Introduction
Acute myeloid leukemia (AML) is a complex
hematological disorder characterized by blockage of differentiation
and high proliferation rates of myeloid progenitors, leading to
bone marrow (BM) failure (1,2).
AML has an incidence of ~20,000 new cases per year in the United
States (3) and is associated with
very high mortality rates (4,5).
AML incidence rates in Brazil vary according to region, mainly due
to socioeconomic inequalities that impact healthcare access and
appropriate diagnosis. Although Brazilian records do not
differentiate the incidence between leukemia types, 5,920 cases in
men and 4,890 in women are expected in 2020-2022 for all types of
leukemia, and AML is the most common type of leukemia in adults
(6). The reduced overall survival
(OS) rate of patients with AML is intrinsically related to
resistance of leukemia cells to therapy, with the majority of
patients initially achieving remission and later progressing to a
more aggressive disease (7,8).
The standard therapy for AML, which includes
anthracycline and cytarabine, has remained unchanged over the last
40 years; however, it is inefficient at prolonging OS and curing a
large majority of patients, except for those with promyelocytic
leukemia (9,10). Pyrimidine analogues, such as
cytarabine, are the key drugs used to treat AML and present a
chemical structure similar to cytosine (11,12). Despite efforts made to understand
the mechanisms underlying cytarabine resistance in AML, only a few
advances have been made (13,14), this may be because it involves
multiple molecular mechanisms (15). Some pathways involved in apoptosis
evasion, including increased levels of the inhibitor of apoptosis
proteins (IAPs) and imbalanced expression of Bcl-2 family proteins,
have been reported to have key roles in cytarabine resistance and
poor prognosis (16-19).
Other pathways involved in chemotherapy resistance
involve dysregulated expression and function of transcription
factors, such as c-Myc and NF-κB (20-22). In addition, c-Myc expression has
been associated with BM stromal cell-mediated resistance in AML
cell lines, and high c-Myc expression has been associated to high
cytogenetic risk and poor prognosis in patients with AML (23,24).
Since most patients develop resistance to AML
standard therapy, the search for alternative treatment options is
essential. The LQB-118 synthetic compound has been reported to
exert antitumoral effects on AML cells with a multidrug resistant
(MDR) profile. The LQB-118 compound has also exhibited cytotoxic
effects on chronic myeloid leukemia (CML) MDR cell lines and
patient samples. Its antitumor activity in AML and CML cells has
been associated with reduced expression of survivin and X-linked
IAP (XIAP) anti-apoptotic proteins, and modulation of NF-κB
subcellular localization (25-29).
The present study developed an in vitro AML
cell line model resistant to cytarabine and investigated the
mechanisms underlying the development of chemoresistance. Moreover,
the potential antitumor effect of LQB-118 on a cytarabine-resistant
AML cell line was assessed; to the best of our knowledge, this has
not been previously investigated. The present study aimed to better
understand the mechanisms underlying cytarabine resistance in an
AML-resistant cell line and investigated the potential antitumor
effect of LQB-118 compound in a cytarabine-resistant cell line.
Materials and methods
Cell culture, generation of a
cytarabine-resistant cell line and drug treatment
HL-60 (ATCC® CCL-240™/P53 null; American
Type Culture Collection) and HL-60R human cell lines (FAB M2) were
cultured in RPMI (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% heat-inactivated fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.) and 2 mM L-glutamine
(Sigma-Aldrich; Merck KGaA). The HL-60R cell line was obtained
through culturing HL-60 parental cells with increasing doses of
cytarabine (Accord Farmacêutica Ltd.). Concentrations ranged
between 0.001 and 50 µM, doubling every three passages, for
6 months. The cell lines were maintained in a humidified incubator
(Thermo Fisher Scientific, Inc.) at 37°C and 5% CO2, and
the HL-60R cell line was continuously cultured with 50 µM
cytarabine. The drug was withdrawn 24 h prior to subsequent assays.
Cell lines were tested for Mycoplasma contamination by PCR
and the genotypes were confirmed by short tandem repeat. The
LQB-118 synthetic compound was developed and produced by Dr Paulo
Costa and Dr Chaquip Netto in the Laboratory of Bioorganic
Chemistry at the Natural Products Research Institute of the Federal
University of Rio de Janeiro (Rio de Janeiro, Brazil) (29). For experiments with the LQB-118
compound, HL-60 and HL-60R cells were incubated with different
concentrations of the compound (1.5, 3, 6 and 9 µM for MTT
assay; 3 µM for other assays) for 24 h at 37°C. Untreated
cells (cultures in drug-free media) or cells treated with the
vehicle dimethyl sulfoxide (DMSO) were used as experimental
controls.
Cytotoxicity assay
HL-60 and HL-60R cell viability was evaluated using
the MTT assay following treatment with cytarabine, idarubicin
(Chemicaltech Farmacêutica), daunorubicin (Farmarin) or with the
LQB-118 compound. A total of 2×104 cells/well were
seeded in 96-well plates and treated with increasing drug
concentrations. After 24, 48 and 72 h, cells were incubated with
the MTT reagent (Sigma-Aldrich; Merck KGaA) in a humidified
incubator at 37°C and 5% CO2 for 4 h. Formazan crystals
were then solubilized in DMSO and the absorbance was measured at
570 nm using a spectrophotometer (EZ Read 400; Biochrom Ltd.).
Cell cycle progression and cell death
detection
For assessment of cell cycle progression and cell
death following treatment with cytarabine or LQB-118, cells were
incubated with 50 µM cytarabine for 48 h or 3 µM
LQB-118 for 24 h. Subsequently, 2×105 cells were
centrifuged (750 × g at room temperature for 3 min), washed with
PBS (pH 7.4) and incubated with Annexin V, Alexa Fluor™ 488
conjugate (cat. no. A13201; Invitrogen; Thermo Fisher Scientific,
Inc.) for 15 min at room temperature. Subsequently, propidium
iodide (PI) was added to the cells and cell death index was
evaluated by assessing the Annexin V/PI-marked cells by flow
cytometry. In order to evaluate the total apoptosis rate induced by
the compounds, early (Annexin V+/PI−) and
late (Annexin V+/PI+) apoptosis was
considered. Cell cycle progression was evaluated by PI DNA
incorporation. After treatment with cytarabine or LQB-118 compound,
3×105 cells were centrifuged (750 × g at room
temperature for 3 min), washed with PBS, and incubated with 100
µg/ml RNAse (100 µg/ml ribonuclease A diluted in 40
mM citrate buffer; Sigma-Aldrich; Merck KGaA) and 50 µg/ml
PI (50 µg/ml diluted in 40 mM citrate buffer with 0.3%
Triton X-100) for 15 min at room temperature. Finally, cells were
analyzed in a flow cytometer. Cells in both assays were assessed
using the CyAn ADP analyzer flow cytometer and Summit v4.3 software
was used for analysis (both from Beckman Coulter, Inc.). Three
independent experiments were performed.
Karyotype evaluation
For karyotyping, cells were initially cultured in a
concentration of 1×107 cells/ml for 24 h. A total of 2 h
before the end of this incubation, cells were treated with
colchicine (0.05 µg/ml) and maintained in a humidified
incubator at 37°C and 5% CO2 for 1 h. Subsequently,
cells were incubated with hypotonic solution (0.075 M KCl) for 15
min for chromosome preparation, followed by a fixation step with
Carnoi solution (3:1, methanol:acetic acid) for 20 min at room
temperature. To obtain GTG banding patterns, the slides were
incubated for 10-14 sec at room temperature in 0.1% trypsin
solution in Dulbecco's solution [0.137 M NaCl, 0.0027 M KCl, 0.0015
M KH2PO4, 0.011 M
NaH2PO4 (pH 6.8)], washed with saline
solution (0.9% NaCl) and stained with 2% Giemsa (Merck KgaA)
solution in phosphate buffer for 15 min at room temperature.
Chromosomes were identified and classified according to the
International System of Nomenclature of Human Cytogenetics 2016
(30) and chromosomal analysis
was performed under optical microscopy using at least 30 metaphases
per cell line. The images were acquired through the Cytovision
Applied Image Karyotyping System (Leica Microsystems, Inc.) for at
least 5 to 10 metaphases for the assembly of karyotypes.
Analysis of FLT3 internal tandem
duplications (ITDs), and CEBPA, DNMT3A, IDH1, IDH2 and NPM1 gene
mutations
DNA was extracted from 1×107 HL-60 and
HL-60R cells using the automated Maxwell® system
(Promega Corporation). Genomic regions of interest were amplified
by PCR using specific primers for CEBPA, DNMT3A exon
23, IDH1 exon 4 and IDH2 exon 4. For amplification of
targeted regions in CEBPA, PCR was performed using 1 unit
Platinum™ Taq DNA Polymerase High Fidelity, 1X PCR buffer, 2 mM
MgSO4, 400 µM dNTPs (all from Thermo Fisher
Scientific, Inc.), 1 M betaine, 400 nM each primer (Integrated DNA
Technologies, Inc.) and 0.1 µg DNA. The thermal profile for
amplification was: 94°C for 5 min; 34 cycles [94°C for 30 sec; 65°C
for 30 sec; 68°C for 1 min]; 68°C for 10 min. For amplification of
targeted regions in DNMT3A and IDH1/2, the PCR reactions were
performed using Taq polymerase (1 unit for DNMT3A or 2.5 units for
IDH1/2), 1X PCR buffer, 1.5 mM MgCl2, 250 µM
dNTPs (all from Thermo Fisher Scientific, Inc.), 400 nM each primer
(Integrated DNA Technologies, Inc.) and 0.1 µg DNA. The
thermal profile for amplification was: 94°C for 3 min; 35 cycles
(94°C for 30 sec; 60°C for 30 sec; 72°C for 1 min); 72°C for 10
min. Primer sequences are provided in Table SI. After purification with
PureLink PCR purification kit (Invitrogen; Thermo Fisher
Scientific, Inc.), amplicons were submitted to direct sequencing
with BigDye Terminator v3.1 Cycle Sequencing kit (Thermo Fisher
Scientific, Inc.) on an ABI 3130xl Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Sequence data files
were analyzed using Mutation Surveyor software V4.0.9
(SoftGenetics, LLC).
For FLT3 and NPM1 gene expression
analysis, RNA was isolated from 5×106 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). For cDNA synthesis, reverse transcription (RT) was performed
using 200 units ImProm II Reverse transcriptase, 1X RT buffer, 3 mM
MgCl2 (all from Promega Corporation), 0.2 µg
random primers, 500 µM dNTPs, 40 units RNaseOUT (all from
Thermo Fisher Scientific, Inc.) and 2 µg RNA. RNA was
initially incubated at 65°C, for 5 min, and RT was performed at
42°C for 40 min and stopped at 65°C for 5 min. FLT3 ITDs or
NPM1 mutations were screened by PCR using specific
fluorescent primers. For amplification of targeted regions in
FLT3 and NMP1, the PCR reactions were performed using
1.5 units Taq Platinum polymerase, 1X PCR buffer, 2.0 mM
MgCl2, 200 µM dNTPs (all from Thermo Fisher
Scientific, Inc.), 400 nM each primer (Integrated DNA Technologies,
Inc.) and 2 µl cDNA. The thermal profile for amplification
was: 94°C for 5 min; 30 cycles (94°C for 30 sec; 56°C for 45 sec;
72°C for 30 sec); 72°C for 20 min, followed by fragment analysis on
ABI 3130xl Genetic Analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Sequences were analyzed using Chimer Marker
software V3.0.2 (SoftGenetics, LLC).
RT-quantitative PCR (RT-qPCR)
HL-60 and HL-60R total RNA was extracted with TRIzol
reagent according to the manufacturer's instructions. For RT-qPCR,
RNA was reverse transcribed into cDNA using SuperScript II Reverse
Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Subsequently, qPCR
(TaqMan® Gene Expression Assay; Applied Biosystems;
Thermo Fisher Scientific, Inc.) was performed using the following
Taqman probes (Applied Biosystems; Thermo Fisher Scientific, Inc.):
MYC (Hs00153408_m1) and β-actin (NM_001101.2).
Thermocycling conditions were as follows: Incubation at 50°C for 2
min and 95°C for 10 min, followed by 40 denaturation cycles at 95°C
for 15 sec, and annealing and extension at 60°C for 1 min in the
StepOne™ System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). β-actin was used as an endogenous reference gene. The
2−ΔΔCq method was used to calculate relative expression
(31).
Western blotting and cell
fractionation
Whole cell lysates were obtained using protein Cell
Extraction Buffer (Invitrogen; Thermo Fisher Scientific, Inc.) and
nuclear/cytoplasm fractionation was performed with the NE-PER™
Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fischer
Scientific, Inc.), according to the manufacturers' instructions.
Protein content was measured using the DC™ Protein Assay (Bio-Rad
Laboratories, Inc.) according to manufacturer's instructions. Total
proteins (20-30 µg) were separated by SDS-PAGE on 10 or 12%
gels and were transferred to Hybond-P membranes (GE Healthcare).
Prior to antibody incubation, membranes were blocked with 5% nonfat
milk for 1 h at room temperature. All primary antibodies were
incubated overnight (16-24 h) at 4°C and secondary antibodies were
incubated for 1 h at room temperature. After all antibody
incubations, membranes were washed with Tris-buffered saline with
0.05% Tween 20 (Sigma-Aldrich; Merck KGaA). Anti-Bcl-2 (1:200
dilution, clone 124; cat. no. IS61430-2; Dako; Agilent
Technologies, Inc.), anti-BAK (1:1,000 dilution, polyclonal; cat.
no. 3814; Cell Signaling Technology, Inc.), anti-Bax (1:1,000
dilution, SPM 336; cat. no. sc-65532; Santa Cruz Biotechnology,
Inc.), anti-XIAP (1:1,000 dilution, 3B6; cat. no. 2045; Cell
Signaling Technology, Inc.), anti-c-Myc (1:1,000 dilution, 9E10;
cat. no. sc-40; Santa Cruz Biotechnology,), anti-NF-κB1 p105/p50
(1:1,000 dilution, polyclonal; cat. no. 3035; Cell Signaling
Technology, Inc.), anti-NF-κB p65 (1:1,000 dilution, C22B4; cat.
no. 4764; Cell Signaling Technology, Inc.), anti-cleaved caspase-3
(1:500 dilution, Asp175; 5A1E; cat. no. 9664; Cell Signaling
Technology, Inc.), anti-pro-caspase 3 (1:500 dilution, CPP32; cat.
no. 610322; BD Biosciences) anti-β-actin (1:1,000 dilution, AC-15;
cat.no. A5441; Sigma-Aldrich; Merck KGaA), anti-lamin B (1:1,000
dilution, cat. no. NA12; Calbiochem; Merck KGaA) and anti-Hsc70
(1:1,000 dilution, B-6; cat. no. sc-7298; Santa Cruz Biotechnology,
Inc.) were used as primary antibodies for western blotting.
Anti-mouse IgG (1:10,000 dilution; cat. no. A9169; Sigma Aldrich;
Merck KGaA) and anti-rabbit IgG-HRP conjugated (1:10,000 dilution;
cat. no. A9044; Sigma Aldrich; Merck KGaA) were used as secondary
antibodies. The Clarity Max kit (Bio-Rad Laboratories, Inc.) was
used to visualize protein expression and blots were detected using
the C-digit digital system (LI-COR Biosciences). Analyses were
performed using ImageJ software version 1.53E (National Institutes
of Health).
Protein phosphorylation profiling
The protein phosphorylation profiles of the HL-60
and HL-60R cell lines were compared using the Human Phospho-Kinase
Antibody Array (cat. no. ARY003B; R&D Systems, Inc.), according
to manufacturer's instruction.
AML xenograft model
BALB/c-nude mice were purchased from Jackson
Laboratory and maintained in specific pathogen-free conditions in
the animal facility of the Brazilian National Cancer Institute
(INCA) all animal experiments were approved by the Animal Ethics
Committees of INCA. For in vivo experiments, 34 male BALB/c
nude mice (age, 8-12 weeks; n=16 mice/HL-60 group; n=18 mice/HL-60R
group) were used. Weight was measured before randomization, and
mean weight was 28.33±0.66 kg (presented as mean ± SEM). Mice were
housed in microisolator cages, with a maximum of five mice per
cage, with sterilized food and water given ad libitum, in an
air-filtered specific pathogen-free (SPF) area. Mice were kept
under a 12-h light/dark cycle, and the SPF area was maintained at
18-23°C with 40-60% humidity. The in vivo growth capacity of
the AML cell lines was tested using a subcutaneous xenograft model.
To establish this model, 5×106 HL-60 or HL-60R cells
were resuspended in 100 µl PBS and injected subcutaneously
into the right flank of male BALB/c nude mice (age, 8-12 weeks;
n=16 mice/HL-60 group; n=18 mice/HL-60R group). The animals were
monitored daily for tumor signal over a 58-day period. After
appearance, tumor dimensions were measured every day using a
digital caliper, and the tumor volume (mm3) was
calculated using the following formula: 0.52× (d2×D),
where d and D refer to the smallest and largest tumor diameters,
respectively. The tumorigenesis ratio (%) was evaluated using the
following formula: N+/N×100, where N+ is the
number of mice with tumor presence and N is the number of injected
mice (32-35).
To evaluate the response of HL-60R cells to
cytarabine in vivo, mice were randomized into two
experimental groups (vehicle or cytarabine; n=4 mice/group) with
the same mean tumor volume (50 mm3) on day 14
post-injection. Cytarabine (100 mg/kg) and vehicle (water) were
then administered every day for 5 days by intraperitoneal injection
with a 22G needle (33). The
welfare and weight of the mice were monitored daily to assess
cytarabine toxicity. On day 15 post-randomization, when the first
mouse reached the largest tumor diameter of 20 mm, all mice were
anesthetized with 2% isoflurane and images were captured. All mice
that presented with a maximum tumor diameter of 20 mm on this day
were euthanized by CO2 asphyxiation (20%/min). Mice that
did not reach the maximum tumor diameter (20 mm) on day 15
post-randomization were followed up until they reached the maximum
tumor diameter and were then euthanized.
Statistical analyses
Statistical analyses were conducted using GraphPad
Prism software (PRISM 5.0; GraphPad Software Inc.). For data
analysis, one-way ANOVA followed by Bonferroni post-hoc test was
applied to the results of the MTT assay, cell death and DNA
fragmentation assays evaluating the effects of the LQB-118
compound. Unpaired Student's t-test was performed to analyze cell
death, DNA fragmentation and MTT assays comparing HL-60 and HL-60R
cells. In addition, Kaplan-Meier and log-rank test was performed to
assess in vivo survival. P<0.05 was considered to
indicate a statistically significant difference.
Results
HL-60R cells demonstrate resistance to
cytarabine but not to anthracyclines used in AML treatment
First, the cytarabine-resistant HL-60R cell line was
generated by continuously exposing the HL-60 parental cell line to
increasing concentrations of this drug. Once the cell line
treatment reached 50 µM and the cells continued to
proliferate in culture, model validation was performed.
The present study assessed whether cytarabine
treatment could induce cell death and inhibit viability of the
HL-60 and HL-60R cell lines. The results demonstrated that
cytarabine impaired cell viability in the HL-60 cell line in a
time-dependent manner, whereas no change was observed in the HL-60R
cell line (Fig. 1A-C). Cell
viability was significantly reduced in the HL-60 cell line in
response to treatment with 2.5 µM cytarabine for 24 h when
compared to HL-60R cells in the same condition (P<0.001;
Fig. 1A). Furthermore HL-60R
cells remained viable even when exposed to 200 µM cytarabine
for 72 h (Fig. S1). Moreover,
treatment with 50 µM cytarabine for 48 h enhanced the
percentage of Annexin V+ cells (P=0.0180; Fig. 2A and B) and DNA fragmentation
(P=0.0004; Figs. 2C and S2B) in the HL-60 cell line, but not in
the HL-60R cell line in the same conditions. In HL-60 cells, cell
cycle profile analysis indicated cell cycle arrest after cytarabine
treatment (Fig. S2a) and high
levels of DNA fragmentation were detected at 48 h. After 24 h of
cytarabine treatment, more cells in the G2/M phase were
detected in the HL-60 group (data not shown). Anthracyclines are
chemotherapeutic agents used alongside cytarabine in AML treatment
(36,37). Thus, the present study
investigated whether the HL-60R cell line exhibited cross
resistance to anthracyclines. Although HL-60 cells were shown to be
more sensitive to idarubicin and daunorubicin treatment at earlier
time-points, HL-60R cell viability was similarly decreased in a
time and dose-dependent manner (Fig.
S3A-F). These data indicated that HL-60R cells exhibited
resistance to cytarabine but sensitivity to anthracyclines used in
AML treatment. Therefore, the HL-60R cell line may be a useful
model to understand specific pathways involved in acquisition of
cytarabine resistance.
HL-60R cells present a complex karyotype
and do not harbor mutations involved in AML development and
chemoresistance
Karyotype complexity and mutation profile are
predictive and prognostic factors for AML, being related to
complete response to induction therapy and OS (36). Therefore, the present study
investigated the karyotype and mutational profile in HL-60 parental
and resistant cell lines. The results demonstrated that both cell
lines presented complex karyotypes, based on the presence of three
or more chromosomal alterations (Fig. S4). The HL-60 cell line comprised
two subpopulations coexisting in the same cell culture, both of
which presented 45 chromosomes, with only one X chromosome. One of
the subpopulations presented the karyotype: Ins (4q), −5, add
(9) (p13), del(10)(p12), add(17)(p13), −18, +2mar, whereas the other
possessed the karyotype: Ins (4q), −5, add(9)(p13), del (10) (p12), add (17)(p13), −18, +3mar (Fig. S4). The HL-60R cell line presented
only one population, demonstrating a karyotype with 48 chromosomes
and the presence of two X chromosomes, −8, −9, + 14, + 15, + 18, +
20, del (9q) (Fig. S4).
Subsequently, possible mutations in CEBPA, DNMT3A,
IDH1, IDH2 and NPM1, as well as FLT3
ITDs were investigated. Notably, no mutations in the genes analyzed
were detected in either cell line (Fig. S5) further suggesting that these
gene alterations may not have a role in the HL-60R resistance
model.
HL-60R cells induce increased
tumorigenicity in vivo and preserve cytarabine resistance in
subcutaneous xenograft models
The present study compared the ability of HL-60 and
HL-60R AML cell lines to generate tumors in xenograft models. HL-60
and HL-60R cell lines were inoculated into 16 and 18 mice,
respectively. The results demonstrated that the HL-60R cell line
presented a higher tumorigenic capacity in vivo compared
with the HL-60 cell line, HL-60R and HL-60 cells induced
tumorigenesis in 44.44 and 18.75% mice, respectively (Table I).
 | Table IHL-60R cells exhibit increased
formation of subcutaneous tumors in vivo. |
Table I
HL-60R cells exhibit increased
formation of subcutaneous tumors in vivo.
| Cell type | Follow-up,
days | Number of injected
mice | Number of mice with
tumor presence | Tumorigenesis
ratio, % |
|---|
| HL-60 | 58 | 16 | 3 | 18.75 |
| HL-60R | 58 | 18 | 8 | 44.44 |
The present study evaluated the response of the
HL-60R cell line to cytarabine treatment in vivo. Due to the
low number of animals with tumors in the HL-60 group, only the
HL-60R group was used in this experiment. After 14 days of HL-60R
inoculation, mice with tumor growth were randomized into two
experimental groups: Vehicle and cytarabine (100 mg/kg as a single
dose, intraperitoneal, for 5 days). Notably, no reduction in tumor
volume was observed in the cytarabine-treated group when compared
with the vehicle group (Fig. 3A and
B). An increase in tumor volume of the animals treated with
cytarabine (day 15: 1,506±468.6 mm3) was detected in
relation to the vehicle group (day 15: 893.2±576 mm3),
but this increase was not statistically significant. Notably, the
treatment protocol exhibited no apparent toxicity, and body weight
and welfare were maintained (Fig.
S6A). The survival curves showed no difference between the two
groups (Fig. S6B), reinforcing
the ability of the HL-60R cell line to retain resistance to
cytarabine in vivo.
Cytarabine resistance is associated with
increasing levels of transcription factors, and anti-apoptotic and
proliferation-inducing proteins
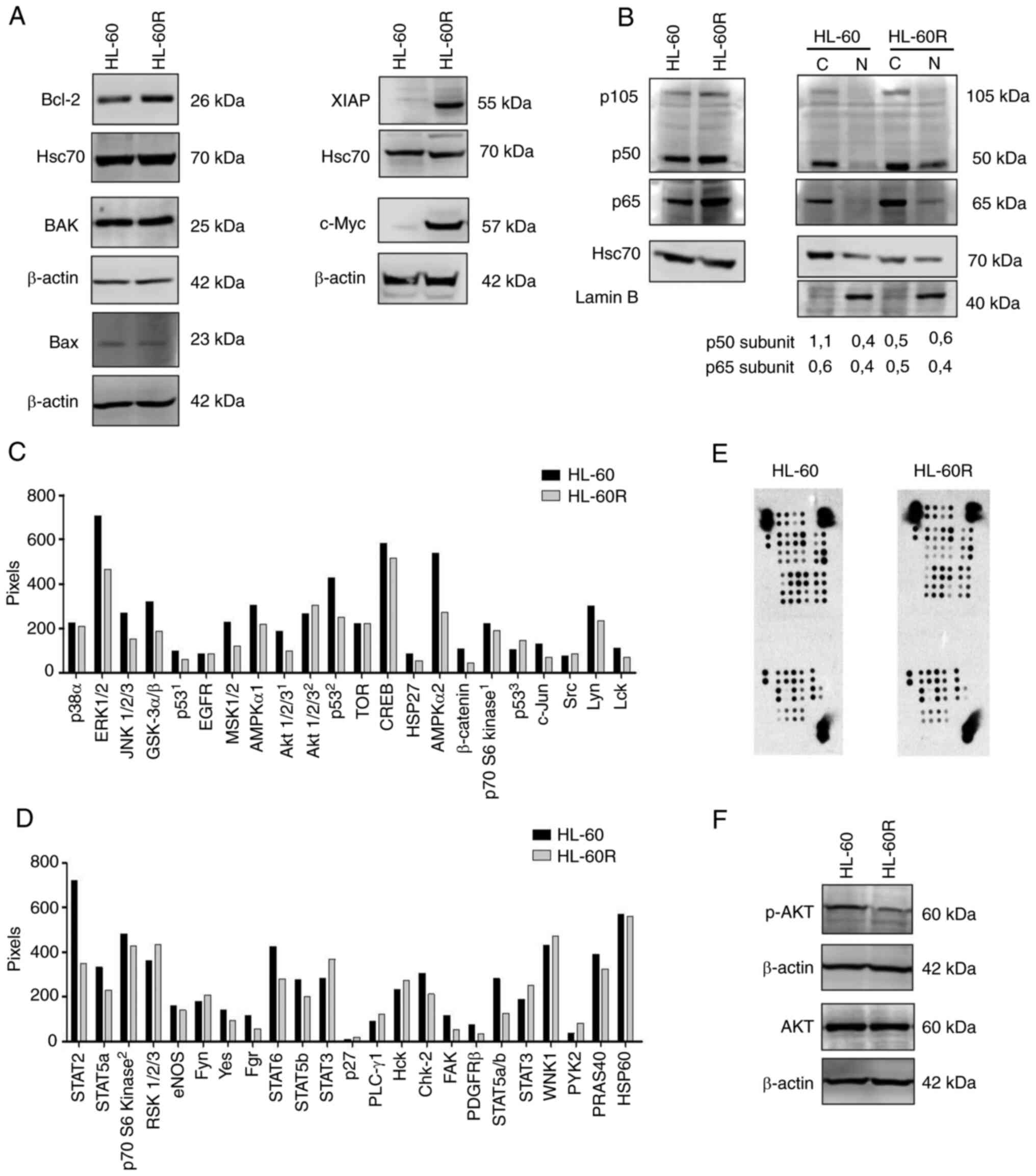
In order to understand the pathways involved in
cytarabine resistance in the HL-60R cell line, the expression
levels of proteins involved in apoptosis signaling, which could be
differentially expressed, were evaluated. Although similar
expression levels of Bax and BAK pro-apoptotic proteins were
detected, the HL-60R cell line exhibited upregulation of Bcl-2 and
XIAP anti-apoptotic proteins compared with in the HL-60 cell line
(Fig. 4A). Furthermore, the
expression levels of c-Myc, which is highly associated with
proliferation in numerous types of cancer (38), was investigated. HL-60R cells
presented higher protein expression levels of c-Myc compared with
the parental cell line (Fig. 4A).
Notably, there was no difference in c-Myc mRNA expression levels
between HL-60 and HL-60R cells (Fig.
S7), suggesting that degradation of the c-Myc protein may be
reduced in HL-60R cells.
NF-κB expression and localization has been
associated with resistance to chemotherapy in certain types of
cancer, including AML (20-22). Therefore, the present study
investigated the localization and expression of p50 and p65
subunits of NF-κB in the HL-60R cell line. The results demonstrated
that the p50 and p65 subunits exhibited enhanced nuclear
localization in the HL-60R cell line compared with in the parental
cell line (Fig. 4B).
Due to the multifactorial profile associated with
cytarabine resistance, a phospho-kinase array was performed to
compare the phosphorylation profile of HL-60R cells and the
parental cell line, HL-60. Notably, the results demonstrated that
HL-60 and HL-60R cells exhibited a different phosphorylation
profile, suggesting the involvement of several signaling pathways
in cytarabine resistance (Fig.
4C-E). The expression of phosphorylated proteins was increased
in both cytarabine-resistant and -sensitive cells. Notably, the
array revealed that AKT oncogenic kinase, which is widely related
to proliferation and survival in several tumor types, including AML
(39), exhibited reduced
phosphorylation in the HL-60R cell line. This result was further
validated by western blotting, where the differential expression of
phosphorylated-AKT was confirmed in cytarabine-sensitive and
cytarabine-resistant cells (Fig.
4F). Taken together, these findings clearly indicated that the
acquisition of cytarabine resistance in AML was a complex process,
involving the modulation of numerous oncogenic signaling
pathways.
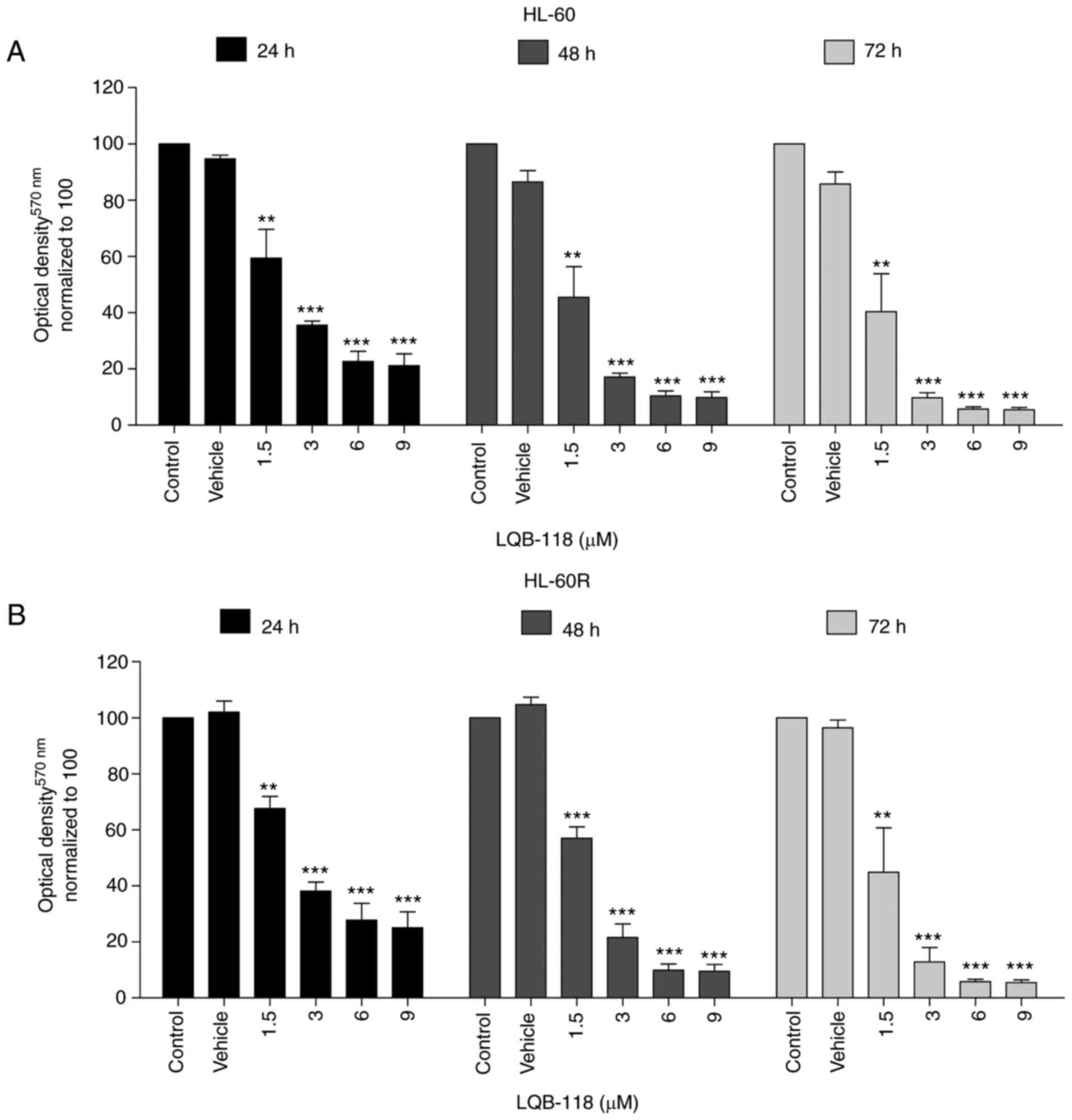
LQB-118 exhibits cytotoxicity against
cytarabine-resistant cells
Our previous studies demonstrated the cytotoxic
activity of the synthetic LQB-118 compound in several types of
cancer, including AML (25-29,40-43). Therefore, the present study aimed
to investigate the antitumor effect of the LQB-118 compound
particularly in cells resistant to cytarabine. Notably, HL-60 and
HL-60R cells treated with different concentrations of LQB-118
exhibited a very similar response after 24, 48 and 72 h, as
determined by MTT assay (Fig. 5A and
B).
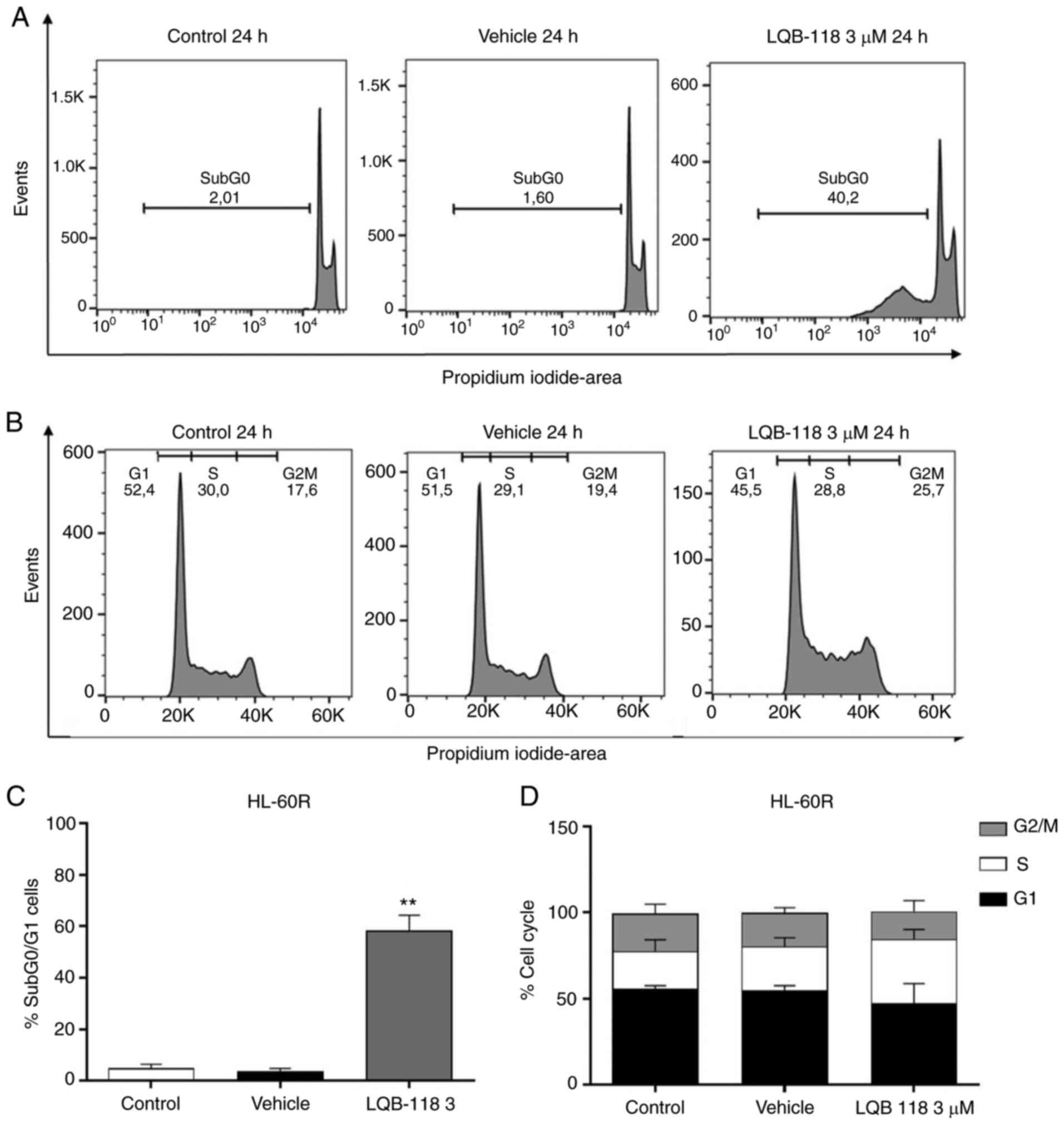
In order to investigate the potential of LQB-118 to
induce cell death in cytarabine-resistant cells, the HL-60R cell
line was treated with 3 µM LQB-118 for 24 h and apoptosis
was evaluated by DNA fragmentation analysis, Annexin V staining,
and analysis of pro-caspase 3 and cleaved caspase 3 protein
expression levels. After treatment, an increase in Annexin
V+ cell percentage was detected (P≤0.01; Fig. 6A and B), and pro-caspase-3
expression was reduced and caspase-3 cleavage was increased
(Fig. 6C) in HL-60R cells
compared with the vehicle group. Furthermore, PI incorporation
analysis revealed that 3 µM LQB-118 treatment for 24 h in
HL-60R cells increased DNA fragmentation compared with in the
vehicle group (P<0.001; Fig. 7A
and C), whereas there was no alteration in cell cycle profile
(Fig. 7B and D). Taken together,
these data demonstrated that LQB-118 treatment induced apoptosis of
HL-60R cells, with little effect on cell cycle progression.
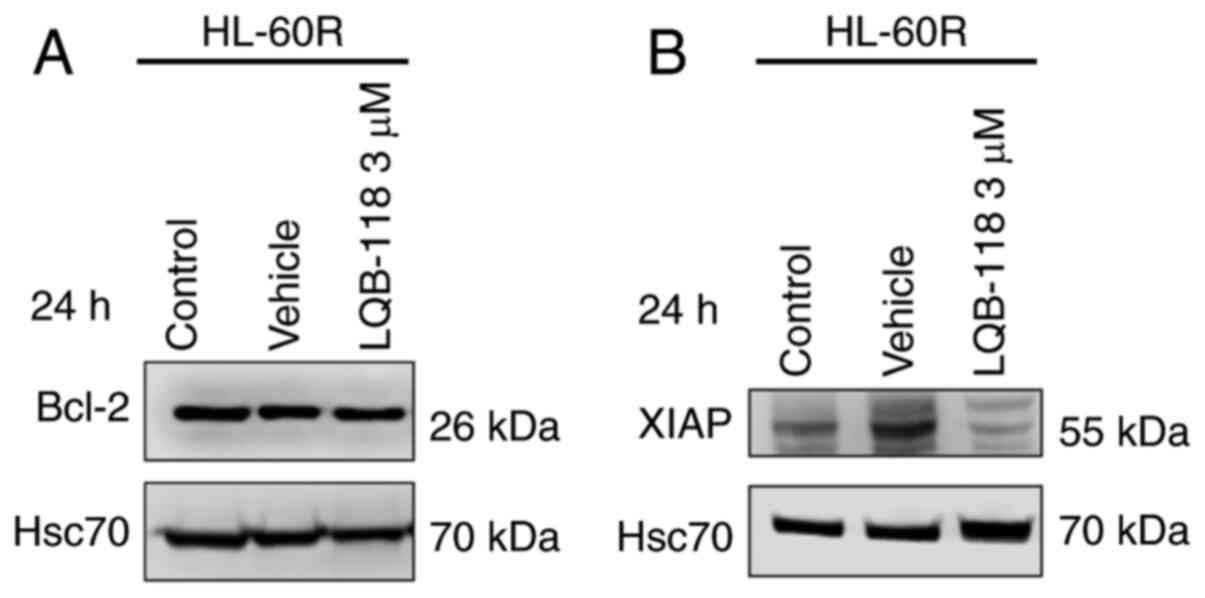
The present study investigated the expression levels
of Bcl-2 and XIAP anti-apoptotic proteins after LQB-118 treatment,
as these proteins were differentially expressed in HL-60R cells
compared with in HL-60 cells. The results demonstrated that
treatment of HL-60R cells with 3 µM LQB-118 for 24 h did not
alter Bcl-2 protein expression levels (Fig. 8A), but it did decrease XIAP
protein expression levels (Fig.
8B), suggesting that XIAP may be involved in the mechanism
underlying the effects of LQB-118 on the induction of apoptosis of
HL-60R cells.
Discussion
The major obstacle for successful treatment of AML,
the most common type of acute leukemia in adults, is intrinsic or
acquired resistance following the induction of remission treatment
(8,44-47). Although some mechanisms have been
implicated in cytarabine resistance, there is no clear
comprehension of this multifactorial phenomenon. The HL-60R cell
line was developed from the HL-60 parental cell line, first
described as a promyelocytic/FAB M3 subtype and later as a FAB M2
subtype due to the lack of promyelocytic characteristics, such as
t(15;17) (48,49). The HL-60 cell line has been widely
used as an in vitro model to understand leukemogenesis and
treatment response (50).
Although some groups have attempted to generate
cytarabine-resistant AML cell lines (51,52), the mechanisms underlying
cytarabine resistance remain unclear. The development of
therapeutic resistance from this in vitro model is an
important tool to understand the pathways underlying AML resistance
acquisition.
Following the induction of cytarabine resistance,
the present study verified that cytarabine treatment did not affect
the HL-60R cell line in vitro and in vivo.
Conversely, the parental cell line was demonstrated to be very
sensitive to cytarabine treatment in vitro. The exposure of
HL-60 cells to low doses of cytarabine was sufficient to decrease
cell viability, increase Annexin V+ cells and DNA
fragmentation, and induce S phase arrest (data not shown). These
effects are in agreement with previous studies describing the
cytotoxic mechanisms underlying cytarabine treatment (53-55). Chen et al (56) demonstrated that mice injected
subcutaneously with HL-60 cells exhibited a reduction in tumor size
following cytarabine treatment. Since the HL-60 cell line
demonstrated decreased capacity to generate tumors in xenograft
models in comparison to the HL-60R cell line, the HL-60R cytarabine
resistance phenotype was validated in vitro and in
vivo. Taken together, the present results indicated that the
HL-60R cell line may be considered an interesting model to study
the specific pathways involved in acquisition of cytarabine
resistance.
AML response to cytarabine depends on several
factors, such as patient mutational profile, age, white-blood cell
count and karyotype (1,3). Complex karyotypes in AML are
associated with poor prognosis and treatment response (57-59). The HL-60 cell line presents two
sub-populations with complex karyotypes co-existing in the same
cell culture, whereas HL-60R presents a single sub-population, also
with a complex aberrant karyotype. Notably, these cell lines do not
harbor mutations highly associated with leukemia development, poor
prognosis and low treatment response in AML, such as CEBPα,
ITD-FLT3, NPM1, c-KIT and others (60-62). Thus, it may be hypothesized that
chromosomal alterations could be a predominant genetic force
underlying the lack of response to cytarabine treatment in HL-60R
cells (58-63).
Resistance induction in HL-60R cells led to several
molecular alterations that are intrinsically linked to the absence
of response to cytarabine-induced apoptosis. Apoptosis evasion is a
significant obstacle in chemotherapy response in several types of
cancer, including AML (64,65). The results of the present study
demonstrated that acquired cytarabine resistance in HL-60R cells
promoted the upregulation of the anti-apoptotic protein Bcl-2;
however, no alterations were observed in the expression levels of
the pro-apoptotic proteins BAK and Bax. Corroborating these data,
high Bcl-2/Bax ratios were previously revealed to be associated
with decreased rates of OS and complete remission in AML (18,65), which might be closely linked to
the acquisition of cytarabine resistance.
IAP family members serve a significant role in
conferring poor prognosis to patients with AML (66). Previous studies revealed that XIAP
upregulation could be associated with AML poor prognosis (65-67), as well as with doxorubicin
resistance in vitro and poor response to chemotherapy in the
first 30 days of patient treatment (67,68). Consistently, XIAP inhibition has
been reported to sensitize AML cells to TRAIL and chemotherapy
(65-69). In addition, high co-expression of
XIAP and survivin proteins has been correlated with poor OS in
childhood de novo AML (66,67). Survivin expression had no
association with acquired cytarabine resistance (data not shown),
whereas XIAP upregulation could potentially have an important role,
which remains to be explored. NF-κB constitutive expression and
activation has been shown to be closely related to chemotherapy
resistance and poor prognosis in patients with AML (70,71). The present study demonstrated that
acquired cytarabine resistance in the HL-60R cell line resulted in
high nuclear localization of p50 and p65 subunits. When activated,
NF-κB binds to DNA, and leads to the transcription of its target
genes, some of which are associated with apoptosis evasion, such as
Bcl-2. A previous study demonstrated that NF-κB activation induced
higher levels of Bcl-2 in prostate cancer, which was related to
poor response to hormone therapy (72). Furthermore, high expression and
activity of NF-κB and elevated levels of Bcl-2 have been reported
to be associated with poor outcome and chemotherapy response in
several tumor types (73-76).
High levels of c-Myc transcription factor in
patients with AML have been shown to be correlated to low OS rate
(77). Thus, the present study
investigated the role of c-Myc in cytarabine-resistant AML cells;
the results revealed that HL-60R cells presented high levels of
c-Myc protein, but not c-Myc mRNA levels when compared with the
parental cell line. In the model used in the present study, it is
likely that elevated expression of c-Myc is not directly related to
an increase of its transcription, and it might reflect an increase
in c-Myc protein stability or regulation by post-translational
modifications. It has already been demonstrated that the NF-κB p50
subunit can inhibit c-Myc degradation through FBW7 suppression
(78). Since HL-60R cells present
high levels of nuclear p50 and c-Myc protein expression, this could
be a mechanism by which c-Myc degradation is inhibited, leading to
the acquisition of cytarabine resistance. Furthermore,
co-expression of c-Myc and Bcl-2 in aggressive B-cell lymphoma
treated with cytarabine was revealed to be correlated to low OS and
response to treatment induction (79). The present study also evaluated
protein kinases, such as AKT, and demonstrated that several of
these proteins were differentially phosphorylated in
cytarabine-resistant cells compared with in cytarabine-sensitive
cells. However, these findings require further investigation to
improve understanding on the role of these kinases in cytarabine
resistance.
Since cytarabine resistance remains an obstacle to
AML treatment, the search for alternative treatment options to
bypass acquired and intrinsic resistance is important. The LQB-118
compound is a pterocarpanquinone with antitumor effects toward
diverse types of hematological and solid tumors in vitro and
in vivo (25-29,40-43). However, to the best of our
knowledge, no studies have assessed its antitumor activity in
cytarabine-resistant established cells. Initially, the present
study demonstrated that LQB-118 treatment decreased cell viability
and induced apoptosis of HL-60R cells, but did not interfere with
cell cycle progression. These data corroborated a previous study
from our group, in which it was demonstrated that LQB-118 exerted
an antitumor effect on the Kasumi-1 AML cell line and on patient
samples with different molecular backgrounds (25). The Kasumi-1 cell line is an AML M2
sub-type with t(8;21), which is intrinsically resistant to
cytarabine (80) and similarly
responsive to LQB-118 treatment as HL-60R cells in vitro,
suggesting that LQB-118 activity in resistant cell lines is
independent of whether the resistance phenotype is acquired or
intrinsic.
Previous studies have already demonstrated the
significance of XIAP in AML cytarabine resistance (19,66-68). The present study revealed that the
expression levels of the anti-apoptotic proteins XIAP and Bcl-2
were increased in the HL-60R cell line after cytarabine exposure,
suggesting that these proteins may be involved in the acquisition
of cytarabine resistance. Moreover, it was demonstrated that the
protein expression levels of XIAP were reduced in response to
LQB-118 treatment in HL-60R cells in vitro, which is
consistent with our previous findings in Kasumi-1 cells (25). In addition, it is well known that
a feedback loop exists between XIAP and NF-κB activation (81,82), which might partially explain the
reason why both proteins were upregulated in HL-60R cells. De Faria
et al (41) observed that
treatment with LQB-118 in CML cell lines induced modulation of
NF-κB subcellular localization. Furthermore the same effect was
also observed in the Kasumi-1 AML cell line treated with LQB-118
(25). Considering the present
data and the results of previous studies, it may be hypothesized
that LQB-118 induces apoptosis of cytarabine-resistant cells
possibly through NF-κB modulation, which in turn may result in
decreased XIAP expression.
In summary, the present data indicated that
acquired cytarabine resistance in AML may be a multifactorial
process, involving chromosomal aberrations and differential
expression of signaling pathways associated with apoptosis and cell
proliferation. Moreover, the present study suggested that LQB-118
might be a promising alternative therapeutic approach for the
treatment of patients with AML that are non-responsive to standard
treatment.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
TH designed the experiments, generated the
cytarabine-resistant cell line, performed the viability and
cellular effects assays (MTT, cell death, DNA fragmentation and
cell cycle profile assays), and protein expression analysis,
analyzed the data and wrote the manuscript. LM analyzed the flow
cytometry experiments. GG performed and analyzed the in vivo
assays. MR performed and analyzed the qPCR experiments. BDSM and
GNEM performed analysis of expression and subcellular localization
of NF-κB family members. BCRM and LMG performed the mutational
screening experiments. LODC performed the karyotype analysis. PRRC
and CDN synthesized the LQB-118 compound. FCCDF designed
experiments, analyzed the data and revised the manuscript draft.
RCM designed the study, provided all the resources and revised the
manuscript draft. TH and RCM confirm the authenticity of all the
raw data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Ethics Committees from INCA.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the Conselho Nacional de
Desenvolvimento Científico e Tecnológico (CNPq; grant nos.
304565/2016-4 and 429687/2018-4) and the Fundação para Amparo à
Pesquisa do Estado do Rio de Janeiro (FAPERJ; grant no.
E26/202-798/2017).
References
|
1
|
Estey EH: Acute myeloid leukemia: 2014
update on risk-stratification and management. Am J Hematol.
89:1063–1081. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Saultz JN and Garzon R: Acute myeloid
leukemia: A concise review. J Clin Med. 5:332016. View Article : Google Scholar :
|
|
3
|
De Kouchkovsky I and Abdul-Hay M: Acute
myeloid leukemia: A comprehensive review and 2016 update. Blood
Cancer J. 6:e4412016. View Article : Google Scholar
|
|
4
|
Burnett AK: Treatment of acute myeloid
leukemia: Are we making progress? Hematology Am Soc Hematol Educ
Program. 2012:1–6. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sakamoto KM, Grant S, Saleiro D, Crispino
JD, Hijiya N, Giles F, Platanias L and Eklund EA: Targeting novel
signaling pathways for resistant acute myeloid leukemia. Mol Genet
Metab. 114:397–402. 2015. View Article : Google Scholar :
|
|
6
|
Feliciano SV, Santos MO, Pombo-de-Oliveira
MS, de Aquino JA, de Aquino TA, Arregi MM, Antoniazzif BN, da Costa
AM, Formigosa LA, Laporte CA, et al: Incidence and mortality of
myeloid malignancies in children, adolescents and Young adults in
Brazil: A population-based study. Cancer Epidemiol. 62:1015832019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schlenk RF and Döhner H: Genomic
applications in the clinic: Use in treatment paradigm of acute
myeloid leukemia. Hematology Am Soc Hematol Educ Program.
2013:324–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yeung CC and Radich J: Predicting
chemotherapy resistance in AML. Curr Hematol Malig Rep. 12:530–536.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coombs CC, Tallman MS and Levine RL:
Molecular therapy for acute myeloid leukaemia. Nat Rev Clin Oncol.
13:305–318. 2016. View Article : Google Scholar
|
|
10
|
Murphy T and Yee KW: Cytarabine and
daunorubicin for the treatment of acute myeloid leukemia. Expert
Opin Pharmacother. 18:1765–1780. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rustum YM and Preisler HD: Correlation
between leukemic cell retention of
1-beta-D-arabinofuranosylcytosine 5′-triphosphate and response to
therapy. Cancer Res. 39:42–49. 1979.PubMed/NCBI
|
|
12
|
Löwenberg B, Pabst T, Vellenga E, van
Putten W, Schouten HC, Graux C, Ferrant A, Sonneveld P, Biemond BJ,
Gratwohl A, et al: Cytarabine dose for acute myeloid leukemia. N
Engl J Med. 364:1027–1036. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rowe JM: AML in 2017: Advances in clinical
practice. Best Pract Res Clin Haematol. 30:283–286. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kayser S and Levis MJ: Advances in
targeted therapy for acute myeloid leukaemia. Br J Haematol.
180:484–500. 2018. View Article : Google Scholar :
|
|
15
|
Negoro E, Yamauchi T, Urasaki Y, Nishi R,
Hori H and Ueda T: Characterization of cytarabine-resistant
leukemic cell lines established from five different blood cell
lineages using gene expression and proteomic analyses. Int J Oncol.
38:911–919. 2011.PubMed/NCBI
|
|
16
|
Tamm I, Richter S, Oltersdorf D, Creutzig
U, Harbott J, Scholz F, Karawajew L, Ludwig WD and Wuchter C: High
expression levels of x-linked inhibitor of apoptosis protein and
survivin correlate with poor overall survival in childhood de novo
acute myeloid leukemia. Clin Cancer Res. 10:3737–3744. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chromik J, Safferthal C, Serve H and Fulda
S: Smac mimetic primes apoptosis-resistant acute myeloid leukaemia
cells for cytarabine-induced cell death by triggering necroptosis.
Cancer Lett. 344:101–109. 2014. View Article : Google Scholar
|
|
18
|
Kulsoom B, Shamsi TS, Afsar NA, Memon Z,
Ahmed N and Hasnain SN: Bax, Bcl-2, and Bax/Bcl-2 as prognostic
markers in acute myeloid leukemia: Are we ready for Bcl-2-directed
therapy? Cancer Manag Res. 10:403–416. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shang J, Chen WM, Wang ZH, Wei TN and Chen
ZZ: Wu WB. CircPAN3 mediates drug resistance in acute myeloid
leukemia through the miR-153-5p/miR-183-5p-XIAP axis. Exp Hematol.
70:42–54.e3. 2019. View Article : Google Scholar
|
|
20
|
Kaltschmidt B, Greiner JFW, Kadhim HM and
Kaltschmidt C: Subunit-specific role of NF-κB in cancer.
Biomedicines. 6:442018. View Article : Google Scholar
|
|
21
|
Colombo F, Zambrano S and Agresti A:
NF-kappaB, the importance of being dynamic: Role and insights in
cancer. Biomedicines. 6:452018. View Article : Google Scholar
|
|
22
|
Guzman ML, Neering SJ, Upchurch D, Grimes
B, Howard DS, Rizzieri DA, Luger SM and Jordan CT: Nuclear
factor-kappaB is constitutively activated in primitive human acute
myelogenous leukemia cells. Blood. 98:2301–2307. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xia B, Tian C, Guo S, Zhang L, Zhao D, Qu
F, Zhao W, Wang Y, Wu X, Da W, et al: c-Myc plays part in drug
resistance mediated by bone marrow stromal cells in acute myeloid
leukemia. Leuk Res. 39:92–99. 2015. View Article : Google Scholar
|
|
24
|
Mughal MK, Akhter A, Street L, Pournazari
P, Shabani-Rad MT and Mansoor A: Acute myeloid leukaemia:
Expression of MYC protein and its association with cytogenetic risk
profile and overall survival. Hematol Oncol. 35:350–356. 2017.
View Article : Google Scholar
|
|
25
|
de Souza Reis FR, de Faria FC, Castro CP,
de Souza PS, da Cunha Vasconcelos F, Bello RD, da Silva AJ, Costa
PR and Maia RC: The therapeutical potential of a novel
pterocarpanquinone LQB-118 to target inhibitor of apoptosis
proteins in acute myeloid leukemia cells. Anticancer Agents Med
Chem. 13:341–351. 2013. View Article : Google Scholar
|
|
26
|
Nestal De Moraes G, Pereira Castro C,
Salustiano EJ, Dumas ML, Costas F, Wing-Fai Lam E, Ribeiro Costa PR
and Maia RC: [Corrigendum] The pterocarpanquinone LQB-118 induces
apoptosis in acute myeloid leukemia cells of distinct molecular
subtypes and targets FoxO3a and FoxM1 transcription factors. Int J
Oncol. 55:13962019.
|
|
27
|
Maia RC, Vasconcelos FC, de Sá Bacelar T,
Salustiano EJ, da Silva LF, Pereira DL, Moellman-Coelho A, Netto
CD, da Silva AJ, Rumjanek VM and Costa PR: LQB-118, a
pterocarpanquinone structurally related to lapachol
[2-hydroxy-3-(3-methyl-2-butenyl)-1,4-naphthoquinone]: A novel
class of agent with high apoptotic effect in chronic myeloid
leukemia cells. Invest New Drugs. 29:1143–1155. 2011. View Article : Google Scholar
|
|
28
|
de Sa Bacelar T, da Silva AJ, Costa PR and
Rumjanek VM: The pterocarpanquinone LQB 118 induces apoptosis in
tumor cells through the intrinsic pathway and the endoplasmic
reticulum stress pathway. Anticancer Drugs. 24:73–83. 2013.
View Article : Google Scholar
|
|
29
|
Netto CD, da Silva AJ, Salustiano EJ,
Bacelar TS, Rica IG, Cavalcante MC, Rumjanek VM and Costa PR: New
pterocarpanquinones: Synthesis, antineoplasic activity on cultured
human malignant cell lines and TNF-alpha modulation in human PBMC
cells. Bioorg Med Chem. 18:1610–1616. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McGowan-Jordan J, Simons A and Schmid M:
ISCN 2016. An International System for Human Cytogenomic
Nomenclature (2016). Karger AG; Basel: 2016
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Faustino-Rocha A, Oliveira PA,
Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG,
Colaco B, Pires MJ, Colaco J, Ferreira R and Ginja M: Estimation of
rat mammary tumor volume using caliper and ultrasonography
measurements. Lab Anim (NY). 42:217–224. 2013. View Article : Google Scholar
|
|
33
|
Tomayko MM and Reynolds CP: Determination
of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother
Pharmacol. 24:148–154. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang Y, Xue K, Li Z, Zheng W, Dong W, Song
J, Sun S, Ma T and Li W: [Corrigendum] cMyc regulates the
CDK1/cyclin B1 dependentG2/M cell cycle progression by histone H4
acetylation in Raji cells. Int J Mol Med. 44:19882019.
|
|
35
|
Zuber J, Radtke I, Pardee TS, Zhao Z,
Rappaport AR, Luo W, McCurrach ME, Yang MM, Dolan ME, Kogan SC, et
al: Mouse models of human AML accurately predict chemotherapy
response. Genes Dev. 23:877–889. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liersch R, Muller-Tidow C, Berdel WE and
Krug U: Prognostic factors for acute myeloid leukaemia in
adults-biological significance and clinical use. Br J Haematol.
165:17–38. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lynch RC and Medeiros BC: Chemotherapy
options for previously untreated acute myeloid leukemia. Expert
Opini Pharmacother. 16:2149–2162. 2015. View Article : Google Scholar
|
|
38
|
Carroll PA, Freie BW, Mathsyaraja H and
Eisenman RN: The MYC transcription factor network: Balancing
metabolism, proliferation and oncogenesis. Front Med. 12:412–425.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Park S, Chapuis N, Tamburini J, Bardet V,
Cornillet-Lefebvre P, Willems L, Green A, Mayeux P, Lacombe C and
Bouscary D: Role of the PI3K/AKT and mTOR signaling pathways in
acute myeloid leukemia. Haematologica. 95:819–828. 2010. View Article : Google Scholar :
|
|
40
|
Martino T, Magalhaes FC, Justo GA, Coelho
MG, Netto CD, Costa PR and Sabino KC: The pterocarpanquinone
LQB-118 inhibits tumor cell proliferation by downregulation of
c-Myc and cyclins D1 and B1 mRNA and upregulation of p21 cell cycle
inhibitor expression. Bioorg Med Chem. 22:3115–3122. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
de Faria FC, Leal ME, Bernardo PS, Costa
PR and Maia RC: NFκB pathway and microRNA-9 and -21 are involved in
sensitivity to the pterocarpanquinone LQB-118 in different CML cell
lines. Anticancer Agents Med Chem. 15:345–352. 2015. View Article : Google Scholar
|
|
42
|
Martino T, Kudrolli TA, Kumar B, Salviano
I, Mencalha A, Coelho MG, Justo G, Costa PR, Sabino KC and Lupold
SE: The orally active pterocarpanquinone LQB-118 exhibits
cytotoxicity in prostate cancer cell and tumor models through
cellular redox stress. Prostate. 78:140–151. 2018. View Article : Google Scholar
|
|
43
|
Bernardo PS, Guimaraes GH, De Faria FC,
Longo G, Lopes GP, Netto CD, Costa PR and Maia RC: LQB118 compound
inhibits migration and induces cell death in glioblastoma cells.
Oncol Rep. 43:346–357. 2020.
|
|
44
|
Veisani Y, Khazaei S and Delpisheh A:
5-year survival rates based on the type of leukemia in Iran, a
meta-analysis. Caspian J Intern Med. 9:316–324. 2018.PubMed/NCBI
|
|
45
|
Lagunas-Rangel FA, Chavez-Valencia V,
Gomez-Guijosa MA and Cortes-Penagos C: Acute myeloid
leukemia-genetic alterations and their clinical prognosis. Int J
Hematol Oncol Stem Cell Res. 11:328–339. 2017.
|
|
46
|
Hackl H, Astanina K and Wieser R:
Molecular and genetic alterations associated with therapy
resistance and relapse of acute myeloid leukemia. J Hematol Oncol.
10:512017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Walter RB, Othus M, Burnett AK, Lowenberg
B, Kantarjian HM, Ossenkoppele GJ, Hills RK, Ravandi F, Pabst T,
Evans A, et al: Resistance prediction in AML: Analysis of 4601
patients from MRC/NCRI, HOVON/SAKK, SWOG and MD anderson cancer
center. Leukemia. 29:312–320. 2015. View Article : Google Scholar
|
|
48
|
Gallagher R, Collins S, Trujillo J,
McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti
F and Gallo R: Characterization of the continuous, differentiating
myeloid cell line (HL-60) from a patient with acute promyelocytic
leukemia. Blood. 54:713–33. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dalton WT Jr, Ahearn MJ, McCredie KB,
Freireich EJ, Stass SA and Trujillo JM: HL-60 cell line was derived
from a patient with FAB-M2 and not FAB-M3. Blood. 71:242–247. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Collins SJ: The HL-60 promyelocytic
leukemia cell line: Proliferation, differentiation, and cellular
oncogene expression. Blood. 70:1233–1244. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yamauchi T, Uzui K, Nishi R, Shigemi H and
Ueda T: Cytarabine-resistant leukemia cells are moderately
sensitive to clofarabine in vitro. Anticancer Res. 34:1657–1662.
2014.PubMed/NCBI
|
|
52
|
Prenkert M, Uggla B, Tidefelt U and Strid
H: CRIM1 is expressed at higher levels in drug-resistant than in
drug-sensitive myeloid leukemia HL60 cells. Anticancer Res.
30:4157–4161. 2010.PubMed/NCBI
|
|
53
|
Furth JJ and Cohen SS: Inhibition of
mammalian DNA polymerase by the 5′-triphosphate of
1-beta-d-arabinofuranosylcytosine and the 5′-triphosphate of
9-beta-d-arabinofuranoxyladenine. Cancer Res. 28:2061–2067.
1968.PubMed/NCBI
|
|
54
|
Kufe DW, Major PP, Egan EM and Beardsley
GP: Correlation of cytotoxicity with incorporation of ara-C into
DNA. J Biol Chem. 255:8997–8900. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shepshelovich D, Edel Y, Goldvaser H,
Dujovny T, Wolach O and Raanani P: Pharmacodynamics of cytarabine
induced leucopenia: A retrospective cohort study. Br J Clin
Pharmacol. 79:685–691. 2015. View Article : Google Scholar :
|
|
56
|
Chen Y, Gan D, Huang Q, Luo X, Lin D and
Hu J: Emodin and its combination with cytarabine induce apoptosis
in resistant acute myeloid leukemia cells in vitro and in vivo.
Cell Physiol Biochem. 48:2061–2073. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Slovak ML, Kopecky KJ, Cassileth PA,
Harrington DH, Theil KS, Mohamed A, Paietta E, Willman CL, Head DR,
Rowe JM, et al: Karyotypic analysis predicts outcome of
preremission and postremission therapy in adult acute myeloid
leukemia: A Southwest Oncology Group/Eastern Cooperative Oncology
Group Study. Blood. 96:4075–4083. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mrozek K: Cytogenetic, molecular genetic,
and clinical characteristics of acute myeloid leukemia with a
complex karyotype. Semin Oncol. 35:365–377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Stölzel F, Mohr B, Kramer M, Oelschlagel
U, Bochtler T, Berdel WE, Kaufmann M, Baldus CD, Schafer-Eckart K,
Stuhlmann R, et al: Karyotype complexity and prognosis in acute
myeloid leukemia. Blood Cancer J. 6:e3862016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dash A and Gilliland DG: Molecular
genetics of acute myeloid leukaemia. Best Pract Res Clin Haematol.
14:49–64. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rubnitz JE, Gibson B and Smith FO: Acute
myeloid leukemia. Hematol Oncol Clin North Am. 24:35–63. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Grove CS and Vassiliou GS: Acute myeloid
leukaemia: A paradigm for the clonal evolution of cancer? Dis Model
Mech. 7:941–951. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kern W, Haferlach T, Schnittger S, Ludwig
WD, Hiddemann W and Schoch C: Karyotype instability between
diagnosis and relapse in 117 patients with acute myeloid leukemia:
Implications for resistance against therapy. Leukemia.
16:2084–2091. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cassier PA, Castets M, Belhabri A and Vey
N: Targeting apoptosis in acute myeloid leukaemia. Br J Cancer.
117:1089–1098. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Del Poeta G, Bruno A, Del Principe MI,
Venditti A, Maurillo L, Buccisano F, Stasi R, Neri B, Luciano F,
Siniscalchi A, et al: Deregulation of the mitochondrial apoptotic
machinery and development of molecular targeted drugs in acute
myeloid leukemia. Curr Cancer Drug Targets. 8:207–222. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tamm I, Kornblau SM, Segall H, Krajewski
S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui YH, Monks A, et al:
Expression and prognostic significance of IAP-family genes in human
cancers and myeloid leukemias. Clin Cancer Res. 6:1796–1803.
2000.PubMed/NCBI
|
|
67
|
Sung KW, Choi J, Hwang YK, Lee SJ, Kim HJ,
Kim JY, Cho EJ, Yoo KH and Koo HH: Overexpression of X-linked
inhibitor of apoptosis protein (XIAP) is an independent unfavorable
prognostic factor in childhood de novo acute myeloid leukemia. J
Korean Med Sci. 24:605–613. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ibrahim AM, Mansour IM, Wilson MM, Mokhtar
DA, Helal AM and Al Wakeel HM: Study of survivin and X-linked
inhibitor of apoptosis protein (XIAP) genes in acute myeloid
leukemia (AML). Lab Hematol. 18:1–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhou J, Lu X, Tan TZ and Chng WJ: X-linked
inhibitor of apoptosis inhibition sensitizes acute myeloid leukemia
cell response to TRAIL and chemotherapy through potentiated
induction of proapoptotic machinery. Mol Oncol. 12:33–47. 2018.
View Article : Google Scholar
|
|
70
|
Bosman MC, Schuringa JJ and Vellenga E:
Constitutive NF-κB activation in AML: Causes and treatment
strategies. Crit Rev Oncol Hematol. 98:35–44. 2016. View Article : Google Scholar
|
|
71
|
Rushworth SA, Zaitseva L, Murray MY, Shah
NM, Bowles KM and MacEwan DJ: The high Nrf2 expression in human
acute myeloid leukemia is driven by NF-κB and underlies its
chemo-resistance. Blood. 120:5188–5198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Catz SD and Johnson JL: Transcriptional
regulation of bcl-2 by nuclear factor kappa B and its significance
in prostate cancer. Oncogene. 20:7342–7351. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Murray S, Briasoulis E, Linardou H,
Bafaloukos D and Papadimitriou C: Taxane resistance in breast
cancer: Mechanisms, predictive biomarkers and circumvention
strategies. Cancer Treat Rev. 38:890–903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Alam M, Kashyap T, Pramanik KK, Singh AK,
Nagini S and Mishra R: The elevated activation of NFκB and AP-1 is
correlated with differential regulation of Bcl-2 and associated
with oral squamous cell carcinoma progression and resistance. Clin
Oral Investig. 21:2721–2731. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Xia Y, Shen S and Verma IM: NF-κB, an
active player in human cancers. Cancer Immunol Res. 2:823–830.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Godwin P, Baird AM, Heavey S, Barr MP,
O'Byrne KJ and Gately K: Targeting nuclear factor-kappa B to
overcome resistance to chemotherapy. Front Oncol. 3:1202013.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ohanian M, Rozovski U, Kanagal-Shamanna R,
Abruzzo LV, Loghavi S, Kadia T, Futreal A, Bhalla K, Zuo Z, Huh YO,
et al: MYC protein expression is an important prognostic factor in
acute myeloid leukemia. Leuk Lymphoma. 60:37–48. 2019. View Article : Google Scholar
|
|
78
|
Huang H, Ma L, Li J, Yu Y, Zhang D, Wei J,
Jin H, Xu D, Gao J and Huang C: NF-κB1 inhibits c-Myc protein
degradation through suppression of FBW7 expression. Oncotarget.
5:493–505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Miura K, Takahashi H, Nakagawa M, Izu A,
Sugitani M, Kurita D, Sakagami M, Ohtake S, Uchino Y, Hojo A, et
al: Clinical significance of co-expression of MYC and BCL2 protein
in aggressive B-cell lymphomas treated with a second line
immunochemotherapy. Leuk Lymphoma. 57:1335–1341. 2016. View Article : Google Scholar
|
|
80
|
Kobune M, Takimoto R, Murase K, Iyama S,
Sato T, Kikuchi S, Kawano Y, Miyanishi K, Sato Y, Niitsu Y and Kato
J: Drug resistance is dramatically restored by hedgehog inhibitors
in CD34+ leukemic cells. Cancer Sci. 100:948–955. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lu M, Lin SC, Huang Y, Kang YJ, Rich R, Lo
YC, Myszka D, Han J and Wu H: XIAP induces NF-kappaB activation via
the BIR1/TAB1 interaction and BIR1 dimerization. Mol Cell.
26:689–702. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hofer-Warbinek R, Schmid JA, Stehlik C,
Binder BR, Lipp J and de Martin R: Activation of NF-kappa B by
XIAP, the X chromosome-linked inhibitor of apoptosis, in
endothelial cells involves TAK1. J Biol Chem. 275:22064–22068.
2000. View Article : Google Scholar : PubMed/NCBI
|