Introduction
Triple-negative breast cancer (TNBC) is defined by a
lack of expression of the estrogen receptor (ER), a lack of
progesterone receptor (PR) expression and the absence of ERBB2 gene
amplification, which encodes human epidermal growth factor receptor
2 (HER2) (1,2). TNBC accounts for ~15% of all breast
cancer cases worldwide and represents a heterogeneous group of
breast tumors (3-5). Numerous patients with TNBC experience
a fast relapse and commonly develop metastases, which results in a
poor prognosis (2). Recent
advances in genomic profiling technologies have provided
significant insights into the pathogenesis of breast cancer,
including TNBC (6-9). However, presently, no individualized
targeted adjuvants or induction treatments for TNBC are available.
Given the lack of recurrent targetable genomic alterations,
functional characterization of the TNBC genome to identify genomic
driver events is of utmost importance.
Somatic copy number alterations (CNAs) are a
universal feature of human cancer (10-12).
Compared with any other type of somatic genetic alteration, CNAs
alter a greater portion of the cancer genome. In general, CNAs are
associated with patient prognoses and therapeutic resistance
(13,14). Different cancer types adopt copy
number changes in different ways to shape their genomes (15,16).
These CNAs can affect the expression of genes and/or influence the
regulation of genes in their vicinity (17,18).
Furthermore, CNAs serve an important role in the classification of
tumor subtypes (9,19,20).
However, comprehensive genomic profiling of TNBC remains to be
adequately assessed. Given the prevalence of CNAs in cancer,
significant progress has been made in understanding the functional
impacts of CNAs, as well as the potential driver genes they contain
(21,22). These studies provide a rich source
of data that allows for performing meta-analysis.
Previously, chromothripsis has been described as a
novel mechanism for cancer initiation and progression (23-25).
In the classic model, tumorigenesis is an evolutionary process in
which the transition of normal cells to neoplastic cells is
mediated by the accumulation of somatic mutations. However, the
phenomenon of chromothripsis reveals a new paradigm of oncogenic
transformation, involving a catastrophic mutational process that is
commonly observed in numerous cancer types, such as colorectal
cancer, neuroblastoma and acute myeloid leukemia (26-28).
Chromothripsis contrasts with the multistep model of cancer
development and is characterized by the shattering of one or
multiple chromosomes in a single catastrophic event. Subsequently,
shattered fragments are randomly stitched together by DNA
double-strand break repair to form a derivative chromosome
(29-31). This process can lead to the
simultaneous acquisition of multiple tumor-promoting lesions. For
example, it may result in a large number of structural genome
variations, including duplications, deletions, inversions and
translocations. In addition, it may give rise to the amplification
of oncogenes or the inactivation of tumor suppressors, which serve
an important role in oncogenesis. To the best of our knowledge, a
comprehensive evaluation of chromothripsis in TNBC has not yet been
performed.
The present study collected 201 TNBC samples from
the National Center for Biotechnology Information (NCBI) Gene
Expression Omnibus (GEO) (32) and
The Cancer Genome Atlas (TCGA) (33) databases to perform a meta-analysis
of genomic CNAs. Statistically significant recurrent CNAs were
identified and the distribution of CNA breakpoints along the cancer
genome was examined. By employing the Genomic Identification of
Significant Targets In Cancer (GISTIC) algorithm, a total of 123
regions of DNA amplification and deletion were obtained (34). In these regions, a number of
potential driver genes for TNBC were revealed. For deletion of the
chro-mothripsis phenomenon, the typical chromothripsis pattern was
identified based on CNA data, and 31 samples with signs of
chromothripsis were detected (35-37).
Further analysis for chromothripsis regions was performed, which
revealed the chromosome pulverization hotspots in TNBC.
Furthermore, unsupervised hierarchical clustering of TNBC samples
demonstrated three different clusters that corresponded to the
tumors with specific CNA profiles (38). The present results extended
characterization of the CNA landscape of TNBC genomes and provided
novel insight into the phenomenon of chromothripsis. Thus, the
present findings provide information that improves understanding of
the mechanisms of TNBC development and may improve targeted
therapies.
Materials and methods
Sample collection and CNA calling
All samples included in the present study were
obtained from the NCBI GEO (32)
and TCGA databases (33). The
selection criteria were as follows: i) The patient was diagnosed
with primary breast cancer, and patients with distant metastasis
were excluded; ii) all samples were hybridized using Affymetrix
Genome-Wide Human SNP Array 6.0 platform to facilitate data
integration; and iii) patients were histologically confirmed as
ER-, PR- and HER2-negative, or the study clearly stated that
patients were diagnosed with TNBC.
To identify somatic CNAs, raw signal intensity files
were downloaded from the GEO website (https://www.ncbi.nlm.nih.gov/geo/) for re-analysis.
The R package aroma. affymetrix (https://aroma-project.org/; version 3.2.0) using the
CRMAv.2 method (39) was employed
for data processing. To call CNA segments, HapMap (40) data were used as a control. The data
annotation was based on human reference genome assembly hg19/GRCh37
(41), and the circular binary
segmentation algorithm (42) was
performed to segment copy number data. Next, the CNA calling
cut-off values for amplifications and deletions were set to 0.2 and
-0.2, respectively. To avoid gender bias, the X and Y chromosomes
were excluded. TCGA data portal (https://portal.gdc.cancer.gov/) was used for
downloading the genomic array data (level 3) and clinical
information. Visual inspection was used for data quality control,
and samples of poor quality were excluded from further analyses. A
total of 201 TNBC cases were collected and are presented in
Table SI. The samples were
collected between October 2011 and August 2018.
Identification of significant recurrent
targets
The GISTIC algorithm (34) was used for the identification of
peak regions that were significantly amplified or deleted in all
samples. The parameters used to run GISTIC 2.0 were as follows: i)
The false discovery rate q-value was set to <0.05; ii) the arm
peel method (34) was used to
reduce data noise; iii) the confidence level used to calculate the
region containing a driver was set to >0.95; iv) the 'Extreme’
method (34) was applied for
reducing marker-level to the gene-level copy number data; and v)
the log2 ratios for calling gains and losses were set to >0.2
and <-0.2, respectively. In total, 719 known cancer consensus
genes were downloaded from the Catalogue of Somatic Mutations in
Cancer (COSMIC) database (43).
Chromothripsis screening
Chromothripsis-like patterns were detected using the
CTLPScanner web server (http://cgma.scu.edu.cn/CTLPScanner/) (35). The pattern of oscillating copy
number changes and localized clustering of breakpoints was screened
based on segmented array data. The parameters and thresholds for
the screening were as follows: i) Copy number status switch times
≥20; ii) log10 of likelihood ratio ≥10; iii) minimum segment size
of 10 Kb; and iv) signal value difference between two adjacent
segments ≥0.4. For visualization of results, signal intensity for
calling genomic gains and losses were set to 0.2 and -0.2,
respectively.
Detection of chromosomal breakpoints
In the present study, both boundaries of each CNA
were defined as chromosomal breakpoints. A stringent definition of
CNA breakpoints was used to reduce the bias caused by technical
noise. If the alteration of log2 signal intensity between two
adjacent genomic segments was >0.4, the related genomic position
was considered to be a breakpoint (44,45).
CNAs <10 Kb were ignored. Breakpoints located in chromosomal
telomeres and centromeres were excluded for further analysis. To
generate a simulated distribution of breakpoints, in-house Perl
scripts were used to bin the genome and randomly shuffle the
positions of breakpoints 10,000 times. Common fragile sites (CFSs)
and non-fragile regions of the human genome were extracted from
previous studies (46-48). All the genome coordinates were
converted from genome assembly hg18/NCBI36 to hg19/GRCh37 by the
LiftOver tool (41). Furthermore,
the copy number based unsupervised hierarchical clustering was
performed based on the Euclidean distance following Ward's
method.
Results
CNA profile in TNBC
In total, 98 and 103 TNBC samples were collected
from the GEO and TCGA databases, respectively. The genome-wide
frequency plot of CNAs is presented in Fig. 1. The CNAs were of varying sizes,
ranging from whole chromosome arms to small focal amplifications
and deletions. The mean number of CNAs per tumor was 414, with
deletions outnumbering amplifications at ~1.7:1. From the
high-resolution CNA data, it was observed that the most common
gains of the entire chromosome arms included 1q, 8q, 10p and 12p,
and losses of 5q, 8p and 17p. Other less frequently affected
chromosomes included 9p and 20q gains and 13q, 14q and 15q losses.
The most frequent focal gains were narrowed down to 3q and 19q.
Focal losses were identified most often in 3q and 12q. Notably,
this profile may reveal TNBC-specific CNAs that have rarely been
reported, such as 9p gain and 5q, 12q loss.
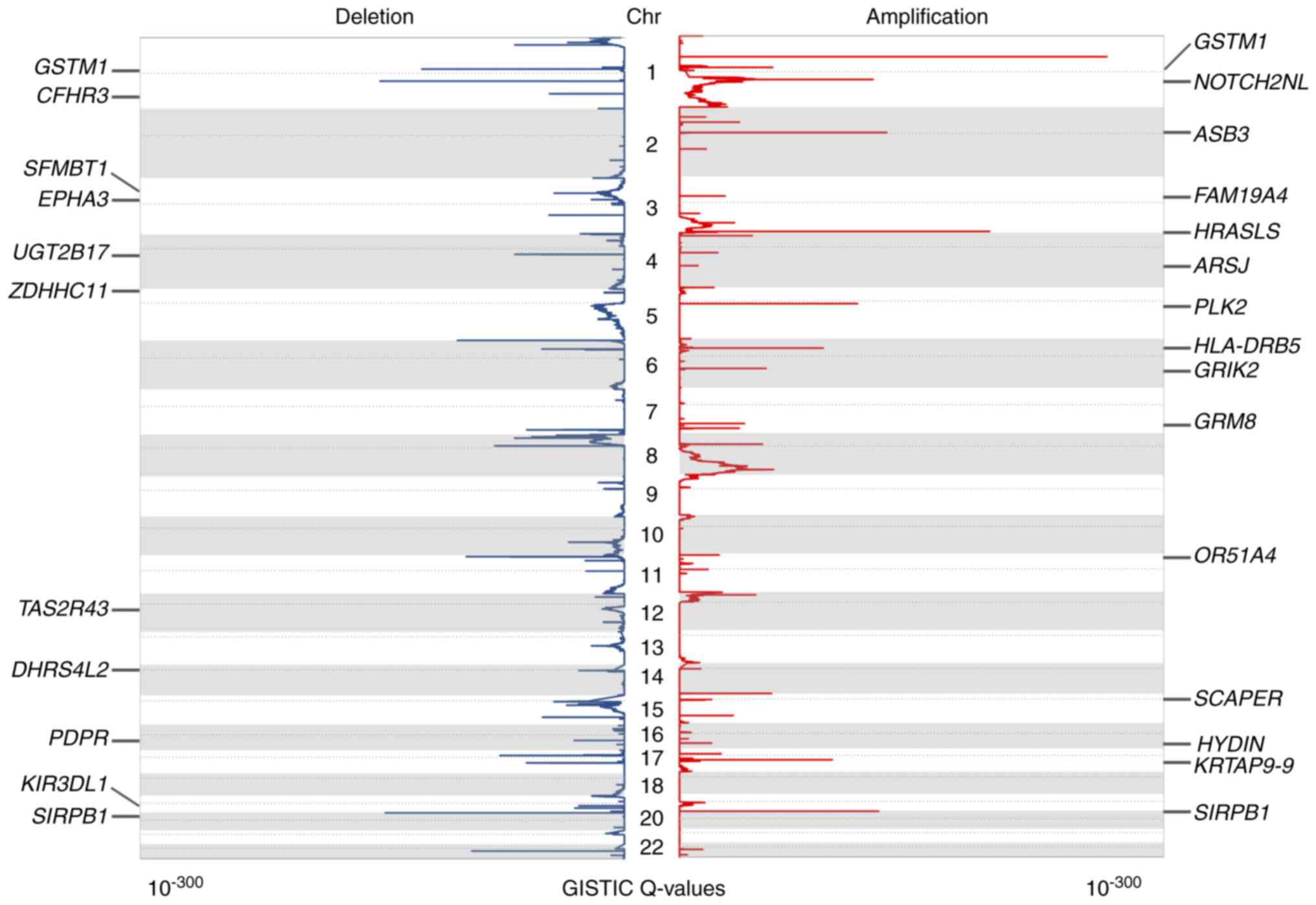
The present study identified statistically
significant recurrent focal CNAs and potential cancer driver genes
by GISTIC 2.0 (34). Using this
algorithm, 49 and 74 amplification and deletion peaks were
identified, respectively (q<0.05). The annotation of these peaks
revealed 993 targeted coding genes. The significantly amplified or
deleted genomic regions as well as identified genes are presented
in Table SII. There were 33
regions of interest that contained only one candidate driver gene.
The aberration score from GISTIC is presented in Fig. 2, in addition to some of the peaks
containing only one candidate gene.
Identification of chromothripsis
events
Using the CTLPScanner algorithm (35), a total of 31 chromothripsis cases
were identified from 201 TNBC samples, with an incidence of ~15%
(Table SIII). Since the overall
chromothripsis incidence in various cancer types is ~5%, TNBC has a
relatively high chromothripsis incidence compared with most other
tumor types, including other subtypes of breast cancer (23,36).
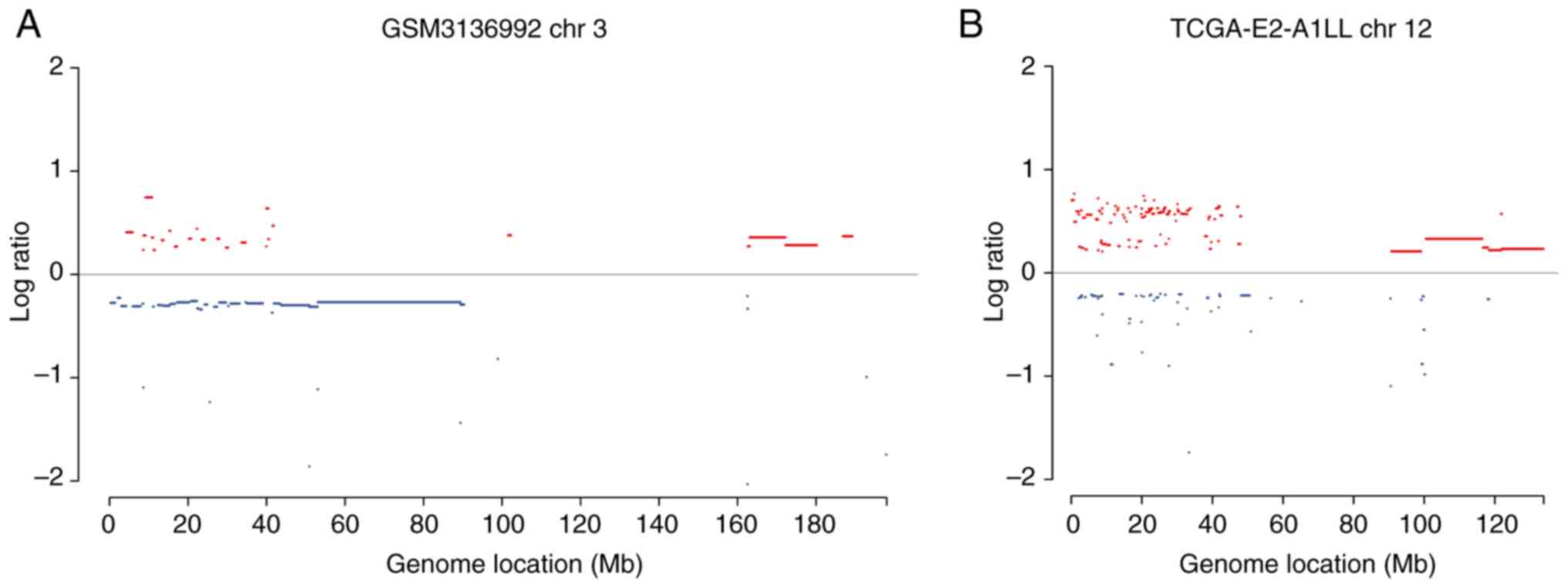
Pulverization regions were identified in various sizes ranging from
dozens of Mb to the entire chromosome arm. Fig. 3 presents examples of identified
chromothripsis cases and pulverized genomic regions. The present
study further investigated the number of affected chromosomes per
tumor sample, and observed that ~22% (7/31) of chromothripsis cases
carried two shattered chromosomes. In other cases, only one
chromosome was affected. The most frequently pulverized focal
genomic regions in TNBC included 6p, 11p and 17q. At the chromosome
level, chromosomes 1, 5 and 12 demonstrated relatively high rates
of pulverization. Fig. 4
illustrates the hotspots of chromothripsis across the cancer genome
and may reveal a TNBC-specific pattern of chromothripsis. Some of
the GISTIC-identified candidate driver genes that were located in
the chromothripsis hotspot regions are also presented. Alterations
in these genes may further contribute to chromosomal instability
and the overall chromothripsis phenotype in TNBC.
Characterization of chromosomal
breakpoints
DNA breakage is an important type of
cancer-associated genomic aberration, and may cause amplifications,
deletions, inversions and translocations. Since array platforms
have reduced the ability for detecting inversion and translocation
(16,39), the landscape of chromosomal
breakage was investigated based on CNA data. In the present study,
the genomic start and end of CNAs were defined as breakpoints.
These breakpoints may contribute to TNBC initiation and
progression. A total of 44,384 CNA breakpoints were identified in
201 TNBC samples. The number of chromosomal breaks per sample
ranged between 30 and 709, with a median value of 193. To
investigate CNA breakpoint hotspots across the genome, each
chromosome was binned into continuous 1 Mb windows, and the density
of breakpoints per bin was calculated. Next, the position of CNAs
was shuffled 10,000 times and the background distribution of DNA
breaks was obtained. The breakpoint-prone genomic regions were
identified by comparing the actual value with the background
distribution of breakpoints. In total, 199 genomic regions that
were significantly enriched for breakpoints of chromosomal
rearrangements were identified (Bonferroni corrected P<0.01;
Table SIV). To compare these
regions with published common fragile sites and non-fragile
regions, related data were downloaded from existing literature
(46). Among the identified
hotspots, only 20 regions (~10%) overlapped with CFS, and the
majority of them were located on chromosome 1 (Fig. 4). A total of 25 (~12%) hotspots
were located within NFS and were relatively evenly distributed
across the genome. Combined, these results possibly reveal the
TNBC-specific genomic instability regions.
Subgroup analysis based on CNAs
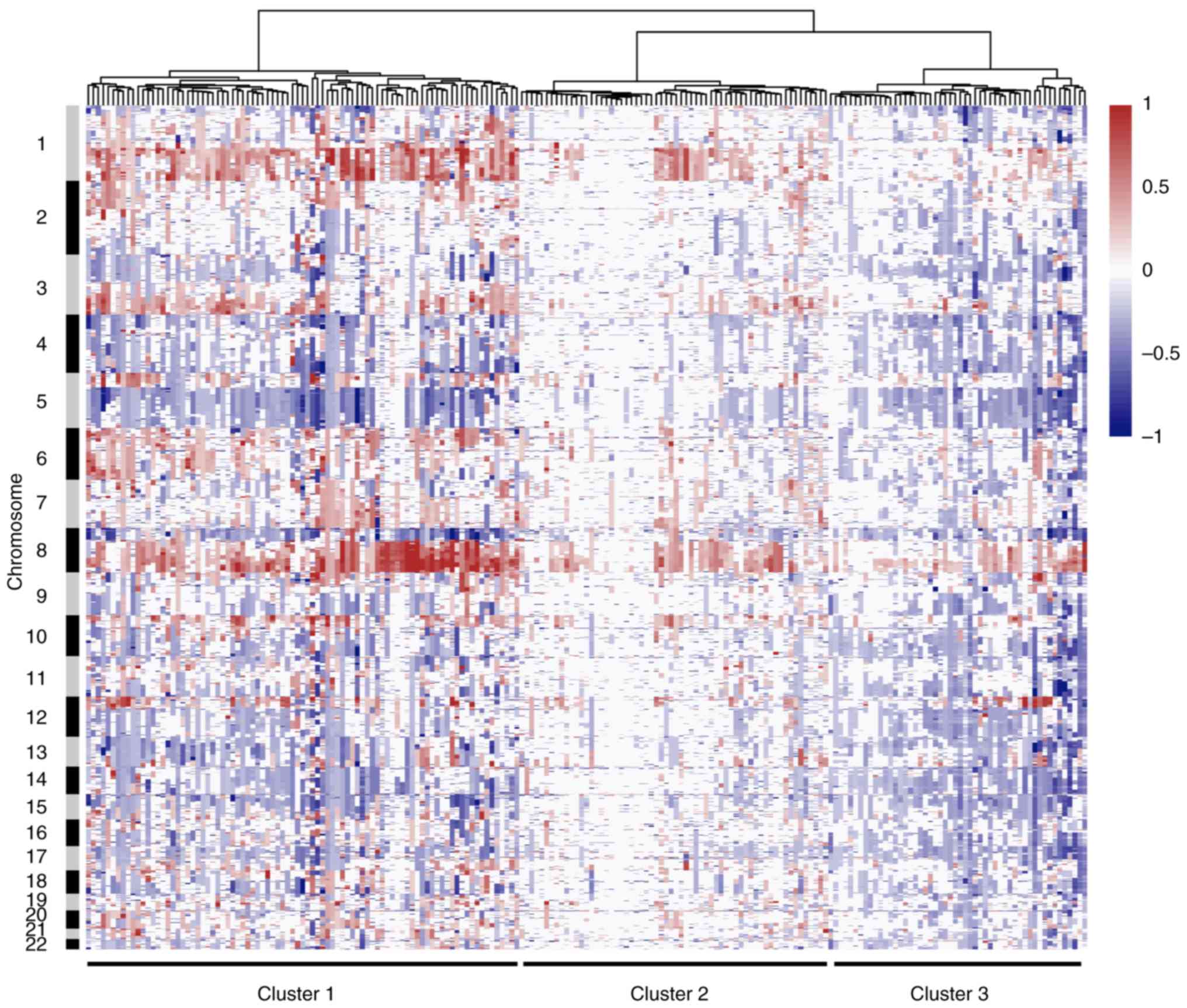
The full dataset underwent copy number based
unsupervised hierarchical clustering. Classification was based on
the Euclidean distance following Ward's method, and three main
clusters were identified. Results of the cluster analysis are
presented as a dendrogram in Fig.
5. Cluster 1 was composed of tumors with extensive arm-level
CNAs. The most common alterations include gains of 1q and 8p, and
losses of 5q and 8p. These tumors were dominated by frequent CNAs,
suggesting an important role of genome instability in the
tumorigenesis of this type of TNBC. By contrast, Cluster 2 was
characterized by few CNAs, and this cluster may represent M class
cancers that were identified in previous studies (38). These DNA copy number stable tumors
may be primarily driven by a mutation rather than by CNA. Tumors in
Cluster 3 are extraordinarily influenced by small focal alterations
rather than by arm-level events, and especially copy number losses.
Furthermore, each of these three main clusters can be further
decomposed into several smaller sub-clusters.
Discussion
TNBC is characterized by its unique molecular
profile, aggressive behavior and lack of targeted therapies.
Currently, treatment options for TNBC are limited when compared to
that of other types of breast cancer. Previous genomic profiling
studies have reported that TNBC is a heterogeneous malignancy
involving diverse genomic alterations (5). Genome-wide meta-analysis of CNAs will
improve the understanding of the mechanisms responsible for TNBC
initiation and progression, and facilitate the detection of more
reliable tumor markers and the development of targeted therapy. The
present study characterized DNA CNAs in TNBC based on published
high-resolution genomic array data. From the overall CNA profile of
all samples, several frequent recurrent arm-level alterations were
observed, which may contribute to the malignant transformation of
TNBC. For the analysis of focal CNA events, the GISTIC algorithm
was utilized to discern significant CNAs, and several candidate
genes were identified, including both known cancer genes and genes
that have not previously been associated with any type of
cancer.
Among the identified driver genes, some were known
cancer genes and were recorded in the COSMIC database (43). For example CCNE1, CD274, epidermal
growth factor receptor (EGFR) and JAK2 were located in genomic gain
regions, while APOBEC3B, CDKN2A, FAT1 and NOTCH1 were identified in
loss regions. Some novel or recently described genes were also
detected from the datasets. For example, the VOPP1 WW domain
binding protein (VOPP1) gene was identified to be located at the
amplified region 7p11.2. It has been reported that VOPP1 is often
co-amplified with EGFR or is the breakpoint location for EGFR
amplification (49,50). The overexpression of VOPP1 can
increase the transcriptional activity of nuclear factor κ B subunit
1 by facilitating its nuclear translocation and associated
apoptotic response. VOPP1 overexpression has been observed in
several tumor types, such as gastric cancer, head and neck squamous
carcinoma and glioma (51-53). A recent study reported that VOPP1
can promote breast cancer development by interacting with the tumor
suppressor WW domain containing oxidoreductase (54). These results suggest that VOPP1 may
serve an important role in TNBC carcinogenesis and could be
exploited to develop therapies for patients with TNBC. Another
candidate gene dehydrogenase/reductase 4 like 2 (DHRS4L2) is
located in the deletion region of 14q11.2 and is a member of the
short-chain dehydrogenases/reductases family. DHRS4L2 produces
multiple transcript variants through alternative splicing (55,56).
The encoded protein may be an NADPH dependent retinol
oxidoreductase. Genomic loss of DHRS4L2 may lead to low expression
and has been demonstrated to be associated with risk of diseases
(56,57). The DHRS4L2 gene has not yet been
reported to be involved in cancer and could be considered a novel
candidate gene for TNBC.
Furthermore, the high degree of genomic instability
is a hallmark of BRCA1-deficient TNBC. This instability is a
prerequisite for the development of large numbers of CNAs, which
can affect tumor suppressor genes and oncogenes. Although the BRCA1
status of tumors in the present cohort was not available, the
significant numbers of CNAs in these samples were indicative of
genomic instability and presented a specific pattern of TNBC.
The present study extensively analyzed the
distribution of the chromothripsis phenomenon in TNBC.
Chromothripsis is a single catastrophic event that generates dozens
of mutations sufficient to produce a malignancy and is distinct
from the progressive accumulation of mutations model of cancer
development (23-25). The incidence of chromothripsis is
heterogeneous across different cancer types. A total of 31
chromothripsis cases were identified out of 201 TNBC samples, with
an incidence rate of ~15%. We previously determined that the
incidence of chromothripsis in breast cancer is ~11% (36). Thus, TNBC has a relatively higher
incidence of chromothripsis and exhibits increased evidence of
genomic instability compared with other breast cancer subtypes.
Currently, the underlying mechanisms leading to chromothripsis
remain largely unknown, although several hypotheses have been
proposed, including the formation of micronuclei (58,59),
premature chromosome condensation (60), abortive apoptosis (61,62)
and breakage-fusion-bridge cycles (63-65).
The present study further evaluated the patterns of chromosomal
pulverization based on the results, in order to provide clues to
identify the underlying mechanisms of chromothripsis in TNBC. In
the present cohort, 22% of chromothripsis cases affected two
chromosomes. Chromothripsis involving more than one chromosome can
result from several chromosomes in a micronucleus or is the
consequence of a process of aborted apoptosis (59). More than one mechanism may be
responsible for the chromothripsis events in TNBC. Several
chromosomal pulverization hotspots were identified across the
genome, which may contain critical genes for genomic instability.
The most frequently affected chromosome regions were identified,
and these results provide a foundation for further analysis. For
example, as presented in Fig. 3,
there were two chromothripsis events in chromosome 3 and chromosome
12, respectively. Distinct cancer genes were located in the
different pulverization regions, which may reflect different
mechanisms that trigger chromothripsis events in these two
samples.
The present results of the chromosomal breaks
analysis provides detailed characterization of recurrent
chromosomal breakpoints and affected genes in the TNBC genome. The
points of copy number level shift in somatic CNA profiles indicate
underlying chromosomal breaks and genomic locations affected by
somatic structural aberrations. Thus, the DNA CNA data generated by
high-resolution genomic arrays enabled a systematic search for
regions and genes that were affected by CNA-associated breakpoints.
These breakpoints may silence tumor suppressor genes or create
novel gene fusions with oncogenic potential. Next, simulation
experiments were performed in which CNA locations were randomly
assigned throughout the genome. The simulation was conducted 10,000
times and 199 recurrent breakpoints were identified that were
clustered more than would be expected. These breakpoint-prone
regions were compared with known common fragile sites and
non-fragile regions of the human genome to reveal TNBC-specific
genome instability regions (46).
Unsupervised hierarchical clustering analysis was
performed to generate a comprehensive view of genome-wide copy
number changes in TNBC. Three main clusters were identified.
Cluster 1 was predominantly represented by tumors with large
chromosomal CNAs, while cluster 3 was primarily influenced by small
focal alterations. These findings may indicate the different
underlying mechanisms that drive tumors in both groups. For
example, the large chromosome or chromosome arm-level CNAs are
usually caused by aneuploidy or abnormal numbers of chromosome,
while focal alterations often cause gene-level mutagenesis
(13). Consistent with previous
studies, the present study identified another cluster with few
CNAs, which may represent the M class cancer that is predominantly
driven by mutations rather than by CNAs (38). Since somatic point mutations cannot
be detected by genomic arrays, this cluster presented a different
CNA pattern compared with the other two clusters. These
observations reveal biologically heterogeneous groups of TNBC,
which may represent different molecular mechanisms that underlie
tumor development. The clustering of these tumor samples will
contribute to the classification and clinical decision making of
TNBC.
Previously, several studies have focused on genomic
alterations in breast cancer. Banerji et al (66) analyzed 103 breast cancer samples of
diverse subtypes using whole exome sequencing, which revealed
several recurrent somatic mutations and fusion genes that
contributed to breast cancer progression, In addition, Kim et
al (67) provided profiles of
longitudinal TNBC samples during neoadjuvant chemotherapy, and
revealed that resistant genotypes were pre-existing and adaptively
selected by therapy. Using single-cell sequencing, Gao et al
(68) identified clonal
subpopulations in individual tumors that shared a common
evolutionary lineage, and demonstrated that most CNAs were acquired
at the earliest stages of tumor evolution. These studies provide
valuable knowledge about genomic alterations in TNBC. However, the
number of tumor samples, particularly TNBC samples, involved in
these studies were limited. The present study focused on TNBC and
included a larger numbers of samples that resulted in the
generation of significant results.
The present study provides valuable new insights
into the mechanisms of genomic instability in TNBC. However, there
remain a number of questions that need to be studied for TNBC. For
example, whether tumors at different stages or metastatic breast
cancers demonstrate significantly distinct CNA profiles, and the
biological or clinical significance of the observed differences is
unknown. Several other types of breast cancer exist, including
inflammatory tumors and HER2-positive tumors. The differences in
CNA profiles between the TNBC-subtype and other tumor types needs
to be further elucidated. Furthermore, the breast cancer genome
evolution during disease progression is not yet fully understood
yet. In addition, the existence of intra-tumor heterogeneity makes
things more complicated. Therefore, in-depth analysis of these
questions will help improve understanding of the etiology of
TNBC.
In conclusion, a comprehensive characterization of
somatic genomic alterations was performed based on a large cohort
of TNBC samples. The current study presented several novel findings
for TNBC, including: i) A total of 123 regions of significant
amplification and deletion were determined; ii) the incidence of
chromothripsis in TNBC was identified as ~15%; ii) the distribution
and hotspots of CNA breakpoints were revealed; and iii) three tumor
clusters and their CNA patterns were identified. The present
findings contribute to an increasingly detailed portrait of genomic
features of TNBC and may accelerate the rate of driver gene
discovery.
Supplementary Data
Funding
No finding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL and XZ performed the experiments and wrote the
original manuscript. CH and YZ performed chromothripsis data
analysis and analyzed the results. JC, HC and YY helped conduct the
experiments and data analyses. JL and NH conceived and supervised
the project, and drafted the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Carey L, Winer E, Viale G, Cameron D and
Gianni L: Triple-negative breast cancer: Disease entity or title of
convenience? Nature Rev Clin Oncol. 7:683–692. 2010. View Article : Google Scholar
|
|
2
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaz-Luis I, Ottesen RA, Hughes ME, Mamet
R, Burstein HJ, Edge SB, Gonzalez-Angulo AM, Moy B, Rugo HS,
Theriault RL, et al: Outcomes by tumor subtype and treatment
pattern in women with small, node-negative breast cancer: A
multi-institutional study. J Clin Oncol. 32:2142–2150. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Metzger-Filho O, Tutt A, de Azambuja E,
Saini KS, Viale G, Loi S, Bradbury I, Bliss JM, Azim HA Jr, Ellis
P, et al: Dissecting the heterogeneity of triple-negative breast
cancer. J Clin Oncol. 30:1879–1887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burstein MD, Tsimelzon A, Poage GM,
Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK,
Hilsenbeck SG, Chang JC, et al: Comprehensive genomic analysis
identifies novel subtypes and targets of triple-negative breast
cancer. Clin Cancer Res. 21:1688–1698. 2015. View Article : Google Scholar :
|
|
7
|
Chin SF, Teschendorff AE, Marioni JC, Wang
Y, Barbosa-Morais NL, Thorne NP, Costa JL, Pinder SE, van de Wiel
MA, Green AR, et al: High-resolution aCGH and expression profiling
identifies a novel genomic subtype of ER negative breast cancer.
Genome Biol. 8:R2152007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chin K, DeVries S, Fridlyand J, Spellman
PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, et
al: Genomic and transcriptional aberrations linked to breast cancer
patho-physiologies. Cancer Cell. 10:529–541. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Melchor L, Honrado E, García MJ, Alvarez
S, Palacios J, Osorio A, Nathanson KL and Benítez J: Distinct
genomic aberration patterns are found in familial breast cancer
associated with different immu-nohistochemical subtypes. Oncogene.
27:3165–3175. 2008. View Article : Google Scholar
|
|
10
|
Beroukhim R, Mermel CH, Porter D, Wei G,
Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J,
Urashima M, et al: The landscape of somatic copy-number alteration
across human cancers. Nature. 463:899–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stratton MR, Campbell PJ and Futreal PA:
The cancer genome. Nature. 458:719–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zack TI, Schumacher SE, Carter SL,
Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J,
Mermel CH, et al: Pan-cancer patterns of somatic copy number
alteration. Nat Genet. 45:1134–1140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim TM, Xi R, Luquette LJ, Park RW,
Johnson MD and Park PJ: Functional genomic analysis of chromosomal
aberrations in a compendium of 8000 cancer genomes. Genome Res.
23:217–227. 2013. View Article : Google Scholar :
|
|
15
|
Cai H, Kumar N, Ai N, Gupta S, Rath P and
Baudis M: Progenetix: 12 years of oncogenomic data curation.
Nucleic Acids Res. 42(Database Issue): D1055–D1062. 2014.
View Article : Google Scholar :
|
|
16
|
Cai H, Gupta S, Rath P, Ai N and Baudis M:
arrayMap 2014: An updated cancer genome resource. Nucleic Acids
Res. 43(Database Issue): D825–D830. 2015. View Article : Google Scholar :
|
|
17
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xue W, Kitzing T, Roessler S, Zuber J,
Krasnitz A, Schultz N, Revill K, Weissmueller S, Rappaport AR,
Simon J, et al: A cluster of cooperating tumor-suppressor gene
candidates in chromosomal deletions. Proc Natl Acad Sci USA.
109:8212–8217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stephens PJ, McBride DJ, Lin ML, Varela I,
Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie
LJ, et al: Complex landscapes of somatic rearrangement in human
breast cancer genomes. Nature. 462:1005–1010. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rakha EA, Elsheikh SE, Aleskandarany MA,
Habashi HO, Green AR, Powe DG, El-Sayed ME, Benhasouna A, Brunet
JS, Akslen LA, et al: Triple-negative breast cancer: Distinguishing
between basal and nonbasal subtypes. Clin Cancer Res. 15:2302–2310.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Waddell N, Arnold J, Cocciardi S, da Silva
L, Marsh A, Riley J, Johnstone CN, Orloff M, Assie G, Eng C, et al:
Subtypes of familial breast tumours revealed by expression and copy
number profiling. Breast Cancer Res Treat. 123:661–677. 2010.
View Article : Google Scholar
|
|
22
|
Jones C, Ford E, Gillett C, Ryder K,
Merrett S, Reis-Filho JS, Fulford LG, Hanby A and Lakhani SR:
Molecular cytogenetic identification of subgroups of grade III
invasive ductal breast carcinomas with different clinical outcomes.
Clin Cancer Res. 10:5988–5997. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stephens PJ, Greenman CD, Fu B, Yang F,
Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA,
et al: Massive genomic rearrangement acquired in a single
catastrophic event during cancer development. Cell. 144:27–40.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu P, Erez A, Nagamani SC, Dhar SU,
Kołodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang
F, Withers MA, et al: Chromosome catastrophes involve replication
mechanisms generating complex genomic rearrangements. Cell.
146:889–903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Korbel JO and Campbell PJ: Criteria for
inference of chromothripsis in cancer genomes. Cell. 152:1226–1236.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kloosterman WP, Hoogstraat M, Paling O,
Tavakoli-Yaraki M, Renkens I, Vermaat JS, van Roosmalen MJ, van
Lieshout S, Nijman IJ, Roessingh W, et al: Chromothripsis is a
common mechanism driving genomic rearrangements in primary and
metastatic colorectal cancer. Genome Biol. 12:R1032011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Molenaar JJ, Koster J, Zwijnenburg DA, van
Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J,
Westerman BA, van Arkel J, et al: Sequencing of neuroblastoma
identifies chromothripsis and defects in neuritogenesis genes.
Nature. 483:589–593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bochtler T, Granzow M, Stölzel F, Kunz C,
Mohr B, Kartal-Kaess M, Hinderhofer K, Heilig CE, Kramer M, Thiede
C, et al: Marker chromosomes can arise from chro-mothripsis and
predict adverse prognosis in acute myeloid leukemia. Blood.
129:1333–1342. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kloosterman WP, Tavakoli-Yaraki M, van
Roosmalen MJ, van Binsbergen E, Renkens I, Duran K, Ballarati L,
Vergult S, Giardino D, Hansson K, et al: Constitutional
chromothripsis rearrangements involve clustered double-stranded DNA
breaks and nonhomologous repair mechanisms. Cell Rep. 1:648–655.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Forment JV, Kaidi A and Jackson SP:
Chromothripsis and cancer: Causes and consequences of chromosome
shattering. Nat Rev Cancer. 12:663–670. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rausch T, Jones DT, Zapatka M, Stütz AM,
Zichner T, Weischenfeldt J, Jäger N, Remke M, Shih D, Northcott PA,
et al: Genome sequencing of pediatric medulloblastoma links
catastrophic DNA rearrangements with TP53 mutations. Cell.
14:59–71. 2012. View Article : Google Scholar
|
|
32
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:Database Issue.
D991–D995. 2013. View Article : Google Scholar
|
|
33
|
Cancer Genome Atlas Research Network;
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mermel CH, Schumacher SE, Hill B, Meyerson
ML, Beroukhim R and Getz G: GISTIC2.0 facilitates sensitive and
confident localization of the targets of focal somatic copy-number
alteration in human cancers. Genome Biol. 12:R412011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang J, Liu J, Ouyang L, Chen Y, Liu B and
Cai H: CTLPScanner: A web server for chromothripsis-like pattern
detection. Nucleic Acids Res. 44:W252–W258. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cai H, Kumar N, Bagheri HC, von Mering C,
Robinson MD and Baudis M: Chromothripsis-like patterns are
recurring but heterogeneously distributed features in a survey of
22,347 cancer genome screens. BMC Genomics. 15:822014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang J, Deng G and Cai H:
ChromothripsisDB: A curated database of chromothripsis.
Bioinformatics. 32:1433–1435. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ciriello G, Miller ML, Aksoy BA,
Senbabaoglu Y, Schultz N and Sander C: Emerging landscape of
oncogenic signatures across human cancers. Nat Genet. 45:1127–1133.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bengtsson H, Wirapati P and Speed TP: A
single-array preprocessing method for estimating full-resolution
raw copy numbers from all Affymetrix genotyping arrays including
GenomeWideSNP 5 & 6. Bioinformatics. 25:2149–2156. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
International HapMap Consortium: The
international HapMap project. Nature. 426:789–796. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rosenbloom KR, Armstrong J, Barber GP,
Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo
L, Haeussler M, et al: The UCSC genome browser database: 2015
update. Nucleic Acids Res. 43(Database Issue): D670–D681. 2015.
View Article : Google Scholar :
|
|
42
|
Olshen AB, Venkatraman ES, Lucito R and
Wigler M: Circular binary segmentation for the analysis of
array-based DNA copy number data. Biostatistics. 5:557–572. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Forbes SA, Beare D, Gunasekaran P, Leung
K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et
al: COSMIC: Exploring the world's knowledge of somatic mutations in
human cancer. Nucleic Acids Res. 43:Database Issue. D805–D811.
2015. View Article : Google Scholar :
|
|
44
|
Zheng S, Fu J, Vegesna R, Mao Y, Heathcock
LE, Torres-Garcia W, Ezhilarasan R, Wang S, McKenna A, Chin L, et
al: A survey of intragenic breakpoints in glioblastoma identifies a
distinct subset associated with poor survival. Genes Dev.
27:1462–1472. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Smida J, Xu H, Zhang Y, Baumhoer D, Ribi
S, Kovac M, von Luettichau I, Bielack S, O'Leary VB, Leib-Mösch C,
et al: Genome-wide analysis of somatic copy number alterations and
chromosomal breakages in osteosarcoma. Int J Cancer. 141:816–828.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fungtammasan A, Walsh E, Chiaromonte F,
Eckert KA and Makova KD: A genome-wide analysis of common fragile
sites: What features determine chromosomal instability in the human
genome? Genome Res. 22:993–1005. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Durkin SG and Glover TW: Chromosome
fragile sites. Annu Rev Genet. 41:169–192. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sarni D and Kerem B: The complex nature of
fragile site plasticity and its importance in cancer. Curr Opin
Cell Biol. 40:131–136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Eley GD, Reiter JL, Pandita A, Park S,
Jenkins RB, Maihle NJ and James CD: A chromosomal region 7p112
transcript map: Its development and application to the study of
EGFR amplicons in glioblastoma. Neuro Oncol. 4:86–94. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Masuda H, Zhang D, Bartholomeusz C,
Doihara H, Hortobagyi GN and Ueno NT: Role of epidermal growth
factor receptor in breast cancer. Breast Cancer Res Treat.
136:331–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gao C, Pang M, Zhou Z, Long S, Dong D,
Yang J, Cao M, Zhang C, Han S and Li L: Epidermal growth factor
receptor-coamplified and overexpressed protein (VOPP1) is a
putative oncogene in gastric cancer. Clin Exp Med. 15:469–475.
2015. View Article : Google Scholar
|
|
52
|
Baras A, Yu Y, Filtz M, Kim B and Moskaluk
CA: Combined genomic and gene expression microarray profiling
identifies ECOP as an upregulated gene in squamous cell carcinomas
independent of DNA amplification. Oncogene. 28:2919–2924. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Baras A and Moskaluk CA: Intracellular
localization of GASP/ECOP/VOPP1. J Mol Histol. 41:153–164. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bonin F, Taouis K, Azorin P, Petitalot A,
Tariq Z, Nola S, Bouteille N, Tury S, Vacher S, Bièche I, et al:
VOPP1 promotes breast tumorigenesis by interacting with the tumor
suppressor WWOX. BMC Biol. 16:1092018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang Q, Li Y, Liu G, Xu X, Song X, Liang
B, Li R, Xie J, Du M, Xiao L, et al: Alternative transcription
initiation and splicing variants of the DHRS4 gene cluster. Biosci
Rep. 29:47–56. 2009. View Article : Google Scholar
|
|
56
|
Su ZJ, Zhang QX, Liu GF, Song XH, Li Q,
Wang RJ, Chen HB, Xu XY, Sui XX and Huang DY: Bioinformatic
analysis of the human DHRS4 gene cluster and a proposed mechanism
for its transcriptional regulation. BMC Mol Biol. 11:432010.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Su Z, Liu G, Song X, Liang B, Chang X and
Huang D: CpG island evolution in the mammalian DHRS4 gene cluster
and its role in the regulation of gene transcription. Genet Mol
Res. 15:2016. View Article : Google Scholar
|
|
58
|
Crasta K, Ganem NJ, Dagher R, Lantermann
AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D and
Pellman D: DNA breaks and chromosome pulverization from errors in
mitosis. Nature. 482:53–58. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang CZ, Spektor A, Cornils H, Francis
JM, Jackson EK, Liu S, Meyerson M and Pellman D: Chromothripsis
from DNA damage in micronuclei. Nature. 522:179–184. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Meyerson M and Pellman D: Cancer genomes
evolve by pulverizing single chromosomes. Cell. 144:9–10. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tubio JM and Estivill X: Cancer: When
catastrophe strikes a cell. Nature. 470:476–477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ichim G, Lopez J, Ahmed SU, Muthalagu N,
Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos
D, et al: Limited mitochondrial permeabilization causes DNA damage
and genomic instability in the absence of cell death. Mol Cell.
57:860–872. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nones K, Waddell N, Wayte N, Patch AM,
Bailey P, Newell F, Holmes O, Fink JL, Quinn MCJ, Tang YH, et al:
Genomic catastrophes frequently arise in esophageal adenocarcinoma
and drive tumorigenesis. Nat Commun. 5:52242014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sorzano CO, Pascual-Montano A, Sánchez de
Diego A, Martínez-A C and van Wely KH: Chromothripsis:
Breakage-fusion-bridge over and over again. Cell Cycle.
12:2016–2023. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li Y, Schwab C, Ryan S, Papaemmanuil E,
Robinson HM, Jacobs P, Moorman AV, Dyer S, Borrow J, Griffiths M,
et al: Constitutional and somatic rearrangement of chromosome 21 in
acute lymphoblastic leukaemia. Nature. 508:98–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Banerji S, Cibulskis K, Rangel-Escareno C,
Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY,
Sougnez C, Zou L, et al: Sequence analysis of mutations and
translocations across breast cancer subtypes. Nature. 486:405–459.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kim C, Gao R, Sei E, Brandt R, Hartman J,
Hatschek T, Crosetto N, Foukakis T and Navin NE: Chemoresistance
evolution in triple-negative breast cancer delineated by
single-cell sequencing. Cell. 173:879–893.e13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gao R, Davis A, McDonald TO, Sei E, Shi X,
Wang Y, Tsai PC, Casasent A, Waters J, Zhang H, et al: Punctuated
copy number evolution and clonal stasis in triple-negative breast
cancer. Nat Genet. 48:1119–1130. 2016. View Article : Google Scholar : PubMed/NCBI
|