Introduction
Liver cancer is estimated to lead to ~750,000
mortalities annually worldwide (1). It is more prevalent in men than in
women, and is the second leading cause of cancer mortality in men.
The most common primary liver cancer is hepatocellular carcinoma
(HCC). The risk factors for HCC include infection with hepatitis B
or hepatitis C virus, alcohol consumption, and non-alcoholic
steatohepatitis (2).
MicroRNAs (miRNAs/miRs) are small non-coding RNAs of
between 18 and 25 nucleotides in length that serve a crucial
function in gene regulation. miRNAs are known to repress thousands
of target genes and co-ordinate normal processes, including
cellular proliferation, differentiation and apoptosis (3–5). The
aberrant expression or alteration of miRNAs contributes to a range
of human pathologies, including cancer (6,7). HCC
is no exception, and various HCC-specific miRNA signatures have
been reported (8). However, the
function of miRNAs in the pathogenesis of HCC remains poorly
understood.
One of the hallmarks of human cancer is intrinsic or
acquired resistance to apoptosis (9). The evasion of apoptosis contributes
to the development and progression of cancer, and to resistance to
treatment. Therefore, a better understanding of the molecular
mechanisms underlying the resistance of tumors to apoptosis is
expected to provide a basis for a rational approach in the
development of molecular targeted therapies.
Caspase-9 is the essential initiator caspase
required in the mitochondrial apoptotic pathway (10). Upon exposure to intracellular
apoptotic stimuli, mitochondria release cytochrome c, which
binds to an adaptor protein known as apoptotic protease-activating
factor 1 (Apaf-1); this binding causes Apaf-1 to oligomerize into a
wheel-like heptamer called an apoptosome. Apaf-1 in the apoptosome
then recruits caspase-9, which is activated in the apoptosome. The
activated caspase-9 is then able to activate downstream executioner
caspases, including caspase-3, -6 and -7, and trigger a cascade of
events leading to apoptosis. As such, caspase-9 is considered to be
a tumor suppressor (11).
To investigate the miRNAs involved in HCC, miRNAs
that were upregulated in primary HCC tumors when compared with
non-tumor tissues were screened for using a miRNA microarray. The
array analysis revealed that miR-96-5p was among the most
significantly upregulated miRNAs in HCC tumors (K. Yasui,
unpublished data).
In the present study, the functions of miR-96-5p
were investigated in HCC. In addition to the forkhead box O1
(FOXO1) gene, which has been identified to be a direct
target of miR-96-5p (12–16), the caspase-9 gene (CASP9)
gene was identified as a potential novel target of miR-96-5p. These
results suggest that increased miR-96-5p expression may contribute
to resistance to apoptosis via the repression of caspase-9
expression in HCC.
Materials and methods
Reagents and antibodies
Antibodies against caspase-9 (cat. no. 9508),
caspase-2 (cat. no. 2224), FOXO1 (cat. no. 2880) and
poly(ADP-ribose) polymerase (PARP; cat. no. 9542) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA). The
antibody against β-actin (cat. no. A1978) was purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Doxorubicin was
obtained from Toronto Research Chemicals Inc. (Toronto, ON,
Canada).
Cell culture and primary tumor
samples
Three HCC cell lines (SNU387, SNU449 and HLF) were
obtained from the American Type Culture Collection (Manassas, VA,
USA) or the Japan Collection of Research Bioresources Cell Bank
(Osaka, Japan). All cell lines were cultured in Dulbecco's modified
Eagle's medium containing 10% fetal bovine serum at 37°C. Primary
HCC tumor samples and non-tumor samples were obtained from patients
who had undergone surgical resection of tumors, as described
previously (17). The present
study, including the collection and analytical methods, was
approved by the Ethics Committees of the Kyoto Prefectural
University of Medicine and was conducted in accordance with the
Declaration of Helsinki. Written informed consent was obtained from
each patient for the use of their tissue samples in the present
study.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) of miRNA and mRNA
Total RNA was obtained using TRIzol®
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Residual
genomic DNA was removed by incubating the RNA samples with
RNase-free DNase I (Takara Bio, Inc., Otsu, Japan) prior to
performing RT-qPCR.
Quantification of human (hsa)-miR-96-5p was
performed using a fluorescence detection method. Single-stranded
cDNA was generated using a Universal cDNA synthesis kit II (Exiqon
A/S, Vedbaek, Denmark), according to the manufacturer's protocol.
RT-qPCR experiments were performed using the StepOnePlus PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) with ExiLENT
SYBR-Green master mix (Exiqon A/S). The hsa-miR-96-5p locked
nucleic acid (LNA) PCR primer set was purchased from Exiqon A/S.
Small nucleolar RNA U49 (Exiqon A/S) was used as an endogenous
control for miRNA levels.
The mRNAs were detected by RT-qPCR as described
previously (18). The expression
level of β-actin was used as an endogenous control for mRNA levels.
The primer sequences used for PCR are presented in Table I.
 | Table IPrimer sequences used for PCR. |
Table I
Primer sequences used for PCR.
| Gene | Forward primer | Reverse primer |
|---|
| FOXO1 |
5′-AAGAGCGTGCCCTACTTCAA-3′ |
5′-CTGTTGTTGTCCATGGATGC-3′ |
| CASP9 |
5′-GCAAGCAGCAAAGTTGTCGA-3′ |
5′-GGACTCACGGCAGAAGTTCA-3′ |
| CASP2 |
5′-TGATGCCTTCTGTGAAGCAC-3′ |
5′-GCTCAACGGTGGGAGTACAT-3′ |
| CDKN1A |
5′-ATGAAATTCACCCCCTTTCC-3′ |
5′-CCCTAGGCTGTGCTCACTTC-3′ |
| FOXO3 |
5′-GGCGGACTTTGTGTTTGTTT-3′ |
5′-AAGCCACCTGAAATCACACC-3′ |
| REV1 |
5′-TTGTGATGAAGCGCTGGTAG-3′ |
5′-TTGGTCACTAGCTGGCCTCT-3′ |
| RAD51 |
5′-CGACTCTCCCTGTCTTCCTG-3′ |
5′-TTTCCCGGAAGCTTTATCCT-3′ |
| BRMS1L |
5′-AGTGAAAACGGAACCACCTG-3′ |
5′-CCATCAGGCCTCTTAAACCA-3′ |
| EFNA5 |
5′-AAACGTATTGTGCCCTGGAG-3′ |
5′-CTGCCACTCCAGAGGAGTTC-3′ |
| AKT1S1 |
5′-AGGGCTCTTTGTGATGGATG-3′ |
5′-ACTTGGCGTACTGCTGTGTG-3′ |
| HBP1 |
5′-CCGTGAAAATGAGGTGGACT-3′ |
5′-GAAGGCTGGTTCACTCTTCG-3′ |
| PAX6 |
5′-CCGGCAGAAGATTGTAGAGC-3′ |
5′-CTCACACATCCGTTGGACAC-3′ |
| IRS1 |
5′-GCTTTCCACAGCTCACCTTC-3′ |
5′-CCTCAGTGCCAGTCTCTTCC-3′ |
| STK17A |
5′-TTTGTCTGAGTCGGCTGTTG-3′ |
5′-GTGCCTTTTCCATCCTGAAA-3′ |
|
PLAGL1/ZAC |
5′-CAAGTGTGTGCAGCCTGACT-3′ |
5′-ACACTCCTCACACCCAAAGG-3′ |
| ACTB |
5′-GTCCACCTTCCAGCAGATGT-3′ |
5′-TGTTTTCTGCGCAAGTTAGG-3′ |
Single-nucleotide polymorphism (SNP)
array analysis
Genomic DNA was isolated as described previously
(18). Changes in DNA copy number
were analyzed in 34 primary HCC tumors using the GeneChip Mapping
250K Sty array (Affymetrix; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol and as described
previously (18,19).
Immunoblotting
Immunoblotting was performed as described previously
(18). The dilutions of the
antibodies were 1:1,000 for anti-caspase-9, anti-caspase-2,
anti-FOXO1 and anti-PARP; and 1:5,000 for β-actin. For
immunodetection, anti-rabbit IgG or anti-mouse IgG (Cell Signaling
Technology, Inc.) was used as the secondary antibody at a dilution
of 1:5,000 or 1:10,000, respectively. Antibody binding was detected
using the enhanced chemiluminescence system (GE Healthcare,
Chicago, IL, USA).
Transfection of miRNA mimics and
inhibitors
Hsa-miR-96-5p mimic (#472140-001), cel-miR-39-3p
(#479902-001), which was used as a miRNA mimic negative control,
hsa-miR-96-5p inhibitor (#4102571-101) and miRNA inhibitor negative
control (#199006-101) were obtained from Exiqon A/S. These mimics
and inhibitors were transfected into cells using
Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.), and were used at a final concentration of 12.5
nmol/l, except for SNU449 cells, which were transfected with 12.5,
25 and 50 nmol/l miR-96-5p inhibitor. After 72 h of incubation,
cells were harvested for RT-qPCR and immunoblotting analyses.
Identification of the target genes of
miR-96-5p
A total of 15 candidate genes were selected
(Table II) that were predicted to
be target genes of miR-96-5p by miRBase (www.mirbase.org), miRTarBase (mirtarbase.mbc.nctu.edu.tw) and microRNA.org (www.microrna.org). miR-96-5p mimic or negative control
mimic was transfected into SNU387 and HLF cells that exhibited low
expression levels of miR-96-5p, and the mRNA and protein levels of
the 15 genes were determined using RT-qPCR and immunoblotting,
respectively, as aforementioned.
| Table IICandidate target genes of
miR-96-5p. |
Table II
Candidate target genes of
miR-96-5p.
| Gene | Expression ratio
|
|---|
| SNU387 | HLF |
|---|
| FOXO1 | 0.30 | 0.44 |
| CASP9 | 0.37 | 0.25 |
| CASP2 | 0.53 | 0.44 |
| CDKN1A | 0.79 | 0.64 |
| FOXO3 | 0.78 | 0.75 |
| REV1 | 0.55 | 0.55 |
| RAD51 | 0.80 | 0.65 |
| BRMS1L | 0.53 | 0.50 |
| EFNA5 | 1.35 | 1.67 |
| AKT1S1 | 0.61 | 0.84 |
| HBP1 | 0.42 | 0.38 |
| PAX6 | 1.40 | 0.55 |
| IRS1 | 0.59 | 0.94 |
| STK17A | 0.33 | 0.24 |
|
PLAGL1/ZAC | 0.60 | 0.64 |
Luciferase reporter assay
The 3′-untranslated region (3′-UTR) sequence of
CASP9 mRNA, inclusive of the miR-96-5p target site, was
cloned downstream of the firefly luciferase gene in the pMirTarget
vector (OriGene Technologies, Inc., Rockville, MD, USA).
Site-specific mutations were generated in the CASP9 3′-UTR
using the KOD Plus Mutagenesis kit (Toyobo Life Science, Osaka,
Japan), according to the manufacturer's protocol. The
luciferase-CASP9 3′-UTR wild-type (WT) vector, the
luciferase-CASP9 3′-UTR mutant vector or empty vector was
transfected into SUN387 cells together with either miR-96-5p mimic
or negative control mimic using Lipofectamine® 3000
(Invitrogen). After 48 h of incubation, cells were harvested for
the measurement of firefly and Renilla luciferase activity
using the Dual-Glo Luciferase assay system (Promega Corporation,
Madison, WI, USA).
RNA interference
Two small interfering RNAs (siRNAs) targeting
CASP9 (#1 and #2; Ambion; Thermo Fisher Scientific, Inc.)
and control (non-silencing) siRNA were delivered into cells using
Lipofectamine RNAiMAX (Invitrogen). After 72 h of incubation, cells
were harvested for immunoblotting.
Cell viability assay
Cell viability was determined using the
water-soluble tetrazolium salt 8 assay (Cell Counting Kit-8;
Dojindo Molecular Technologies, Inc., Kumamoto, Japan), according
to the manufacturer's protocol.
Apoptosis assay
Cells were transfected with miR-96-5p mimic or
negative control mimic. After 48 h of incubation, the cells were
treated with doxorubicin or exposed to ultraviolet (UV) irradiation
at 40 J/m2 (SNU387 cells) or 10 J/m2 (HLF
cells) using a UV HybriLinker HL-2000 (BM Equipment Co., Ltd.,
Tokyo, Japan). Apoptosis was evaluated from the expression level of
cleaved PARP by immunoblotting and the levels of caspase-3/7
activities using the Caspase-Glo 3/7 assay (Promega
Corporation).
Statistical analysis
Statistical analyses were performed using SPSS
Statistics (version 24.0; IBM Corp., Armonk, NY, USA). Comparisons
were made using the Wilcoxon signed-rank test, Mann-Whitney U test,
Student's t-test or analysis of variance (ANOVA) followed by post
hoc tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression of miR-96-5p is upregulated in
primary HCC tumors
Using RT-qPCR to analyze 27 paired tumor and
non-tumor tissues, it was identified that miR-96-5p expression was
significantly upregulated in the HCC tumors compared with their
non-tumorous counterparts (Fig.
1A). To investigate the molecular mechanisms by which miR-96-5p
is upregulated in the HCC tumors, aberrations in DNA copy number
were examined in primary HCC tumors. High-resolution SNP array
analysis revealed a frequent copy number gain at chromosomal region
7q32.2, where the MIR96 locus is located; it was identified
in 17/34 (50%) HCC tumors (Fig.
1B). The presence of a copy number gain at the MIR96
locus was significantly associated with increased expression levels
of miR-96-5p in the HCC tumors (Mann-Whitney U test; Fig. 1C).
The potential association between the increased
expression levels of miR-96-5p in HCC tumors and various
clinicopatho-logical parameters was analyzed. However, no
significant association was apparent between the levels of
miR-96-5p and any of the parameters, including age, sex, tumor
size, clinical stage, hepatitis B or hepatitis C virus infection
and α-fetoprotein level. No association was observed between the
levels of miR-96-5p and overall or disease-free survival (data not
shown).
CASP9 is identified as a novel direct
target of miR-96-5p
To identify the target genes of miR-96-5p, it was
investigated whether or not enforced expression of miR-96-5p was
able to decrease the expression levels of the 15 candidate genes.
These genes were selected because they were predicted to be target
genes of miR-96-5p using an in silico approach, and they
were potential tumor suppressor genes. Results of the RT-qPCR
analysis are presented in Table
II and Fig. 2A. Representative
images of immunoblotting are presented in Fig. 2B. Of the 15 candidate genes,
FOXO1, CASP9 and CASP2 exhibited decreased
expression at the mRNA and protein levels following transfection
with the miR-96-5p mimic compared with transfection with the
control mimic in SNU387 and HLF cells (Fig. 2A and B). To confirm these results,
rescue experiments were performed using the miR-96-5p inhibitor in
combination with the miR-96-5p mimic. Immunoblot analyses revealed
that transfection with the miR-96-5p inhibitor attenuated the
suppression of FOXO1, caspase-9 and caspase-2 protein expression by
the miR-96-5p mimic (Fig. 2C).
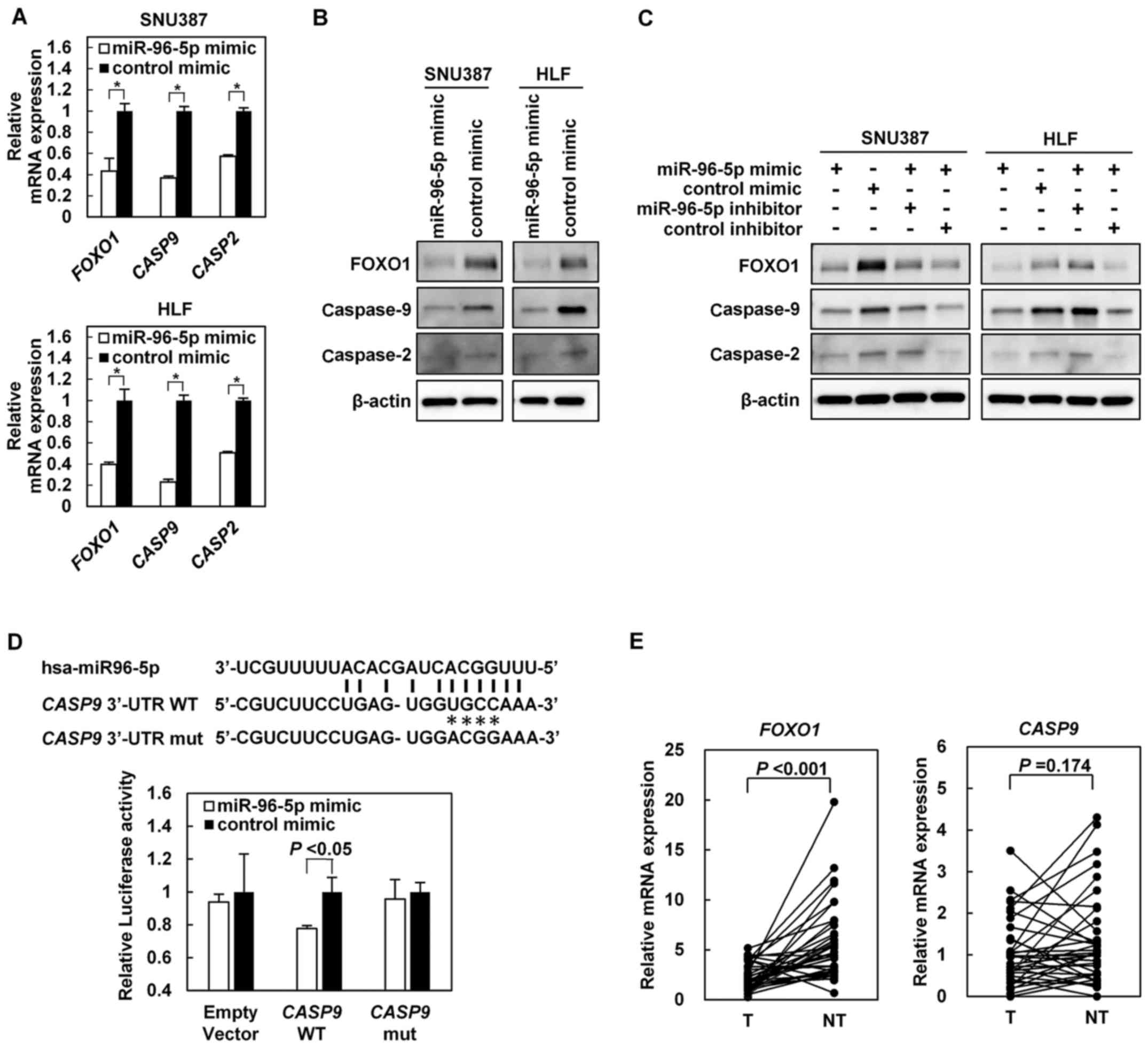 | Figure 2(A) Relative mRNA expression levels
of FOXO1, CASP9 and CASP2 in SNU387 and HLF
cells transfected with the miR-96-5p mimic or control mimic. (B)
Immunoblot analyses of the indicated proteins in SNU387 and HLF
cells transfected with the miR-96-5p mimic or control mimic.
β-actin was used as an internal control. (C) Rescue experiments
using the miR-96-5p inhibitor in combination with the miR-96-5p
mimic. SNU387 and HLF cells were transfected with the miR-96-5p
mimic or control mimic in combination with the miR-96-5p inhibitor
or control inhibitor. Immunoblot analyses of the FOXO1, caspase-9
and caspase-2 proteins are shown. (D) Upper panel: A putative
binding site for miR-96-5p in the CASP9 3′-UTR. Point
mutations (*) were introduced by site-directed mutagenesis to
destroy the base-paring with the miRNA. Lower panel: Luciferase
assay. A luciferase reporter plasmid bearing the CASP9
3′-UTR WT, CASP9 3′-UTR mut or empty vector was co-transfected with
the miR-96-5p mimic or control mimic into SNU387 cells. (E)
Relative mRNA expression levels of FOXO1 and CASP9 in
paired tumor and non-tumor tissues from 35 patients with primary
HCC as determined using reverse transcription-quantitative
polymerase chain reaction. FOXO1, forkhead box O1; CASP,
caspase; miR, microRNA; UTR, untranslated region; mut, mutant; WT,
wild-type; T, tumor; NT, non-tumor. |
A luciferase assay was performed to investigate
whether or not CASP9 is a direct target of miR-96-5p using a
CASP9 3′-UTR-luciferase reporter construct. A putative
binding site of miR-96-5p was identified in the CASP9 3′-UTR
(Fig. 2D). SNU387 cells that were
co-transfected with the luciferase-CASP9 3′-UTR WT vector
and miR-96-5p mimic exhibited significantly decreased luciferase
activity compared with the cells co-transfected with the
luciferase-CASP9 3′-UTR WT vector and control mimic
(Fig. 2D). In addition, no
decrease in luciferase activity was observed when the empty
luciferase reporter vector or the luciferase-CASP9 3′-UTR
mutant vector was co-transfected with the miR-96-5p mimic (Fig. 2D). Regarding CASP2, the
luciferase assay was not able to demonstrate that CASP2 was
a direct target of miR-96-5p (data not shown).
Furthermore, the mRNA levels of FOXO1 and
CASP9 were determined using RT-qPCR in paired tumor and
non-tumor liver tissues from 35 patients with primary HCC. The mRNA
levels of FOXO1 and CASP9 were decreased in the
tumors compared with in their non-tumorous counterparts, although
the difference was only statistically significant for FOXO1
(Fig. 2E). However, contrary to
expectations, there was no inverse association between the
expression levels of miR-96-5p and the mRNA levels of FOXO1
or CASP9 in the HCC tumors (data not shown).
miR-96-5p-mediated suppression of
caspase-9 inhibits apoptosis
To examine whether miR-96-5p inhibits apoptosis by
decreasing caspase-9 expression, caspase-9 expression was knocked
down in SNU387 and HLF cells by transfection with two different
siRNAs targeting CASP9, followed by treatment with
doxorubicin, an anticancer agent that causes DNA strand breaks.
Caspase-9 knockdown was confirmed by immunoblotting (Fig. 3A). Doxorubicin decreased the cell
viability of SNU387 and HLF cells in a dose-dependent manner.
However, knockdown of caspase-9 attenuated the doxorubicin-induced
decrease in cell viability in SNU387 and HLF cells, when SNU387
cells were treated with doxorubicin at 0.3, 1.2 and 5.0
µmol/l and HLF cells were treated with doxorubicin at 0.15
and 0.3 µmol/l (Fig.
3B).
 | Figure 3(A) Immunoblot analysis of caspase-9
in SNU387 and HLF cells transfected with CASP9 siRNA (#1 or
#2) or with control siRNA. (B) Effect of knockdown of caspase-9 on
the cytotoxicity of doxorubicin. SNU387 and HLF cells were
transfected with CASP9 siRNA (#1 or #2) or control siRNA,
then treated with doxorubicin 48 h later at the indicated
concentrations. Cell viability was measured 48 h after doxorubicin
treatment. *P<0.01 and **P<0.05 vs.
control siRNA. SNU387 and HLF cells were transfected with the
miR-96-5p mimic or control mimic, then treated with doxorubicin 48
h later. (C) SNU387 and HLF cells were treated with doxorubicin at
the indicated concentrations. Cell viability was measured 96 h
after doxorubicin treatment. *P<0.01 and
**P<0.05 vs. control mimic. (D) SNU387 and HLF cells
were treated with doxorubicin at 0.6 and 1.0 µmol/l,
respectively. Cell viability was measured at the indicated times.
*P<0.01 vs. control mimic. Effect of the miR-96-5p
mimic on apoptosis induced by doxorubicin and UV. SNU387 and HLF
cells were transfected with the miR-96-5p mimic or control mimic,
then treated with doxorubicin or UV 48 h later. (E) SNU387 cells
were treated or not treated with 0.6 µmol/l doxorubicin for
6 h. HLF cells were treated or not treated with 2.5 µmol/l
doxorubicin for 12 h. (F) SNU387 cells were exposed or not exposed
to UV irradiation at 40 J/m2. HLF cells were exposed or
not exposed to UV irradiation at 10 J/m2. Subsequently,
the cells were cultured for 18 h. Immunoblot analyses of caspase-9,
PARP and cleaved PARP as a marker of apoptosis are shown. (G)
Measurement of caspase-3/7 activity. HLF cells were transfected
with the miR-96-5p mimic or control mimic, then treated with 2.5
µmol/l doxorubicin 48 h later. Caspase-3/7 activity was
measured 12 h after doxorubicin treatment. CASP9, caspase-9; siRNA,
small interfering RNA; UV, ultraviolet; miR, microRNA; PARP,
poly(ADP-ribose) polymerase. |
SNU387 and HLF cells were transfected with the
miR-96-5p mimic or control mimic, followed by treatment with
doxorubicin. The rate of the doxorubicin-induced decrease in cell
viability was significantly lower in cells transfected with the
miR-96-5p mimic compared with in cells transfected with the control
mimic in a dose-dependent and time-dependent manner (Fig. 3C and D).
Finally, SNU387 and HLF cells were transfected with
the miR-96-5p mimic or control mimic, followed by treatment with
doxorubicin or UV to induce apoptosis. Immunoblotting of caspase-9
and cleaved PARP revealed that, in SNU387 and HLF cells,
transfection with the miR-96-5p mimic decreased caspase-9
expression and attenuated the apoptosis induced by doxorubicin or
UV compared with the control mimic (Fig. 3E and F). Results of the caspase-3/7
activity assay indicated that transfection with the miR-96-5p mimic
significantly decreased the caspase-3/7 activity in HLF cells
treated with doxorubicin compared with similarly treated HLF cells
transfected with the control mimic (Fig. 3G).
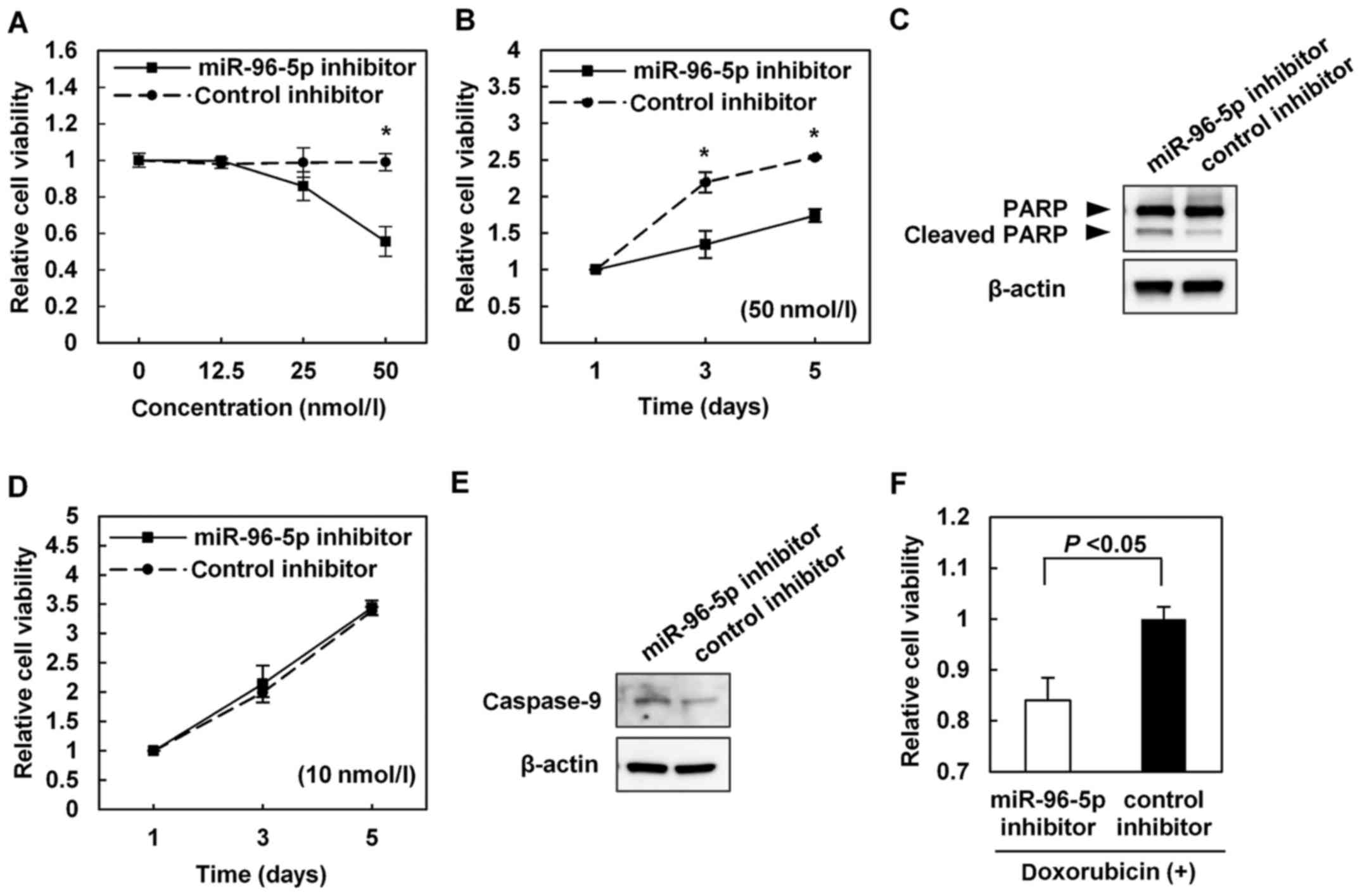
Apoptosis is induced by the inhibition of
miR-96-5p
To evaluate miR-96-5p as a potential therapeutic
target for HCC, miR-96-5p inhibitor or control inhibitor was
transfected into SNU449 cells, which exhibited increased expression
of miR-96-5p. SNU449 cells transfected with a high concentration
(i.e. 50 nmol/l) of miR-96-5p inhibitor exhibited decreased cell
viability compared with those transfected with the same
concentration of the control inhibitor (Fig. 4A and B). Immunoblotting of cleaved
PARP identified that the miR-96-5p inhibitor induced apoptosis
(Fig. 4C).
Although SNU449 cells transfected with a low
concentration (i.e. 10 nmol/l) of miR-96-5p inhibitor did not
exhibit any changes in cell viability (Fig. 4A and D), they did exhibit increased
caspase-9 expression, as revealed by immunoblotting (Fig. 4E). In addition, SNU449 cells
transfected with 10 nmol/l miR-96-5p inhibitor prior to treatment
with doxorubicin exhibited significantly lower cell viability
compared with the cells transfected with the control inhibitor
(Fig. 4F).
Discussion
In the present study, it was identified that
miR-96-5p was significantly upregulated in the HCC tumors compared
with in the non-tumor tissues; this result was consistent with the
results of previous studies (13,20).
miR-96-5p is a putative oncogenic miRNA that is upregulated in
different types of cancer, including breast (14), colorectal (21), prostate (22), bladder (23) and non-small cell lung (24) cancer. However, the underlying
molecular mechanism of miR-96-5p upregulation in cancer remains
unclear. Although previous studies have identified that
transforming growth factor-β1 (25), hypoxia (26) and mitochondrial dysfunction
(27) induced the expression of
miR-96-5p, these factors did not increase the expression of
miR-96-5p in HCC cells in the present study (data not shown). The
reason for this difference is unclear, but it may be due to the
different types of cell used. The results of the present study
suggest that the copy number gain at the MIR96 locus is one
of the molecular mechanisms by which miR-96-5p expression is
increased in HCC.
miR-96 belongs to the miR-183/-96/-182 cluster, a
conserved polycistronic miRNA cluster. The miR-183/-96/-182 cluster
is transcribed into the same primary miRNA. Thus, miR-183, miR-96
and miR-182 are likely to have similar expression levels. However,
despite the upregulation of miR-96, unpredictably, the microarray
and RT-qPCR analyses revealed that miR-183 and miR-182 were not
upregulated in the HCC tumors compared with in the non-tumor
tissues. Although the reason for this is unknown, it is possible
that there is a difference in stability between the three miRNAs in
HCC cells.
Next, target genes of miR-96-5p were identified.
Each miRNA is predicted to negatively regulate tens to hundreds of
target genes. Several previous studies have identified that
FOXO1 is a direct target of miR-96-5p in HCC (12,13),
and in other types of malignancy, including breast (14), prostate (15) and endometrial (16) cancer. FOXO1 orchestrates the
regulation of genes involved in the apoptotic response, cell cycle
checkpoints and cellular metabolism (14,28).
FOXO1 is a putative tumor suppressor (29) and the expression of this gene is
downregulated in various types of cancer. In addition, it appears
that FOXO1-dependent cell cycle arrest and apoptosis are critical
for its tumor-suppressive effect. The results of the present study
indicated that the miR-96-5p mimic decreased the expression of
FOXO1 at the mRNA and protein levels in HCC cells, and that
the mRNA levels of FOXO1 were significantly decreased in the
primary HCC tumors compared with in their non-tumorous
counterparts, confirming that FOXO1 is a target of miR-96-5p
in HCC. Although miR-183 and miR-182 were not upregulated in the
HCC tumors in the present study, it has been reported that miR-183,
miR-96 and miR-182 concordantly downregulated FOXO1 in HCC
cells (13). In addition to
FOXO1, previous studies have identified that several genes,
including those encoding forkhead box O3 (30), glypican 3 (31), insulin receptor substrate 1
(27), reversion inducing
cysteine-rich protein with Kazal motifs (32), metastasis suppressor 1 (33) and protein tyrosine phosphatase
non-receptor 9 (34), are
potential targets of miR-96-5p in cancers.
The present study attempted to identify novel target
genes of miR-96-5p in HCC. Using an in silico approach, the
putative binding site of miR-96-5p was identified in the
CASP9 3′-UTR. RT-qPCR and immunoblot analyses revealed that
the miR-96-5p mimic decreased CASP9 expression at the mRNA
and protein levels, respectively. Results of the luciferase assay
using the CASP9 3′-UTR-luciferase reporter construct
suggested that CASP9 is a potential direct target of
miR-96-5p. Furthermore, the mRNA levels of CASP9 were
decreased in the primary HCC tumors compared with in their
non-tumorous counterparts. Collectively, the results of the present
study suggested that CASP9 is a novel direct target of
miR-96-5p, and that CASP9 expression is decreased by the
upregulation of miR-96-5p in HCC. Since caspase-9 is the essential
initiator caspase required for apoptosis signaling and it is
considered to be a tumor suppressor, downregulation of caspase-9 by
miR-96-5p may be beneficial to cancers.
Resistance to apoptosis contributes to the
development and progression of cancer, and to resistance to
chemotherapy. Indeed, the present study demonstrated that knockdown
of caspase-9 resulted in resistance to doxorubicin-induced
apop-tosis. Furthermore, it was identified that enforced expression
of miR-96-5p led to resistance to doxorubicin- and UV-induced
apoptosis through the downregulation of caspase-9. These results
suggested that miR-96-5p inhibits apoptosis by decreasing caspase-9
expression. Therefore, upregulation of miR-96-5p may be involved in
the development and progression of HCC, and may contribute to
resistance to chemotherapy by inhibiting apoptosis through a
decrease in caspase-9 expression in HCC. Furthermore, because FOXO1
also induces apoptosis, the miR-96-5p-mediated decrease in FOXO1
and caspase-9 expression may have a synergistic effect on
inhibiting apoptosis.
Finally, it was investigated whether miR-96-5p may
be a potential therapeutic target in HCC. A high concentration
(i.e. 50 nmol/l) of miR-96-5p inhibitor decreased the viability of
HCC cells by inducing apoptosis. Although the molecular mechanism
underlying this apoptosis is unclear and the high concentration of
the inhibitor used may have caused off-target effects, these
results suggest that miR-96-5p may be a potential therapeutic
target. Interestingly, a low concentration (i.e. 10 nmol/l) of
miR-96-5p inhibitor, which did not affect the viability of the HCC
cells, enhanced the cytotoxic effect of doxorubicin by increasing
caspase-9 expression. These results suggested that HCC cells may be
sensitized to doxorubicin by pretreatment with a low concentration
of miR-96-5p inhibitor, and that there is a synergistic effect
between the miR-96-5p inhibitor and doxorubicin. Of note, in
previous studies in which small molecules against miR-96-5p were
designed, it was demonstrated that the drugs induced apoptosis in
breast cancer cells while leaving healthy cells unaffected
(35,36). Taken together, these results and
those of the present study indicate that miR-96-5p inhibitor, alone
or together with a cytotoxic drug, may provide a promising strategy
for the treatment of HCC.
In conclusion, the results of the present study
suggest that miR-96-5p, which is frequently upregulated in HCC,
inhibits apoptosis by targeting CASP9. Therefore, miR-96-5p
may be a potential therapeutic target for HCC.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Japan Society
for the Promotion of Science KAKENHI (grant no. 15K09017).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to confidentiality but
are available from the corresponding author on reasonable
request.
Authors' contributions
NI performed laboratory experiments with the help of
AT, YG, KT, TK, TS, NY, OD, YS, AU, TN, KY, MM, HK, YN and YI, and
prepared the manuscript. KY designed the present study and prepared
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study, including the collection and
analytical methods, was approved by the Ethics Committees of the
Kyoto Prefectural University of Medicine, and was conducted in
accordance with the Declaration of Helsinki. Written informed
consent was obtained from each patient for the use of their tissue
samples in the present study.
Consent for publication
Written informed consent was obtained from each
patient.
Competing interests
The authors have no conflicts of interest to
disclose.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar
|
|
3
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al:
Frequent deletions and down-regulation of micro-RNA genes miR15 and
miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci
USA. 99:15524–15529. 2002. View Article : Google Scholar
|
|
7
|
Lujambio A and Lowe SW: The microcosmos of
cancer. Nature. 482:347–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mao B and Wang G: MicroRNAs involved with
hepatocellular carcinoma (Review). Oncol Rep. 34:2811–2820. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohammad RM, Muqbil I, Lowe L, Yedjou C,
Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, et al:
Broad targeting of resistance to apoptosis in cancer. Semin Cancer
Biol. 35(Suppl): S78–S103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Crawford ED, McNew JA, Nagata S and Wells
J: Cell death. Molecular Biology of the Cell. 6th edition. Alberts
B, Johnson A, Lewis J, Morgan D, Raff M, Roberts K and Walter P:
Garland Science; New York: pp. 1021–1034. 2015
|
|
11
|
Soengas MS, Alarcón RM, Yoshida H, Giaccia
AJ, Hakem R, Mak TW and Lowe SW: Apaf-1 and caspase-9 in
p53-dependent apoptosis and tumor inhibition. Science. 284:156–159.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu D, He X, Chang Y, Xu C, Jiang X, Sun S
and Lin J: Inhibition of miR-96 expression reduces cell
proliferation and clonogenicity of HepG2 hepatoma cells. Oncol Rep.
29:653–661. 2013. View Article : Google Scholar
|
|
13
|
Leung WK, He M, Chan AW, Law PT and Wong
N: Wnt/β-catenin activates MiR-183/96/182 expression in
hepatocellular carcinoma that promotes cell invasion. Cancer Lett.
362:97–105. 2015. View Article : Google Scholar
|
|
14
|
Guttilla IK and White BA: Coordinate
regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast
cancer cells. J Biol Chem. 284:23204–23216. 2009. View Article : Google Scholar :
|
|
15
|
Fendler A, Jung M, Stephan C, Erbersdobler
A, Jung K and Yousef GM: The antiapoptotic function of miR-96 in
prostate cancer by inhibition of FOXO1. PLoS One. 8:e808072013.
View Article : Google Scholar :
|
|
16
|
Myatt SS, Wang J, Monteiro LJ, Christian
M, Ho KK, Fusi L, Dina RE, Brosens JJ, Ghaem-Maghami S and Lam EW:
Definition of microRNAs that repress expression of the tumor
suppressor gene FOXO1 in endometrial cancer. Cancer Res.
70:367–377. 2010. View Article : Google Scholar
|
|
17
|
Yasui K, Konishi C, Gen Y, Endo M, Dohi O,
Tomie A, Kitaichi T, Yamada N, Iwai N, Nishikawa T, et al: EVI1, a
target gene for amplification at 3q26, antagonizes transforming
growth factor-β-mediated growth inhibition in hepatocellular
carcinoma. Cancer Sci. 106:929–937. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zen K, Yasui K, Nakajima T, Zen Y, Zen K,
Gen Y, Mitsuyoshi H, Minami M, Mitsufuji S, Tanaka S, et al: ERK5
is a target for gene amplification at 17p11 and promotes cell
growth in hepatocellular carcinoma by regulating mitotic entry.
Genes Chromosomes Cancer. 48:109–120. 2009. View Article : Google Scholar
|
|
19
|
Endo M, Yasui K, Zen Y, Gen Y, Zen K,
Tsuji K, Dohi O, Mitsuyoshi H, Tanaka S, Taniwaki M, et al:
Alterations of the SWI/SNF chromatin remodelling subunit-BRG1 and
BRM in hepatocellular carcinoma. Liver Int. 33:105–117. 2013.
View Article : Google Scholar
|
|
20
|
Pineau P, Volinia S, McJunkin K, Marchio
A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM and
Dejean A: miR-221 overexpression contributes to liver
tumorigenesis. Proc Natl Acad Sci USA. 107:264–269. 2010.
View Article : Google Scholar :
|
|
21
|
Ress AL, Stiegelbauer V, Winter E,
Schwarzenbacher D, Kiesslich T, Lax S, Jahn S, Deutsch A,
Bauernhofer T, Ling H, et al: MiR-96-5p influences cellular growth
and is associated with poor survival in colorectal cancer patients.
Mol Carcinog. 54:1442–1450. 2015. View Article : Google Scholar
|
|
22
|
Schaefer A, Jung M, Mollenkopf HJ, Wagner
I, Stephan C, Jentzmik F, Miller K, Lein M, Kristiansen G and Jung
K: Diagnostic and prognostic implications of microRNA profiling in
prostate carcinoma. Int J Cancer. 126:1166–1176. 2010.
|
|
23
|
Han Y, Chen J, Zhao X, Liang C, Wang Y,
Sun L, Jiang Z, Zhang Z, Yang R, Chen J, et al: MicroRNA expression
signatures of bladder cancer revealed by deep sequencing. PLoS One.
6:e182862011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li C, Yin Y, Liu X, Xi X, Xue W and Qu Y:
Non-small cell lung cancer associated microRNA expression
signature: Integrated bioinformatics analysis, validation and
clinical significance. Oncotarget. 8:24564–24578. 2017.PubMed/NCBI
|
|
25
|
Siu MK, Tsai YC, Chang YS, Yin JJ, Suau F,
Chen WY and Liu YN: Transforming growth factor-β promotes prostate
bone metastasis through induction of microRNA-96 and activation of
the mTOR pathway. Oncogene. 34:4767–4776. 2015. View Article : Google Scholar
|
|
26
|
Ma Y, Yang HZ, Dong BJ, Zou HB, Zhou Y,
Kong XM and Huang YR: Biphasic regulation of autophagy by miR-96 in
prostate cancer cells under hypoxia. Oncotarget. 5:9169–9182. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jeong HJ, Park SY, Yang WM and Lee W: The
induction of miR-96 by mitochondrial dysfunction causes impaired
glycogen synthesis through translational repression of IRS-1 in
SK-Hep1 cells. Biochem Biophys Res Commun. 434:503–508. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Calnan DR and Brunet A: The FoxO code.
Oncogene. 27:2276–2288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dong XY, Chen C, Sun X, Guo P, Vessella
RL, Wang RX, Chung LW, Zhou W and Dong JT: FOXO1A is a candidate
for the 13q14 tumor suppressor gene inhibiting androgen receptor
signaling in prostate cancer. Cancer Res. 66:6998–7006. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin H, Dai T, Xiong H, Zhao X, Chen X, Yu
C, Li J, Wang X and Song L: Unregulated miR-96 induces cell
proliferation in human breast cancer by downregulating
transcriptional factor FOXO3a. PLoS One. 5:e157972010. View Article : Google Scholar
|
|
31
|
Jalvy-Delvaille S, Maurel M, Majo V,
Pierre N, Chabas S, Combe C, Rosenbaum J, Sagliocco F and Grosset
CF: Molecular basis of differential target regulation by miR-96 and
miR-182: The Glypican-3 as a model. Nucleic Acids Res.
40:1356–1365. 2012. View Article : Google Scholar :
|
|
32
|
Zhang J, Kong X, Li J, Luo Q, Li X, Shen
L, Chen L and Fang L: miR-96 promotes tumor proliferation and
invasion by targeting RECK in breast cancer. Oncol Rep.
31:1357–1363. 2014. View Article : Google Scholar
|
|
33
|
Xu L, Zhong J, Guo B, Zhu Q, Liang H, Wen
N, Yun W and Zhang L: miR-96 promotes the growth of prostate
carcinoma cells by suppressing MTSS1. Tumour Biol. 37:12023–12032.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hong Y, Liang H, Uzair-Ur-Rehman, Wang Y,
Zhang W, Zhou Y, Chen S, Yu M, Cui S, Liu M, et al: miR-96 promotes
cell proliferation, migration and invasion by targeting PTPN9 in
breast cancer. Sci Rep. 6:374212016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Velagapudi SP, Gallo SM and Disney MD:
Sequence-based design of bioactive small molecules that target
precursor microRNAs. Nat Chem Biol. 10:291–297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Velagapudi SP, Cameron MD, Haga CL,
Rosenberg LH, Lafitte M, Duckett DR, Phinney DG and Disney MD:
Design of a small molecule against an oncogenic noncoding RNA. Proc
Natl Acad Sci USA. 113:5898–5903. 2016. View Article : Google Scholar : PubMed/NCBI
|