Introduction
Antitumor nucleosides, particularly the
deoxycytidine analog, are key anticancer drugs which play an
important role in the treatment of patients with solid tumors and
leukemia. For instance, gemcitabine (GEM) and cytosine arabinoside
(Ara-C) have been clinically used as a standard of care treatment
in patients with pancreatic and non-small cell lung cancers
(1–3), as well as in patients with and acute
myeloid leukemia (AML) (4–8). A well-accepted cytotoxic mechanism of
these drugs involves the inhibition of DNA biosynthesis through
their incorporation into DNA molecules or the inhibition of DNA
polymerases following the conversion to their tri-phosphate forms
in cancer cells. However, these drugs are rapidly inactivated by
the cytidine deaminase (CDA) in normal and tumor tissues (8). Accordingly, relatively high-doses of
these drugs via bolus injection is common clinical practice in
patients to avoid such an inactivation; 1,000–1,500
mg/m2 of GEM and 2–3 g/m2 of Ara-C as
induction therapy are frequently administered to patients with
pancreatic cancer and AML, respectively.
2′-C-Cyano-2′-deoxy-1-β-D-arabino-pentofranocylcytosine (DFP-10917,
CNDAC) was developed in the early 1990s as a deoxycytidine analog
(9,10), which has shown potent antitumor
effects on various murine and human tumors in vitro and
in vivo (11). Similar to
other cytidine nucleosides, such as GEM and Ara-C, DFP-10917 has
been demonstrated to be efficiently phosphorylated to its mono-,
di- and tri-phosphate forms and is incorporated into the DNA of
tumor cells in vitro (12).
As regards the behavior of the triphosphate form of DFP-10917,
following incorporation into DNA, Hayakawa et al speculated
that it would chemically terminate an enzymatic DNA-chain
elongation (13) and for this,
there are some supporting data that DFP-10917 induces DNA
double-strand breaks and G2 cell cycle arrest in tumor cells in
vitro (14–17). On the other hand, a major
functional mechanism of GEM and Ara-C has been estimated to be the
direct inhibition of DNA polymerases, rather than their
incorporation into DNA in tumor cells, affecting DNA synthesis
(18–20).
However, such reports mentioned above, including
those on GEM, Ara-C and DFP-10917, have focused on the
clarification of their drug-induced functional mechanisms in tumor
cells in vitro. However, the exact clinically available
treatment regimen using these compounds which would prove to be
most effective for cancer patients remains to be established. In
addition, the functional mechanisms related to the drug schedule
need to be elucidated.
Thus, the aim of this study was to investigate the
optimal application dose and the treatment schedule for DFP-10917,
which would be useful for the treatment of hospitalized patients
with malignant tumors, including AML and advanced lung and
pancreatic cancer. For this purpose, we developed human tumor
xenograft models and aimed to confirm the association between the
dose intensity with the dosing schedule, and elucidate the novel
functional mechanisms of DFP-10917 compared to other deoxycytidine
analogs.
Materials and methods
Chemicals
DFP-10917 (CNDAC) was manufactured by Delta-Fly
Pharma Inc. (Tokushima, Japan). Ara-C and GEM were purchased from
Sigma-Aldrich Inc. (St. Louis, MO, USA). Paclitaxel and cisplatin
(CDDP) were purchased from Wako Pure Chemical Industries, Ltd.
(Osaka, Japan). Decitabine, an inhibitor of DNA methyltransferase,
was kindly provided by Otsuka Pharmaceutical Co. Ltd. (Tokyo,
Japan). The comet assay™ kit was obtained from Trevigen Inc.
(Gaitherburg, MD, USA). All other chemical and biochemical
materials were commercial products.
Tumor cells
The human MV-4-11 and CCRF-CEM leukemia cells, and
the U937 lymphoma cells were purchased from Dai-Nippon Sumitomo
Pharmaceutical Co. Ltd. (Osaka, Japan). The human colon cancer
KM20C cells, lung cancer Lu-99 and Lu-61 cells were obtained from
JCRB Cell Bank (National Institutes of Biomedical Innovation,
Health and Nutrition, Japan). The human pancreatic cancer PAN-4
cells were obtained from the Central Institute for Experimental
Animals (Kawasaki, Japan). Although the KM20C cells are considered
to have been contaminated and are mixed recto-sigmoid
adenocarcinoma cells, and the data regarding the PAN-4 cells have
not been disclosed, we decided to use these cells in our study, as
it was considered that this contamination and these undisclosed
data would have no influence on the comparative experiments. Human
cervical cancer HeLa cells were obtained from ATCC (Global
BioSource Center, Manassas, VA, USA). The SKOV-3 human ovarian
cancer cells were kindly provided by Dr Mitsuaki Suzuki at
Jichi-Medical University (Tochigi, Japan). These tumor cells were
cultivated and maintained in RPMI-1640 medium containing 10% fetal
bovine serum. Human solid tumor xenografts were prepared by the
serial implantation of cells in vitro into the right axilla
of nude mice at 3-week intervals until analysis.
Animals
A total of 135 male BALB/cA Jcl-nu mice and 190 male
C.B-17/Icr-scid Jcl mice (5 weeks old, weighing 17.2–24.6 g), were
purchased from KREA Japan, Inc. (Tokyo, Japan) and maintained on a
commercial diet and autoclaved water, made available ad
libitum. The care and treatment of the animals were in
accordance with the guidelines issued by the Science and
International Affairs Bureau of the Japanese Ministry of Education,
Science, Culture and Sports. The experimental protocol was carried
out following the approval of the Institutional Animal Ethics
Committee at the research facility of Delta-Fly Pharma Inc.
Antitumor experiments
Groups of 6 or 7 nude mice were used. The KM20C,
Lu-99, MV-4-11 and U937 tumor xenografts were prepared,
respectively, by the subcutaneous implantation (~2×2 mm fragments
of tumor slices) into the right axilla of BALB/cA Jcl-nu mice. When
the tumor volume reached ~200 mm3, DFP-10917 (30, 8 or
4.5 mg/kg/day) was continuously infused by an Alzet osmotic pump
for 24 consecutive hours on days 1 and 8, for 3 consecutive days on
days 1 and 15, or for 14 consecutive days, or DFP-10917 (500
mg/kg/day) was administered via bolus injection on days 1 and 8.
Ara-C (100 mg/kg) was administered via intravenous (i.v.) injection
on days 1–5 and 8–12, and GEM (300 mg/kg) was administered via i.v.
injection on days 1 and 8. Following drug treatment, the condition
of the mice was monitored daily for 30 days. The longest tumor
diameter formed by the KM20C cells was 18.51 mm and the maximum
tumor volume was 2750.75 mm3. None of the mice developed
multiple tumors. The tumor volume [1/2 × (the major axis) × (the
minor axis)2] was measured twice a week throughout the
treatment period (14 days), and the relative tumor volume (RTV) was
calculated as follows: RTV = (mean tumor volume during
therapy)/(mean tumor volume at the start of therapy). The antitumor
effects of DFP-10917, Ara-C and GEM were estimated by the following
equation: Mean inhibition rate of tumor growth (IR, %) = [1−(mean
RTV of drug-treated group/mean RTV of control group) ×100].
Survival experiments
For the U937 lymphoma cells, groups of 10
C.B-17/Icr-scid Jcl mice were used. Prior to tumor cell
implantation, 0.2 ml of anti-mouse Asialo GM1 antibody [antibody to
natural killer (NK) cells] was injected intraperitoneally into all
mice. The following day (day 1), the U937 lymphoma cells
(1×107 cells /0.5 ml/mouse) were implanted into the
intraperitoneal cavity of the mice. From day 2, DFP-10917 (4.5
mg/kg/day) was continuously infused for 14 days, Ara-C (100
mg/kg/day) was administered via i.v. injection on days 3–7 and days
10–14, and decitabine (1.0 mg/kg/day) was administered via
intraperitoneal (i.p.) injection on days 3–5 and days 10–12.
Following drug treatments, the survival of the mice was monitored
daily for 90 days. The antitumor activity of the drug was evaluated
as the survival effect (ILS, %) by the following equation: ILS (%)
= [mean survival days in drug-treated group/mean survival days in
control (no treatment) group − 1] ×100. As one of the endpoints,
the survival times with the respective agents were comparatively
investigated.
For another tumor model using intraperitoneally
disseminated ovarian cancer (SKOV-3) cells, the tumor cells
(2×107 cells/0.5 ml/mouse) were implanted, and after 24
h of implantation the mice (C.B-17/Icr-SCID Jcl) were treated with
DFP-10917 (4.5 mg/kg/day, 14 days), GEM (300 mg/kg/day, days 1 and
8), CDDP (7 mg/kg/day, days 1 and 8) and paclitaxel (50 mg/kg/day,
day 1) as a positive control under the scheduled time periods.
Evaluation of drug-related toxicity
The body weights of the tumor-bearing mice were
measured as an index of drug-induced toxicity, and the rate (%) of
changes in body weight (BWC) was calculated by the following
equation: BWC (%) = [(body weight on day n) − (body weight on day
0)]/(body weight on day 0) ×100. The maximum body weight of the
mice with tumors derived from the Lu-99 cells was 25.0 g at the
beginning of the experiment and 29.9 g upon sacrifice.
Comet assay
According to the manual provided with the Comet
assay kit (21), the drug-treated
cells were fixed on a glass slide, and treated with lysis solution
for >1 h. The slide glass containing the drug-treated cells was
then moved to an electrophoresis instrument and the cells were
exposed to 33 V and 300 mA electrophoresis for 20 min, and then
soaked in 0.4 M Tris-HCl buffer (pH 7.5) for ~10 min. This
procedure was repeated once more. After soaking in 70% ethanol for
5 min, the glass slide was dried to yield a thin-layer film.
Finally, 50 μl aliquots of ethidium bromide solution (20
μg/ml) were dropped onto the glass slide. After a coverglass
was placed on the glass slide, the migration rate of the
drug-treated cells (50 cells) was observed with an analysis
apparatus for Comet assay.
Evaluation of DNA fragmentation
The drug-induced fragmentation of DNA in the
CCRF-CEM cells was evaluated by the rate of electrophorated DNA (%
tail DNA) and olive tail moment calculated by the following
equation: Olive tail moment = (tail.mean−head.mean) × % tail
DNA/100.
Cell cycle analysis
The HeLa cells (1×106) were inoculated
into 6-well plates and treated with either 1 to 30 μM of
DFP-10917 or 0.03 to 0.3 μM of GEM. Untreated cells were
used as the control group. Viable cells were counted 24, 48 and 72
h after the inoculation to analyze the effects of the drug on cell
cycle progression using a commercially available cell-cycle
analyzer, FACSCalibur flow cytometer (Becton-Dickinson, Franklin
Lakes, NJ, USA).
Statistical analysis
The significance of differences between groups with
or without drug treatment was assessed using the generalized
Wilcoxon test. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of infusion time on the antitumor
activity of DFP-10917 in vivo
It is well known that the dosing schedule of drugs
influences their antitumor activity and has adverse events on
cancer patients (22,23). In a preliminary experiment using
KM20C tumor-bearing mice to determine the maximum tolerance dose
(MTD) of DFP-10917 at various doses and administration schedules
without death and a −20% change in body weight, we found that the
MTD was thus determined to be 500 mg/kg/day for i.v. bolus
injection on days 1 and 8, 30 mg/kg/day for 1-day continuous
infusions on days 1 and 8, 8 mg/kg/day for 3-day continuous
infusions on days 1 and 15, and 4.5 mg/kg/day for 14-day continuous
infusion (data not shown). Based on the results, we evaluated the
antitumor activity and toxicity of the infusion time of DFP-10917
on the same KM20C human cancer xenografts in nude mice. As shown in
Fig. 1A, no re-growth of tumors or
body weight loss were observed during days 15–29 following the
initiation of treatment with DFP-10917 for the 14-day continuous
infusion schedule (4.5 mg/kg) compared to the other schedules [i.v.
bolus injection (500 mg/kg, days 1 and 8), 1-day continuous
infusion (30 mg/kg, days 1 and 8), and 3-day continuous infusion (8
mg/kg, days 1 and 15)]; the prolonged (at least over 7 days)
administration of DFP-10917 at a low-dose of 4.5 mg/kg/day resulted
in the most potent antitumor activity with an >70% inhibition of
tumor growth (IR) compared to its shorter (1 to 3 days)
administrations. These results thus suggest that in oder to achieve
the maximal response against human cancers, DFP-10917 should be
administered by long-term infusion (14 days) at a lower dose (4.5
mg/kg).
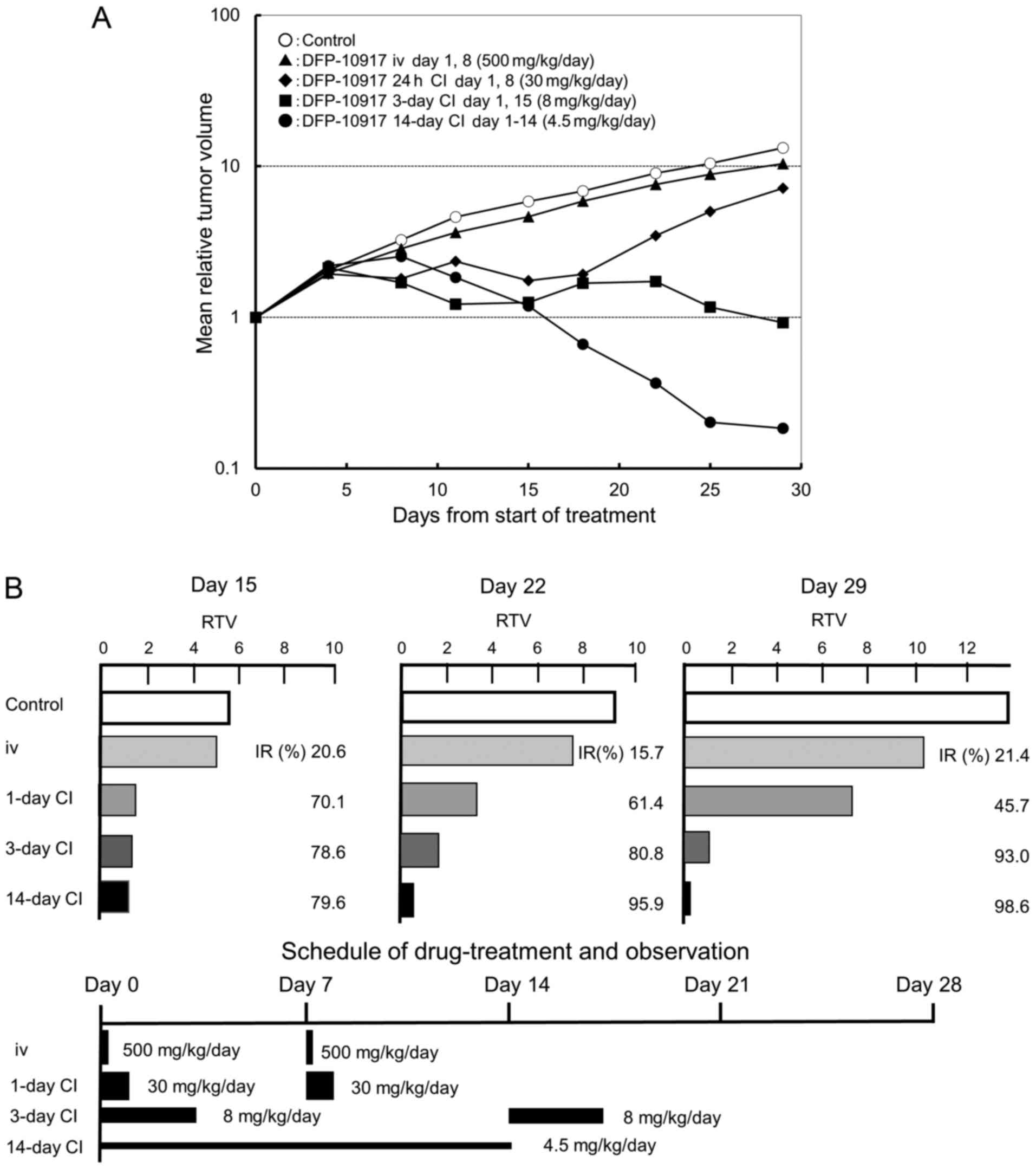 | Figure 1Effect of infusion time on the
antitumor activity of DFP-10917 in KM20C human tumor xenografts in
nude mice. The maximum tolerance dose (MTD) dose of DFP-10917 was
infused for 1 day (30 mg/kg/day) ×2, 3 days (8 mg/kg/day) ×2, and
14 days (4.5 mg/kg/day), respectively, to KM20C tumor-bearing mice
from 7 days after implantation. Relative tumor volume (RTV) was
calculated as follows: RTVn = (TV on day n)/(TV on day 0), n=4, 8,
11, 15, 18, 22, 25 and 29. (A) On day 15, the tumor growth
inhibitory effects of DFP-10917 were evaluated for each treatment
group, and thereafter, (B) the regrowth rate of the tumors was
measured on days 15, 22 and 29, respectively. Data represent mean
values of relative tumor volume for 7 mice. |
Re-growth rates of colon tumors following
treatment with DFP-10917 in various dosing schedules
In the same anti-tumor experiment, we observed the
re-growth rates of KM20C tumors on days 15, 22 and 29 after the
infusion of DFP-10917 with the 1- to 14-day schedules. As shown in
Fig. 1B, the IR of tumor growth
after a 1-day infusion with high-dose (30 mg/kg/day) DFP-10917
significantly decreased from 70.1% (on day 15) to 45.7% on day 29.
By contrast, the 14-day infusion of 4.5 mg/kg/day DFP-10917
resulted in an increased IR from 79.6% (on day 15) to 98.6% (on day
29), suggesting that the prolonged infusion with low-dose DFP-10917
prevented tumor re-growth after the terminated administration.
Effect of the prolonged infusion of
DFP-10917 on human lung cancer xenografts
GEM is mainly used to treat patients with advanced
pancreatic and lung cancer as one of the first-line treatment
regimens (24,25). However, such a treatment is
generally ineffective and patients become resistant to this drug
(26,27). Therefore, there is an urgent need
for the development of a novel treatment regimen with which to
control the progression of pancreatic and lung cancer. In this
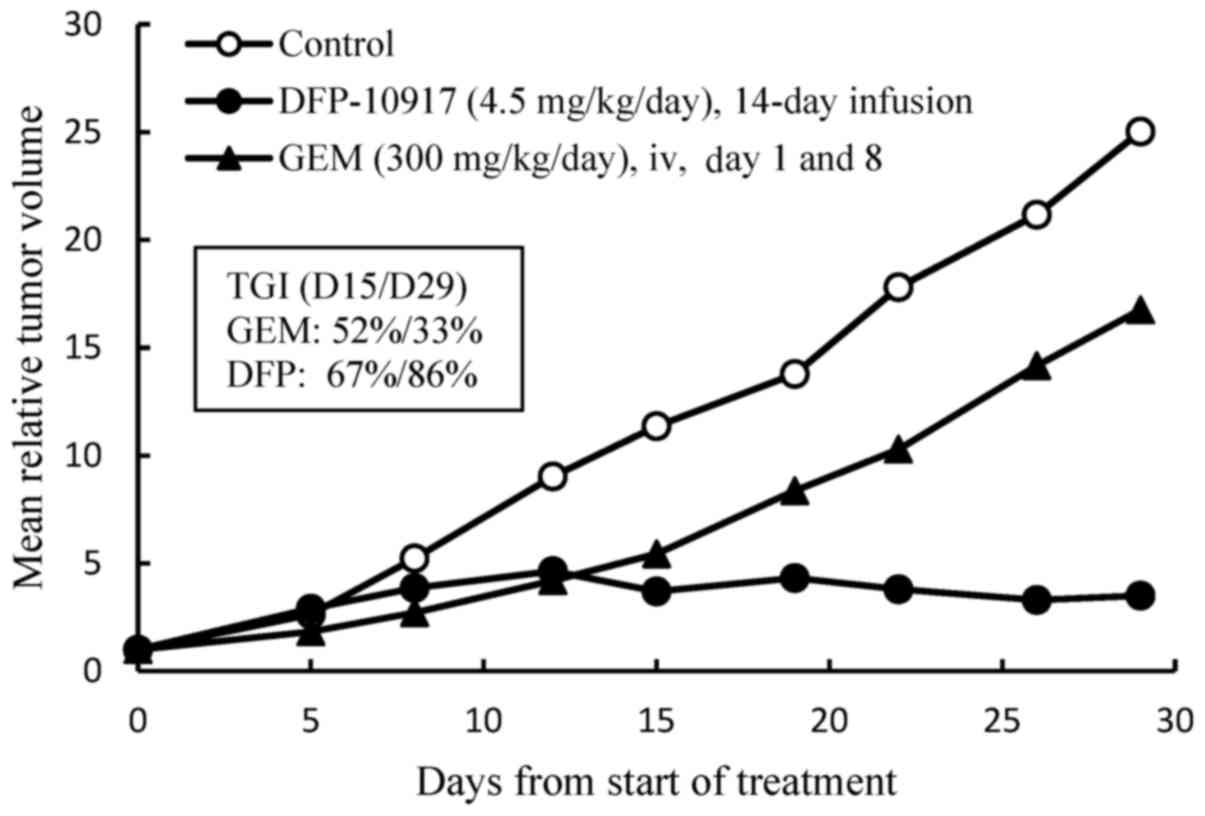
study, we examined the efficacy of DFP-10917 in comparison to GEM
of one of the deoxycytidine analogs by using Lu-99 human lung
cancer xenografts. DFP-10917 was found to control and/or suppress
the growth of Lu-99 tumor throughout the therapeutic periods up to
29 days, while GEM exhibited limited efficacy on this tumor, as was
expected (Fig. 2). In separate
experiments for comparing DFP-10917 and GEM by using PAN-4
pancreatic and Lu-61 lung cancer xenografts, the same dose rates
and treatment schedules for DFP-10917 and GEM resulted in an 84 and
59% IR, respectively in PAN-4-derived tumors, and in 91 and 17% IR,
respectively, in Lu-61-derived tumors (data not shown). Throughout
these experiments, the prolonged 14-day infusion of DFP-10917 was
suggested to have an antitumor activity comparative to that of
GEM.
Effect of the long-term infusion
DFP-10917 on hematological tumor cells in mice
Ara-C has been a standard of care for the treatment
of patients with AML. A bolus injection of high-doses of Ara-C has
been applied due to the rapid inactivation by cytidine deaminase.
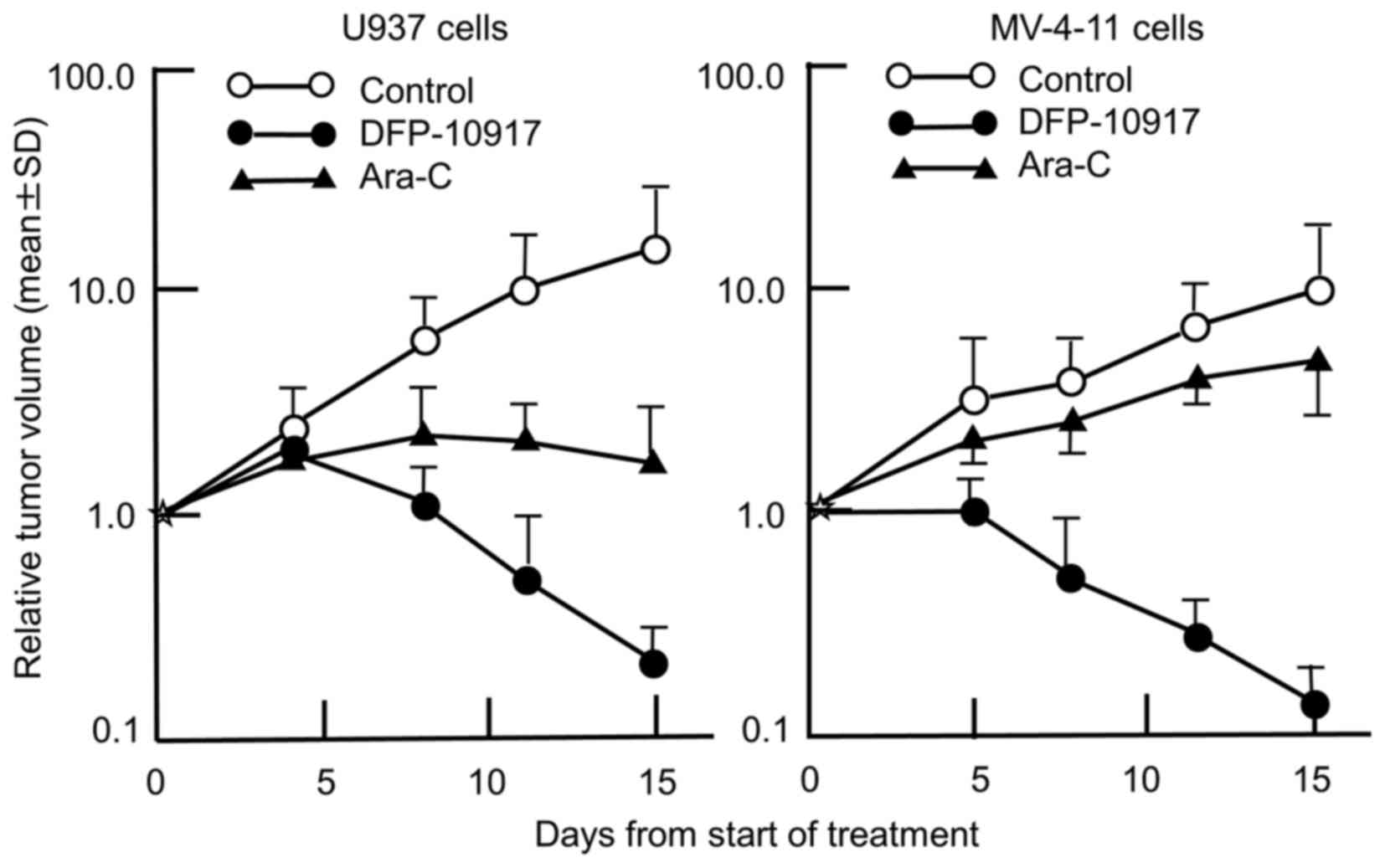
We thus comparatively evaluated the anticancer activity of
DFP-10917 and Ara-C in the U937 and MV-4-11 cells tumors (solid
forms in both cases). As shown in Fig.
3, the prolonged infusion of DFP-10917 markedly affected both
U937 and MV-4-11 tumor xenografts with a 98.8 and 98.7% IR,
respectively, compared to treatment with Ara-C which yieled an 83
and 43.6% IR, respectively. It is worth mentioning that DFP-10917
markedly abolished tumor growth over the 14-day therapeutic
period.
Comparative survival effect of DFP-10917
on ascitic U937 and SKOV-3 tumor xenografts in mice
As the ultimate objective of drug treatment in
advanced cancers is the achievement of prolonged survival with a
good quality of life (QOL), the effects of DFP-10917 by 14-day
infusion on survival were examined on ascitic U937 leukemia cells.
As shown in Table I, DFP-10917,
Ara-C and decitabine led to a 165, 127 and 33% increase in lifespan
(ILS), respectively; this suggests a favorable pro-survival effect
of DFP-10917 compared with Ara-C and decitabine. We further
evaluated the effects of DFP-10917, GEM, CDDP and paclitaxel as
standards of care for ovarian cancer, in a disseminated ascitic
form of SKOV-3 ovarian cancer cells in mice (Table I). The prolonged infusion of
DFP-10917 was also demonstrated to significantly increase the
long-term survival by 234% compared to GEM (12%), CDDP (39%) and
paclitaxel (47%). Throughout two therapeutic experiments, the
prolonged infusion of DFP-10917 at a low-dose clearly prolonged the
survival time in both leukemia cells and intraperitoneally
disseminated solid tumors, suggesting its potential clinical
benefit for patients with cancer.
 | Table IEffects of DFP-10917 on the survival
of mice with U937 or SKOV-3 human tumor xenografts. |
Table I
Effects of DFP-10917 on the survival
of mice with U937 or SKOV-3 human tumor xenografts.
| Cell line | Drug | Dose
(mg/kg/day) | Treatment | No. of mice | Survival
time
(days, mean ± SD) | ILSa (%) |
|---|
| U937
(lymphoma) | Control | – | – | 10 | 32.9±30.5 | – |
| DFP-10917 | 4.5 | s.c., days
3–17 | 10 | 87.2±6.1b,c,d | 165.0 |
| Ara-C | 100 | i.v., days 3–7,
10–14 | 9 | 74.4±15.5b | 127.0 |
| Decitabine | 1.0 | i.p., days 3–5,
10–12 (bid) | 10 | 43.8±24.7 | 33.1 |
| SKOV-3 (ovarian
carcinoma) | Control | – | – | 10 | 22.0±2.2 | – |
| DFP-10917 | 4.5 | s.c., days
1–14 | 10 | 73.5±4.6e,f,g,h | 234.1 |
| Paclitaxel | 50 | i.v., day 1 | 10 | 32.3±7.9e | 46.8 |
| CDDP | 7 | i.v., days 1,
8 | 10 | 30.6±4.7e | 39.1 |
| Gemcitabine | 300 | i.v., days 1,
8 | 10 | 24.7±1.3e | 12.3 |
DNA double-strand breaks in human cancer
cells following treatment with DFP-10917
To investigate the cytotoxic mechanisms of the
prolonged infusion of DFP-10917 compared to other deoxycytidine
analogs, such as Ara-C and GEM, DNA strand-breakage following the
continuous exposure to DFP-10917 and Ara-C or GEM was investigated
by comet assay (20) in human
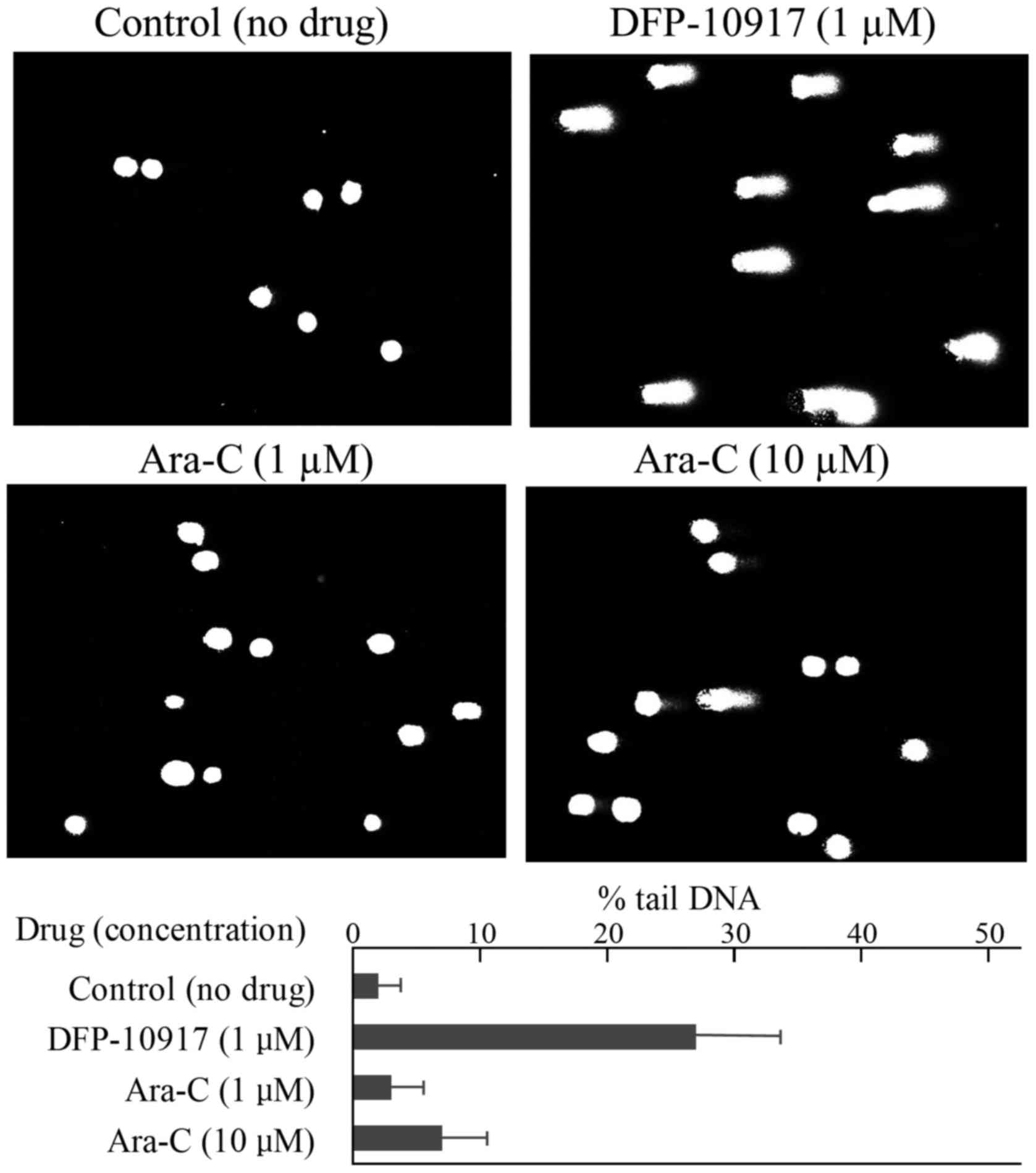
leukemia CCRF-CEM cells and solid cervical cancer HeLa cells. In
the CCRF-CEM cells, 1 μM DFP-10917 (IC50 value)
induced marked DNA fragmentation with 26.88±12.84 of % tail, while
1 to 10 μM (IC50 and IC70 values) Ara-C induced
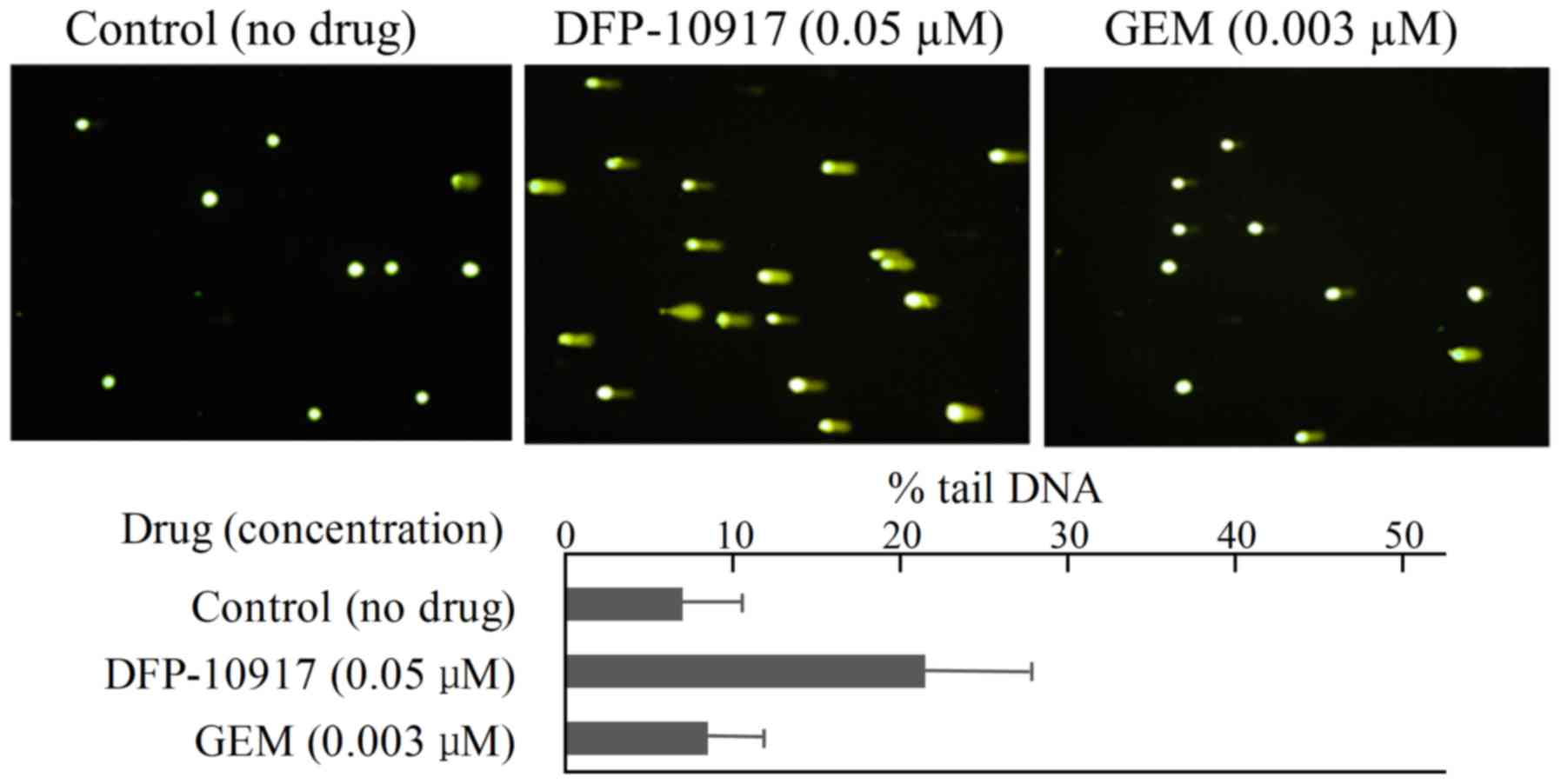
limited DNA fragmentation with ~6.95±6.65 of % tail DNA (Fig. 4). Similarly, DNA damage as an
indicator of DNA strand breaks by 0.05 μM DFP-10917 and 0.03
μM GEM (both IC50 values) was evaluated and
calculated as % tail DNA using HeLa cells. The rate of DNA
fragmentation (% tail DNA) for 72-h incubation was 7.16±6.86 for
the control group, 21.69±12.67 for the DFP-10917 group and
8.37±6.45 for the GEM group (Fig.
5). These result suggest that DFP-10917 induces potent DNA
damage as evaluated by DNA fragmentation (comet assay) compared to
Ara-C and GEM.
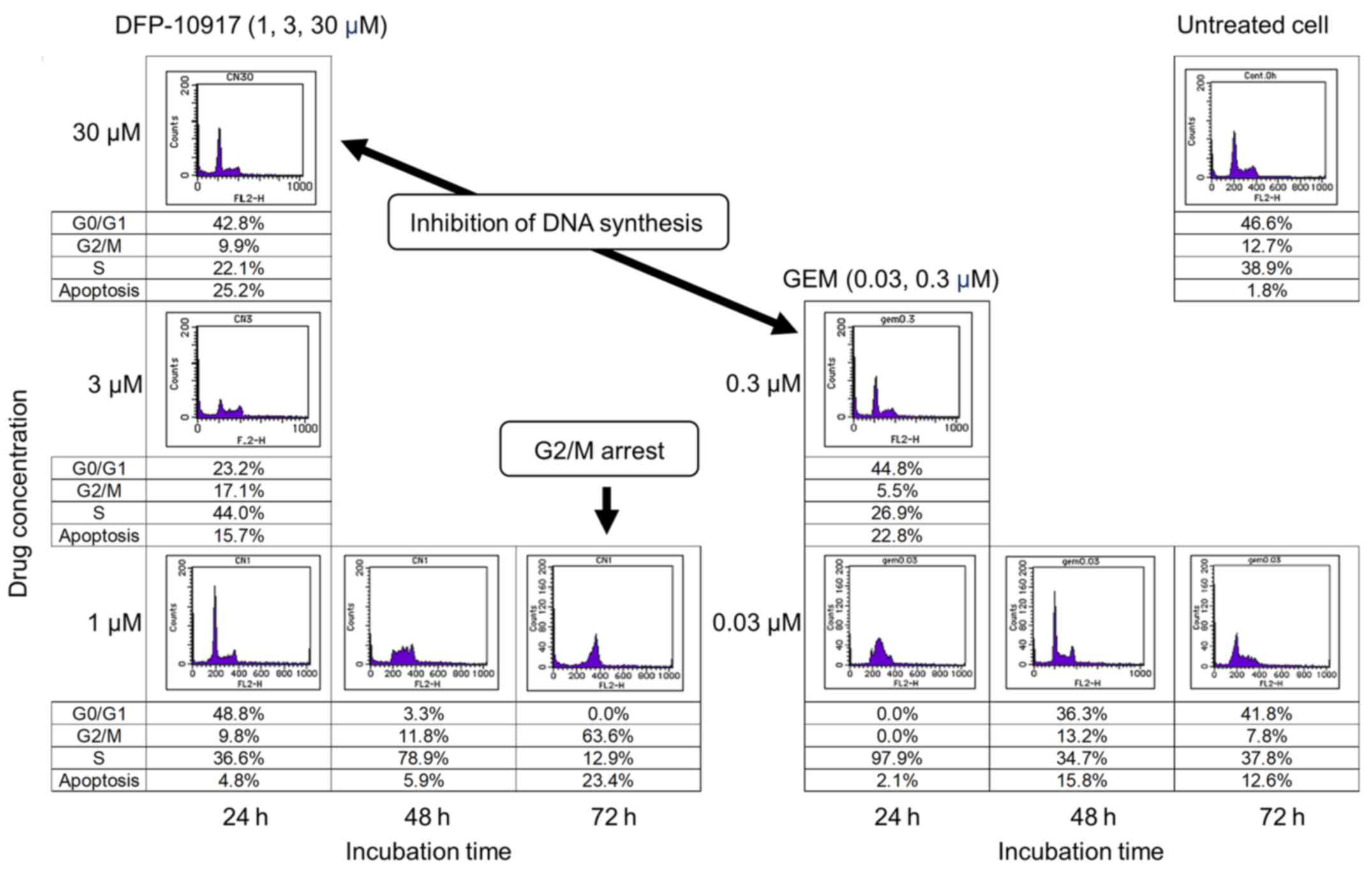
Effect of DFP-10917 and GEM on cell-cycle
of human tumor cells
To investigate whether DSFP-10917 and GEM, similar
cytidine analogs, have similar functions in tumor cells, HeLa cells
was exposed for various amounts of time to various concentrations
of both drugs, and the numbers of treated cells in the G0/G1, S and
G2M of the cell cycle were respectively determined by a cell cycle
analyzer. As shown in Fig. 6,
long-term exposure (72 h) to 1 μM DFP-10917 significantly
increased the numbers of cells in the G2/M phase of the cell cycle
(G2/M arrest), whereas a high-dose (30 μM) and a shorter
incubation time (24 h) with this drug resulted in a decrease in the
numbers of cells in the S phase, suggesting the inhibition of DNA
synthesis under such conditions. By striking contrast, GEM caused
only a decrease in the numbers of cells in the S phase when used
for 24 h at high (0.3 μM) concentrations. GEM also did not
increase the cell number in the G2/M phase of the cell cycle. These
results suggest that the functional mechanism of the prolonged
exposure to low-dose DFP-10917 clearly differs from that of
exposure to GEM or other deoxycytidine analogs.
Discussion
Of the anti-metabolites widely used in the treatment
of cancers, antitumor nucleosides, such as Ara-C, GEM and
decitabine have been recognized to play a vital role in the
treatment of hematological and solid cancers as single agent and/or
combined therapeutic regimens. The major mechanisms for the
antitumor activity of these cytosine nucleosides is the inhibition
of DNA replication or repair, which is much higher in tumor cells
than in normal cells via the inhibition of key enzymes, such as DNA
polymerases and ribonucleotide reductase or direct incorporation
into DNA. However, due to the unfavorable intracellular metabolism
(particularly catabolism) of these nucleosides, it would be
important to establish suitable exposure conditions, such as dosage
and the administration time, in order to reach a desirable
antitumor activity with limited toxicity which leads to the
objective exhibition of their DNA regulation mentioned above.
DFP-10917 (also kown as CNDAC) is a unique
synthesized promising deoxycytidine analog which has been shown to
exert potent anticancer activity against various murine and human
tumors in vitro and in vivo (9,11).
Different from GEM and Ara-C, the cytotoxic mechanism of DFP-10917
has been speculated to be the induction of DNA strand breakage
following its incorporation into DNA and the arrest in the G2/M
phase of the cell cycle in treated tumor cells (14,15).
However, the association between the cumulative dosage and
treatment schedule of DFP-10917 in order to exert a maximal
antitumor activity has not been yet defined in vivo human
tumor models. In this study, we thus evaluated the effects of
cumulative dose and infusion schedule on the antitumor efficacy of
DFP-10917 using KM20C human tumor xenografts in mice. As shown in
Fig. 1, when a total of 63 mg/kg
of DFP-10917 was administered over a 14-day therapeutic period, the
prolonged continuous infusion (14 days) with the lowest dose (4.5
mg/kg/day) resulted in the maximal tumor growth inhibition (tumor
regression) with no drug-related toxicity in compared to 1-day
infusion with the high-doses (30 mg/kg/day, total of 60 mg/kg) or
3-day infusion with the intermediate doses (8 mg/kg/day, total of
48 mg/kg).
Furthermore, the prolonged (14-day) infusion of
DFP-10917 was comparatively evaluated in human lung cancer Lu-99
xenografts in which GEM showed limited antitumor activity. As shown
in Fig. 2, the 14-day infusion of
low-dose (4.5 mg/kg/day) DFP-10917 reduced tumor volume, while the
weekly intravenous injection of high-dose GEM (300 mg/kg) resulted
in only a 33% decrease on day 14 and 14% of tumor growth inhibition
(TGI) on day 29 in the same therapeutic periods.
We were interested in investigating whether the
prolonged continuous infusion of DFP-10197 at a lower dose also
plays the same role in human leukemia cells in vivo compared
to Ara-C which is a standard therapeutic drug used in the treatment
of patients with AML, and comparatively evaluated the growth
inhibitory effect of the 14-day infusion of low-dose DFP-10917 (4.5
mg/kg/day) and consecutive i.v. injection (days 1–5 and 8–12) of
high-dose Ara-C (100 mg/kg/day) on U937 and MV-4-11 human leukemia
tumor xenografts in nude mice, respectively. The prolonged infusion
of low-dose DFP-10917 markedly suppressed tumor volume in both
leukemia xenografts, and compared to DFP-10917, the repeated 5-day
consecutive administration of Ara-C was less effective on these
tumors (Fig. 3), suggesting that
DFP-10917 may be useful for the treatment of leukemia.
In terms of finding the optimal dose of GEM in
preclinical and clinical reports, there are various reports over
the past 2–3 decades. Veerman et al (28) suggested that the prolonged infusion
of GEM led to a better antitumor activity than bolus injections
in vivo and that it showed promise of being active in
clinical trials. On the other hand, Kirstein et al (29) showed that long-term survival was
significantly diminished following continuous infusion compared
with the short-term infusion of GEM, although treatment induced
apoptosis following both short-term and continuous infusions in
non-small cell lung cancer cells in vitro. In clinical
trials, Rajdev et al (30)
performed a phase I trial of GEM administered as a 96-h continuous
intravenous infusion in patients with advanced carcinoma and
lymphoma and concluded that the administration of GEM as a 96-h
infusion resulted in a markedly different toxicity profile than
when administered by a conventional 30-min infusion. In addition, a
number of clinical studies have supported a short-time (30-min)
infusion rather than a long-term (over 2.5 h) infusion of GEM from
the viewpoint of the survival and GEM-induced toxicity profiles in
cancer patients (31,32).
Ara-C, the other deoxycytidine analog, is the
standard of care for the treatment of patients with AML. Although
there are few reports of the optimization of the dosing schedule of
Ara-C in preclinical studies using leukemia cells, various clinical
trials have been carried out to determine a suitable or optimal
dosing schedules of Ara-C for patients with AML (33–44).
Currently, for the treatment of patients with AML, the 7-day
infusion of standard-dose Ara-C (100–200 mg/m2) or the
6-day infusion of the 12-h high-dose Ara-C (2,000 mg/m2)
in combination with anthracycline (idarubicin or daunorubicin) are
conducted as induction therapy, and/or 3-h infusion of high-dose
Ara-C (HiDAC, 3,000 mg/m2) every 12 h/day for 3 to 4
cycles is the consolidation therapy. Accordingly, the dose and
schedule of GEM and Ara-C used in this in vivo study would
reflect the clinical treatment regimen and the prolonged continuous
infusion of low-dose DFP-10917 and may contribute to the treatment
of patients with solid tumors or leukemia cells resistant or
insensitive to standard of deoxycytidine analogs in future clinical
studies.
As regards the molecular and pharmacological
mechanisms of DFP-10917 (also known as CNDAC), although Azuma et
al (14) and Liu et al
(15) reported that DFP-10917
caused the DNA strand breaks and subsequent G2/M phase arrest of
the cell cycle in DFP-10917-treated tumor cells, we evaluated the
strength of DNA damage in solid tumor (HeLa) and leukemia
(CCRF-CEM) cells, respectively, induced by low-dose and the
prolonged exposure to DFP-10917 compared to treatment with GEM and
Ara-C. By comet assay, DFP-10917 was confirmed to induce DNA damage
by inducing DNA strand breaks (mainly double-strand breaks) in both
cells. On the other hand, GEM and Ara-C did not cause such events
at the concentrations and exposure times used, which suggests a
marked difference in the mechanisms of action between DFP-10917 and
the two deoxycytidine analogs, as regards the pharmacological
mechanism of the drugs (Figs. 4
and 5). Furthermore, we
investigated the influence of DFP-10917-induced DNA damage on the
cell cycle of HeLa cells in vitro and compared the effects
to those of GEM. We found that only low-dose (1 μM) and
long-term exposure (72 h) to DFP-10917 fairly increased the
population of cells in the G2/M phase, while 0.003 to 0.03
μM GEM used in this study did not lead to such an
accumulation of cells in the G2/M phase of the cell cycle (Fig. 6). Our data are consistent with
those of previous studies on several leukemia cells in vitro
(14–17). Importantly, our in vitro
mechanistic experiments were performed based on the finding of
which the prolonged infusion of low-dose DFP-10917 attained the
regression of tumor growth without any toxicities on human solid
and hematological tumor xenografts compared to clinically available
deoxycytidine analogs. Accordingly, DFP-10917, when infused
consecutively for a long-term period at a low-dose, may be a
beneficial therapy, not only for hospitalized, but also for
outpatients with advanced and inoperable tumors, including AML and
pancreatic cancer. Another clinical phase I/II trial for DFP-10917
administered by continuous infusion is ongoing in patients with
relapsed or refractory AML, which failed to respond to treatment
with an Ara-C-containing standard regimen (https://www.clinical-trials.gov/ct2/show/NCT01702155?term=DFP-10917&rank=1).
In conclusion, based on the cellular metabolism of
DFP-10917 and its possible utility strategy, various treatment
schedules were investigated using human tumor xenografts in mice
and found that the prolonged continuous infusion rather than the
short-term administration provided the best outcome for the
treatment of the rapid growth of tumor cells in vivo. Such
an antitumor activity by DFP-10917 was suggested to be depend on
the induction of DNA damage and the subsequent accumulation of
cells in the G2/M phase (namely G2/M arrest) of the cell cycle in
tumor cells, which is markedly different from the functional
mechanisms of other antitumor nucleosides.
Acknowledgments
The authors would like to express their gratitude to
Dr Tatsuhiro Ishida, Tokushima University, for his valuable advice
regarding technical issues.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Noble S and Goa KL: Gemcitabine A review
of its pharmacology and clinical potential in non-small cell lung
cancer and pancreatic cancer. Drugs. 54:447–472. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barton-Burke M: Gemcitabine: A
pharmacologic and clinical overview. Cancer Nurs. 22:176–183. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Toschi L, Finocchiaro G, Bartolini S,
Gioia V and Cappuzzo F: Role of gemcitabine in cancer therapy.
Future Oncol. 1:7–17. 2005. View Article : Google Scholar
|
|
4
|
Cole N and Gibson BE: High-dose cytosine
arabinoside in the treatment of acute myeloid leukaemia. Blood Rev.
11:39–45. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kern W and Estey EH: High-dose cytosine
arabinoside in the treatment of acute myeloid leukemia: Review of
three randomized trials. Cancer. 107:116–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reese ND and Schiller GJ: High-dose
cytarabine (HD araC) in the treatment of leukemias: A review. Curr
Hematol Malig Rep. 8:141–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li W, Gong X, Sun M, Zhao X, Gong B, Wei
H, Mi Y and Wang J: High-dose cytarabine in acute myeloid leukemia
treatment: A systematic review and meta-analysis. PLoS One.
9:e1101532014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Somasekaram A, Jarmuz A, How A, Scott J
and Navaratnam N: Intracellular localization of human cytidine
deaminase. Identification of a functional nuclear localization
signal. J Biol Chem. 274:28405–28412. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Azuma A, Nakajima Y, Nishizono N, Minakawa
N, Suzuki M, Hanaoka K, Kobayashi T, Tanaka M, Sasaki T and Matsuda
A: Nucleosides and nucleotides. 122
2′-C-cyano-2′-deoxy-1-β-D-arabinofuranosylcytosine and its
derivatives A new class of nucleoside with a broad antitumor
spectrum. J Med Chem. 36:4183–4189. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Azuma A, Hanaoka K, Kurihara A, Kobayashi
T, Miyauchi S, Kamo N, Tanaka M, Sasaki T and Matsuda A:
Nucleosides and nucleotides. 141. Chemical stability of a new
antitumor nucleoside,
2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine in
alkaline medium: Formation of
2′-C-cyano-2′-deoxy-1-β-D-ribo-pentofuranosylcytosine and its
antitumor activity. J Med Chem. 38:3391–3397. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tanaka M, Matsuda A, Terao T and Sasaki T:
Antitumor activity of a novel nucleoside,
2′-C-cyano-2′-deoxy-1-β-D-arabinofuranosylcytosine (CNDAC) against
murine and human tumors. Cancer Lett. 64:67–74. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Azuma A, Huang P, Matsuda A and Plunkett
W: Cellular pharmacokinetics and pharmacodynamics of the
deoxycytidine analog
2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine (CNDAC).
Biochem Pharmacol. 61:1497–1507. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hayakawa Y, Kawai R, Otsuki K, Kataoka M
and Matsuda A: Evidence supporting the activity of
2′-C-cyano-2′-deoxy-1-β-D-arabino-pentafuranosylcytosine as a
terminator in enzymatic DNA-chain elongation. Bioorg Med Chem Lett.
8:2559–2562. 1998. View Article : Google Scholar
|
|
14
|
Azuma A, Huang P, Matsuda A and Plunkett
W: 2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine: A
novel anticancer nucleoside analog that causes both DNA strand
breaks and G(2) arrest. Mol Pharmacol. 59:725–731. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu X, Guo Y, Li Y, Jiang Y, Chubb S,
Azuma A, Huang P, Matsuda A, Hittelman W and Plunkett W: Molecular
basis for G2 arrest induced by
2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine and
consequences of checkpoint abrogation. Cancer Res. 65:6874–6881.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y and Liu X: M<atsuda A and
Plunkett W: Repair of
2′-C-cyano-2′-deoxy-1-β-D-arabino-pantofuranosylcytosine-induced
DNA single-starand breaks by transcription-coupled nucleotide
excision repair. Cancer Res. 68:3881–2889. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu X, Wang Y, Benaissa S, Matsuda A,
Kantarjian H, Estrov Z and Plunkett W: Homologous recombination as
a resistance mechanism to replication-induced double-strand breaks
caused by the antileukemia agent CNDAC. Blood. 116:1737–1746. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang P, Chubb S, Hertel LW, Grindey GB
and Plunkett W: Action of 2′,2′-difluorodeoxycytidine on DNA
synthesis. Cancer Res. 51:6110–6117. 1991.PubMed/NCBI
|
|
19
|
Jiang HY, Hickey RJ, Abdel-Aziz W and
Malkas LH: Effects of gemcitabine and araC on in vitro DNA
synthesis mediated by the human breast cell DNA synthesome. Cancer
Chemother Pharmacol. 45:320–328. 2000. View Article : Google Scholar
|
|
20
|
Miura S and Izuta S: DNA polymerases as
targets of anticancer nucleosides. Curr Drug Targets. 5:191–195.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liao W, McNutt MA and Zhu WG: The comet
assay: A sensitive method for detecting DNA damage in individual
cells. Methods. 48:46–53. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Food and Drug Administration HHS: HHS
International conference on harmonisation; guidance on S9
nonclincal evaluation for anticancer pharmaceuticals; availability.
Fed Regist. 75:10487–10488. 2010.
|
|
23
|
Cook N, Hansen AR, Siu LL and Abdul Razak
AR: Early phase clinical trials to identify optimal dosing and
safety. Mol Oncol. 9:997–1007. 2015. View Article : Google Scholar :
|
|
24
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sandler AB, Nemunaitis J, Denham C, von
Pawel J, Cormier Y, Gatzemeier U, Mattson K, Manegold C, Palmer MC,
Gregor A, et al: Phase III trial of gemcitabine plus cisplatin
versus cisplatin alone in patients with locally advanced or
metastatic non-small-cell lung cancer. J Clin Oncol. 18:122–130.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spratlin J, Sangha R, Glubrecht D, Dabbagh
L, Young JD, Dumontet C, Cass C, Lai R and Mackey JR: The absence
of human equilibrative nucleoside transporter 1 is associated with
reduced survival in patients with gemcitabine-treated pancreas
adenocarcinoma. Clin Cancer Res. 10:6956–6961. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ho CC, Kuo SH, Huang PH, Huang HY, Yang
CH, Yang PC and Ho CC1: Caveolin-1 expression is significantly
associated with drug resistance and poor prognosis in advanced
non-small cell lung cancer patients treated with gemcitabine-based
chemotherapy. Lung Cancer. 59:105–110. 2008. View Article : Google Scholar
|
|
28
|
Veerman G, Ruiz van Haperen VW, Vermorken
JB, Noordhuis P, Braakhuis BJ, Pinedo HM and Peters GJ: Antitumor
activity of prolonged as compared with bolus administration of
2′,2′-difluorodeoxycytidine in vivo against murine colon tumors.
Cancer Chemother Pharmacol. 38:335–342. 1996. View Article : Google Scholar
|
|
29
|
Kirstein MN, Wieman KM, Williams BW,
Fisher JE, Marker PH, Le CT, Yee D and Kratzke RA: Short versus
continuous gemcitabine treatment of non-small cell lung cancer in
an in vitro cell culture bioreactor system. Lung Cancer.
58:196–204. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rajdev L, Goldberg G, Hopkins U and
Sparano JA: A phase I trial of gemcitabine administered as a 96-h
continuous intravenous infusion in patients with advanced carcinoma
and lymphoma. Med Oncol. 23:369–376. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tempero M, Plunkett W, Ruiz Van, Haperen
V, Hainsworth J, Hochster H, Lenzi R and Abbruzzese J: Randomized
phase II comparison of dose-intense gemcitabine: Thirty-minute
infusion and fixed dose rate infusion in patients with pancreatic
adenocarcinoma. J Clin Oncol. 21:3402–3408. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cappuzzo F, Novello S, De Marinis F,
Selvaggi G, Scagliotti GV, Barbieri F, Maur M, Papi M, Pasquini E,
Bartolini S, et al: A randomized phase II trial evaluating standard
(50 mg/min) versus low (10 mg/min) infusion duration of gemcitabine
as first-line treatment in advanced non-small-cell lung cancer
patients who are not eligible for platinum-based chemotherapy. Lung
Cancer. 52:319–325. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ho DH, Brown NS, Benvenuto J, McCredie KB,
Buckels D and Freireich EJ: Pharmacologic studies of continuous
infusion of arabinosylcytosine by liquid infusion system. Clin
Pharmacol Ther. 22:371–374. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kreis W, Chaudhri F, Chan K, Allen S,
Budman DR, Schulman P, Weiselberg L, Freeman J, Deere M and
Vinciguerra V: Pharmacokinetics of low-dose
1-beta-D-arabinofuranosylcytosine given by continuous intravenous
infusion over twenty-one days. Cancer Res. 45:6498–6501.
1985.PubMed/NCBI
|
|
35
|
Spriggs DR, Robbins G, Takvorian T and
Kufe DW: Continuous infusion of high-dose
1-beta-D-arabinofuranosylcytosine: A phase I and pharmacological
study. Cancer Res. 45:3932–3936. 1985.PubMed/NCBI
|
|
36
|
Donehower RC, Karp JE and Burke PJ:
Pharmacology and toxicity of high-dose cytarabine by 72-hour
continuous infusion. Cancer Treat Rep. 70:1059–1065.
1986.PubMed/NCBI
|
|
37
|
Spriggs DR, Sokal JE, Griffin J and Kufe
DW: Low-dose ara-C administered by continuous subcutaneous
infusion: A pharmacologic evaluation. Cancer Drug Deliv. 3:211–216.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bolwell BJ, Cassileth PA and Gale RP:
Low-dose cytosine arabinoside in myelodysplasia and acute
myelogenous leukemia: A review. Leukemia. 1:575–579.
1987.PubMed/NCBI
|
|
39
|
Stentoft J: The toxicity of cytarabine.
Drug Saf. 5:7–27. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stone RM, Spriggs DR, Dhawan RK, Arthur
KA, Mayer RJ and Kufe DW: A phase I study of intermittent
continuous infusion high-dose cytosine arabinoside for acute
leukemia. Leukemia. 4:843–847. 1990.PubMed/NCBI
|
|
41
|
Schiller G, Gajewski J, Nimer S, Territo
M, Ho W, Lee M and Champlin R: A randomized study of intermediate
versus conventional-dose cytarabine as intensive induction for
acute myelogenous leukaemia. Br J Haematol. 81:170–177. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fleming RA, Capizzi RL, Rosner GL, Oliver
LK, Smith SJ, Schiffer CA, Silver RT, Peterson BA, Weiss RB, Omura
GA, et al: Clinical pharmacology of cytarabine in patients with
acute myeloid leukemia: A cancer and leukemia group B study. Cancer
Chemother Pharmacol. 36:425–430. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bishop JF, Matthews JP, Young GA, Szer J,
Gillett A, Joshua D, Bradstock K, Enno A, Wolf MM, Fox R, et al: A
randomized study of high-dose cytarabine in induction in acute
myeloid leukemia. Blood. 87:1710–1717. 1996.PubMed/NCBI
|
|
44
|
Löwenberg B, Pabst T, Vellenga E, van
Putten W, Schouten HC, Graux C, Ferrant A, Sonneveld P, Biemond BJ,
Gratwohl A, et al Dutch-Belgian Cooperative Trial Group for
Hemato-Oncology (HOVON) and Swiss Group for Clinical Cancer
Research (SAKK) Collaborative Group: Cytarabine dose for acute
myeloid leukemia. N Engl J Med. 364:1027–1036. 2011. View Article : Google Scholar : PubMed/NCBI
|