Introduction
Gastric cancer is a malignant tumor that is common
worldwide and has a poor prognosis (1,2). The
5-year survival rate of patients with gastric cancer is <10%
(3). The majority of patients are
diagnosed at an advanced stage (4), and few efficacious treatment options
are available for patients with this late stage of the disease
(5). Surgical therapy combined
with adjuvant chemotherapy is the primary treatment option for
gastric cancer. It has been demonstrated that the single
administration of traditional chemotherapeutic drugs, such as
cisplatin and fluorouracil is only 10–20% efficacious in the
treatment of gastric cancer (6).
Even when combined with new drugs, such as docetaxel, irinotecan
and oxaliplatin, the optimum reaction rate is <50% (7). Currently, an early diagnosis coupled
with a good treatment strategy is considered an effective approach
for the treatment of gastric cancer. The use of biomarkers has been
confirmed to be a less invasive method for gastric cancer diagnosis
(8). Moreover, targeted therapies
for the treatment of gastric cancer have attracted increasing
attention (9). However, there is
still a lack of effective targeted therapies for the treatment of
this disease.
Polo-like kinases (PLKs) are associated with
oncogenesis in several types of cancer (10). PLKs exist in 4 isoforms, PLK1-4;
however, only one of these isoforms, PLK1, is involved in
centrosome maturation, chromosome segregation, bipolar spindle
formation and cytokinesis execution (11). It has been reported that PLK1
exhibits oncogenic potential in gastric cancer (12). The inhibition of PLK1 following
transfection with PLK1 siRNA and folate deficiency have been shown
to synergistically inhibit the growth of gastric cancer cell lines
(13). Moreover, a high PLK1
expression and DNA aneuploidy have been shown to correlate with a
poor prognosis in patients with gastric cancer (14). PLK1 plays a key role in
carcinogenesis and represents a promising target in the treatment
of cancer (15,16). PLK1 inhibitors have recently
emerged as a feasible strategy for the treatment of cancer
(11). BI2536 is a highly
selective and potent inhibitor of PLK1, which always participates
in mitotic progression (17).
Preclinical studies have indicated that BI2536 can disrupt spindle
assembly, leading to mitotic arrest and the apoptosis of human
cancer cell lines (18,19). However, the effects of BI2536 on
the regulation of gastric cancer development have not yet been
documented, at least to the best of our knowledge.
In the present study, the pivotal roles of BI2536
and cisplatin in regulating gastric cancer cell viability,
migration, invasion and apoptosis were investigated. Differentially
expressed proteins in gastric cancer cells treated with BI2536
(IC50) for 48 h, as well as the signaling pathways of
these differentially expressed proteins were analyzed by protein
pathway array (PPA). The aim of this study was to determine whether
BI2536 exerts an antitumor effect on gastric cancer and whether it
can synergistically inhibit the malignant behavior of gastric
cancer cells when used in combination with cisplatin. Our findings
may provide new insight into the targeted therapy for this
disease.
Materials and methods
Drugs and treatments
BI2536 (cat. no. 50-873-3) and cisplatin (cat. no.
50-901-13218) were purchased from Thermo Fisher Scientific
(Waltham, MA, USA) and diluted in dimethyl sulfoxide (DMSO) in
accordance with the manufacturer's instructions.
Cell culture
The human gastric cancer cell lines, AGS, BGC-823,
Hs746T, N87, KATOIII, SGC-7901 and SGC-7901/DDP (a
cisplatin-resistant cell line), were obtained from the Molecular
Pathology Laboratory at Mount Sinai Medical Center (New York, NY,
USA). The BGC-823, SGC-7901 and SGC-7901/DDP cells were cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS;
Gibco, Grand Island, NY, USA). The AGS cells were grown in Ham's
F12 medium. The Hs746T cells were cultured in DMEM containing 10%
FBS. The KATOIII cells were maintained in IMDM mixed with 20% FBS.
All media contained penicillin (100 U/ml) and streptomycin (100
U/ml), and all cells were cultured at 37°C in a humidified
incubator at 5% CO2.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT)
assay
MTT assay was used to evaluate cell viability. In
brief, the cells (5,000 cells/well) at the logarithmic growth phase
were seeded in 96-well plates. Following 24 h of incubation, the
cells were treated with various concentrations of cisplatin (1, 2,
4, 8, 16, 32 and 64 µM) and BI2536 (1, 2, 4, 8, 16, 32 and
64 nM) for 72 h at 37°C. Subsequently, 20 µl of MTT solution
(5 mg/ml, pH 7.4) were added to each well, followed by incubation
of the cells at 37°C for a further 4 h. After terminating the
reaction, some of the supernatant was discarded, and 150 µl
of DMSO were added to dissolve the crystals. The absorbance (570
nm) was then measured using a microplate reader (serial no. 155489;
Bio-Tek Instruments, Inc., Winooski, VT, USA). Each experiment was
performed in triplicate. Furthermore, the half maximal inhibitory
concentration (IC50) of cisplatin and BI2536 was further
calculated by the modified Kou-type method (20): lgIC50 = Xm-I
[P-(3-Pm-Pn)/4], in which Xm indicates lg maximum dose, I indicates
lg (maximum dose/adjacent dose), P indicates the sum of positive
response rate, Pm indicates the largest positive response rate and
Pn indicates the smallest positive response rate.
Colony formation assay
The cells were digested with 0.25% trypsin and split
into individual cells. Subsequently, 50, 100 and 200 cells were
seeded into 10-ml culture dishes and maintained under standard
culture conditions for 2–3 weeks. When the colonies were visible to
the naked eye, the culture dish was washed twice with
phosphate-buffered saline (PBS). The colonies were then fixed with
4% paraformaldehyde for 15 min, followed by staining with crystal
violet (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 20
min. Under a microscope (Nikon Eclipse TS100; Nikon Instruments,
Badhoevedorp, The Netherlands) the colonies that comprised at least
10 cells were counted.
Cell invasion assay
Cell invasion was evaluated using Transwell chambers
(8-µm pore size; Corning Inc., Corning, NY, USA) coated with
serum-free RPMI-1640 medium containing Matrigel (Sigma-Aldrich,
Shanghai, China). In brief, the SGC-7901 and SGC-7901/DDP cells
(5×104 cells) were grown in the upper chamber containing
medium with 10% FBS, and BI2536 (IC10) and cisplatin
(IC50) were then added to treat the cells. The lower
chamber was filled with RPMI-1640 medium containing 20% FBS as a
chemoattractant. Following incubation for 24 h at 37°C, the
non-invading cells were removed using cotton swabs, and the
invading cells were stained with 1% crystal violet for 30 min. The
invading cells in different fields were then counted using a light
microscope (Nikon Model Eclipse TS100LED MV; Nikon Corp., Tokyo,
Japan).
Cell cycle analysis
The cells (1×105 cells/ml) were
collected, washed twice with ice-cold PBS, and fixed with 75%
ice-cold ethanol. After washing with ice-cold PBS again, the cells
were suspended in 300 µl of PBS and 20 µl of RNase A
was then added, followed by incubation of the cells for 30 min at
37°C. Subsequently, the cells were stained with 400 µl of
propidium iodide (PI) for 45 min in the dark. Cell cycle analysis
at 488 nm was performed using a FACSCalibur flow cytometer (BD
Biosciences, San Jose, CA, USA).
Cell apoptosis analysis
Cell apoptosis was assessed by flow cytometry after
Annexin V and PI staining (BD Pharmingen, San Diego, CA, USA). In
brief, the cells (1×106 cells/ml) were harvested and
resuspended in 1X Annexin V-binding buffer. Subsequently, 5
µl of Annexin V-FITC was added, and the cells were incubated
for 15 min away from light, followed by the addition of 10
µl of PI and incubation of the cells for 5 min at 4°C. Cell
apoptosis was then analyzed using a FACSCalibur flow cytometer (BD
Biosciences).
Western blot analysis
The cells were lysed with 1X cell lysis buffer (Cell
Signaling Technology, Danvers, MA, USA). Using a Pierce BCA protein
assay kit (Pierce, Rochford, IL, USA), the protein concentration
was adjusted to 1 µg/µl. An equal amount of protein
extract was separated by 10% sodium dodecyl sulfate polyacrylamide
gel electrophoresis (SDS-PAGE). The blots were then transferred
onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). The
membranes were then blocked in 5% non-fat milk in 1X TBST
containing 100 mM NaCl, 20 mM Tris-HCl (pH 7.5), and 0.1% Tween-20
for 1 h. Primary antibodies to PLK1 (1:1,000; cat. no. sc-5585;
Santa Cruz Biotechnology, Inc.), p-Cdc2 (1:1,000; cat. no. 9111;
Cell Signaling Technology), cyclin B1 (1:1,000; cat. no. sc-594),
p-cdc25c (1:1,000; cat. no. sc-327) and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) (1:1,000; sc-32233) (all from Santa Cruz
Biotechnology, Inc.) were added, followed by incubation of the
membranes overnight at 4°C. GAPDH served as an internal control.
Subsequently, the membranes were probed with horseradish peroxidase
(HRP)-labeled secondary antibodies (1:10,000; cat. no. sc-2370 or
sc-2371, Santa Cruz Biotechnology, Inc.) at room temperature for 1
h. After washing with 1X TBST buffer, the bands were detected with
a chromogenic substrate using the enhanced chemiluminescence (ECL)
method and analyzed using the Quantity One software package
(Bio-Rad).
Protein pathway array (PPA) analysis
The cells were lysed with 1X cell lysis buffer, and
equal amounts of protein extracts were separated by 10% SDS-PAGE,
as described above. The blots were then transferred onto
nitrocellulose membranes (Bio-Rad). After blocking in 3% bovine
serum albumin (BSA) for 1 h, the membranes were fixed on a western
blotting manifold (Mini-PROTEAN II Multiscreen apparatus, cat. no.
170-4017; Bio-Rad) containing 20 channels. A total of 286
protein-specific or phosphorylation-specific antibodies (Table I) were used in the multiplex
immunoblot. To each channel (1–19), a
mixture of two antibodies dissolved in the blocking buffer was
added, followed by incubation of the membranes overnight at 4°C;
BSA without any antibody was added to channel 20. Following
incubation with HRP-conjugated secondary anti-rabbit (1:10,000;
cat. no. sc-2371) or anti-goat (1:10,000; cat. no. sc-2370) or
anti-mouse antibodies (1:10,000; cat. no. sc-2345) (all from Santa
Cruz Biotechnology, Inc.) for 1 h, Immun-Star™ HRP Peroxide Buffer
and Immun-Star™ HRP Luminol Enhancer (cat. no. 94547; Bio-Rad) were
added followed by incubation of the membranes for 4 min.
Chemiluminescence signals were then analyzed with the ChemiDoc XRS
system (Bio-Rad). The same membranes was then washed twice with 1X
TBST buffer and used to detect other primary antibodies, as
described above. The signal intensity of each protein was analyzed
using Quantity One software 4.5.0 (Bio-Rad). To reduce the
variations caused by total protein loading amount, transferring and
blotting efficiency, 'global median subtraction' was used to
normalize the background subtracted intensity. The normalized
expression of each protein = the average intensity of each protein
in all samples × (the signal intensity of each protein/the total
intensity of all proteins in the same blot membrane).
 | Table IList of antibodies included in the
protein pathway array. |
Table I
List of antibodies included in the
protein pathway array.
| Antibodies specific
for phosphorylation |
| p-AKT (Ser473) | p-ERK5
(Thr218/Tyr220) | p-p44/42 MAPK
(Erk1/2)
(Thr202/Tyr204) | p-PKCα/βII
(Thr638/641) | p-STAT3
(Ser727) |
| p-β-catenin
(Ser33/37/Thr41) | p-FAK (Tyr397) | p-p53 (Ser392) | p-PKCδ
(Thr505) | p-STAT5
(Tyr694) |
| p-CDC2 (Tyr15) | p-GSK-3α/β
(Ser21/9) | p-p70 S6 kinase
(Thr389) | p-PTEN
(Ser380) | |
| p-c-Jun
(Ser73) | p-JNK(G-7) | p-P90RSK
(Ser380) | p-Rb (Ser780) | |
| p-CREB
(Ser133) | p-Met
(Tyr1234) | p-PDK1
(Ser241) | p-Rb
(Ser807/811) | |
| p-eIF4B
(Ser422) | p-p38 MAPK
(Thr180/Tyr182) | p-PKCα
(Ser657) | p-Smad1/5
(Ser463/465) | |
| Antibodies specific
for non-phosphorylation |
| 14-3-3 β | cSHMT | HER2/ErbB2 | MMP-13 | Rap1 |
| α-tubulin | CTGF | HES1 | MSR | Reg IV |
| ADAM8 | CTLA-4 | HGF | MTA1 | RHAMM |
| ADAM10 | CUL-1 | HIF-1α | MTHFD1 | RhoA |
| ADH | CX3CR1 | HIF-2α | MTHFD2 | Ribosomal protein
L6 |
| AIM2 | Cyclin B1 | HIF-3α | MTHFR | RIP |
| Akt | Cyclin D1 | Hint | NALP1 | RUNX3 |
| ALG-2 | Cyclin E | HMG-1 | N-cadherin | SK3 |
| Annexin I | Cytokeratin 5 | HNF-3α | NFATc1 | SLUG |
| ASCL1 | Cytokeratin 18 | HoxC11 | NF-κB p50 | Smad4 |
| ASC-R | Cytokeratin 19 | H-Ras | NF-κB p52 | Smad7 |
| ATF-1 | DACH1 | HSL | NF-κB p65 | Snail |
| Aurora A/AIK | DARPP-32 | HSP27 | NHERF-2 | SOD-1 |
| Autotaxin | DDB2 | HSP70 | Nkx-3.1 | SPAK |
| Axin | DHFR | Hsp90 | nm23-H1/2/3 | SRC-1 |
| β3-tubulin | Dnmt1 | ICAM-1 | NMT1 | Stat1 |
| β-catenin | DPYD | IDO | NOS2 | Stat3 |
| Bad | DRG1 | IFN-γ | Notch4 | SUGT1 |
| Bak | E2A | IGFBP5 | NQO1 | Survivin |
| Bax | E2F1 | IGF-Irβ | ODC | Syk |
| Bcl-2 | E-cadherin | IL-1β | OPN | Tak1 |
| Bcl-6 | Eg5 | IL-3Rα | p14 | Tau |
| Bcl-xL | EGFR | IL-6 | p16 | TCF-1 |
| BECN1 | eIF4B | IL-8 | p27 | TDP1 |
| BID | Endoglin | IL-8RA | P2X7 | TFIIH p89 |
| BMP-2 | ENT1 | IL-11 | p38α/β | TGF-β |
| Calpain 2 | Ep-CAM | IL-18 | p44/42 MAPK
(Erk1/2) | TIMP-3 |
| Calpastatin | EphB2 | Integrin α4 | P504S | TIP30 |
| Calretinin | Epo | IRF-1 | p53 | TIRAP |
| CaMKKα | ERCC1 | ITF | p63 | TNF-R2 |
| CARD12 | ERα | Jagged1 | p73 | TNFα |
| Caspase-1 | ERβ | JAK2 | Pannexin-1 | tPA |
| Cathepsin B | E-Selectin | JNK1 | Patched | TRAF6 |
| CD10 | Factor XIII B | KAI1 | Pax-2 | TS |
| CD33 | FAH | Keratin 10 | PC2 | tsg101 |
| Cdc2 p34 | FAS | KiSS-1 | P-cadherin | TTF-1 |
| Cdc25B | FEN-1 | KLF6 | PCNA | Twist |
| Cdc25C | FGF-8 | K-Ras | PDEF | Tyro3 |
| Cdc42 | FGFR-4 | LKB1 | PEDF | uPA |
| Cdk2 | FKHR | LSD1 | PERK | uPAR |
| Cdk4 | FLIPS/L | L-Selectin | PKCα | VAP-1 |
| Cdk6 | Flt-3/Flk-2 | Lyn | PKCε | V-ATPase H |
| Cdx2 | FOXM1 | Maspin | PLK | VCAM-1 |
| c-Fms/CSF-1R | FTα | MAT IIβ | PRL-3 | VEGF |
| Chk1 | FUS/TLS | MDM2 | PSCA | Vimentin |
| c-IAP2 | Fusin | Mesothelin | PSM | VSV-G |
| CKR-7 | Galectin-3 | MetAP-2 | PSTPIP1 | Wnt-1 |
| Clusterin | GLP-1R | MetRS | PTEN | WT1 |
| COL1A2 | Glutamine
synthetase | MGr1-Ag | Rab 7 | XIAP |
| Connexin 43 | GSTP1 | MMP-2 | Raf-B | YB-1 |
| Cox-2 | HCAM | MMP-7 | RAGE | |
| CREB | HDAC1 | MMP-9 | RANKL | |
Statistical analysis
All in vitro experiments were repeated 3
times and PPA was performed twice. All measurement data are
expressed as the means ± SD. The differences between groups were
calculated using the Student's t-test or one-way ANOVA. Further
comparison between groups was performed using a Tukey post-hoc
test. Statistical analyses were performed using SPSS 17.0 (SPSS
Inc., Chicago, IL, USA). Unsupervised hierarchical clustering
analysis was performed using the BRB ArrayTools Software V3.3.0.
The significant pathway for the differentially expressed proteins
was analyzed using Ingenuity Pathway Analysis (IPA) software. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
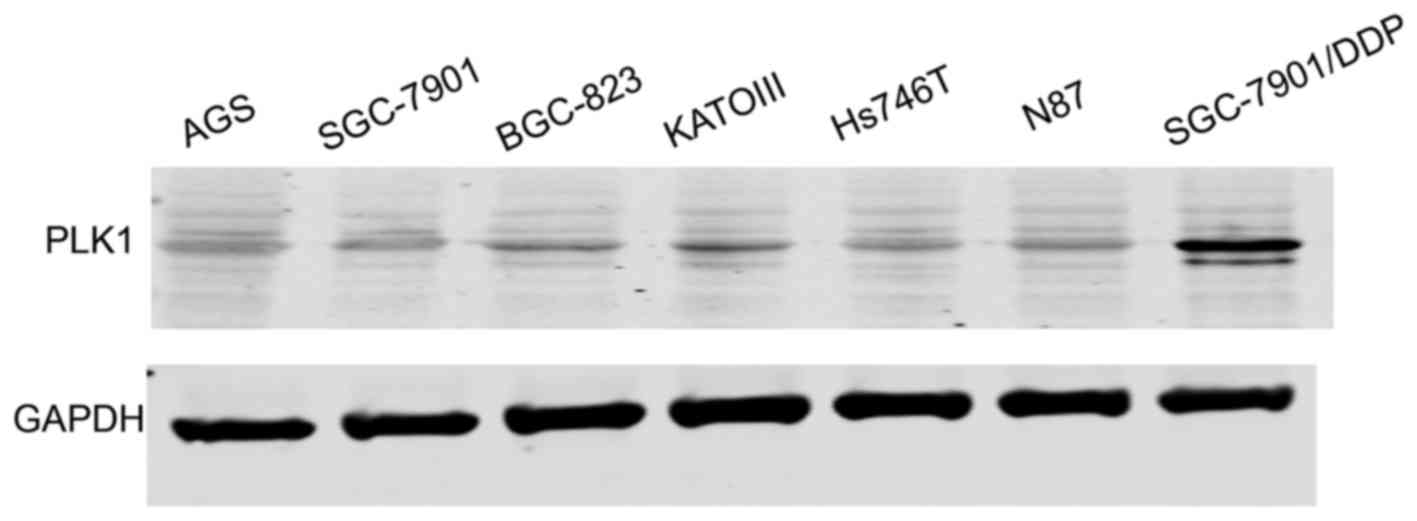
PLK1 is upregulated in SGC-7901/DDP
gastric cancer cells
As shown in Fig. 1,
PLK1 was upregulated in the SGC-7901. DDP (cisplatin-resistant)
gastric cancer cells compared with the SGC-7901 cells. Thus, we
further explored the function of the PLK1 inhibitor, BI2536, in
gastric cancer cells.
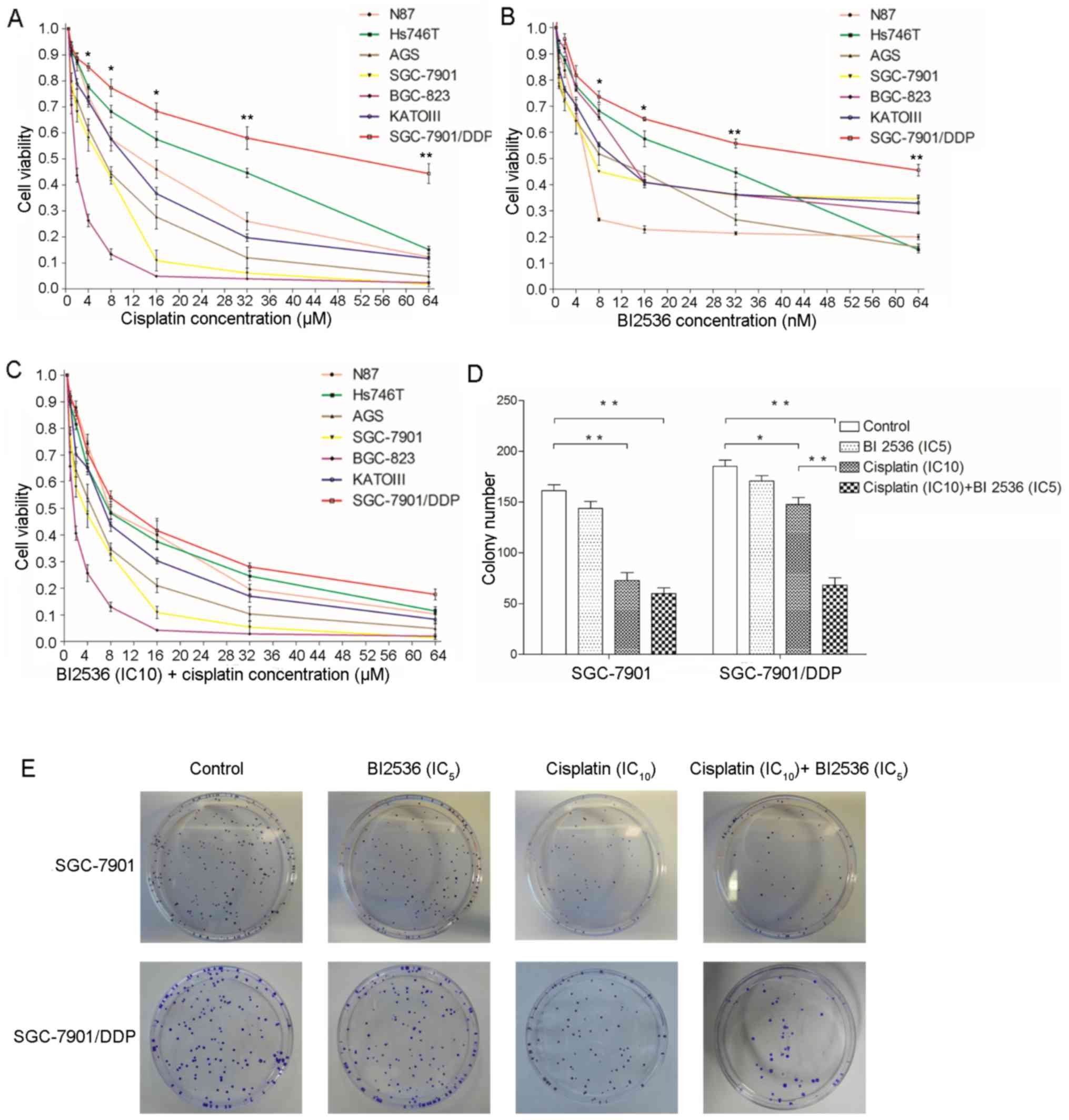
BI2536 enhances the inhibitory effects of
cisplatin on the viability and colony-forming ability of the
SGC-7901/DDP cells
As shown in Fig. 2A and
B, cisplatin and BI2536 significantly inhibited the viability
of the 7 gastric cancer cell lines in a dose-dependent manner. The
highest chemosensitivity to cisplatin was observed in the BGC-823
and SGC-7901 cells, the IC50 values of which were 2 and
6 µM, respectively. The least chemosensitivity to cisplatin
was exhibited by the Hs746T and SGC-7901/DDP cells, the
IC50 values of which were 30 and 60 µM,
respectively. Notably, BI2536 (IC10) significantly
enhanced the inhibitory effects of cisplatin on the viability of
the gastric cancer cells, particularly by improving the
chemosensitivity of SGC-7901/DDP to cisplatin (Fig. 2C). Therefore, a colony formation
assay was then performed using the SGC-7901 and SGC-7901/DDP cells
in order to verify the effects of BI2536 and cisplatin on cell
viability. As shown in Fig. 2D and
E, BI2536 (IC5) alone did not inhibit colony
formation compared with the controls (P>0.05); however,
cisplatin (IC10) significantly inhibited colony
formation (P<0.05), particularly in the SGC-7901 cells
(P<0.01). Following co-treatment with BI2536 (IC5)
and cisplatin (IC10), the results revealed that BI2536
(IC5) significantly enhanced the inhibitory effects of
cisplatin on the colony-forming ability of the SGC-7901/DDP cells
(P<0.01), but not that of the SGC-7901 cells (P>0.05).
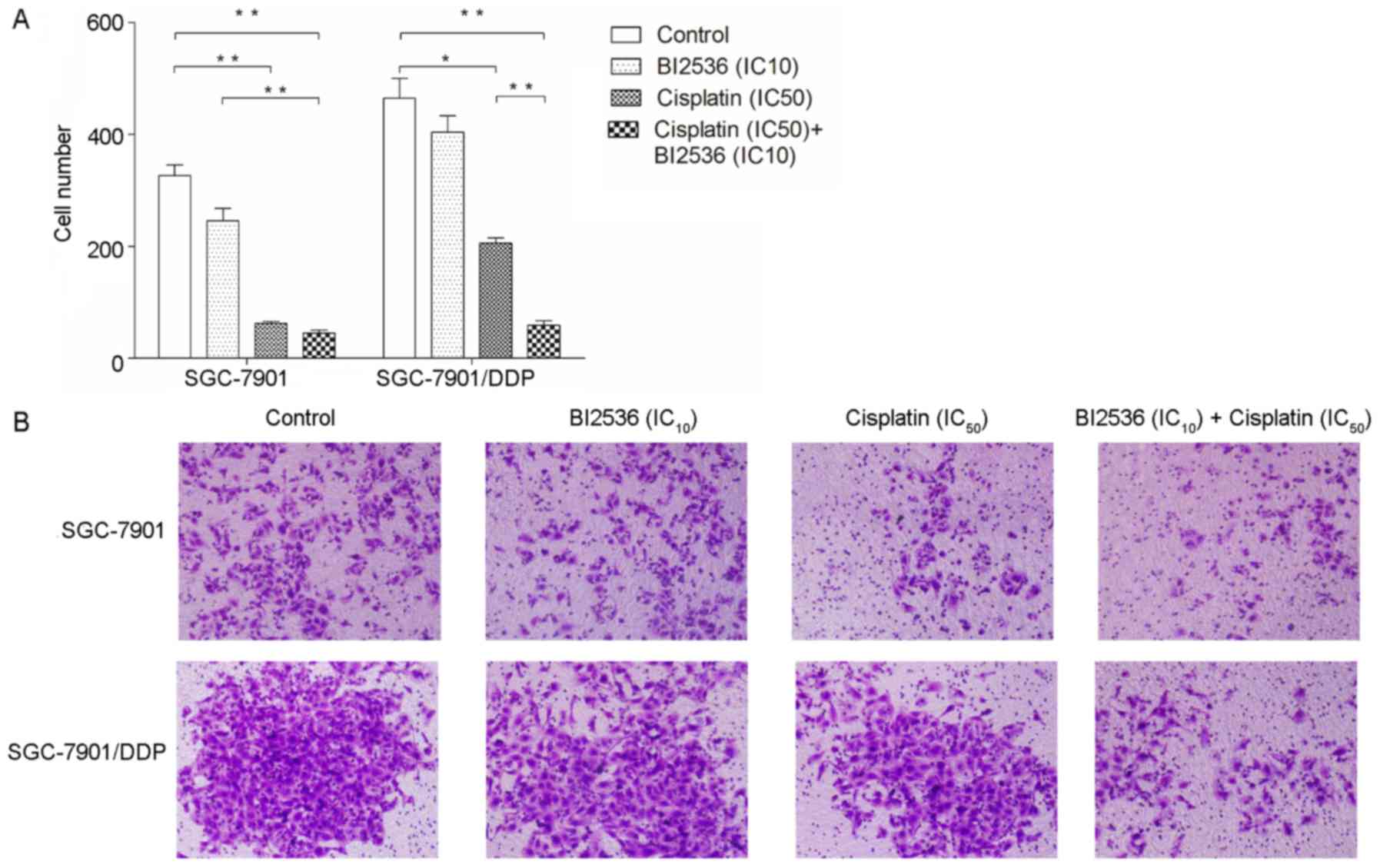
BI2536 enhances the inhibitory effects of
cisplatin on the invasive ability of the SGC-7901/DDP cells
We further determined the effects of BI2536 and
cisplatin on gastric cancer cell invasion (Fig. 3). The results revealed that BI2536
(IC10) did not inhibit the invasive ability of the
SGC-7901 and SGC-7901/DDP cells (P>0.05), although cisplatin
(IC50) significantly inhibited the invasive ability of
the cells (P<0.05). Moreover, following treatment with a
combination of BI2536 (IC10) and cisplatin
(IC50), only the inhibitory effects of cisplatin on the
invasiveness of the SGC-7901/DDP cells, but not that of the
SGC-7901 cells (P>0.05), were enhanced (P<0.01).
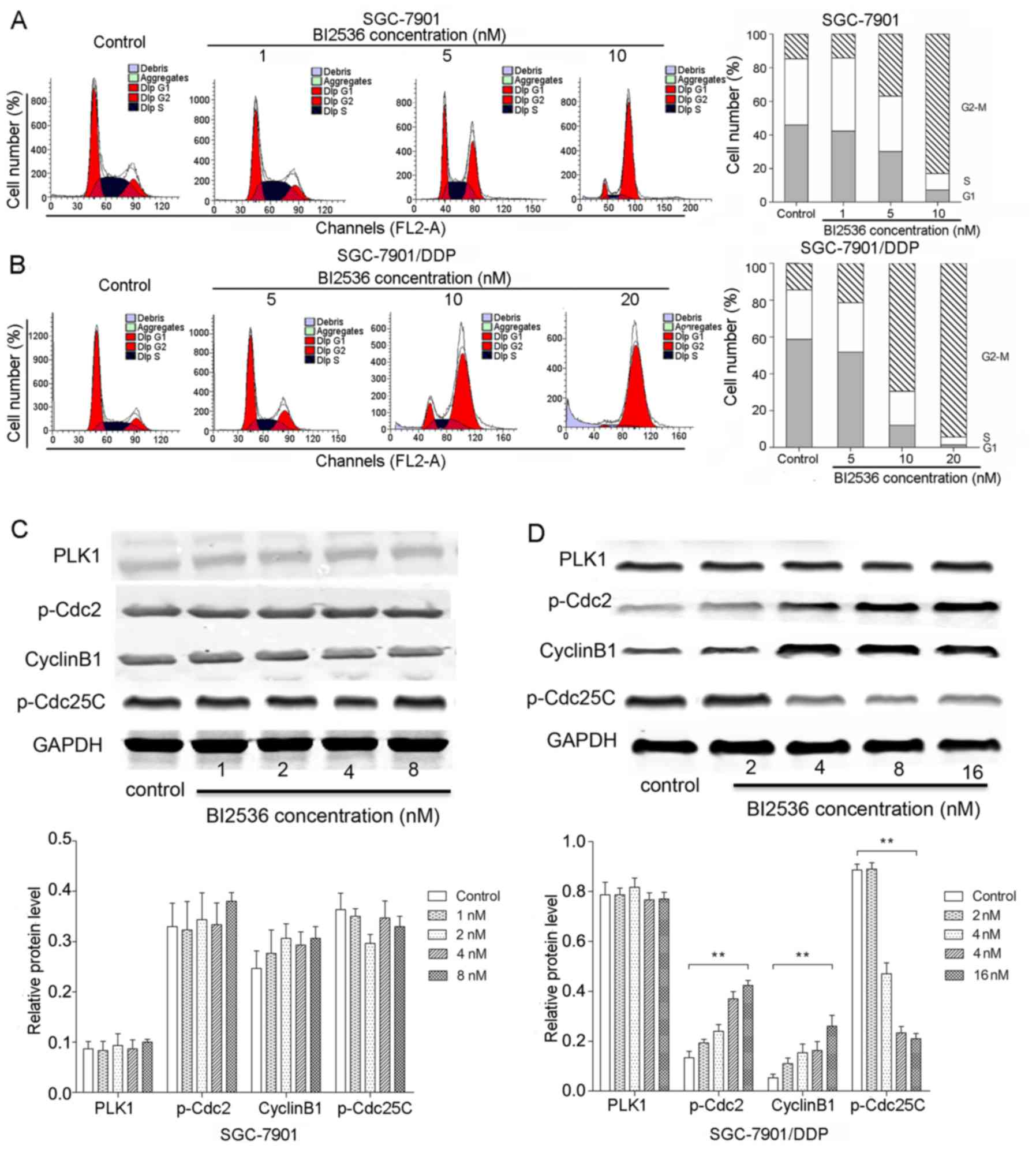
BI2536 significantly induces
G2/M arrest in the SGC-7901/DDP cells
In the cell cycle analysis, the SGC-7901 cells were
treated with 1, 5 and 10 nM BI2536 for 72 h, and the SGC-7901/DDP
cells were treated with 5, 10 and 20 nM BI2536 for 24 h. The
results of flow cytometry revealed that BI2536 significantly
induced G2/M arrest in both the SGC-7901 and
SGC-7901/DDP cells (P<0.05) (Fig.
4A and B). We further determined the expression of key proteins
involved in the G2/M cell cycle, including p-Cdc2,
cyclin B1 and p-Cdc25C by western blot analysis (Fig. 4C and D). We found that PLK1
expression was not significantly altered following treatment with
various concentrations of BI2536 in both the SGC-7901 and
SGC-7901/DDP cells (P>0.05). Notably, compared with the control
group, BI2536 treatment resulted in the decreased expression of
p-Cdc25C and in the increased expression of p-Cdc2 and cyclin B1 in
the SGC-7901/DDP cells in a dose-dependent manner (P<0.01)
(Fig. 4D), while the expression
levels of these proteins exhibited no significant changes in the
SGC-7901 cells (P>0.05).
BI2536 promotes cisplatin-induced
SGC-7901/DDP cell apoptosis
Flow cytometry was also performed to determine the
effects of BI2536 on gastric cancer cell apoptosis. Following
treatment with various concentrations of BI2536 for 24 h, the
proportions of SGC-7901 and SGC-7901/DDP cells undergoing early
apoptosis were all significantly increased (P<0.05) (Fig. 5A and B). Furthermore, we found that
cisplatin significantly induced SGC-7901 and SGC-7901/DDP cell
apoptosis when used in combination with BI2536 (IC20)
(P<0.05) (Fig. 5C and D).
Notably, BI2536 (IC20, 20 nM) significantly promoted
cisplatin-induced SGC-7901/DDP cell apoptosis (P<0.05) (Fig. 5D).
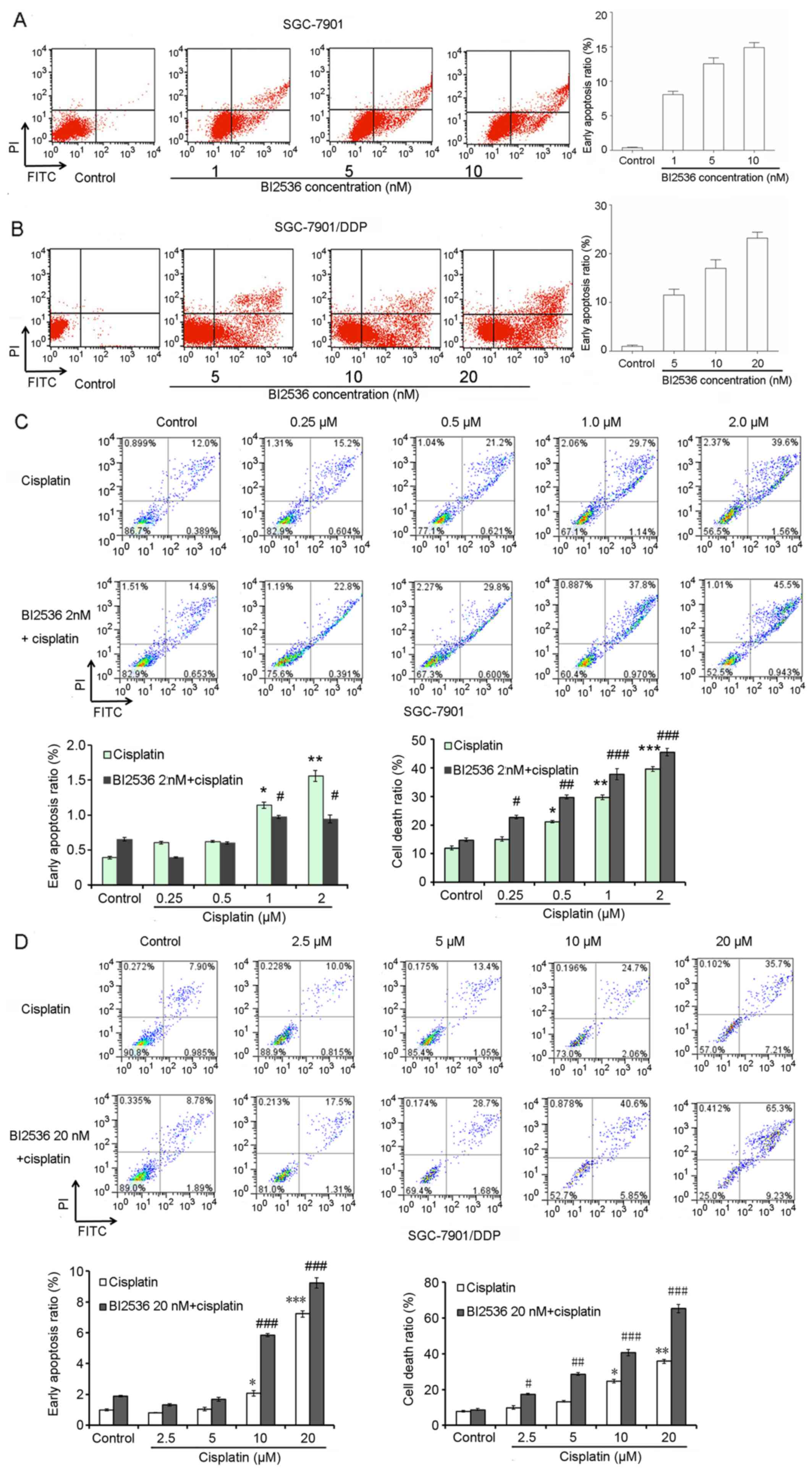 | Figure 5BI2536 promotes cisplatin-induced
SGC-7901/DDP gastric cancer cell apoptosis. (A and B) Flow
cytometry demonstrated the effects of BI2536 on SGC-7901 and
SGC-7901/DDP cell apoptosis. (C) Flow cytometry demonstrated the
effects of the combination of various concentrations of cisplatin
(0, 0.25, 0.5, 1 and 2 µM) and BI2536 (2 nM) on SGC-7901 and
SGC-7901/DDP cell apoptosis. (D) Flow cytometry demonstrated the
effects of the combination of various concentrations of cisplatin
(0, 2.5, 5, 10 and 20 µM) and BI2536 (20 nM) on SGC-7901 and
SGC-7901/DDP cell apoptosis Error bars indicate the means ± SD, and
the symbols * and # indicate a statistically
significant difference compared with the corresponding control
group. *,#p<0.05, **,##p<0.01 and
***,###p<0.001. |
BI2536 induces the differential
expression of signaling proteins between the SGC-7901 and
SGC-7901/DDP cells
We applied PPA analysis to analyze the
differentially expressed proteins between the SGC-7901 and
SGC-7901/DDP cells following treatment with BI2536
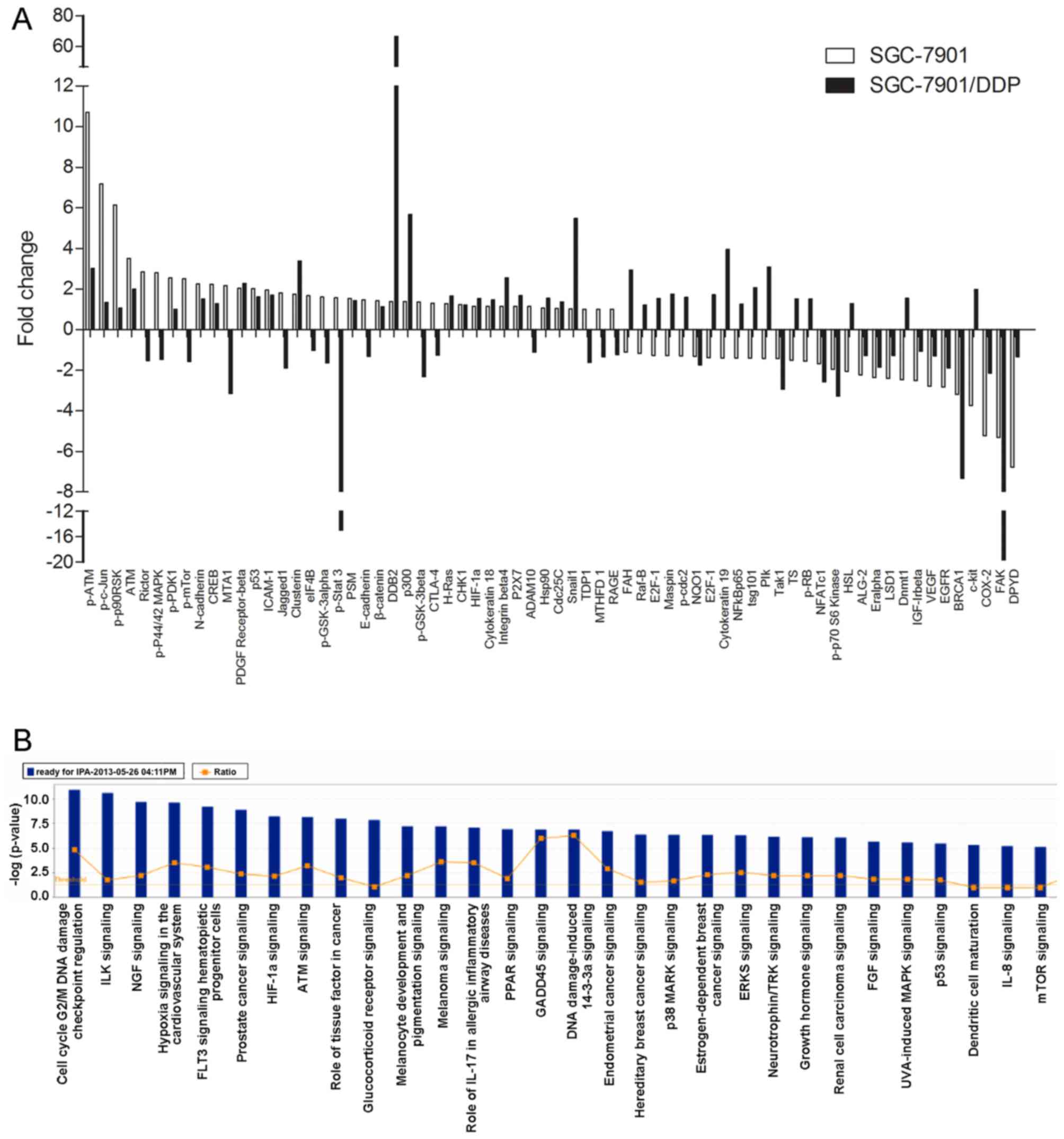
(IC50) for 48 h. We found that 68 proteins were
differentially expressed when compared with the controls (Fig. 6A). IPA analysis also revealed that
the differentially expressed proteins induced by BI2536 treatment
were involved in many cell functions and signaling pathways, such
as cell death, cell development, tumorigenesis, the cell cycle, DNA
duplication/recombination/repair, cellular movement, and in the
Wnt/β-catenin and mitogen-activated protein kinase
(MEK)/extracellular signal-regulated kinase (ERK)/ribosomal S6
kinase 1 (RSK1) signaling pathways (Fig. 6B).
Discussion
Cisplatin is a common and effective anticancer drug;
however, its use is limited due to its related side-effects, such
as renal, gastrointestinal and neurological toxicities (21). Therefore, to improve the antitumor
efficacy of cisplatin and reduce cisplatin-induced side-effects,
further studies are warranted in order to aid the develelopment of
small-molecule drugs. In the present study, we combined the PLK1
inhibitor, BI2536, with cisplatin to treat gastric cancer cells and
to determine whether BI2536 and cisplatin can synergistically
inhibit the malignant behavior of gastric cancer cells. The results
revealed that BI2536 enhanced the cisplatin-induced inhibitory
effects on SGC-7901/DDP cell viability and invasion. BI2536 induced
G2/M arrest in the SGC-7901/DDP cells by decreasing the
expression of p-Cdc25C and increasing the expression of p-Cdc2 and
cyclin B1. BI2536 promoted cisplatin-induced SGC-7901/DDP cell
apoptosis. Moreover, BI2536 induced the differential expression of
68 proteins between the SGC-7901 and SGC-7901/DDP cells, and these
differentially expressed proteins were involved in sevral cell
functions and signaling pathways, such as the Wnt/β-catenin and
MEK/ERK/RSK1 signaling pathways.
In many anticancer treatments, the G2/M
checkpoint is an effective target site for molecular targeted
therapy and chemotherapy sensitization (22,23).
There are data to suggest that mammalian PLK1 plays a regulatory
role at the cell cycle G2 checkpoint (24,25).
PLK1 has been implicated in mitotic entry via the activation of
Cdc25C (26). PLK1 has also been
identified as a target that can sensitize cells to traditional
chemotherapeutic drugs in the treatment of cancer (27,28).
In addition, a high degree of G2/M arrest induced by
PLK1 inhibition has been found to be associated with
radiosensitization in various cancer cell lines (29). The combination of MS275 and BI2536
has been shown to synergistically inhibit cell growth and to induce
G2/M phase arrest in A549 non-small cell lung cancer
cells (30). Gleixner et al
demonstrated that the inhibitory effect of BI2536 on CML cell
growth was associated with mitotic arrest, particularly
G2/M arrest, and consecutively resulted in apoptosis
(31). In this study, BI2536
enhanced the cisplatin-induced inhibitory effects on SGC-7901 cell
viability and invasive ability. BI2536 induced G2/M
arrest in the SGC-7901/DDP cells by decreasing the expression of
p-Cdc25C and increasing the expression of p-Cdc2 and cyclin B1.
BI2536 promoted cisplatin-induced SGC-7901/DDP cell apoptosis.
Taken together, we speculate that the combination of cisplatin and
BI2536 can synergistically inhibit cell growth, induce
G2/M phase arrest, and consecutively induce the
apoptosis of SGC-7901/DDP cells.
Furthermore, we applied PPA analysis to examine the
differentially expressed proteins between the SGC-7901 and
SGC-7901/DDP cells following treatment with BI2536
(IC50) for 48 h. A total of 68 proteins were found to be
differentially expressed, which were involved in signaling
pathways, such as the Wnt/β-catenin and MEK/ERK/RSK1 signaling
pathways. It has been reported that Wnt/β-catenin signaling plays a
key role in regulating the self-renewal of gastric cancer stem
cells, and salinomycin treatment may be used for the treatment of
gastric cancer by targeting Wnt/β-catenin signaling (32). The inhibition of the Wnt/β-catenin
pathway by niclosamide has been shown to result in decreased
cellular proliferation and increased cell death in gastric cancer
(33). In addition, ERK/RSK1
activation by growth factors can delay the cell cycle at the
G2 phase, thus reducing mitotic aberrations and
maintaining genomic integrity (34). Notably, PLK1 is involved in mitotic
arrest via the inhibition of the MEK/ERK/RSK1 cascade (35). Although the association between
BI2536 and the Wnt/β-catenin or MEK/ERK/RSK1 signaling pathways has
not yet been verified experimentally, our results provide an
important indication pertaining to BI2536 likely promoting the
chemotherapeutic sensitivity of SGC-7901/DDP cells to cisplatin via
the involvement of the Wnt/β-catenin or MEK/ERK/RSK1 signaling
pathways.
The strengths of our study were that BI2536 and
cisplatin synergistically inhibited the malignant behavior of the
SGC-7901/DDP (cisplatin-resistant) gastric cancer cells, which may
provide a broader perspective for improving the chemotherapeutic
sensitivity of cancer cells to cisplatin. Despite the clear
strength of our study, however, some limitations merit further
consideration. Firstly, there were no significant effects of BI2536
treatment alone on cell viability, migration and apoptosis, which
limited the clinical application of BI2536. Secondly, the
synergistic effects of BI2536 and cisplatin were not verified using
gastric cancer primary cells or an in vivo xenograft model
of SGC7901 and SGC7901/DDP cells. Further research is still
required in order to verify the synergistic interaction between
BI2536 and cisplatin in gastric cancer primary cells. Thirdly, we
did not analyze PLK1 expression according to the information of the
The Cancer Genome Atlas (TCGA) and Cancer Cell Line Encyclopedia
(CCLE) databases. Further studies are required to investigate the
role of PLK1 in SGC7901 and SGC7901/DDP gastric cancer cells using
siRNA-mediated gene knockdown. Fourthly, signaling pathways were
only analyzed by PPA. The expression of Wnt/β-catenin and
MEK/ERK/RSK1 signaling pathway-related proteins were not determined
by qPCR or western blot analysis in treated samples. Fifthly, we
only used MTT assay to determine changes in cell viability, which
only monitored the ATP-dependent metabolic activity. To better
detect the synergisstic effects of BI2536 and cisplatin on cell
proliferation, BrdU DNA proliferation assay should also be
performed to monitor the number of cellular divisions and DNA
synthesis. Finally, we only analyzed the differentially expressed
proteins between the SGC-7901 and SGC-7901/DDP cells following
treatment with BI2536 (IC50) for 48 h. The key
mechanisms involved in the combined effects of BI2536 and cisplatin
treatment in regulating the malignant behavior of gastric cancer
cells remain largely unknown. Therefore, further studies are still
required in order to verify our observations.
In conclusion, the findings of the present study
suggest that BI2536 and cisplatin synergistically inhibit the
malignant behavior of SGC-7901/DDP (cisplatin-resistant) gastric
cancer cells. BI2536 may enhance the chemotherapeutic sensitivity
of SGC-7901/DDP cells to cisplatin via the involvement of the
Wnt/β-catenin or MEK/ERK/RSK1 signaling pathways. The development
of a PLK1 inhibitor may thus be an effective strategy for the
treatment of gastric cancer.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81572355 and 81702363).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
de Martel C, Forman D and Plummer M:
Gastric cancer: Epidemiology and risk factors. Gastroenterol Clin
North Am. 42:219–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Daniyal M, Ahmad S, Ahmad M, Asif HM,
Akram M, Ur Rehman S and Sultana S: Risk factors and epidemiology
of gastric cancer in Pakistan. Asian Pac J Cancer Prev.
16:4821–4824. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Orditura M, Galizia G, Sforza V,
Gambardella V, Fabozzi A, Laterza MM, Andreozzi F, Ventriglia J,
Savastano B, Mabilia A, et al: Treatment of gastric cancer. World J
Gastroenterol. 20:1635–1649. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yasui W, Oue N, Ito R, Kuraoka K and
Nakayama H: Search for new biomarkers of gastric cancer through
serial analysis of gene expression and its clinical implications.
Cancer Sci. 95:385–392. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lordick F, Kang YK, Chung HC, Salman P, Oh
SC, Bodoky G, Kurteva G, Volovat C, Moiseyenko VM, Gorbunova V, et
al: Arbeitsgemeinschaft Internistische Onkologie and EXPAND
Investigators: Capecitabine and cisplatin with or without cetuximab
for patients with previously untreated advanced gastric cancer
(EXPAND): A randomised, open-label phase 3 trial. Lancet Oncol.
14:490–499. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Köhne CH, Wils JA and Wilke HJ:
Developments in the treatment of gastric cancer in Europe. Oncology
(Williston Park). 14(Suppl 14): 22–25. 2000.
|
|
7
|
Catalano V, Labianca R, Beretta GD, Gatta
G, de Braud F and Van Cutsem E: Gastric cancer. Crit Rev Oncol
Hematol. 71:127–164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tong W, Ye F, He L, Cui L, Cui M, Hu Y, Li
W, Jiang J, Zhang DY and Suo J: Serum biomarker panels for
diagnosis of gastric cancer. Onco Targets Ther. 9:2455–2463.
2016.PubMed/NCBI
|
|
9
|
Ngeow J, Tan IB and Choo SP: Targeted
therapies in the treatment of gastric cancer. Asia Pac J Clin
Oncol. 7:224–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takai N, Hamanaka R, Yoshimatsu J and
Miyakawa I: Polo-like kinases (Plks) and cancer. Oncogene.
24:287–291. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chopra P, Sethi G, Dastidar SG and Ray A:
Polo-like kinase inhibitors: An emerging opportunity for cancer
therapeutics. Expert Opin Investig Drugs. 19:27–43. 2010.
View Article : Google Scholar
|
|
12
|
Jang YJ, Kim YS and Kim WH: Oncogenic
effect of Polo-like kinase 1 expression in human gastric
carcinomas. Int J Oncol. 29:589–594. 2006.PubMed/NCBI
|
|
13
|
Zha X, Huang L, Yang M, Jingmin OU, Chen D
and Fei Z: Study on folate deficiency and Polo-like kinase-1
(PLK-1) siRNA in synergistically inhibiting the growth of gastric
carcinoma cell lines. Modern J Integrated Trad Chin Western Med.
24:917–920. 2015.
|
|
14
|
Otsu H, Iimori M, Ando K, Saeki H, Aishima
S, Oda Y, Morita M, Matsuo K, Kitao H, Oki E, et al: Gastric cancer
patients with high PLK1 expression and DNA aneuploidy correlate
with poor prognosis. Oncology. 91:31–40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weiss L and Efferth T: Polo-like kinase 1
as target for cancer therapy. Exp Hematol Oncol. 1:382012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cholewa BD, Liu X and Ahmad N: The role of
polo-like kinase 1 in carcinogenesis: Cause or consequence. Cancer
Res. 73:6848–6855. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mross K, Frost A, Steinbild S, Hedbom S,
Rentschler J, Kaiser R, Rouyrre N, Trommeshauser D, Hoesl CE and
Munzert G: Phase I dose escalation and pharmacokinetic study of BI
2536, a novel Polo-like kinase 1 inhibitor, in patients with
advanced solid tumors. J Clin Oncol. 26:5511–5517. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Steegmaier M, Hoffmann M, Baum A, Lénárt
P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant
J, et al: BI 2536, a potent and selective inhibitor of polo-like
kinase 1, inhibits tumor growth in vivo. Curr Biol. 17:316–322.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lénárt P, Petronczki M, Steegmaier M, Di
Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N and Peters JM: The
small-molecule inhibitor BI 2536 reveals novel insights into
mitotic roles of polo-like kinase 1. Curr Biol. 17:304–315. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chou T and Martin N: CompuSyn for drug
combinations: PC software and user's guide: A computer program for
quantitation of synergism and antagonism in drug combinations, and
the determination of IC50 and ED and LD NJ, 50 50
values. ComboSyn, Paramus. 2005.
|
|
21
|
Pace A, Savarese A, Picardo M, Maresca V,
Pacetti U, Del Monte G, Biroccio A, Leonetti C, Jandolo B, Cognetti
F, et al: Neuroprotective effect of vitamin E supplementation in
patients treated with cisplatin chemotherapy. J Clin Oncol.
21:927–931. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kawabe T: Kawabe. Mol Cancer Ther.
3:513–519. 2004.PubMed/NCBI
|
|
23
|
Anderson HJ, Andersen RJ and Roberge M:
Inhibitors of the G2 DNA damage checkpoint and their potential for
cancer therapy. Prog Cell Cycle Res. 5:423–430. 2003.PubMed/NCBI
|
|
24
|
Smits VA, Klompmaker R, Arnaud L, Rijksen
G, Nigg EA and Medema RH: Polo-like kinase-1 is a target of the DNA
damage checkpoint. Nat Cell Biol. 2:672–676. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
van Vugt MA, Smits VA, Klompmaker R and
Medema RH: Inhibition of Polo-like kinase-1 by DNA damage occurs in
an ATM- or ATR-dependent fashion. J Biol Chem. 276:41656–41660.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van Vugt MA and Medema RH: Getting in and
out of mitosis with Polo-like kinase-1. Oncogene. 24:2844–2859.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim SA, Kwon SM, Yoon JH and Ahn SG: The
antitumor effect of PLK1 and HSF1 double knockdown on human oral
carcinoma cells. Int J Oncol. 36:867–872. 2010.PubMed/NCBI
|
|
28
|
Jimeno A, Rubio-Viqueira B, Rajeshkumar V,
Chan A, Solomon A and Hidalgo M: A fine-needle aspirate-based
vulnerability assay identifies polo-like kinase 1 as a mediator of
gemcitabine resistance in pancreatic cancer. Mol Cancer Ther.
9:311–318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wong N and Khan M: Abstract 4915: High
degree of G2/M arrest induced by Polo-like kinase 1 (PLK1)
inhibition is associated with radiosensitization. Cancer Res.
74:49152014. View Article : Google Scholar
|
|
30
|
Liu YF, Chen YJ and Chen CL: MS275
synergistically enhances the growth inhibitory effects of BI2536 in
non-small-cell lung cancer cells. Pharm Biotechnol. 18:308–312.
2011.
|
|
31
|
Gleixner KV, Ferenc V, Gruze A, Kneidinger
M, Baumgartner C, Mayerhofer M, et al: The Plk-1 Inhibitor BI 2536
Counteracts Proliferation and Viability of CML Cells and Synergizes
with Imatinib and Nilotinib (AMN107) in Producing Growth
Inhibition. Blood. 110:317A2007.
|
|
32
|
Mao J, Fan S, Ma W, Fan P, Wang B, Zhang
J, Wang H, Tang B, Zhang Q, Yu X, et al: Roles of Wnt/β-catenin
signaling in the gastric cancer stem cells proliferation and
salinomycin treatment. Cell Death Dis. 5:e10392014. View Article : Google Scholar
|
|
33
|
Shrivastava S, Kumar P, Jeengar MK and
Naidu VG: T3038 - Inhibition of Wnt/β-catenin pathway by
niclosamide: A therapeutic target for gastric cancer. National
Institute of Pharmaceutical Education and Research (NIPER).
2014.
|
|
34
|
Nam HJ, Kim S, Lee MW, Lee BS, Hara T,
Saya H, Cho H and Lee JH: The ERK-RSK1 activation by growth factors
at G2 phase delays cell cycle progression and reduces mitotic
aberrations. Cell Signal. 20:1349–1358. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li R, Chen DF, Zhou R, Jia SN, Yang JS,
Clegg JS and Yang WJ: Involvement of polo-like kinase 1 (Plk1) in
mitotic arrest by inhibition of mitogen-activated protein
kinase-extracellular signal-regulated kinase-ribosomal S6 kinase 1
(MEK-ERK-RSK1) cascade. J Biol Chem. 287:15923–15934. 2012.
View Article : Google Scholar : PubMed/NCBI
|