Introduction
The E2F transcription factors are involved in cell
cycle progression, DNA synthesis and cellular proliferation, and
are key regulators of cell cycle-dependent gene expression. The E2F
family contains 8 different genes encoding transcriptional
activators and repressors, and their activity is regulated by the
pathway that controls the activity of the retinoblastoma tumor
suppressor protein (RB), an essential regulator of cell cycle
progression, apoptosis, senescence and differentiation. E2F
activity is frequently altered in human malignancies, due to the
dysregulation of the RB/E2F pathway, i.e., the overexpression of
cyclins or cyclin-dependent kinases (CDKs) and the inactivation of
RB or CDK inhibitors (1-4).
AT-rich interacting domain 3A (ARID3A/Bright/DRIL1/
E2FBP1, hereafter referred to as ARID3A) (5-8)
was identified as a B cell-specific transcription factor that
transactivates the immunoglobulin heavy chain (IgH) enhancer.
ARID3A was also identified as a protein that interacts with E2F1.
ARID3B/Bdp/DRIL2 (hereafter referred to as ARID3B) is closely
related to ARID3A and was initially identified as a protein that
physically interacts with RB (9).
ARID3A and ARID3B share a highly conserved ARID DNA-binding domain
and REKLES, a common domain required for self-association and
tetramerization (6,10-13). ARID3A forms heteromeric complexes
with ARID3B through its REKLESb domain. ARID3B predominantly
localizes in the nucleus, while its heterodimerization with ARID3A
increases in the nuclear retention of ARID3A that shuttles between
the nucleus and cytoplasm (12,13). ARID3A rescues the oncogenic
RAS-induced premature senescence in mouse embryonic fibroblasts
(MEFs) (8) and immortalizes MEFs,
and collaborates with oncogenic RAS to transform MEFs. Furthermore,
ARID3A can increase the transcriptional activity of E2F1 (7) and induce cyclin E1, a target of E2F,
which in turn triggers escape from senescence and induces malignant
transformation. A recent study reported that ARID3A binds to RB in
chromatin and sequesters the RB-HDAC1 complex from the E2F1
promoter (14). Similar to
ARID3A, ARID3B has also been shown to immortalize MEFs and confer
malignancy to MEFs in collaboration with N-Myc oncogene (15). ARID3B expression is increased in
human malignancies, including neuroblastoma, ovarian cancer and
thyroid cancer (15-17). Furthermore, accumulating evidence
has indicated that ARID3A and ARID3B are involved in the regulation
of stem cell genes in embryonic stem cells (ESCs) and cancer stem
cells (17-21). ARID3B regulates the expression of
stemness genes to promote cancer stemness in ovarian and head and
neck squamous carcinoma cells (21-26).
ARID3A and ARID3B have similar DNA-binding
properties and recognize specific AT-rich DNA sequences
(ARID3-binding sites, ARID3 BSs) in the matrix attachment region
(MAR) located in the IgH enhancer region (5,9).
The authors have previously reported that both ARID3A and ARID3B
bind to ARID3 consensus BSs in p53 target genes and activate
transcription of p53 target genes (27,28). Although ARID3A and ARID3B have
been shown to bind to E2F1 and RB, respectively, their role in
regulating E2F target gene expression remains largely unknown. The
present study identified putative ARID3 BSs in E2F target genes,
including Cdc2, cyclin E1 and p107. In
addition, the role of ARID3A and ARID3B in E2F-dependent gene
expression and cell cycle progression was examined by using normal
human cells, normal human dermal fibroblasts (NHDFs).
Materials and methods
Cells and cell culture
Neonatal NHDFs (CC-2511) and 293A (R70507) cells
were purchased from Lonza Group AG and Invitrogen; Thermo Fisher
Scientific, Inc., respectively. T98G (CRL-1690), H1299 (CRL-5803),
and Saos-2 (HTB-85) cells were obtained from the American Type
Culture Collection (ATCC). The cells were maintained in Dulbecco's
modified Eagle's medium (DMEM) (Sigma-Aldrich; Merck KGaA)
supplemented with 10% fetal bovine serum (FBS) (Gibco; Thermo
Fisher Scientific, Inc.) and 50 µg/ml gentamicin (Gibco;
Thermo Fisher Scientific, Inc.), and incubated at 37°C in a
humidified atmosphere containing 5% CO2. To induce
quiescence in NHDFs the cells were cultured in DMEM containing 0.1%
FBS for 48 h, and then either left unstimulated or stimulated with
DMEM containing 10% FBS and cultured for an additional 24 h.
Plasmids and transfection
To generate Cdc2-Luc reporter plasmids, the
Cdc2 promoter fragments (-848 to +30) containing either
wild-type, mutated ARID3 (mARID3), or mutated E2F (mE2F) BSs were
synthesized (Genscript Japan Inc.) and then cloned into pGL2
(Promega Corp.). To generate expression vectors for ARID3A and
ARID3B short hairpin RNA (shRNA) (pshARID3A and pshARID3B,
respectively), double-stranded oligonucleotides targeting ARID3A
and ARID3B were cloned into the pcPURU6β (Takara Bio Inc.) vector
downstream of the human U6 promoter. The recombinant plasmids were
confirmed by DNA sequencing. The shRNA sequences are available upon
request. A control shRNA (shCon) expression vector targeting the
luciferase gene was obtained from Takara Bio Inc. Expression
vectors for Xpress-ARID3A, V5-ARID3B and E2F1 were previously as
described (28). T98G, 293A,
H1299 and Saos-2 cells were transfected using the Gene Juice
transfection reagent (Sigma-Aldrich; Merck KGaA) according to the
manufacturer's instructions.
Small interfering RNA (siRNA)
Stealth small interfering RNA (siRNA) duplexes
targeting ARID3A (5′-AACAGAACUCCUG UGUACAUGAUGC-3′) and ARID3B
(5′-UUUCCUUUCAGG AUCACCGUCCAGU-3′) were purchased from Invitrogen,
Thermo Fisher Scientific, Inc. MISSION siRNA universal negative
control (Sigma-Aldrich; Merck KGaA) was used for negative control
duplexes. NHDFs and T98G cells were reverse transfected with 15 nM
siRNA using Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Recombinant adenoviruses
Ad-ARID3A, Ad-E2F1 and Ad-con viruses have been
previously described (29,30).
To generate the Ad-ARID3B virus, pEF-ARID3B was recombined with
pDONR221 vector (Invitrogen; Thermo Fisher Scientific, Inc.) using
BP clonase enzyme mix (Invitrogen; Thermo Fisher Scientific, Inc.).
The resulting ARID3B entry vector was recombined with the
destination vector pAd/CMV/V5-DEST (Invitrogen; Thermo Fisher
Scientific, Inc.) using the ViraPower Adenoviral Gateway Expression
kit (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Amplification, purification and titer
determination of the recombinant adenoviruses were performed as
previously described (29). Cells
were infected at the indicated multiplicity of infection (MOI) in
OPTI-MEM (Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h at
37°C with brief agitation every 5 min, after which the cells were
cultured in DMEM supplemented with either 0.1 or 10% FBS.
Reverse transcription-quantitative PCR
(RTq-PCR)
Total RNA was prepared using the the Direct-zol RNA
miniprep kit (Zymo Research Corp.) according to the manufacturer's
instructions. Complementary DNA was synthesized from 0.5 µg
of total RNA with the ReverTra Ace qPCR RT Master Mix with gDNA
Remover (Toyobo Life Science). qPCR was performed on the
LightCycler 480 real-time PCR instrument (Roche Applied Science) in
triplicate using TaqMan GeneExpression Assays for Cdc2,
cyclin E1, p107 and 18S rRNA (Hs00938778_m1,
Hs01026536_m1, Hs00765700_m1, and Hs03003631_g1, respectively)
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and the
LightCycler 480 Probes Master kit (Roche Applied Science) under the
following conditions: Initial denaturation at 95°C for 2 min,
followed by 50 cycles at 95°C for 15 sec and 60°C for 60 sec,
according to the manufacturer's instructions. The relative
expression of mRNA, normalized to 18S rRNA, was calculated using
the ΔΔCq method (31).
Semiquantitative RT-PCR
PCR was performed with GoTaq Hot Start Green Master
Mix (Promega Corp.) under the following conditions: Initial
denaturation at 95°C for 1 min, followed by 28 cycles for GAPDH, 30
cycles for ARID3A, 34 cycles for ARID3B at 95°C for 40 sec, 55°C
for 40 sec, 72°C for 10 sec, with a final extension at 72°C for 3
min. PCR products were resolved on 2% agarose gels, stained with
ethidium bromide, and visualized by UV transillumination. The
primer sequences used for RT-PCR are presented in Table SI.
Electrophoretic mobility shift assays
(EMSAs)
Reticulocyte lysate was programmed with
Xpress-ARID3A or V5-ARID3B cDNAs using the TNT Coupled Reticulocyte
Lysate System (Promega Corp.) and then incubated with
32P-labeled double-stranded oligonucleotides in 10
µl of a reaction mixture containing 20 mM HEPES (pH 7.9), 40
mM KCl, 6 mM MgCl2, 1 mM EGTA, 0.1% Nonidet P-40, 0.2 mM
DTT, 10% glycerol, 1 µg of Poly(dI-dC), and 30 µg of
bovine serum albumin, and resolved using electrophoresis as
described previously (32). The
reaction mixtures were incubated for 20 min at 25°C and resolved in
a 5% polyacrylamide gel containing 5% glycerol in 0.5X TBE (50 mM
Tris-borate, 1 mM EGTA) for 2 h at 300 V at 4°C.
Chromatin immunoprecipitation (ChIP)
assays
T98G cells were cross-linked with 1% formaldehyde
for 10 min in PBS (pH 7.4). Nuclei preparation and chromatin
digestion were performed using the SimpleChIP Enzymatic Chromatin
IP kit (Cell Signaling Technology, Inc.) according to the
manufacturer's instructions. Nuclei that were digested with
micrococcal nuclease were sonicated with a sonicator (VP-5S; Taitec
Corp.) in wet ice (3 cycles of a 20-sec pulse with a 30-sec
interval). DNA concentration of soluble chromatin was measured
using the Qubit Fluorometer (Invitrogen; Thermo Fisher Scientific,
Inc.). Chromatin immunoprecipitation was performed using the ChIP
Kit Magnetic-One Step kit (Abcam) according to the manufacturer's
instructions. Briefly, soluble chromatin (5 mg) was incubated with
an immunoglobulin G (IgG), anti-ARID3A, or anti-ARID3B antibodies.
qPCR analysis was performed using the BRYT Green and GoTaq qPCR
Master Mix (Promega Corp.) on the LightCycler 480 real-time PCR
instrument. For input control, 2% of the chromatin sample was
amplified. Data were calculated using the ΔΔCq method (31) and presented as fold enrichment
relative to the input.
Luciferase assay
For transient reporter assays, T98G cells in 12-well
plates were co-transfected with 50 ng of reporter constructs and 10
ng of pRL-TK (Promega Corp.), along with 10 ng of E2F1, or 100 ng
of ARID3A or ARID3B expression vectors by GeneJuice Transfection
Reagent (Sigma-Aldrich; Merck KGaA). The total amount of
transfected DNA was adjusted to 500 ng per well by adding control
vectors. Cells were cultured for 48 h in DMEM supplemented with
either 0.1 or 10% FBS before reporter assays. According to the
manufacturer's instructions, luciferase and Renilla
luciferase activities were measured using the Dual-Luciferase
reporter assay system (Promega Corp.). The relative Firefly
luciferase activities were normalized to those of the control
Renilla luciferase.
Western blot analysis and
immunoprecipitation
Whole-cell extract preparation and
immunoprecipitation and the western blot analysis procedures were
performed as previously described (32). T98G cells were collected, lysed in
5 times packed cell volume of whole cell extraction buffer [50 mM
Tris Cl (pH 8.0)/350 mM NaCl/0.5% Nonidet P-40/10% (vol/vol)
glycerol/1 mM dithiothreitol/proteinase inhibitors (Complete; Roche
Biochemicals,), kept on ice for 60 min, and centrifuged at 14,000 ×
g for 10 min at 4°C. The protein concentration of the supernatant
was measured using Bio-Rad protein assay kit (Bio-Rad Laboratories,
Inc.). A total of 80 µg of the lysates was loaded onto an
SDS-PAGE gel (7.5 or 10%), electrophoresed, transferred onto a
polyvinylidene difluoride membrane, blocked with 5% skimmed milk in
TBS-0.1% Tween-20 buffer (TBST) at 4°C overnight, and incubated
with the following primary antibodies at 4°C overnight: Rabbit
polyclonal anti-ARID3A (1:3,000; ref. 29;), anti-ARID3B (1:3,000; A302-564A;
Bethyl Laboratories), anti-E2F-1 (1:3,000; sc-193; Santa Cruz
Biotechnology, Inc.) antibodies, and mouse monoclonal anti-Xpress
(1:5,000; R910-25; Invitrogen, Thermo Fisher Scientific, Inc.),
anti-V5 (1:5,000; 37-7500; Invitrogen, Thermo Fisher Scientific,
Inc.), anti-Cdc2 p34 (1:2,000; sc-54; Santa Cruz Biotechnology,
Inc.), anti-cyclin E (1:2,000; sc-247; Santa Cruz Biotechnology,
Inc.), anti-p107 (1:3,000; sc-250; Santa Cruz Biotechnology, Inc.)
and anti-β-actin (1:1,5,000; A1978; Sigma-Aldrich; Merck KGaA)
antibodies. The membrane was washed with TBST 6 times, reacted with
Amersham ECL HRP-Linked anti-rabbit (1:50,000; NA934; GE
Healthcare) or anti-mouse (1:50,000; NA931; GE Healthcare) antibody
at room temperature for 60 min, washed with TBST buffer 6 times,
and visualized by Amersham ECL Prime Western blotting detection
reagent (GE Healthcare).
Caspase-3/7 assays
Cells were infected with adenoviruses and then
cultured in DMEM containing 0.1% FBS for 48 h, after which
caspase-3/7 activity was determined using the Caspase-Glo 3/7 assay
kit (Promega Corp.) according to the manufacturer's
instructions.
EdU (5-ethynyl-2′-deoxyuridine)
incorporation
NHDFs were reverse transfected with siRNA on
4-chamber glass slides (BD Falcon, Thermo Fisher Scientific, Inc.)
and cultured in DMEM containing 0.1% FBS for 48 h. The cells were
induced to re-enter cell cycle by adding DMEM containing 10% FBS.
The cells were cultured for 24 h, and EdU was added at a 10
µM final concentration 4 h before harvesting the cells. The
detection of incorporated EdU and Hoechst 33342 staining was
performed using the Click-iT EdU Alexa Fluor 488 Imaging Kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions.
Flow cytometric analysis
T98G cells reverse transfected with siRNA were
subjected to serum starvation in DMEM/F-12 medium (Sigma-Aldrich;
Merck KGaA) without FBS for 60 h, then stimulated with fresh DMEM
medium containing 10% FBS. After 24 h, the cells were fixed in 70%
ethanol on ice, re-suspended in PBS containing RNase A (100
µg/ml; Sigma-Aldrich; Merck KGaA), propidium iodide (50
µg/ml) (Sigma-Aldrich; Merck KGaA) and 0.05% Triton X-100,
and then incubated for an additional 15 min at 37°C. Cellular DNA
content was detected using a MoFlo XDPflow cytometer (Beckman
Coulter Inc.) and analyzed using FCS Express 7 software (De Novo
Software Inc.).
Colony formation assay
Colony formation was assayed as previously described
(33). Briefly, since puromycin
susceptibility varies between cell lines, its optimal concentration
of puromycin for 293A and Saos-2 cells were initially determined
(2.5 µg/ml for the 293A cells and 1 µg/ml for the
Saos-2 cells). The cells were transfected with 5 µg plasmid
DNA consisting of either a control, pshARID3A, or pshARID3B
vectors, along with a vector expressing the puromycin resistance
gene at a ratio of 3:1, passaged at a split ratio of 1:3 48 h
following transfection, and then selected with puromycin for 3
weeks. Puromycin-resistant colonies were stained with Giemsa for 10
min at room temperature, and then visualized using a GF-X900
scanner (Epson Inc.).
Statistical analysis
Results are presented as the means ± standard
deviation. Statistical tests were analyzed using both one-way and
two-way ANOVA followed by Tukey's multiple comparisons test, using
GraphPad Prism7 software (GraphPad Software, Inc.). P-values
<0.05 were considered to indicate statistically significant
differences.
Results
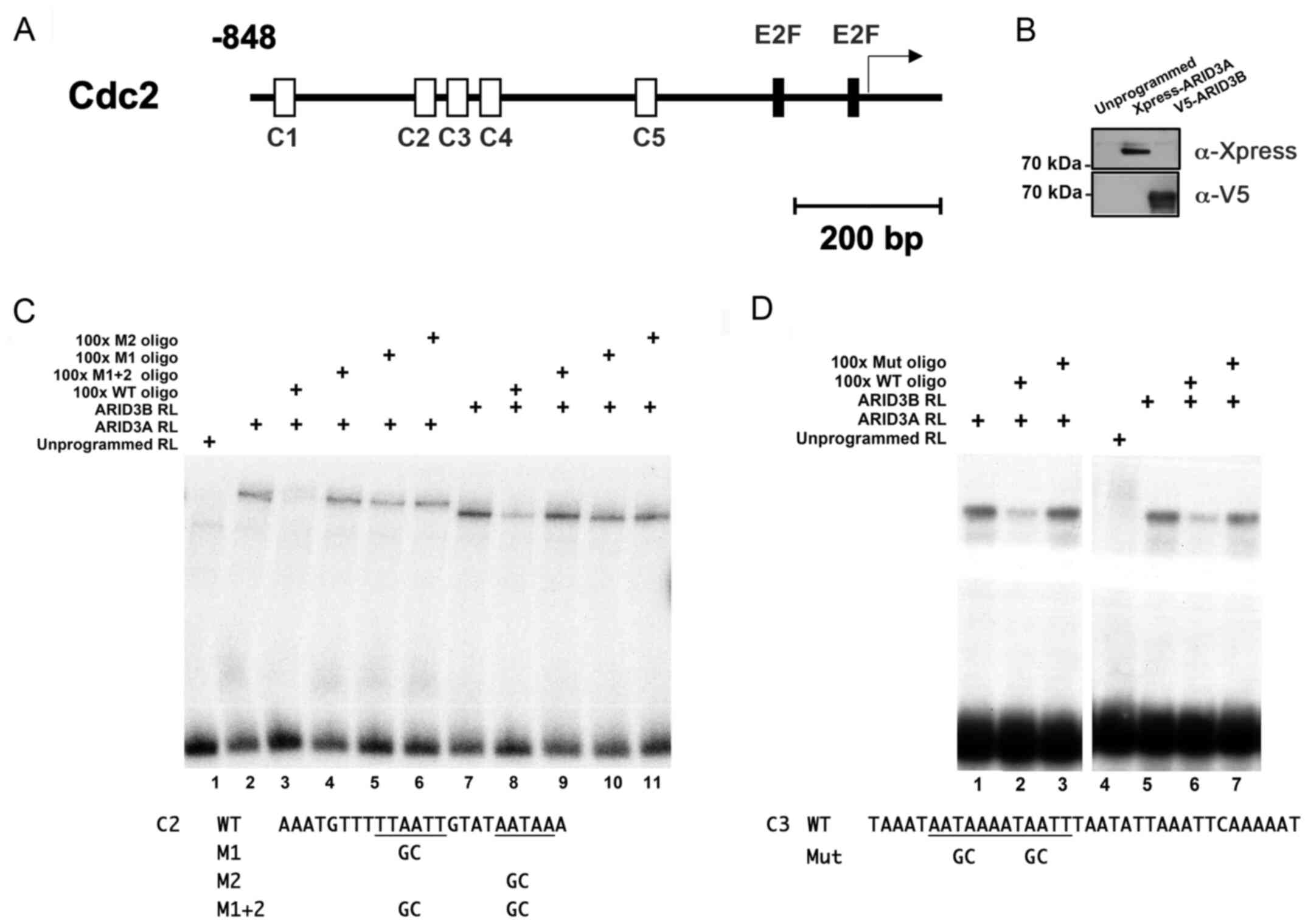
Binding of ARID3A and ARID3B proteins to
ARID3 BSs on the Cdc2 promoter in vitro
To investigate whether ARID3A and ARID3B are
directly involved in E2F target gene expression, the present study
sought to identify BSs for ARID3A and ARID3B in E2F target genes.
Both ARID3 proteins have been shown to bind to the consensus
ARID3A/Bright DNA-binding sites (ARID3 BSs) that contain a core
hexamer ((G/A)AT(T/A)AA) within a region of over 12 bp of
AT/ATC-rich sequences, together with a second AT dimer located near
the hexamer (5,9,27,28). Additionally, ARID3A has been
implicated in spatially distant interactions with regulatory
elements (5). Herein, a cluster
of ARID3 consensus BSs (C1-C5, containing 7 core hexamers) was
found between -833 and -303 upstream the transcription start site
(TSS) of the human Cdc2 gene (Table I and Fig. 1A).
 | Table ILocations of the AT-rich sequences
that matched consensus ARID3-binding sites in humans. |
Table I
Locations of the AT-rich sequences
that matched consensus ARID3-binding sites in humans.
| Cdc2 promoter
designation | 5′-Sequence-3′ | Position |
|---|
| C1 | CTTAAATATAATTAAAACACAAAAATTCACAATTCTAT | -796 to -833 |
| C2 | AAATGTTTTTAATTGTATAATAAA | -620 to -597 |
| C3 | TAAATAATAAAATAATTTAATATTAAATTCAAAAAT | -589 to -554 |
| C4 | ATATAAATAATAAATTTTCCTTTACATTTTT | -531 to -501 |
| C5 | TATTTAGAGTATAATAAATTTGAA | -326 to -303 |
To determine whether ARID3A and ARID3B bind to these
sequences, we performed EMSAs using oligonucleotide probes
corresponding to C2 and C3 (−620 to −597 and −589 to −554,
respectively) with recombinant proteins produced by rabbit
reticulocyte lysates programmed with Xpress-ARID3A and V5-ARID3B
cDNAs (Fig. 1B). The incubation
of ARID3A and ARID3B lysates with C2 and C3 probes, each containing
2 core hexamers, generated shifted bands, whereas the unprogrammed
lysate did not (Fig. 1C and D,
respectively). Competitive EMSA experiments revealed that the
addition of a 100-fold molar excess of unlabeled oligonucleotides
corresponding to the wild-type ARID3 BS inhibited the interaction
between ARID3A/ARID3B proteins and both C2 and C3 sites. By
contrast, competition with unlabeled oligonucleotides in which both
hexamers had been mutated did not affect ARID3A/ARID3B binding to
both sites. Furthermore, ARID3A/ARID3B failed to bind to the C2
probe in which only either one of the hexamers had been mutated (M1
and M2), indicating that the binding to one hexamer depended on the
interactions with the other one (Fig.
1C). Consistent with previous studies, these results indicate
that ARID3A and ARID3B bind to ARID3 BSs in the Cdc2 promoter with
similar DNA-binding properties in vitro (5,9,27,28).
ARID3A and ARID3B activate the Cdc2
promoter depending on both ARID3 and E2F BSs in transient reporter
assays
To investigate the role of ARID3 BSs in regulating
the Cdc2 promoter activity, a luciferase reporter gene
driven by the Cdc2 promoter (-848 relative to the transcription
start site, WT) was constructed and mutations were introduced into
core hexamers in ARID3 BSs in the reporter construct (mARID3)
(Fig. 2A). Mutations were also
introduced to the 2 E2F-binding sites (E2F-BSs), corresponding to
positive- and negative-acting E2F elements (mE2F) (34,35). The T98G cells transfected with the
WT or mutant reporter constructs were incubated under serum-starved
conditions, and then either left unstimulated or stimulated with
serum prior to the reporter assays. The WT Cdc2 reporter
activity increased following serum stimulation, whereas the
mutations in the ARID3 BSs impaired the reporter activity (a 74.8%
reduction relative to the WT control) (Fig. 2B). To examine the effects of E2F1
overexpression on the mutations of ARID3 BSs in the Cdc2
promoter, the reporter constructs were co-transfected with either
an empty or E2F1 expression vector under serum-starved conditions
to reduce endogenous E2F activity. E2F1 overexpression led to a 20-
and 10-fold activation of the reporter activity of WT and mARID3
reporters, respectively, indicating that ARID3 BSs are not
essential for E2F1 to transactivate the Cdc2 promoter;
however, ARID3 BS mutations (mARID3) resulted in a reduction
(43.9%) in the reporter activity relative to the WT (Fig. 2C). These results indicate that
ARID3 BSs play an important role in activating the Cdc2
promoter in response to endogenous and ectopic E2F activity. By
contrast, the E2F BS mutation increased the Cdc2 promoter
activity in quiescent cells, which was not altered in either
serum-stimulated or E2F1-transfected cells, in line with previous
findings demonstrating that the E2F BSs can mediate both the
activation and repression of the Cdc2 promoter (34,35).
To examine the effects of the ectopic expression of
ARID3A and ARID3B on the Cdc2 promoter, cells were
co-transfected with the reporter constructs, along with empty,
ARID3A, or ARID3B expression vector, under either quiescent or
growing conditions. Ectopic ARID3A and ARID3B increased the WT
promoter activity (3.8- and 7.2-fold, respectively) in growing
cells, but not in quiescent cells, and mutations in ARID3 BSs
abolished their transactivation activities (Fig. 2D and E). Of note, the ectopic
expression of ARID3A and ARID3B did not transactivate the mE2F
promoter in both growing and quiescent cells. Furthermore, they
failed to activate the ARID3 BS-containing Cdc2 promoters (WT and
mARID3) under quiescent conditions, where endogenous E2F activities
are low, indicating that ARID3A and ARID3B activate the Cdc2
promoter in an E2F-dependent manner.
ARID3B overexpression induce the
transcription of E2F target genes
The present study then determined whether ARID3A and
ARID3B can transactivate Cdc2 expression in NHDFs using
recombinant adenoviruses expressing ARID3A, ARID3B and E2F1
(Ad-ARID3A, Ad-ARID3B and Ad-E2F1, respectively). The infection of
Ad-ARID3A or Ad-ARID3B exerted no or minimal effects on Cdc2
expression in exponentially growing NHDFs with a high endogenous
E2F activity (data not shown). Therefore, serum-starved quiescent
NHDFs were infected with these adenoviruses and subjected to
RT-qPCR analysis at 24 h post-infection. As shown in Fig. 3, the overexpression of ARID3B and
E2F1 activated Cdc2 expression. The cyclin E1 and
p107 genes were further examined, which have been shown to
be regulated by E2F via E2F BSs on these genes (36-38). Clusters of ARID3 BSs were found on
the cyclin E1 and p107 promoters (-2,481 to -1,105
and -1,568 to -931, respectively). Similar to that observed in
Cdc2, the ectopic expression of ARID3B and E2F1
transactivated cyclin E1 and p107 expression under
quiescent conditions. Furthermore, ARID3B cooperated with E2F1 to
synergistically activate cyclin E1 transcription (Fig. 3E), indicating that ARID3B can also
enhance E2F1 transcriptional activity. Under quiescent conditions,
ARID3B activated endogenous Cdc2 gene expression, but not in
transient reporter assays (Fig.
2E), suggesting that ARID3B may play an important role in
expression of genes integrated into chromatin structure, as
previously reported for ARID3A (39). By contrast, no apparent
upregulation was detected following Ad-ARID3A infection in
quiescent NHDFs, which may be due to cytoplasmic sequestration of
exogenous ARID3A in quiescent cells (12).
Binding of ARID3A and ARID3B to
E2F-responsive genes in living cells
To confirm the implication of ARID3A and ARID3B in
the regulation of E2F-responsive genes in living cells, ChIP assays
we performed using the T98G cells. As shown in Fig. 4A, the cross-linked Cdc2
promoter-ARID3B complexes immunoprecipitated with the antibody
against ARID3B, but not with the control IgG, were detected by qPCR
amplification with primers spanning the ARID3 BSs (-717 to -482) on
the Cdc2 gene. The levels of ARID3B complexes were
significantly higher than those of control ChIPs using primers
spanning a region of the Cdc2 gene which recognizably lacks
ARID3 BSs (+350 to +589). However, significantly higher ARID3A
complexes were not detected with the ARID3 BS primers than the
control primers. A significantly higher ARID3A and ARID3B binding
to the cyclin E1 and p107 promoters was detected with
primers spanning the ARID3 BSs (cyclin E1; −2,517 to −2,332,
p107; −1,590 to −1,344, respectively), compared to the
control primers (Fig. 4B and C).
Lower levels of ARID3A binding were detected on the p107
promoters compared to that of ARID3B. These results suggested that
the role of ARID3A and ARID3B in controlling gene expression may
differ depending on the E2F target genes.
 | Figure 4Roles of ARID3A and ARID3B in E2F
target gene expression. (A-C) Binding of ARID3A and ARID3B to E2F
target genes in living cells. T98G cells were fixed and processed
for ChIP assay using control IgG, anti-ARID3A, or anti-ARID3B
antibodies. Signals amplified by the control and ARID3 primers that
cover the putative ARID3 BSs were measured using qPCR. (D-G and
I-K) The knockdown of ARID3A and ARID3B inhibited transcription of
E2F target genes. NHDFs were transfected with either control,
ARID3A, or ARID3B siRNA (siControl, siARID3A, and siARID3B,
respectively) (D-F). RT-qPCR analysis of the expression levels of
the indicated transcripts at 48 h post-transfection (D-F). (G)
Lysates were immunoprecipitated with ARID3A or ARID3B antibodies,
followed by western blot analysis with the same antibodies. β-actin
was used as a control. (H) The ARID3A and ARID3B knockdown
inhibited the expression of E2F target gene products. T98G cells
were transfected with the indicated siRNA, followed by western blot
analysis to determine the expression levels of the indicated
proteins at 48 h post-transfection. (I-K) ARID3B and ARID3A
knockdown inhibited Cdc2 and cyclin E expression
induced by E2F1. NHDFs were transfected with either control,
ARID3A, or ARID3B siRNA. At 48 h following transfection, the cells
were infected with either Ad-Con or Ad-E2F1 and cultured in DMEM
containing 0.1% FBS for an additional 24 h. (I and J) RT-qPCR
analysis for expression levels of Cdc2 and cyclin E1
and transcripts. (K) Western blot analysis for E2F1 at 24 h
post-infection. β-actin was used as the loading control (upper
panel). Semiquantitative RT-PCR analysis of ARID3A and ARID3B
transcripts at 24 h post-infection. GAPDH was used as an internal
control (lower panel). Data represent the average of 3 independent
experiments, each performed in duplicate and are shown as mean ±
SD. *P<0.05, ***P<0.001; ns, not
significant. ARID, AT-rich interacting domain; NHDFs, normal human
dermal fibroblasts. |
ARID3B knockdown reduces the
transcription of E2F-responsive genes
To examine the role of ARID3A and ARID3B in the
expression of E2F target genes, the effects of the siRNA-mediated
knockdown of endogenous ARID3A or ARID3B on the expression of E2F
target genes were examined. The NHDFs were transfected with either
control siRNA (siCon), specific siRNA against ARID3A (siARID3A), or
ARID3B (siARID3B). The RNA levels of these genes were examined by
RT-qPCR. As shown in Fig. 4D-G,
ARID3B knockdown suppressed the transcription of all tested E2F
target genes, whereas that of ARID3A inhibited the transcription of
cyclin E1 and p107, but not that of Cdc2, in
line with the results of the ChIP assays (Fig. 4A-C), and western blot analysis of
the E2F target gene products following ARID3A or ARID3B knockdown
in T98G cells (Fig. 4H).
To further examine the difference between ARID3A and
ARID3B in controlling the expression of E2F target genes, the
effects of ARID3A and ARID3B knockdown on the E2F1-induced
expression of Cdc2 and cyclin E1 were compared, the
latter of which is a crucial target for ARID3A to rescue cellular
senescence in MEFs (8). NHDFs
that had been transfected with siCon, siARID3A, or siARID3B for 48
h were infected with Ad-con or Ad-E2F1 and then cultured for an
additional 24 h under serum-starved conditions (Fig. 4J). Both ARID3A and ARID3B
knockdown (Fig. 4K) blocked
cyclin E1 transcription following Ad-E2F1 infection, whereas ARID3A
knockdown inhibited Cdc2 transcription to a lesser extent than
ARID3B knockdown did (Fig. 4I and
J). These results suggest that ARID3A and ARID3B are directly
involved in E2F1-mediated transcriptional regulation and that the
roles of ARID3A and ARID3B may differ depending on the E2F target
genes.
Role of ARID3A and ARID3B in cell
proliferation and cell death
ARID3A knockdown has been shown to cause premature
senescence in normal human fibroblasts (40). The data presented above suggested
that ARID3B also plays a critical role in cell cycle progression.
To examine this possibility, quiescent NHDFs transfected with
siCon, siARID3A or siARID3B for 48 h were stimulated to re-enter
the cell cycle by the addition of 10% FBS. At 24 h following growth
stimulation, DNA synthesis was determined by EdU incorporation
assay. Growth stimulation of siCon-transfected quiescent NHDFs
resulted in 49.8% of the cells becoming EdU-positive (Fig. 5A and B). siARID3A and siARID3B
transfection reduced the number of EdU-positive cells to 17.2 and
30.1%, respectively, indicating that ARID3A and ARID3B knockdown
attenuated the ability of quiescent NHDFs to enter the S phase.
Furthermore, cell cycle analysis revealed that the growth
stimulation of siCon-transfected quiescent T98G cells increased the
population of cells in the S and G2/M phases (9.7-fold). However,
the knockdown of ARID3A and ARID3B reduced the S and G2/M cell
population by approximately half (Fig. 5C), indicating that ARID3A and
ARID3B knockdown attenuated the cell cycle entry of T98G cells.
 | Figure 5Role of ARID3A and ARID3B in cell
proliferation and death. (A and B) The knockdown of ARID3A and
ARID3B inhibited DNA synthesis. NHDFs were reverse transfected with
the indicated siRNA. At 16 h following transfection, the cells were
cultured in DMEM containing 0.1% FBS for 48 h, after which the
cells were either left unstimulated or stimulated with 10% FBS and
cultured for an additional 24 h. EdU was added 4 h before
harvesting the cells. (A) Cells were visualized for EdU
incorporation or DNA staining with Hoechst 33342 using fluorescence
microscopy. (B) Quantification of EdU incorporation. More than 200
Hoechst 33342-stained nuclei per sample were scored. Data represent
the mean percentages of EdU-positive cells obtained from three
independent experiments. Data are shown as means ± SD. (C) The
ARID3A and ARID3B knockdown inhibited cell cycle progression. T98G
cells reverse transfected with the indicated siRNA were
serum-starved for 60 h, after which they were either left
unstimulated or stimulated with 10% FBS and cultured for an
additional 24 h, followed by flow cytometric analysis. (D) ARID3A
and ARID3B knockdown inhibited tumor cell growth. 293 and Saos-2
cells were transfected with vectors expressing either control
shCFF, shARID3A, or shARID3B and then cultured in the presence of
puromycin for three weeks. Images of plates showing colony
formation assays in 293 and Saos-2 cells. Puromycin-resistant
colonies were visualized using Giemsa staining (left panel).
Semi-quantitative RT-PCR for ARID3A and ARID3B in H1299 cells 48 h
following transfection. GAPDH was used as an internal control
(right panel). (E) NHDFs were infected with the indicated
combination of adenoviruses at 80 MOI and then cultured in DMEM
containing 0.1% FBS. At 48 h post-transfection, cells were
subjected to caspase-3/7 analysis. Data present the average of 3
independent experiments, each performed in duplicate, and are shown
as the means ± SD. *P<0.05, ***P<0.001;
ns, not significant. ARID, AT-rich interacting domain; NHDFs,
normal human dermal fibroblasts. |
To further confirm the role of ARID3A and ARID3B in
cell proliferation, colony formation assays were performed using
shRNA expression vectors that target ARID3A and ARID3B sequences
(shARID3A and shARID3B, respectively). Both ARID3A and ARID3B have
been shown to be involved in transcriptional regulation of the Rb
and p53 tumor suppressor pathways (7-7,14,27-29). To determine RB- and
p53-independent effects of ARID3A and ARID3B knockdown on cell
proliferation, the Saos-2 and 293A cell lines were used, in which
both Rb and p53 pathways are inactivated due to gene deletions and
adenovirus EIA/EIB expression, respectively. The cells were
transfected with plasmids expressing shARID3B and shARID3B and then
cultured in the presence of puromycin for 3 weeks. A control shRNA
vector targeting the luciferase gene (CFF) was used as a negative
control. As shown in Fig. 5D,
ARID3A and ARID3B knockdown resulted in a substantial reduction in
the colony formation of both cell lines.
Previous studies have demonstrated that ARID3B has a
unique function in inducing cell death (28,44). The present study
thus analyzed the effects of ARID3A and ARID3B expression on
E2F1-induced cell death. As shown in Fig. 5E, infection with Ad-E2F1 and
Ad-ARID3B, but not Ad-ARID3A, induced caspase-3/7 activity.
Furthermore, ARID3B and E2F1 co-operatively induced caspase-3/7
activity. These results demonstrate the overlapping and distinct
roles of these ARID3 proteins in promoting cell proliferation and
inducing cell death, respectively.
Discussion
The cooperative interactions of E2F with other
transcription factors regulates the expression of E2F-dependent
genes, facilitating the tighter regulation of gene expression
(34,35,41,42). ARID3A and ARID3B have been
originally identified as E2F1 and RB binding proteins,
respectively. However, their direct involvement in controlling the
E2F target gene has not been clarified. The present study
demonstrated the cooperation of E2F with ARID3A and ARID3B to
activate E2F-dependent transcription via the direct binding to E2F
target genes. Clusters of ARID3 BSs upstream of the transcription
start sites of certain E2F target genes were found, including
Cdc2, cyclin E1 and p107. Both ARID3 proteins
activated the Cdc2 promoter depending on not only on ARID3
BSs, but also on E2F BSs, indicating that ARID3A and ARID3B require
cooperation with E2F to activate the Cdc2 promoter. The
requirement of their interaction with specific transcription
factors to activate transcription is supported by a previous study
reporting that ARID3A was unable to activate a reporter gene driven
by tandemly repeated ARID3 consensus BSs (39). In B lymphocytes, ARID3A functions
as a tether to bring TFII-I and Btk adjacent to the IgH promoter
thereby activating transcription. By contrast, E2F1 activated the
Cdc2 promoter without ARID3 BSs, since E2F BSs alone are
sufficient for E2F1 to activate transcription. In the absence of
ARID3 BSs, however, the promoter activity was significantly
reduced, indicating that ARID3 BSs play a vital role in the
complete activation of the Cdc2 promoter in response to E2F
activity.
ARID3B induced endogenous E2F target genes, but not
the transiently transfected Cdc2 promoter under quiescent
conditions, suggesting that ARID3B is involved in controlling gene
expression by altering chromatin structure. ARID3A is activated
only when the MAR-containing IgH S107 promoter is integrated into
chromatin (39). Similarly, the
authors have previously reported that ARID3A did not activate the
transiently transfected promoter of p21, a p53 target gene,
but activated the stably transfected p21 promoter integrated
into the chromatin (27). ARID3A
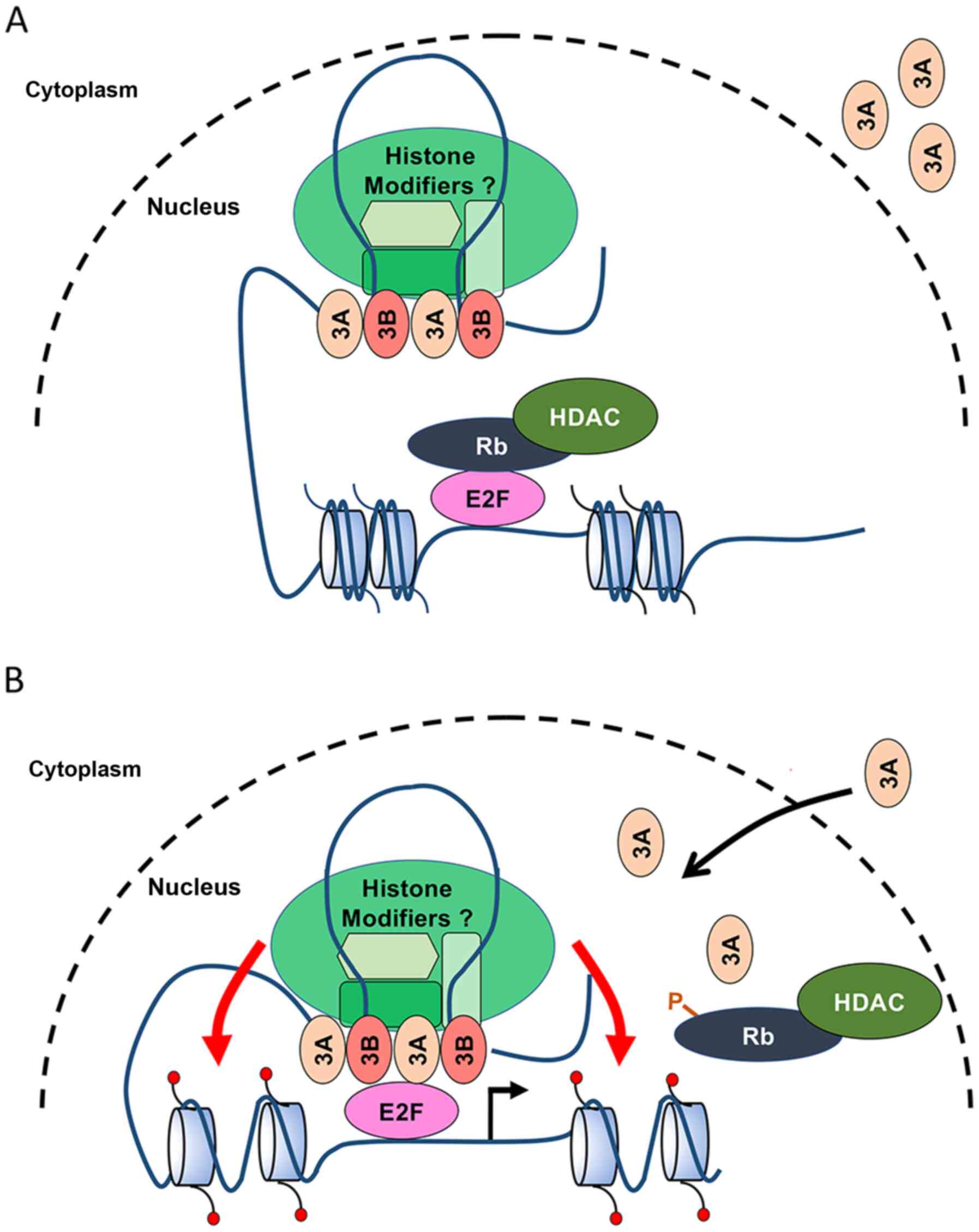
and ARID3B have been shown to bind to histone-modifying enzymes,
including histone deacetylase (HDAC1 and HDAC2) and histone
demethylase 4C (KDM4C) (Fig. 6)
(14,21,43). ARID3A also binds to histone
deacetylase HDAC1 and sequester the RB-HDAC1 complexes from the
E2F1 promoter (14). RB
controls local promoter activity and also chromatin structure
through physical interactions between histone modifiers and
chromatin-bound proteins. Further investigations are warranted for
determining whether ARID3B regulates E2F target genes by disrupting
the RB-E2F repressor complexes or by hitherto unrecognized
epigenetic functions in chromatin remodeling.
It was also found that ARID3 consensus BSs in the
E2F target genes are clustered >300 bp upstream of TSS (Cdc2,
-303 to -833; cyclin E1, -1,105 to -2,481; p107, -931 to -1,568),
which is in contrast to the previously reported mechanism of
E2F-dependent gene activation and repression that involve other
transcription factor BSs located adjacent to E2F BSs (34,40-42). It has been reported that ARID3A
distorts the DNA structure by bending DNA up to 90°, allowing it to
interact with spatially distant regulatory elements of the
IgH gene (5,39). Moreover, ARID3A exists as a stable
tetramer that could bind to 2 ARID3 BSs. These findings suggest
that ARID3A can interact with spatially separated BSs and bring the
enhancer close to the promoter by interacting with additional
DNA-binding proteins (Fig. 6).
Considering the highly conserved functional domains and
heterodimerization between ARID3A and ARID3B, both ARID3 proteins
may promote E2F target gene expression by bringing ARID3 BSs close
to E2F BSs by interacting with E2Fs and other chromatin
modifiers.
The present study demonstrated that the roles of
ARID3A and ARID3B in controlling E2F target gene expression
differed, depending on the E2F target gene. It appears that ARID3B
may play a more important role in the expression of Cdc2
compared to ARID3A. By contrast, both ARID3A and ARID3B were
indispensable for cyclin E1 expression, a critical target
for ARID3A to rescue MEFs from replicative senescence or
RAS-induced premature senescence (8,14).
The fact that ARID3B cooperates with E2F1 to activate cyclin
E1 transcription and immortalizes MEFs (15), suggests that ARID3B may have a
similar function in senescence rescue. The unique functions of
ARID3B are indicated by the fact that ARID3B, but not ARID3A,
cooperates with E2F1 to induce cell death, which is consistent with
the findings of previous studies (28,44). In line with this result,
phenotypes of ARID3A and ARID3B knockout mice differ. Furthermore,
ARID3A is strongly expressed in the lung and spleen and at lower
levels in various other tissues, whereas ARID3B is expressed
ubiquitously (9,17). These observations support the
notion that ARID3A and ARID3B have overlapping and distinct
functions.
Additionally, the regulation of subcellular
localization of ARID3A and ARID3B may contribute to the differences
in their function. Although stably transfected ARID3A induces E2F1
transcriptional activity (8,14),
transient ARID3A overexpression failed to activate cyclin E1, as
well as Cdc2 and p107 genes under quiescent conditions. Unlike
ARID3B, which is localized in the nucleus, ARID3A actively shuttles
between the nucleus and the cytoplasm in the S phase. Thus,
overexpressed ARID3A protein may be retained in the cytoplasm under
quiescent conditions. The regulation of ARID3A nucleocytoplasmic
shuttling may regulate E2F target gene expression during the cell
cycle. Alternatively, epigenetic changes induced by ARID3A, such as
remodeling of chromatin structures, may require to activate gene
expression, as suggested by previous studies (8,14).
ARID3A and ARID3B have been shown to regulate stem
cell genes in ovarian cancer and head and neck squamous carcinoma
cells (18,20-26). ARID3A binds to the OCT4/SOX2
binding site and an A/T-rich sequence, in addition to ARID3 BSs in
the promoter/enhancer regions of pluripotency factors, Oct4, Sox2,
and Nanog in MEFs (20). In
ARID3B-overexpressing ovarian cancer cells, ChIP-seq experiments
have shown that ARID3B binds to a consensus binding motif of
5′-TGGGATTACAG-3′ (23). This
differs from the previously established ARID3 consensus BSs,
possibly due to differences in cell types or ARID3A/ARID3B binding
partners. Furthermore, both ARID3A and ARID3B are regulated by
lethal-7 (let-7), a tumor-suppressor microRNA, and recruit KDM4C
onto target genes, contributing to H3K9 demethylation and promotion
of transcription of stemness genes (21). These studies, together with the
results presented herein, indicate that the dysregulation of ARID3A
and ARID3B in ARID3B-overexpressing or let-7-deficient cancer cells
may disrupt the regulation of the RB-E2F and stem cell pathways,
which contributes to their oncogenic activities in human
malignancies.
There are some limitations as regards the number of
E2F target genes examined in the present study that must be
addressed in future research. The authors observed the presence of
consensus BSs for ARID3 proteins at a distance from TSS in many E2F
target genes, even though their functional roles in transcriptional
regulation of E2F target genes remain undetermined. Given the
implication of ARID3A and ARID3B in spatially distant interactions
with regulatory elements, further studies are warranted to analyze
the formation of tertiary structures of both ARID3 proteins and E2F
target genes, along with other chromatin modifiers, and elucidate
the extent to which ARID3A and ARID3B are involved in the
regulation of E2F target genes. Furthermore, the present study
highlights the need for investigations directed at a comprehensive
understanding of the mechanisms underlying epigenetic regulation by
the ARID3, E2F and RB family of proteins, which could provide
deeper insight into the regulation of cell proliferation,
differentiation and tumorigenesis.
Supplementary Data
Availability of data and materials
The data used and/or analyzed during the current
study are presented in the manuscript or are available from the
corresponding author on reasonable request.
Authors' contributions
KASMS and MAI conceived and designed the
experiments. KASMS, TM, MT and MAI performed the experiments.
KASMS, WL, EP and MAI analyzed the data. WL and SI interpreted the
data. MAI wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by JSPS KAKENHI (grant nos.
JP21390502, JP24659870 and JP19K10259).
Abbreviations:
|
ARID
|
AT-rich interacting domain
|
|
BSs
|
binding sites
|
|
NHDFs
|
normal human dermal fibroblasts
|
|
RB
|
retinoblastoma tumor suppressor
protein
|
|
MEFs
|
mouse embryonic fibroblasts
|
|
MAR
|
matrix attachment region
|
|
siRNA
|
small interfering RNA
|
|
shRNA
|
short hairpin RNA
|
|
MOI
|
multiplicity of infection
|
|
TSS
|
transcription start site
|
|
EdU
|
5-ethynyl-2′-deoxyuridine
|
References
|
1
|
Dimova DK and Dyson NJ: The E2F
transcriptional network: Old acquaintances with new faces.
Oncogene. 24:2810–2826. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
DeGregori J and Johnson DG: Distinct and
overlapping roles for E2F family members in transcription,
proliferation and apoptosis. Curr Mol Med. 6:739–748.
2006.PubMed/NCBI
|
|
3
|
Iaquinta PJ and Lees JA: Life and death
decisions by the E2F transcription factors. Curr Opin Cell Biol.
19:649–657. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kent LN and Leone G: The broken cycle: E2F
dysfunction in cancer. Nat Rev Cancer. 19:326–338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Herrscher RF, Kaplan MH, Lelsz DL, Das C,
Scheuermann R and Tucker PW: The immunoglobulin heavy-chain
matrix-associating regions are bound by Bright: A B cell-specific
trans-activator that describes a new DNA-binding protein family.
Genes Dev. 9:3067–3082. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kortschak RD, Reimann H, Zimmer M, Eyre
HJ, Saint R and Jenne DE: The human dead ringer/bright homolog,
DRIL1: cDNA cloning, gene structure, and mapping to D19S886, a
marker on 19p13.3 that is strictly linked to the Peutz-Jeghers
syndrome. Genomics. 51:288–292. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suzuki M, Okuyama S, Okamoto S, Shirasuna
K, Nakajima T, Hachiya T, Nojima H, Sekiya S and Oda K: A novel E2F
binding protein with Myc-type HLH motif stimulates E2F-dependent
transcription by forming a heterodimer. Oncogene. 17:853–865. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Peeper DS, Shvarts A, Brummelkamp T, Douma
S, Koh EY, Daley GQ and Bernards R: A functional screen identifies
hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat Cell
Biol. 4:148–153. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Numata S, Claudio PP, Dean C, Giordano A
and Croce CM: Bdp, a new member of a family of DNA-binding
proteins, associates with the retinoblastoma gene product. Cancer
Res. 59:3741–3747. 1999.PubMed/NCBI
|
|
10
|
Webb C, Zong RT, Lin D, Wang Z, Kaplan M,
Paulin Y, Smith E, Probst L, Bryant J, Goldstein A, et al:
Differential regulation of immunoglobulin gene transcription via
nuclear matrix-associated regions. Cold Spring Harb Symp Quant
Biol. 64:109–118. 1999. View Article : Google Scholar
|
|
11
|
Kortschak RD, Tucker PW and Saint R: ARID
proteins come in from the desert. Trends Biochem Sci. 25:294–299.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim D and Tucker PW: A regulated
nucleocytoplasmic shuttle contributes to Bright's function as a
transcriptional activator of immunoglobulin genes. Mol Cell Biol.
26:2187–2201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim D, Probst L, Das C and Tucker PW:
REKLES is an ARID3-restricted multifunctional domain. J Biol Chem.
282:15768–15777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmidt C, Kim D, Mathur S, Covarrubias D,
Das C, Brown MA, Storsberg J and Tucker H: The Arid3a transcription
factor rescues natural and RAS-V12-induced senescence via a
Rb-dependent pathway. Am J Immunol. 13:216–232. 2017. View Article : Google Scholar
|
|
15
|
Kobayashi K, Era T, Takebe A, Jakt LM and
Nishikawa S: ARID3B induces malignant transformation of mouse
embryonic fibroblasts and is strongly associated with malignant
neuroblastoma. Cancer Res. 66:8331–8336. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cowden Dahl KD, Dahl R, Kruichak JN and
Hudson LG: The epidermal growth factor receptor responsive miR-125a
represses mesenchymal morphology in ovarian cancer cells.
Neoplasia. 11:1208–1215. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Samyesudhas SJ, Roy L and Cowden Dahl KD:
Differential expression of ARID3B in normal adult tissue and
carcinomas. Gene. 543:174–180. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kobayashi K, Jakt LM and Nishikawa SI:
Epigenetic regulation of the neuroblastoma genes, Arid3b and Mycn.
Oncogene. 32:2640–2648. 2013. View Article : Google Scholar :
|
|
19
|
Wang J, Rao S, Chu J, Shen X, Levasseur
DN, Theunissen TW and Orkin SH: A protein interaction network for
pluripotency of embryonic stem cells. Nature. 444:364–368. 2006.
View Article : Google Scholar : PubMed/NCBI
Popowski M, Templeton TD, Lee BK, Rhee C,
Li H, Miner C, Dekker JD, Orlanski S, Bergman Y, et al:
Bright/Arid3A acts as a barrier to somatic cell reprogramming
through direct regulation of Oct4, Sox2, and Nanog. Stem Cell
Reports. 2:26–35. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liao TT, Hsu WH, Ho CH, Hwang WL, Lan HY,
Lo T, Chang CC, Tai SK and Yang MH: let-7 modulates chromatin
configuration and target gene repression through regulation of the
ARID3B complex. Cell Rep. 14:520–533. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roy L, Samyesudhas SJ, Carrasco M, Li J,
Joseph S, Dahl R and Cowden Dahl KD: ARID3B increases ovarian tumor
burden and is associated with a cancer stem cell gene signature.
Oncotarget. 5:8355–8366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bobbs A, Gellerman K, Hallas WM, Joseph S,
Yang C, Kurkewich J and Cowden Dahl KD: ARID3B directly regulates
ovarian cancer promoting genes. PLoS One. 10:e01319612015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chien CS, Wang ML, Chu PY, Chang YL, Liu
WH, Yu CC, Lan YT, Huang PI, Lee YY, Chen YW, et al: Lin28B/Let-7
regulates expression of Oct4 and Sox2 and reprograms oral squamous
cell carcinoma cells to a stem-like state. Cancer Res.
75:2553–2565. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roy L, Bobbs A, Sattler R, Kurkewich JL,
Dausinas PB, Nallathamby P and Cowden Dahl KD: CD133 promotes
adhesion to the ovarian cancer metastatic niche. Cancer Growth
Metastasis. 11:11790644187678822018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dausinas P, Pulakanti K, Rao S, Cole JM,
Dahl R and Cowden Dahl KD: ARID3A and ARID3B induce stem promoting
pathways in ovarian cancer cells. Gene. 738:1444582020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lestari W, Ichwan SJ, Otsu M, Yamada S,
Iseki S, Shimizu S and Ikeda MA: Cooperation between ARID3A and p53
in the transcriptional activation of p21WAF1 in response to DNA
damage. Biochem Biophys Res Commun. 417:710–716. 2012. View Article : Google Scholar
|
|
27
|
Pratama E, Tian X, Lestari W, Iseki S,
Ichwan SJ and Ikeda MA: Critical role of ARID3B in the expression
of pro-apoptotic p53-target genes and apoptosis. Biochem Biophys
Res Commun. 468:248–254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma K, Araki K, Ichwan SJ, Suganuma T,
Tamamori-Adachi M and Ikeda MA: E2FBP1/DRIL1, an AT-rich
interaction domain-family transcription factor, is regulated by
p53. Mol Cancer Res. 1:438–444. 2003.PubMed/NCBI
|
|
29
|
Schwarz JK, Bassing CH, Kovesdi I, Datto
MB, Blazing M, George S, Wang XF and Nevins JR: Expression of the
E2F1 transcription factor overcomes type beta transforming growth
factor-mediated growth suppression. Proc Natl Acad Sci USA.
92:483–487. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
31
|
Ikeda MA, Jakoi L and Nevins JR: A unique
role for the Rb protein in controlling E2F accumulation during cell
growth and differentiation. Proc Natl Acad Sci USA. 93:3215–3220.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ichwan SJ, Yamada S, Sumrejkanchanakij P,
Ibrahim-Auerkari E, Eto K and Ikeda MA: Defect in serine 46
phosphorylation of p53 contributes to acquisition of p53 resistance
in oral squamous cell carcinoma cells. Oncogene. 25:1216–1224.
2006. View Article : Google Scholar
|
|
33
|
Freedman JA, Chang JT, Jakoi L and Nevins
JR: A combinatorial mechanism for determining the specificity of
E2F activation and repression. Oncogene. 28:2873–2881. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu W, Giangrande PH and Nevins JR: hu W,
Giangrande PH and Nevins JR: E2Fs link the control of G1/S and G2/M
transcription. EMBO J. 23:4615–4626. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ohtani K, DeGregori J and Nevins JR:
Regulation of the cyclin E gene by transcription factor E2F1. Proc
Natl Acad Sci USA. 92:12146–12150. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Smith EJ, Leone G and Nevins JR: Distinct
mechanisms control the accumulation of the Rb-related p107 and p130
proteins during cell growth. Cell Growth Differ. 9:297–303.
1998.PubMed/NCBI
|
|
37
|
Takahashi Y, Rayman JB and Dynlacht BD:
Analysis of promoter binding by the E2F and pRB families in vivo:
Distinct E2F proteins mediate activation and repression. Genes Dev.
14:804–816. 2000.PubMed/NCBI
|
|
38
|
Kaplan MH, Zong RT, Herrscher RF,
Scheuermann RH and Tucker PW: Transcriptional activation by a
matrix associating region-binding protein. contextual requirements
for the function of bright. J Biol Chem. 276:21325–21330. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fukuyo Y, Takahashi A, Hara E, Horikoshi
N, Pandita TK and Nakajima T: E2FBP1 antagonizes the p16(INK4A)-Rb
tumor suppressor machinery for growth suppression and cellular
senescence by regulating promyelocytic leukemia protein stability.
Int J Oral Sci. 3:200–208. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Araki K, Nakajima Y, Eto K and Ikeda MA:
Distinct recruitment of E2F family members to specific E2F-binding
sites mediates activation and repression of the E2F1 promoter.
Oncogene. 22:7632–7641. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Schlisio S, Halperin T, Vidal M and Nevins
JR: Interaction of YY1 with E2Fs, mediated by RYBP, provides a
mechanism for specificity of E2F function. EMBO J. 21:5775–5786.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rhee C, Lee BK, Beck S, Anjum A, Cook KR,
Popowski M, Tucker HO and Kim J: Arid3a is essential to execution
of the first cell fate decision via direct embryonic and
extraembryonic transcriptional regulation. Genes Dev. 28:2219–2232.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Joseph S, Deneke VE and Cowden Dahl KD:
ARID3B induces tumor necrosis factor alpha mediated apoptosis while
a novel ARID3B splice form does not induce cell death. PLoS One.
7:e421592012. View Article : Google Scholar : PubMed/NCBI
|