radic Biol Med. 85:45–55. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sarsour EH, Kalen AL and Goswami PC:
Manganese superoxide dismutase regulates a redox cycle within the
cell cycle. Antioxid Redox Signal. 20:1618–1627. 2014. View Article : Google Scholar :
|
|
44
|
Hart PC, Mao M, de Abreu AL,
Ansenberger-Fricano K, Ekoue DN, Ganini D, Kajdacsy-Balla A,
Diamond AM, Minshall RD, Consolaro ME, et al: MnSOD upregulation
sustains the Warburg effect via mitochondrial ROS and
AMPK-dependent signalling in cancer. Nat Commun. 6:60532015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dhar SK, Tangpong J, Chaiswing L, Oberley
TD and St Clair DK : Manganese superoxide dismutase is a
p53-regulated gene that switches cancers between early and advanced
stages. Cancer Res. 71:6684–6695. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun Y, St Clair DK , Xu Y, Crooks PA and
St Clair WH : A NADPH oxidase-dependent redox signaling pathway
mediates the selective radiosensitization effect of parthenolide in
prostate cancer cells. Cancer Res. 70:2880–2890. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Becuwe P, Ennen M, Klotz R, Barbieux C and
Grandemange S: Manganese superoxide dismutase in breast cancer:
From molecular mechanisms of gene regulation to biological and
clinical significance. Free Radic Biol Med. 77:139–151. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dhar SK and St Clair DK : Manganese
superoxide dismutase regulation and cancer. Free Radic Biol Med.
52:2209–2222. 2012. View Article : Google Scholar : PubMed/NCBI
|