Introduction
Colorectal cancer (CRC) is one of the leading global
causes of cancer-associated mortality (1,2).
Although improvements have been made to the diagnosis and treatment
of CRC, the effects of surgery and chemotherapy are still not
satisfactory, and the 5-year survival rate for patients with CRC
remains low (3,4). Traditional clinicopathological
parameters, including TNM stage and serum carcinoembryonic antigen
levels, have been widely used in prognostic evaluation; however,
most of these parameters cannot fully predict individual clinical
results (5). Therefore, novel
diagnosis and treatment methods are required to improve the
prognosis of patients with CRC.
Polo-like kinase 4 (PLK4), which is a member of the
polo family of serine/threonine protein kinases, localizes to
centrioles, which are complex microtubule-based structures present
in centrosomes (6,7). PLK4 functions primarily as a
regulator that mediates centriole duplication during the cell cycle
(8). In addition, the oncogenic
role of PLK4 has been studied in various types of cancer (9-15).
The expression of PLK4 has been detected in breast cancer (11,14),
lung cancer (12), neuroblastoma
(13) and prostate cancer
(15); however, its expression
pattern differs among various types of cancer. Increased PLK4
expression is correlated with higher rates of metastasis in breast
cancer (11,14). Furthermore, PLK4 knockdown
significantly inhibits invasion of neuroblastoma cells (13). However, the exact function of PLK4
in CRC development and metastasis remains unclear and requires
further investigation.
Epithelial-mesenchymal transition (EMT) is a
developmental process, which enhances invasion and metastasis in
several types of cancer (16).
During EMT, E-cadherin, and occludin are the most commonly detected
epithelial markers, and N-cadherin, vimentin and snail are the most
commonly detected mesenchymal markers, respectively. The multi-step
process of EMT involves numerous regulatory mechanisms, including
activation of the Wnt/β-catenin signaling pathway (17,18).
However, to the best of our knowledge, the association between PLK4
and EMT in CRC has yet to be investigated.
To determine the expression pattern of PLK4 in CRC,
the expression status of PLK4 was profiled in CRC tissues.
Furthermore, its role in colorectal carcinogenesis was explored by
silencing the PLK4 expression in CRC cell lines. Finally, it was
revealed that PLK4 promoted proliferation and invasion of CRC cells
through the Wnt/β-catenin signaling pathway.
Materials and methods
Patients and tissue specimens
CRC tissues were obtained from patients who received
surgical resection at the Tongji Hospital of Huazhong University of
Science and Technology (HUST) (Wuhan, China) between January 2014
and December 2016. All patients were diagnosed by two pathologists.
Briefly, 39 paired freshly frozen CRC and corresponding
noncancerous tissues were immersed in RNAlater overnight at 4°C for
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR), or were lysed using radioimmunoprecipitation assay
(RIPA) buffer (Beyotime Institute of Biotechnology, Shanghai,
China) at 4°C for western blotting. All patients provided written
informed consent prior to study enrollment. This study was approved
by the Ethics Committee of Tongji Hospital, HUST and the study was
conducted according to the principles of the Declaration of
Helsinki.
Online PLK4 expression analysis
The transcript levels of PLK4 in CRC samples were
analyzed using the online UALCAN program: http://ualcan.path.uab.edu (19). Sample data from The Cancer Genome
Atlas (TCGA) database were analyzed using the program. A total of
41 normal tissues and 286 primary tumour tissues were included in
the analysis.
Cell culture
The SW48, SW480, HCT116, SW620, RKO, LoVo and Caco-2
human CRC cell lines were purchased from American Type Culture
Collection (Manassas, VA, USA) and were maintained in our
laboratory. The cell lines were cultured in Dulbecco’s modified
Eagle’s medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) at 37°C in an atmosphere
containing 5% CO2.
Plasmid construction and lentivirus
infection
The pLKO.1-TRC cloning vector (cat. no. 10878; from
Addgene, Inc., Cambridge, MA, USA) was a gift from Professor David
Root (Broad Institute of Massachusetts Institute of Technology and
Harvard, Cambridge, MA, USA). pMD2.G and psPAX2 (cat. nos. 12259
and 12260; Addgene, Inc.) were gifts from Professor Didier Trono
(School of Life Sciences, EPFL, Lausanne, Switzerland). cDNA
encoding full-length human PLK4 was obtained from Addgene, Inc.
(cat. no. 41165) and subcloned into the BamHI/EcoRI
sites of the pCDNA3.1 vector (cat. no. V79020; Invitrogen; Thermo
Fisher Scientific, Inc.), for overexpression by transient
transfection (2 µg, 36 h) into the HCT116 and SW480 cells
(1×106 cells/well in 6-well plates) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Short hairpin (sh)RNAs against human PLK4 were
subcloned into the AgeI/EcoRI sites of the pLKO.1
vector. The sequences were as follows: shRNA#1, TRCN0000002371;
sense 5′-CCGGCGTTGGTTGCTCACAGGTTAACTCGA
GTTAACCTGTGAGCAACCAACGTTTTT-3′; shRNA#2, TRCN0000121260: sense
5′-CCGGGACCTTATTCACCA GTTACTTCTCGAGAAGTAACTGGTGAATAAGGTCTTT TTG-3′;
and control vector (cat. no. SHC005). shRNA and control vectors
were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
For plasmid transfection, 293T cells (China Center for Type Culture
Collection, Wuhan, China) were plated into 6-cm dishes
(2×106 cells) and were cotransfected with pLKO.1-shRNA
plasmids (1 µg) and virus packaging plasmids (pMD2.G, 0.25
µg; psPAX2, 0.75 µg) using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) or Xtreme HP
(Roche Diagnostics, Basel, Switzerland) at 37°C. A total of 8 h
post-transfection, cells were transferred to fresh medium
containing 10% FBS and were incubated for 48-72 h at 37°C.
Collected lentiviral supernatants were filtered through a
0.45-µm filter (Pall Life Sciences, Port Washington, NY,
USA), concentrated with Centricon Plus 70 (Merck KGaA), according
to the manufacturer’s protocol, and used to infect LoVo and SW620
cells (1×106 cells/well in 6-well plates). Lentiviruses
were infected into CRC cells (multiplicity of infection=10) in the
presence of polybrene (8 µg/ml). A total of 48 h
post-infection, cells were selected with growth medium containing 5
µg/ml puromycin for 7 days. Knockdown/overexpression
efficiency was verified by RT-qPCR or western blotting.
RT-qPCR
Total RNA was extracted from CRC cell lines using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer’s protocol. Equal amounts of
RNA were used to generate first strand cDNA using the
PrimeScript® RT reagent kit (Takara Bio, Inc., Otsu,
Japan), according to the manufacturer’s protocol. qPCR
amplification was conducted using the CFX96 Touch™ Real-Time PCR
Detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and the SYBR-Green PCR Master Mix-PLUS (Toyobo Life Science, Osaka,
Japan), according to the manufacturers’ protocols. The qPCR
experiment was performed in a volume (10 µl) comprising 1
µl cDNA, 0.6 µl primers, 3.4 µl
ddH2O and 5 µl SYBR. The cycling parameters were
as follows: Initial denaturation at 95°C for 3 min, followed by 35
cycles at 95°C for 10 sec, 55-60°C for 20 sec and 72°C for 15 sec,
and a final extension step at 72°C for 5 min. The primer sequences
were as follows: PLK4, forward 5′-AGACCACCCTTCGACACTGA-3′, reverse
5′-GTCCTTGGCCTCTATTGACAAA-3′; c-Myc, forward
5′-GGCTCCTGGCAAAAGGTCA-3′, reverse 5′-CTGCGTAGTTGTGCTGATGT-3′;
cyclin D1, forward 5′-GCTGCGAAGTGGAAACCATC-3′, reverse
5′-CCTCCTTCTGCACACATTTGAA-3′; p21, forward
5′-TGTCCGTCAGAACCCATGC-3′, reverse 5′-AAAGTCGAAGTTCCATCGCTC-3′; and
GADPH, forward 5′-GACAAGCTTCCCGTTCTCAG-3′ and reverse
5′-GAGTCAACGGATTTGGTCGT-3′. Subsequently, the relative expression
levels of each gene were analyzed using the 2−ΔΔCq
method (20). The experiments were
conducted in triplicate. All primers were synthesized and purchased
from Tsingke Biological Technology (Beijing, China).
Cell proliferation assay
To determine the effects of PLK4 on the viability of
CRC cells, 1,000 cells/well were cultured in 96-well plates; each
well contained 200 µl medium. Cell viability was detected
using a Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan), according to the
manufacturer’s protocol. After 24, 48, 72 and 96 h, 10 µl
CCK-8 assay reagent was added to each well mixed with 90 µl
serum-free medium. The absorbance was measured 2 h later using a
microplate reader at a test wavelength of 450 nm.
Colony formation assay
CRC cells (500/well) were cultured in 6-well plates
to investigate the effects of PLK4 on the efficiency of colony
formation. After 10 days, each well was washed with PBS three times
at room temperature. The cells were then fixed in each well using
anhydrous ethanol (99.5%) for 10 min and stained for 20 min using
crystal violet dye (0.1% in PBS) at room temperature. After washing
with PBS, colonies (≥50 cells/colony) in each well were manually
counted using a ChemiDoc™ Imaging system (Bio-Rad Laboratories,
Inc.). The experiments were repeated three times.
Wound scratch assay
A confluent monolayer of CRC cells (confluence, 95%)
was cultured overnight and a scratch was introduced using a pipette
tip. Cell migration was recorded under a phase contrast microscope
(Nikon Digital Eclipse C1 system; magnification, ×40; Nikon
Corporation, Tokyo, Japan) with white light at 0 and 24 h after
scratch generation. Images of five random fields across three
replicate wells were captured for semi-quantification using
Image-Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MD,
USA).
Transwell cell migration and invasion
assays
Cell migration assays were performed using a 24-well
Transwell plate (pore size, 8 µm; Corning Incorporation,
Corning, NY, USA), according to the manufacturer’s protocol. For
the Matrigel invasion assay, filters were precoated with 50
µl 1:4 mixture of Matrigel (BD Biosciences, Franklin Lakes,
NJ, USA) and DMEM for 4 h at room temperature. Briefly, for
invasion and migration assays, culture medium containing 10% FBS
was added to the lower chambers and aliquots of 5×104
cells in 100 µl serum-free medium were seeded into the upper
chambers. After a 24 h incubation at 37°C, non-migrated or
non-invaded cells were removed by scraping the upper surface of the
membranes with a cotton swab. Cells on the lower surface of the
membranes were fixed with 4% paraformaldehyde at room temperature
for 15 min and stained with 0.1% crystal violet at room temperature
for 20 min. Cell numbers were counted under an optical microscope.
Each experiment was repeated at least three times.
Immunohistochemistry
Surgically excised tumor specimens and xenograft
tumour were fixed in 4% neutral formalin at room temperature for 24
h, embedded in paraffin and cut into 4 µm sections. The
sections were then deparaffinized in xylene, rehydrated in a graded
alcohol series and treated with boiling 0.01 mol/l citrate buffer
15 min for antigen retrieval. Endogenous peroxidase activity was
blocked with hydrogen peroxide (0.3%) at room temperature for 15
min, and the sections were incubated with 5% bovine serum albumin
(R&D Systems, Minneapolis, MN, USA) at 37°C for 45 min to
reduce non-specific binding. Immunostaining with PLK4 rabbit
polyclonal antibodies (Table I)
was carried out at 4°C for 16 h, followed by incubation with a
horseradish peroxidase (HRP)-conjugated secondary antibody from the
Envision kit (Dako; Agilent Technologies, Inc., Santa Clara, CA,
USA; Table I) for 45 min at room
temperature. Antibody binding was detected by DAB (Dako; Agilent
Technologies, Inc.), according to manufacturer’s protocol, at room
temperature for 1 min and the reaction was terminated by immersion
of tissue sections in distilled water once brown staining appeared.
Tissue sections were counterstained with 1% hematoxylin at room
temperature for 3 min and dehydrated in a graded series of ethanol.
The xenograft tumour tissues were also immunostained with PLK4,
Ki-67 and β-catenin primary antibodies (Table I) using the same protocol.
Immunohistochemical scores were obtained by multiplying the
percentage score to the intensity score of positively stained
cells; images of representative fields were obtained from Nikon
Digital ECLIPSE C1 microscope (Nikon Corporation) (21). Analysis was independently performed
by two certified pathologists, which were blinded to the clinical
and demographic characteristics of the patients. The expression
status represents the average of the independent readings. An
overall score of >6 and ≤6 was defined as high and low
expression, respectively.
 | Table IAntibodies used in the present
study. |
Table I
Antibodies used in the present
study.
| Antibody/kit
name | Cat. no. and
manufacturer | Application |
|---|
| PLK4 | 12952-1-AP;
Proteintech Group, Inc., Chicago, IL, USA | 1:100 for IHC;
1:1,000 for WB |
| β-catenin | #8480; Cell
Signaling Technology, Inc., Danvers, MA, USA | 1:100 for IHC;
1:1,000 for WB |
| Ki-67 | ab15580; Abcam,
Cambridge, UK | 1:100 for IHC |
| pGSK3β | #5558; Cell
Signaling Technology, Inc. | 1:1,000 for WB |
| GSK3β | #12456; Cell
Signaling Technology, Inc. | 1:1,000 for WB |
| GAPDH | KC-5G4; KangChen
BioTech Co., Ltd., Shanghai, China | 1:10,000 for
WB |
| p21 | #2947; Cell
Signaling Technology, Inc. | 1:1,000 for WB |
| Cyclin D1 | #2978; Cell
Signaling Technology, Inc. | 1:1,000 for WB |
| c-Myc | #13987; Cell
Signaling Technology, Inc. | 1:2,000 for WB |
| MMP1 | ab137332; Abcam,
Cambridge, MA, USA | 1:1,000 for WB |
| MMP2 | ab37150; Abcam | 1:1,000 for WB |
| E-cadherin | ab76055; Abcam | 1:1,000 for WB |
| N-cadherin | ab98952; Abcam | 1:1,000 for WB |
| Occludin | 13409-1-AP;
Proteintech Group, Inc. | 1:1,000 for WB |
| Snail | ab82846; Abcam | 1:1,000 for WB |
| HRP-conjugated
anti-rabbit IgG | 111-035-003;
Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA | 1:5,000 for WB |
| HRP-conjugated
anti-mouse IgG | 115-035-003;
Jackson ImmunoResearch Laboratories, Inc. | 1:5,000 for WB |
| Secondary antibody
in the Envision kit (HRP, rabbit/mouse, DAB+) | Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA | Ready-to-use for
IHC |
Western blotting
Cells were lysed with RIPA buffer (Beyotime
Institute of Biotechnology) containing a protease inhibitor mixture
on ice for 30 min. Cell lysates were then quantified using the
bicinchoninic acid protein assay prior to separation (20
µg/lane) by 10% SDS-PAGE and transfer to polyvinylidene
fluoride membranes (Roche Diagnostics). The membranes were blocked
with 5% non-fat milk at 37°C for 1 h and were incubated with
primary antibodies (Table I) at
4°C overnight. Subsequently, the membranes were incubated with
HRP-conjugated goat anti-rabbit or goat anti-mouse immunoglobulin G
secondary antibodies (Table I) for
at 37°C for 1 h. Finally, the enhanced chemiluminescence detection
system (Bio-Rad Laboratories, Inc.) was used for visualization.
Image Lab™ 3.0 software (Bio-Rad Laboratories, Inc.) was used to
semi-quantify blots.
Animal study
A total of 15 male BALB/c nude mice (age, 4 weeks;
weight, 18-19 g) were purchased from the Animal Center of East
China Normal University (Shanghai, China), after signing the
animal-raising agreement. The mice were maintained under the
following conditions: Room temperature, 20-26°C; humidity, 40-60%;
12-h light/dark cycle; free access to food and water. Mice were
randomly divided into three groups (n=5/group). After anesthesia,
mice in the control group were injected with 2×106 LoVo
cells transduced with the empty vector in 100 µl DMEM,
whereas mice in the treatment groups were injected with
2×106 LoVo cells transduced with PLK4 knockdown viruses
in 100 µl DMEM. Tumor length (L) and width (W) were manually
monitored using a Vernier caliper twice a week, and tumor volume
(V) was calculated according to the following equation: V
(mm3) = 0.5 × L (mm) × W2 (mm2). A
total of 4 weeks post-injection and after the last tumor volume was
measured all mice were sacrificed. For sacrifice, the mice were
placed into a sealed container and the concentration of
CO2 in the container was increased gradually (flow rate,
20% of the chamber volume/min). Subsequently, tumor tissues were
removed and tumor weight was measured; in the present study, each
mouse bore a single tumor, the largest weight of which was 0.35 g,
and the percentage of all tumors to the body weight of the animals
was <1.5%. All of the animal experiments met the National
Institutes of Health (NIH) guidelines (NIH publication no. 86-23,
revised 1985) (22) and the animal
studies were approved by the Committee on the Ethics of Animal
Experiments of the Tongji Medical College, HUST.
Statistical analysis
All experiments were repeated at least twice with
consistent results. If the variance was homogenous, the difference
between two groups was analyzed using Student’s t-test, and the
difference between more than two groups was analyzed by one-way
analysis of variance. If the variance was not homogeneous, the
difference between two groups was analyzed using the Mann-Whitney U
test, and the difference between more than two groups was analyzed
by Kruskal-Wallis H test. The least-significant difference post hoc
test was used to compare datasets containing multiple groups.
Categorical data in Table II were
analyzed by χ2 test or Fisher’s exact test. All
statistical analyses were performed using SPSS Statistics version
20 (IBM Corp., Armonk, NY, USA) and GraphPad Prism 6 (GraphPad
Software, Inc., San Diego, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
| Table IIAssociation between PLK4 levels in
CRC tissues and clinicopathological features (n=39). |
Table II
Association between PLK4 levels in
CRC tissues and clinicopathological features (n=39).
| Clinical
characteristic | n (%) | PLK4 expression
| P-value |
|---|
| Low (%) | High (%) |
|---|
| Overall | 39 | 14 (35.9) | 25 (64.1) | |
| Age | | | | |
| <65 years | 27 (69.2) | 11 (28.2) | 16 (41.0) | 0.283 |
| ≥65 years | 12 (30.8) | 3 (7.7) | 9 (23.1) | |
| Sex | | | | |
| Male | 18 (46.2) | 5 (12.8) | 13 (33.3) | 0.261 |
| Female | 21 (53.9) | 9 (23.1) | 12 (30.8) | |
| Tumor size | | | | |
| ≤5 cm | 16 (41.0) | 9 (23.1) | 7 (17.9) | 0.031 |
| >5 cm | 23 (59.0) | 5 (12.8) | 18 (46.2) | |
| Lymph node
metastasis | | | | |
| Absent | 15 (38.5) | 9 (23.1) | 6 (15.4) | 0.016 |
| Present | 24 (61.5) | 5 (12.8) | 19 (48.7) | |
| Location | | | | |
| Rectum | 19 (48.7) | 8 (20.5) | 11 (28.2) | 0.325 |
| Colon | 20 (51.3) | 6 (15.4) | 14 (35.9) | |
| TNM stage
(AJCC) | | | | |
| Stage I-II | 16 (41.0) | 11 (28.2) | 5 (12.8) | 0.001 |
| Stage III | 23 (59.0) | 3 (7.7) | 20 (51.3) | |
Results
PLK4 expression is significantly
upregulated in CRC tissues and is associated with
clinicopathological features
To evaluate the expression status of PLK4 in human
CRC tissues, UALCAN (http://ualcan.path.uab.edu/) was used to analyze the
microarray dataset from TCGA; the results revealed that the mRNA
expression levels of PLK4 were upregulated in the majority of tumor
tissues compared with in adjacent normal tissues (Fig. 1A). Subsequently, 39 paired CRC and
adjacent normal tissues were analyzed by western blotting. The
results demonstrated that the protein expression levels of PLK4
were higher in the majority of CRC tissues compared with in paired
noncancerous colorectal tissues (Fig.
1B and C). Furthermore, immunohistochemistry was used to assess
the protein expression levels of PLK4 in 39 paraffin-embedded CRC
specimens; 64.1% (25/39) of CRC samples exhibited high PLK4
expression, whereas the remaining 35.9% (14/39) of samples
exhibited low PLK4 expression (Fig. 1D
and E). The association between PLK4 protein expression and
clinicopathological parameters was then analyzed. Upregulation of
PLK4 was significantly associated with tumor size (≤5 cm vs. >5
cm; P=0.031), lymph node metastasis (absent vs. present; P=0.016)
and TNM stage (I-II vs. III-IV; P=0.001). However, no significant
association was observed between PLK4 expression and the other
clinicopathological factors, including sex and age (Table II).
Expression of PLK4 is upregulated in CRC
cells, and can be regulated by shRNA and plasmids
To detect the expression levels of PLK4 in CRC
cells, seven CRC cell lines (Caco2, SW480, SW48, HCT116, SW620,
LoVo and RKO) were analyzed by RT-qPCR and western blot analysis;
SW620 and LoVo exhibited high endogenous PLK4 expression, whereas
HCT116 and SW480 exhibited low endogenous PLK4 expression (Fig. 2A). Subsequently, PLK4 was knocked
down in SW620 and LoVo cells using shRNA-PLK4 and was overexpressed
in HCT116 and SW480 cells using plasmids. Knockdown/overexpression
efficiency was confirmed by RT-qPCR and western blot analysis
(Fig. 2B and C).
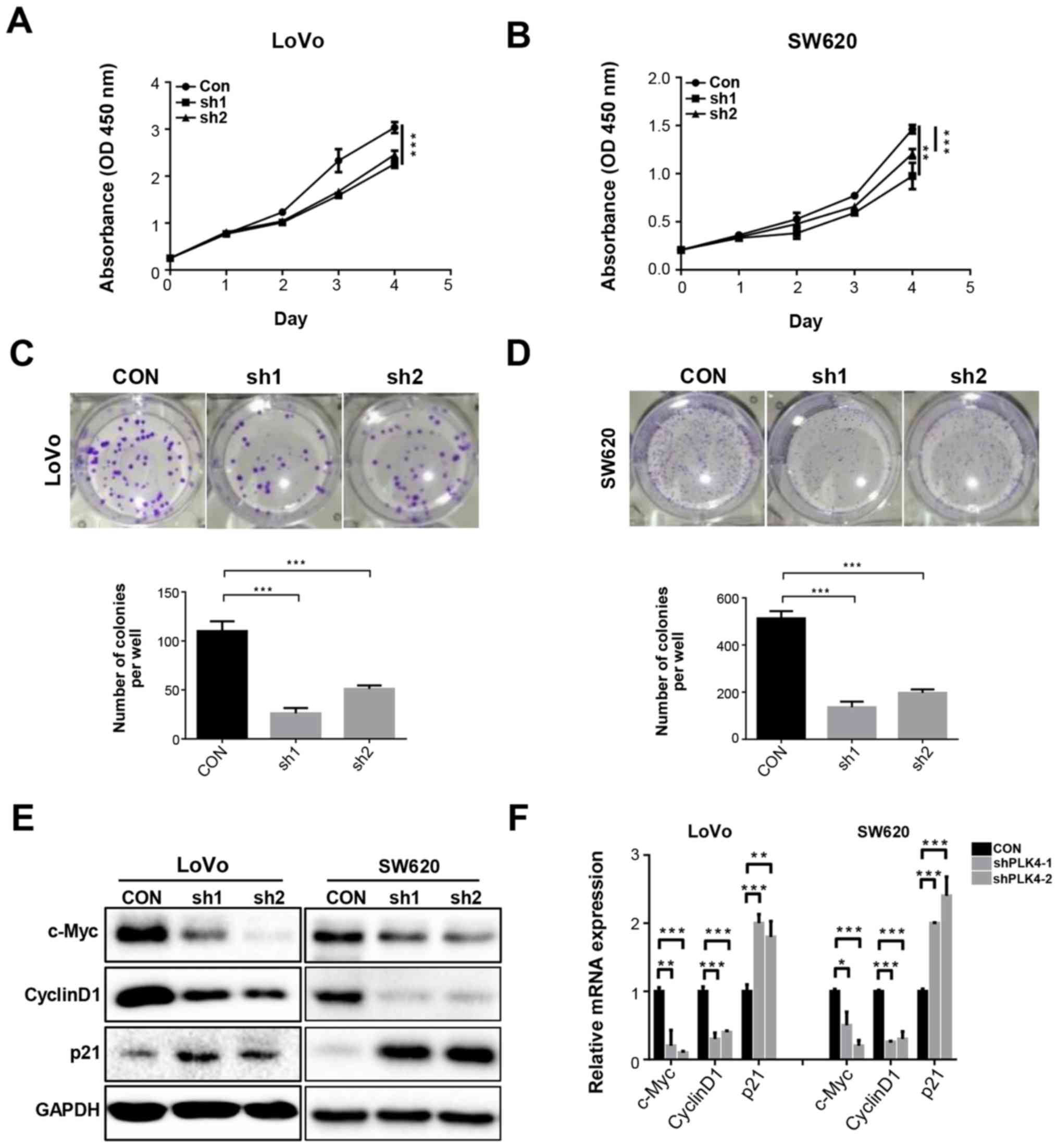
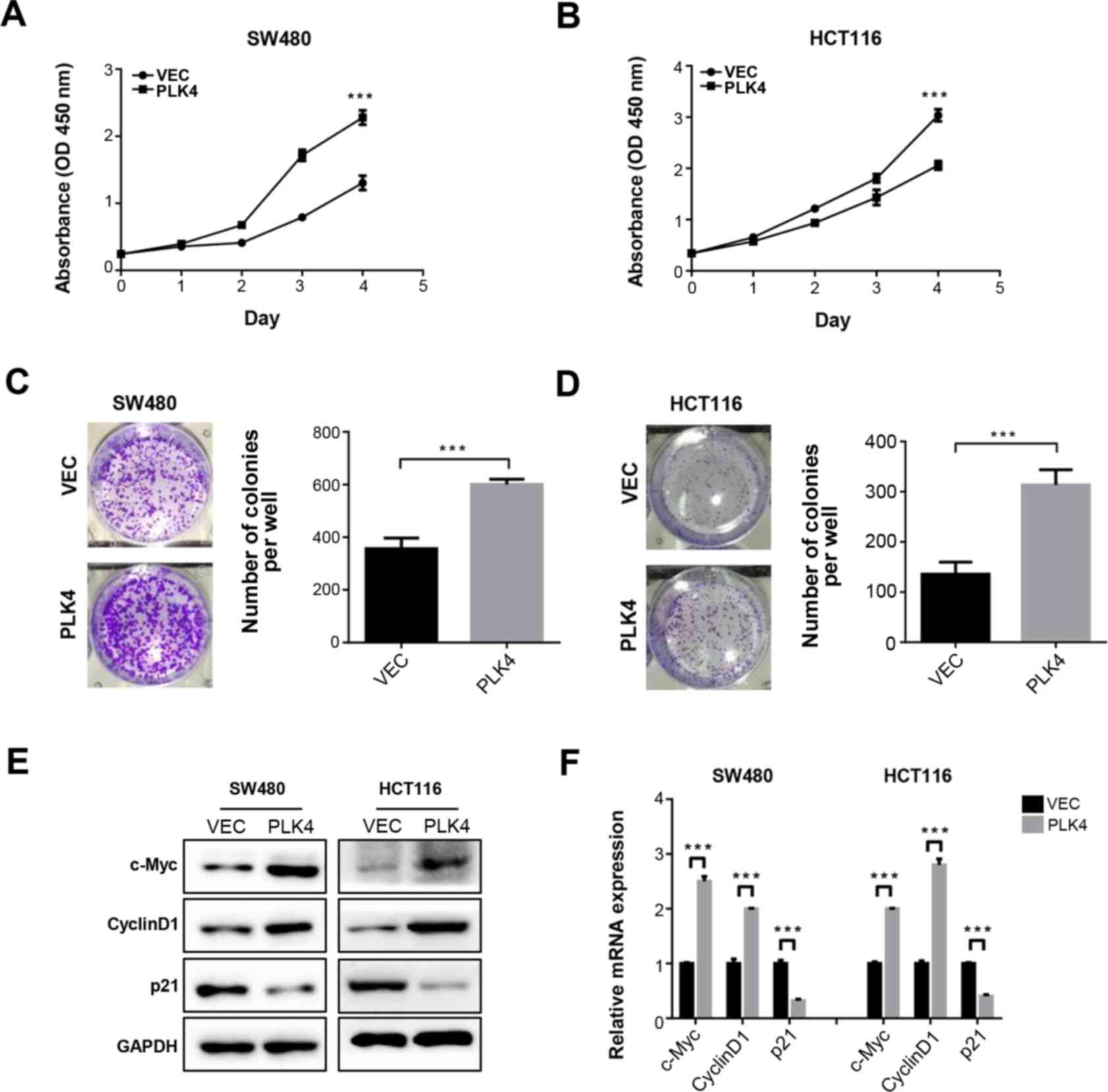
PLK4 significantly increases CRC cell
proliferation and colony formation in vitro
CCK-8 and colony formation assays were conducted to
determine the effects of PLK4 on CRC cell proliferation (Figs. 3 and 4). It was revealed that knockdown of PLK4
significantly inhibited the proliferation of SW620 and LoVo cells
(Fig. 3A and B). Conversely,
proliferation was promoted in SW480 and HCT116 cells overexpressing
PLK4 (Fig. 4A and B).
Consistently, the results of the colony formation assay
demonstrated that PLK4 shRNA-infected SW620 and LoVo cells
exhibited reduced colony formation (Fig. 3C and D), whereas
PLK4-overexpressing plasmid-transfected SW480 and HCT116 cells
exhibited opposite effects (Fig. 4C
and D). Analysis of the mRNA and proteins involved in cell
proliferation and cell cycle progression indicated that cyclin D1
and c-Myc expression was downregulated, whereas p21 expression was
upregulated by PLK4 knockdown (Fig. 3E
and F), whereas PLK4 overexpression had the opposite effects
(Fig. 4E and F).
Regulation of PLK4 affects the migration
and invasion of CRC cells in vitro
Wound-healing and Transwell assays were conducted to
detect migration and invasion of LoVo and SW480 cells. The results
of the wound-healing assay demonstrated that the wound area was
wider in LoVo cells in which PLK4 was knocked down (Fig. 5A). Conversely, the wound area was
narrower in SW480 cells transfected with the PLK4-overexpressing
plasmid (Fig. 5B). The results of
the Transwell assay revealed that the number of cells that
traversed the membrane and Matrigel was significantly reduced by
PLK4 knockdown in LoVo and SW620 cells (Fig. 5C and D), whereas overexpression of
PLK4 in SW480 cells increased the number of cells that traversed
the membrane and Matrigel (Fig.
5E). These findings indicated that PLK4 may promote invasion
and metastasis of CRC cells.
 | Figure 5PLK4 promotes migration and invasion
of colorectal cancer cells. (A) Effects of PLK4 knockdown on LoVo
cell migration, as evaluated by wound scratch assay. (B) Effects of
PLK4 overexpression on SW480 cell migration, as evaluated by wound
scratch assay. (C) Effects of PLK4 knockdown on LoVo cell migration
and invasion, as evaluated by Transwell assay. (D) Effects of PLK4
knockdown on SW620 cell migration and invasion, as evaluated by
Transwell assay. (E) Effects of PLK4 overexpression on SW480 cell
migration and invasion, as evaluated by Transwell assay. Results
are presented as the means ± standard error of the mean of
triplicate repeats from three independent experiments.
*P<0.05, **P<0.01,
***P<0.001. CON, control; CRC, colorectal cancer;
PLK4, polo-like kinase 4; sh/shRNA, short hairpin RNA; VEC, empty
vector. |
PLK4 facilitates EMT and activates the
Wnt/β-catenin pathway in CRC cells
To determine whether PLK4 was associated with EMT in
CRC cells, the present study examined the expression levels of
EMT-associated proteins, including MMP1, MMP2, N-cadherin, occludin
and snail, by western blotting. The results revealed that knockdown
of PLK4 significantly increased the expression of occludin, and
decreased the expression of MMP1, MMP2, N-cadherin and snail
(Fig. 6). These results suggested
that PLK4 may induce EMT in CRC cells. In addition, the expression
levels of pGSK3β, GSK3β and β-catenin were detected in LoVo, SW620
and SW480 cells. It was revealed that the protein expression levels
of pGSK3β and β-catenin were increased when PLK4 was overexpressed,
but were decreased when PLK4 was knocked down. However, no
significant differences were detected in GSK3β expression among the
groups. These findings suggested that PLK4 promoted activation of
the Wnt/β-catenin pathway in CRC (Fig.
6).
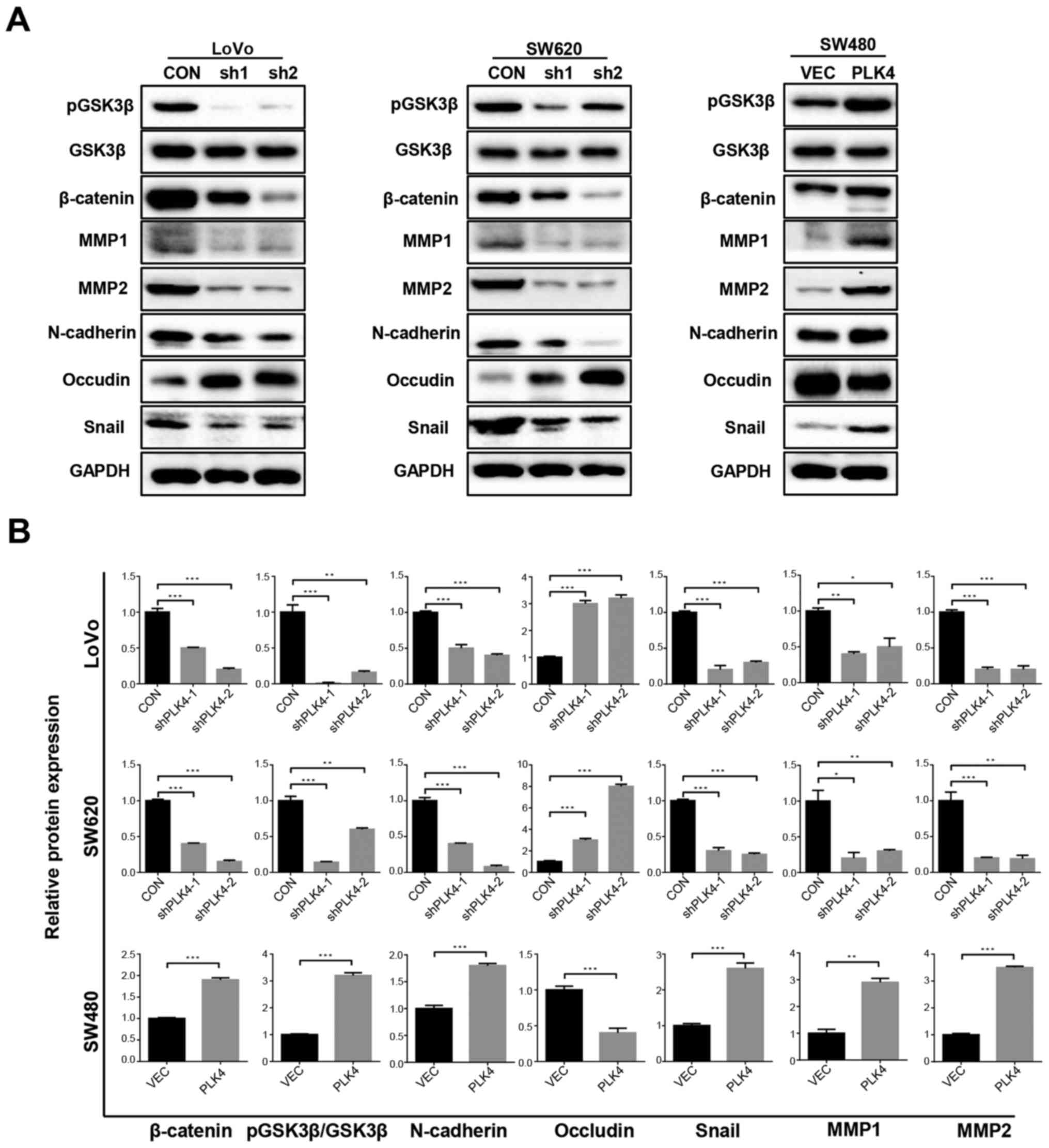 | Figure 6PLK4 promotes activation of the
Wnt/β-catenin signaling pathway and epithelial-mesenchymal
transition in colorectal cancer cells. (A) Expression levels of
pGSK3β, β-catenin, MMP1, MMP2, N-cadherin, occludin and snail in
LoVo, SW620 and SW480 cells following PLK4 knockdown and
overexpression were detected using western blot analysis. (B)
Relative protein expression levels of β-catenin, N-cadherin,
occludin, snail, MMP1 and MMP2 were semi-quantified in LoVo, SW620
and SW480 cells. Data are presented as the means ± standard
deviation from three independent experiments.
**P<0.01 and ***P<0.001. CON, control;
GSK3β, glycogen synthase kinase 3β; MMP, matrix metalloproteinase;
p, phosphorylated; PLK4, polo-like kinase 4; sh/shRNA, short
hairpin RNA; VEC, empty vector. |
Downregulation of PLK4 significantly
suppresses CRC tumorigenesis in vivo
To verify the effects of PLK4 expression on
tumorigenesis in vivo, mice were transplanted with LoVo
cells transduced with a PLK4 knockdown virus or empty vector. Tumor
growth was markedly decreased in the shPLK4 groups compared with in
the vector group (Fig. 7A). A
total of 4 weeks post-inoculation, tumor volume (Fig. 7B) and weight (Fig. 7C) were significantly smaller in the
shPLK4 groups compared with in the vector group. Furthermore,
Ki67-positive signals in the xenograft tumors were decreased in the
shPLK4 groups (Fig. 7D). To
further detect whether PLK4 was associated with EMT, the expression
levels of EMT-associated proteins were detected in tumor tissues
using western blot analysis. Following PLK4 knockdown, it was
revealed that the expression levels of the epithelial markers
E-cadherin and occludin were increased, whereas the expression
levels of the mesenchymal markers N-cadherin and snail were
decreased (Fig. 7F and G). In
addition, the expression levels of pGSK3β, GSK3β and β-catenin were
detected. The results demonstrated that lower levels of pGSK3β and
β-catenin were detected in the shPLK4 group (Fig. 7E and G). Taken together, these
results indicated that PLK4 may serve a crucial role in CRC
progression and may activate the Wnt/β-catenin pathway in
vivo.
 | Figure 7Knockdown of PLK4 inhibits tumor
growth in vivo. (A) Representative images of isolated tumors
in the control group (first row), shPLK4-1 group (second row) and
shPLK4-2 group (third row). (B) Xenograft tumor volume was smaller
in the shPLK4 groups compared with in the control group. (C)
Xenograft tumor weight was reduced in the shPLK4 groups compared
with in the control group. (D) Ki67-positive cells in subcutaneous
xenograft tumor tissues developed from CRC cells. (E and F)
Relative protein expression levels of pGSK3β, β-catenin, c-Myc,
cyclin D1, p21 and epithelial-mesenchymal transition markers were
examined in CRC xenograft tissues. (G) Relative protein expression
levels of PLK4, β-catenin, pGSK3β, c-Myc, cyclin D1, p21, MMP1,
MMP2, E-cadherin, N-cadherin, occludin and snail were
semi-quantified. Results are presented as the means ± standard
error of the mean of triplicate repeats from three independent
experiments. *P<0.05, **P<0.01,
***P<0.001. CRC, colorectal cancer; GSK3β, glycogen
synthase kinase 3β; MMP, matrix metallopro-teinase; p,
phosphorylated; PLK4, polo-like kinase 4; sh/shRNA, short hairpin
RNA; VEC, empty vector. |
Discussion
The present study indicated that PLK4 was highly
expressed in CRC tissues. Stepwise investigation demonstrated that
PLK4 activated the Wnt/β-catenin pathway to promote cell
proliferation and invasion in CRC. These findings suggested that
PLK4 may be a novel biomarker for patients with CRC.
Abnormal PLK4 expression may regulate centroid
replication, abnormal mitosis, centrosome amplification (CA) and
chromosomal instability (CIN) (6,23,24),
which are common causes of cancer development (23,25,26),
thus we speculated that PLK4 may be involved in CRC development.
Furthermore, it has been reported that PLK4 is overexpressed in
various types of tumor (13-15)
and is closely associated with the prognosis of patients with
cancer. Furthermore, it has been confirmed that the mRNA expression
levels of PLK4 are associated with breast cancer aggression and
resistance to traditional therapy (14). Furthermore, Kazazian et al
indicated that PLK4 enhances migration and invasion of HeLa and
U2OS cells (11). PLK4 also
facilitates aggressiveness of neuroblastoma, and is correlated with
adverse clinical features and poor survival (13). These studies indicated that PLK4
may be an oncogenic factor in these cancer types, and may promote
tumor development and progression. Conversely, knockdown of PLK4
inhibits cell apoptosis, and low PLK4 expression is associated with
poor prognosis in hepatocellular carcinoma (HCC) (27). These findings suggested that PLK4
may be a tumor suppressor in HCC; these discrepancies in findings
suggested that PLK4 may exert tumour promoter and suppressor
functions according to cancer type.
Consistent with the findings of previous studies,
PLK4 expression was upregulated in human CRC tissues in the present
study. Furthermore, PLK4 promoted the proliferation and invasion of
CRC cells. PLK4 also affected the expression of several
EMT-associated proteins; N-cadherin and snail were upregulated, and
occludin was downregulated in cells overexpressing PLK4, thus
suggesting a novel mechanism underlying the role of PLK4 in cancer.
The present study investigated the function of PLK4 and suggested
that it may act as an oncogene in CRC. Previous studies revealed
that PLK4 manipulates numerous signaling pathways, including the
transforming growth factor β and epidermal growth factor receptor
signaling pathways. In addition, the Wnt/β-catenin pathway is a
critical mediator of carcinogenic signals in various tumor types
(17,28-30).
The Wnt/β-catenin signaling pathway induces the EMT process and
inhibits the transcription of E-cadherin, which is commonly
considered an activator of cancer progression (31-33).
β-catenin directly influences the biological characteristics of
cells, and has been reported to downregulate E-cadherin expression,
and to promote EMT-like transition and invasiveness in carcinoma
cells (34). In addition,
activation of the Wnt/β-catenin pathway contributes to EMT through
upregulation of EMT-associated factors, including snail and slug
(34-36). In the present study, the expression
levels of β-catenin were significantly increased in response to
PLK4 overexpression in CRC cells. These findings indicated that
PLK4 may mediate EMT in CRC cells via its effects on the
Wnt/β-catenin signaling pathway, ultimately improving the migratory
and invasive potential of the cells.
In conclusion, the present study demonstrated that
PLK4 promoted the proliferative and invasive phenotype of CRC cells
via the Wnt/β-catenin signaling pathway. These findings suggested
that PLK4 may serve as a biomarker or a target for treatment of
patients with CRC.
Funding
The present study was supported by the State Key
Project on Infectious Diseases of China (grant no.
2018ZX10723204-003); the National Natural Science Foundation of
China (grant no. 81572855 to XC, grant no. 81572427 to BZ, grant
no. 81502530 to ZZ and grant no. 81400653 to LC); and the
Graduates’ Innovation Fund, Huazhong University of Science and
Technology (grant no. 5003540055 to ZL).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors’ contributions
ZL, ZZ and BZ designed the experiments. ZL performed
experiments and generated the data. HZ and KD collected
clinicopathological data. PF, QH, YQ, JS, LC, HL and XC analyzed
the results. HZ prepared the panels for Fig. 1, and KD prepared the panels for
Figs. 2-7; KD also assembled the tables. ZZ and BZ
wrote the manuscript. All authors reviewed the final version of the
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Tongji Hospital, HUST. Written informed consent was
obtained from each patient. Animal studies were approved by the
Committee on the Ethics of Animal Experiments of the Tongji Medical
College, HUST.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Changshu Ke and
Dr Jing Xiong (Department of Pathology, Tongji Hospital) for
assistance in immunohistochemistry scoring; Miss Lanping Ding
(Institute of Organ Transplantation, Tongji Hospital) and Mr.
Shunchang Zhou (Department of Experimental Zoology, Tongji Medical
College) for animal care; and Dr Xiaolong Tan (Department of
Geriatrics, Tongji Hospital) for kindly providing technical
assistance in the animal experiment.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar
|
|
3
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
4
|
Tan Z, Liu X, Yu E, Wang H, Tang L, Wang H
and Fu C: Lentivirus-mediated RNA interference of tripartite motif
68 inhibits the proliferation of colorectal cancer cell lines
SW1116 and HCT11 in vitro. Oncol Lett. 13:2649–2655. 2017.
View Article : Google Scholar :
|
|
5
|
Ribero D, Viganò L, Amisano M and
Capussotti L: Prognostic factors after resection of colorectal
liver metastases: From morphology to biology. Future Oncol.
9:45–57. 2013. View Article : Google Scholar
|
|
6
|
Nakamura T, Saito H and Takekawa M: SAPK
pathways and p53 cooperatively regulate PLK4 activity and
centrosome integrity under stress. Nat Commun. 4:17752013.
View Article : Google Scholar
|
|
7
|
Xu X, Huang S, Zhang B, Huang F, Chi W, Fu
J, Wang G, Li S, Jiang Q and Zhang C: DNA replication licensing
factor Cdc6 and Plk4 kinase antagonistically regulate centrosome
duplication via Sas-6. Nat Commun. 8:151642017. View Article : Google Scholar :
|
|
8
|
Rosario CO, Ko MA, Haffani YZ, Gladdy RA,
Paderova J, Pollett A, Squire JA, Dennis JW and Swallow CJ: Plk4 is
required for cytokinesis and maintenance of chromosomal stability.
Proc Natl Acad Sci USA. 107:6888–6893. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fischer M, Quaas M, Wintsche A, Müller GA
and Engeland K: Polo-like kinase 4 transcription is activated via
CRE and NRF1 elements, repressed by DREAM through CDE/CHR sites and
deregulated by HPV E7 protein. Nucleic Acids Res. 42:163–180. 2014.
View Article : Google Scholar
|
|
10
|
Rosario CO, Kazazian K, Zih FS,
Brashavitskaya O, Haffani Y, Xu RS, George A, Dennis JW and Swallow
CJ: A novel role for Plk4 in regulating cell spreading and
motility. Oncogene. 34:3441–3451. 2015. View Article : Google Scholar
|
|
11
|
Kazazian K, Go C, Wu H, Brashavitskaya O,
Xu R, Dennis JW, Gingras AC and Swallow CJ: Plk4 Promotes Cancer
Invasion and Metastasis through Arp2/3 Complex Regulation of the
Actin Cytoskeleton. Cancer Res. 77:434–447. 2017. View Article : Google Scholar
|
|
12
|
Kawakami M, Mustachio LM, Zheng L, Chen Y,
Rodriguez-Canales J, Mino B, Kurie JM, Roszik J, Villalobos PA, Thu
KL, et al: Polo-like kinase 4 inhibition produces polyploidy and
apoptotic death of lung cancers. Proc Natl Acad Sci USA.
115:1913–1918. 2018. View Article : Google Scholar
|
|
13
|
Tian X, Zhou D, Chen L, Tian Y, Zhong B,
Cao Y, Dong Q, Zhou M, Yan J, Wang Y, et al: Polo-like kinase 4
mediates epithelial-mesenchymal transition in neuroblastoma via
PI3K/Akt signaling pathway. Cell Death Dis. 9:542018. View Article : Google Scholar
|
|
14
|
Li Z, Dai K, Wang C, Song Y, Gu F, Liu F
and Fu L: Expression of Polo-Like Kinase 4(PLK4) in Breast Cancer
and Its Response to Taxane-Based Neoadjuvant Chemotherapy. J
Cancer. 7:1125–1132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Korzeniewski N, Hohenfellner M and
Duensing S: CAND1 promotes PLK4-mediated centriole overduplication
and is frequently disrupted in prostate cancer. Neoplasia.
14:799–806. 2012. View Article : Google Scholar
|
|
16
|
Li L and Li W: Epithelial-mesenchymal
transition in human cancer: Comprehensive reprogramming of
metabolism, epigenetics, and differentiation. Pharmacol Ther.
150:33–46. 2015. View Article : Google Scholar
|
|
17
|
Qi J, Yu Y, Akilli Öztürk Ö, Holland JD,
Besser D, Fritzmann J, Wulf-Goldenberg A, Eckert K, Fichtner I and
Birchmeier W: New Wnt/β-catenin target genes promote experimental
metastasis and migration of colorectal cancer cells through
different signals. Gut. 65:1690–1701. 2016. View Article : Google Scholar
|
|
18
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar
|
|
19
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Ding ZY, Jin GN, Wang W, Chen WX, Wu YH,
Ai X, Chen L, Zhang WG, Liang HF, Laurence A, et al: Reduced
expression of transcriptional intermediary factor 1 gamma promotes
metastasis and indicates poor prognosis of hepatocellular
carcinoma. Hepatology. 60:1620–1636. 2014. View Article : Google Scholar
|
|
22
|
Guide for the Care and Use of Laboratory
Animals. NIH Publication No. pp. 85–23. National Academies Press
(US); Washington, DC: 1985
|
|
23
|
Godinho SA, Picone R, Burute M, Dagher R,
Su Y, Leung CT, Polyak K, Brugge JS, Théry M and Pellman D:
Oncogene-like induction of cellular invasion from centrosome
amplification. Nature. 510:167–171. 2014. View Article : Google Scholar
|
|
24
|
Coelho PA, Bury L, Shahbazi MN,
Liakath-Ali K, Tate PH, Wormald S, Hindley CJ, Huch M, Archer J,
Skarnes WC, et al: Overexpression of Plk4 induces centrosome
amplification, loss of primary cilia and associated tissue
hyperplasia in the mouse. Open Biol. 5:1502092015. View Article : Google Scholar
|
|
25
|
Bakhoum SF, Ngo B, Laughney AM, Cavallo
JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et
al: Chromosomal instability drives metastasis through a cytosolic
DNA response. Nature. 553:467–472. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gulluni F, Martini M, De Santis MC, Campa
CC, Ghigo A, Margaria JP, Ciraolo E, Franco I, Ala U, Annaratone L,
et al: Mitotic Spindle Assembly and Genomic Stability in Breast
Cancer Require PI3K-C2 α Scaffolding Function. Cancer Cell.
32:444–459.e7. 2017. View Article : Google Scholar
|
|
27
|
Pellegrino R, Calvisi DF, Ladu S, Ehemann
V, Staniscia T, Evert M, Dombrowski F, Schirmacher P and Longerich
T: Oncogenic and tumor suppressive roles of polo-like kinases in
human hepatocellular carcinoma. Hepatology. 51:857–868. 2010.
|
|
28
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar
|
|
29
|
Nusse R and Clevers H: Wnt/β-Catenin
Signaling, Disease, and Emerging Therapeutic Modalities. Cell.
169:985–999. 2017. View Article : Google Scholar
|
|
30
|
Ferrarelli LK: Treating WNT-driven
colorectal cancer. Science. 356:1346–1348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar
|
|
32
|
Singh M, Yelle N, Venugopal C and Singh
SK: EMT: Mechanisms and therapeutic implications. Pharmacol Ther.
182:80–94. 2018. View Article : Google Scholar
|
|
33
|
Sun Y, Ji B, Feng Y, Zhang Y, Ji D, Zhu C,
Wang S, Zhang C, Zhang D and Sun Y: TRIM59 facilitates the
proliferation of colorectal cancer and promotes metastasis via the
PI3K/AKT pathway. Oncol Rep. 38:43–52. 2017. View Article : Google Scholar
|
|
34
|
Brabletz T, Jung A, Reu S, Porzner M,
Hlubek F, Kunz-Schughart LA, Knuechel R and Kirchner T: Variable
beta-catenin expression in colorectal cancers indicates tumor
progression driven by the tumor environment. Proc Natl Acad Sci
USA. 98:10356–10361. 2001. View Article : Google Scholar
|
|
35
|
Xie SL, Fan S, Zhang SY, Chen WX, Li QX,
Pan GK, Zhang HQ, Wang WW, Weng B, Zhang Z, et al: SOX8 regulates
cancer stem-like properties and cisplatin-induced EMT in tongue
squamous cell carcinoma by acting on the Wnt/β-catenin pathway. Int
J Cancer. 142:1252–1265. 2018. View Article : Google Scholar
|
|
36
|
Zhang Q, Li Y, Zhao R, Wang X, Fan C, Xu
Y, Liu Y, Li J and Wang S: The gain-of-function mutation E76K in
SHP2 promotes CAC tumorigenesis and induces EMT via the
Wnt/β-catenin signaling pathway. Mol Carcinog. 57:619–628. 2018.
View Article : Google Scholar : PubMed/NCBI
|