Reversible post-translational modifications (PTMs),
including acetylation, phosphorylation, methylation and
ubiquitination, are crucial molecular regulatory mechanisms to
control the specific function of proteins in almost all
physiological processes of eukaryotic cells (1). Ubiquitin is the most well-known
protein modifier. Protein modification via the small
ubiquitin-related modifier (SUMO) system has become a hot topic of
research amongst PTMs, as this pathway regulates hundreds of
proteins that are closely associated with biological processes,
such as DNA repair, macromolecular assembly, chromatin
organization, transcription, transport and intracellular signaling
(2-4). High throughput
mass-spectrometry-based studies revealed that >6,000 proteins
could be SUMOylated (5,6). The interaction between SUMOylation
and other PTMs can significantly expand the specificity and
regulatory potential of each PTM (7). Moreover, SUMOylation has been
reported to regulate a diverse range of physiological and cellular
processes (8), and it is highly
sensitive to various stressors that alter the cellular homeostasis,
such as nutrient deprivation, hypoxia, heat, oxidative stress and
genotoxic stressors (9). A
previous bioinformatics analysis of 13 frequent PTMs, based on the
co-occurrence of sites within proteins across eight eukaryotes,
revealed that SUMOylation was the fastest evolving PTM type, and in
humans alone these >50,000 residues within ≥6,000 proteins form
a vast interconnected network that involve various PTMs, including
SUMOylation (10). Thus, it was
suggested that SUMOylation may be involved in the clonal evolution
of tumor cells and should be considered during the selection of
anticancer-drug treatments.
Accumulating evidence has demonstrated the critical
roles of the SUMO pathway in both carcinogenesis and responses to
therapies. Previous reviews have provided details on the
dysregulation of SUMO cascade enzymes in various solid tumors
(11-13). Thus, the present review focused on
hematological malignancies (leukemias, lymphomas and myelomas),
with an emphasis on multiple myeloma (MM), a genetically
heterogeneous plasma cell malignancy characterized by massive
secretion of (in-)complete monoclonal antibodies, which is prone to
recurrence and relapse (14,15). Balancing protein homeostasis and
post-translational regulation is vital for the survival of MM cells
(16). This review describes the
known functions of the SUMOylation and deSUMOylation pathways and
summarizes the mechanisms of SUMOylation-mediated tumorigenesis and
drug resistance. At present, promising methods and drugs targeting
the SUMOylation pathway have been discovered and provide potential
future directions of therapeutic research regarding the SUMO
pathway.
SUMO modification is an evolutionarily conserved
pathway that is predominantly found within the nucleus of all
eukaryotes. SUMOs are a class of structurally ubiquitin like
proteins (~12 kDa), and the amino acid sequence of SUMO1 is only
18% identical to ubiquitin. SUMO1, 2 and 3 are the best-studied
members of this class. It has been shown that SUMO1 has ~50%
identity with SUMO2/3, which are 97% identical and most often
indistinguishable (8). SUMO1
primarily participates in normal cellular physiology, whereas SUMO2
and SUMO3 are primarily associated with the cell stress response
(17). Other less well
characterized SUMOs are SUMO4, which may be associated with
diabetes (18,19), and SUMO5, which appears to be
expressed specifically in certain tissues, such as the testes and
peripheral blood leukocytes (20), and these remain enigmatic as they
may be unconjugated under normal physiological conditions and,
thus, should be further examined.
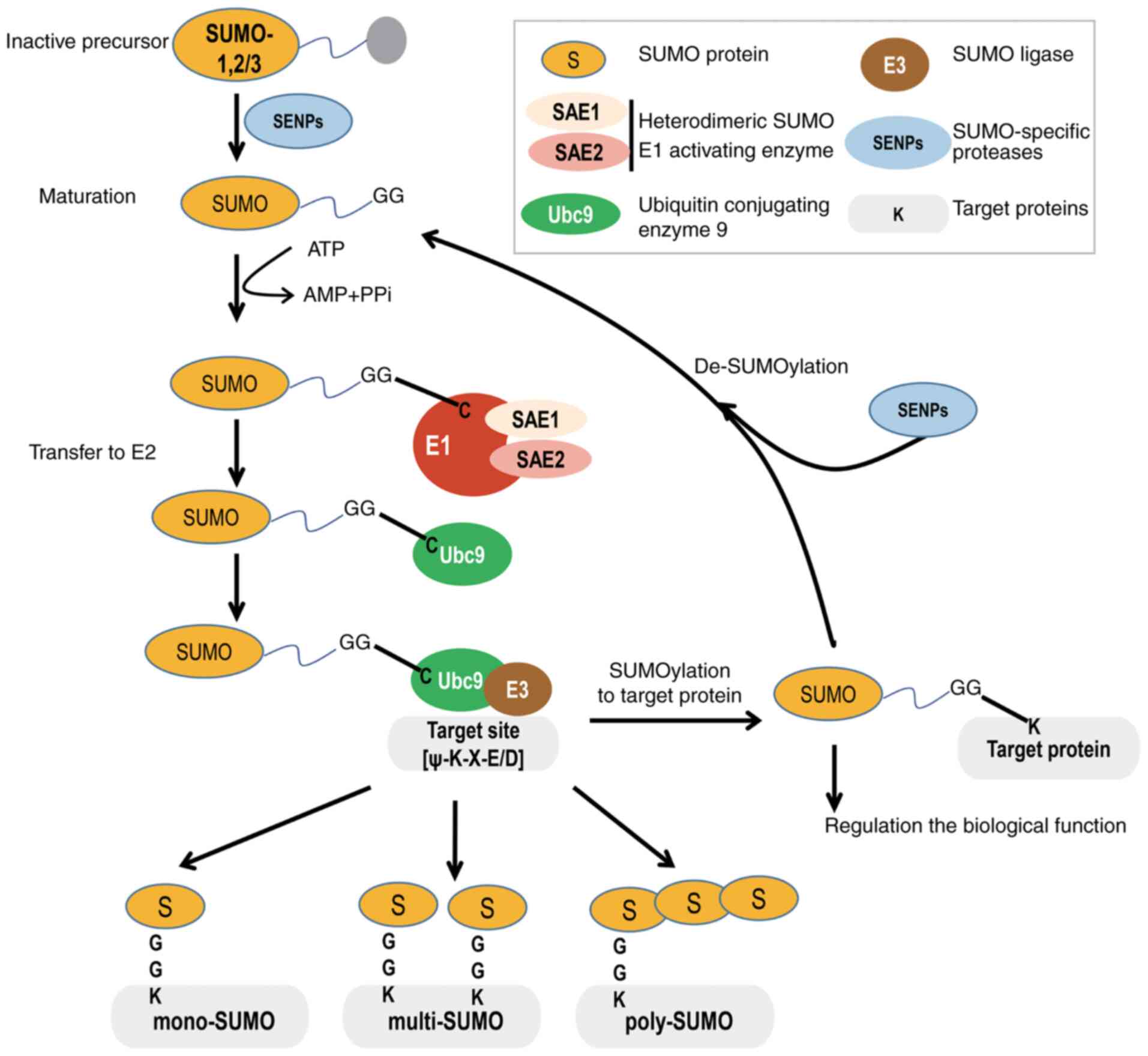
In the SUMOylation pathway, SUMO precursors are
maturated as a result of the action of SUMO isopeptidases cleaving
the C-terminus of SUMO to reveal a glycine (Gly)-Gly (GG) motif.
Then, the mature SUMO protein is activated by the heterodimeric
SUMO1 activating enzyme subunit 1 (SAE1)/ubiquitin like modifier
activating enzyme 2 (UBA2) to form an ATP-dependent thioester bond
between the C-terminal carboxyl GG group of SUMO and the catalytic
cysteine (Cys) residue of UBA2. Next, activated SUMO is shifted
from UBA2 to the catalytic Cys of the only known E2-conjugated
enzyme, ubiquitin conjugating enzyme E2 I (Ubc9), by forming a
thioester bond. Finally, Ubc9 transfers SUMO to the substrate
protein, either alone or with the help of a SUMO E3 ligase, to form
an isopeptide bond between the C-terminal Gly residue of SUMO and
the lysine (Lys/K) residue of the side chain of the target within
the consensus motif of ψKxD/E (where 'ψ' denotes a large
hydrophobic residue and 'x' is any residue) (Fig. 1) (21,22).
Similar to the ubiquitin pathway, SUMOylation is a
cascade process involving SUMO processing and coupling by multiple
enzymes, such as (Conjugation) E1, (Activation) E2 and (Ligation)
E3, to form an isopeptide bond between the C-terminal Gly residue
of SUMO and the ε-amino group of the Lys residue of the substrate
(23,24). Indeed, the target protein could be
mono-, multi- and/or polySUMOylated. SUMO2 and SUMO3 are able to
form homopolymerized or heteropolymerized chains, mainly via
conjugation to their conserved N-terminally located residue at K11,
which is absent in SUMO1. Additionally, SUMO2/3 can form mixed
chains with SUMO1 (25). However,
SUMO1 acts as a chain terminator, as none of its Lys residues can
be further conjugated by any SUMO (26). Importantly, similar to
ubiquitination, most SUMOylated substrate proteins can undergo
constant cycles of conjugation and deconjugation. In contrast to
deubiquitinating enzymes, only one class of the
sentrin/SUMO-specific protease (SENP) family deSUMOylases is
reported in mammals, including SENP-1, 2, 3, 5, 6 and 7, which
cleave the isopeptide bonds and thereby release free SUMO (27). Of note, SUMOylation also conveys
information via a non-covalent interaction with other proteins that
harbor the specific SUMO-interacting motif (SIM), either masking
certain domains, generating a new peptidic moiety or inducing
intraprotein conformational changes (28).
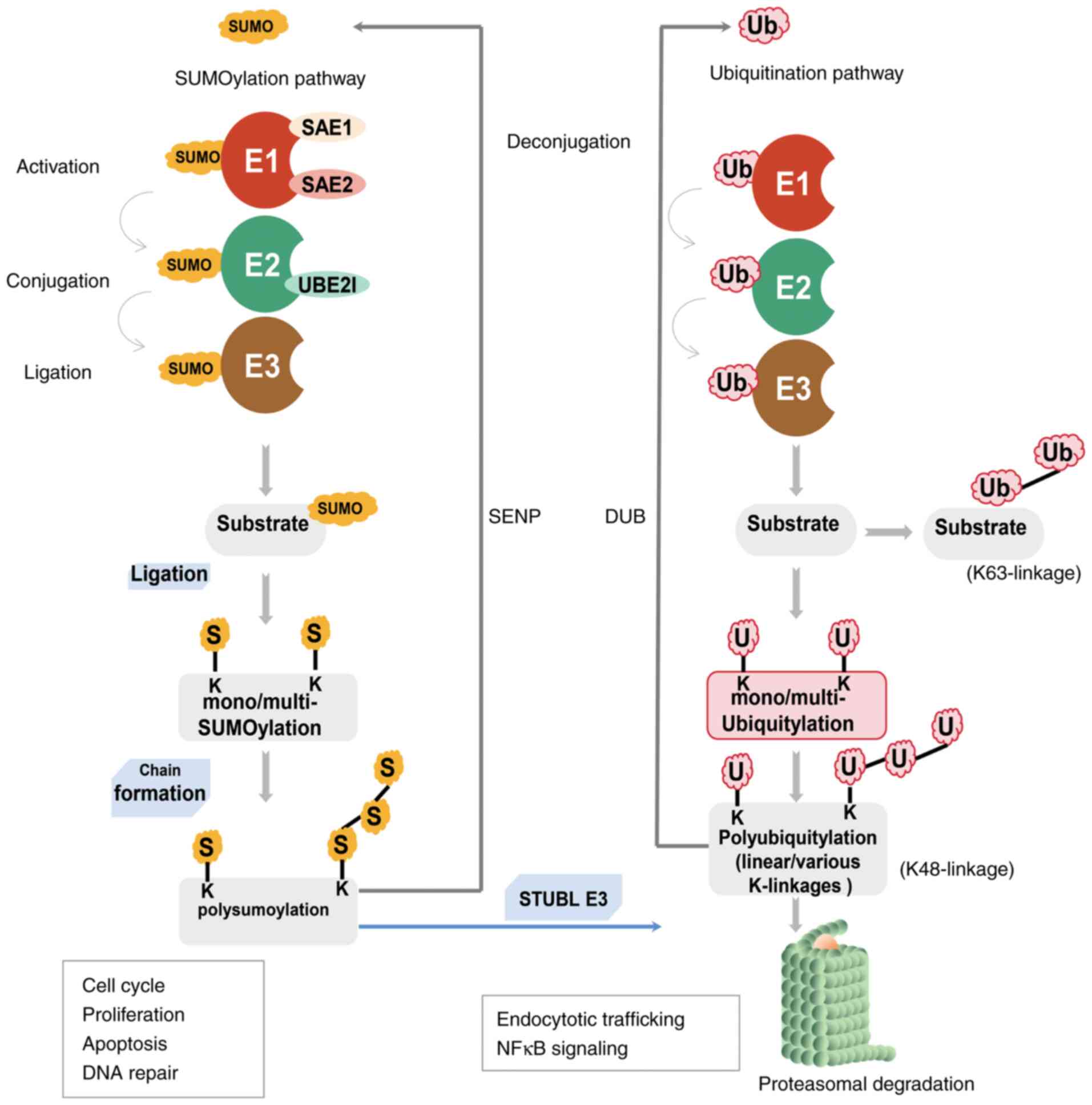
Although both SUMOylation and ubiquitination are
important reversible PTMs that occur at Lys residues, the key
difference between them is that ubiquitination can mark proteins
for proteolytic degradation via proteasomes or have other signaling
functions, whereas SUMOylation is not used to mark proteins for
degradation (29,30). The SUMOylation pathway exhibits
crosstalk with ubiquitination via SUMO targeted ubiquitin ligases
(STUbL) to ubiquitinate and tag SUMOylated proteins for proteasomal
degradation (Fig. 2) (31,32).
The SUMO E1-activating enzyme is a heterodimer
consisting of two subunits: SAE1 and UBA2, which perform
independent adenylation and thioesterification, respectively
(21,32). SAE1 resembles the N-terminal
portion of the ubiquitin E1 enzyme, and UBA2 is similar to the
C-terminal region (33). At
present, only a few E3 ligases have been characterized, such as the
protein inhibitor of activated STAT (PIAS) family, the polycomb
protein PC2 and RAS-related nuclear binding protein 2 (RanBP2). It
has been shown that other ligases, such as zinc finger protein 451,
can elongate SUMO2/3 chains (34-36).
SUMO is a necessary pathway for almost all
eukaryotic cells, and aberrant expression or activity of different
components in the SUMO pathway may alter the nature of the cell
completely, impacting processes such as cell proliferation,
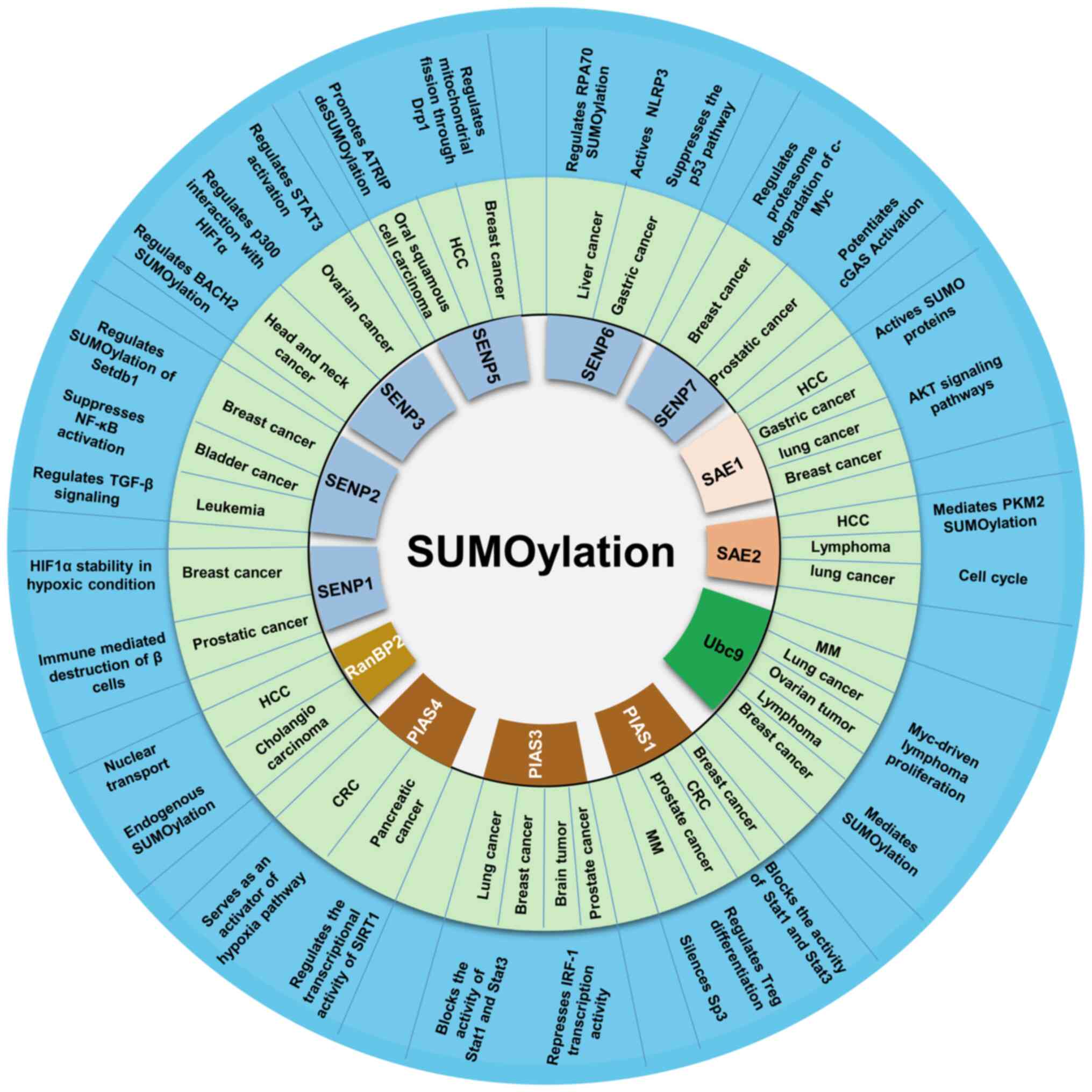
senescence, metastasis and apoptosis (37,38). Abnormalities in SUMOylation
signaling and levels of its targeted proteins can lead to the
development of a variety of diseases, including cancer (39,40) (summarized in Fig. 3). In fact, SUMOylation enzyme
upregulation and/or increased activity have been shown to be
essential in the development of numerous types of tumors, including
various hematological malignancies, such as leukemia, lymphoma and
myeloma (41).
B-cell lymphomas are a group of blood cell
malignancies that affect B cells, and SUMO1 has been shown to be
upregulated in all lymphomas (42). In 2010, a study using bone marrow
samples from newly diagnosed patients with MM observed that the
SUMO-conjugating E2 Ubc9, the SUMO-E3 PIAS1 and the
SUMOylation-inducer tumor suppressor ARF were markedly increased
and associated with adverse patient outcomes compared with healthy
individuals (43). Moreover,
overexpression of the dominant-negative mutant Ubc9 promoted
apoptosis, with unfavorable survival observed in MM cell lines
under γ-irradiation to induce DNA damage, indicating that
SUMOylation induced by Ubc9 and PIAS1 has a protective effect
against DNA-damaging agents (44).
It has been reported that SAE1 and UBA2 knockdown
using RNA interference suppressed the proliferation of lymphoma
cells via disruption of the G2/M transition, and
inhibition of E1 and E2 activity (45). Furthermore, downregulation of UBA2
significantly increased the number of cells in the G1
and G2/M phase, decreased the number of cells in the S
phase, and significantly inhibited the expression of poly
[ADP-ribose] polymerase 1 and micro chromosome maintenance 7
(46). Inhibition of the SUMO E1
complex SAE1/UBA2 with ginkgolic acid can impair the proliferation
of mammary epithelial cells activated by Notch receptor 1 (NOTCH1).
Moreover, activation of the NOTCH1 signaling pathway leads to SUMO
depletion and is more sensitive to SUMOylation inhibition (47), which is discussed in the following
sections.
Ubc9 is the only SUMO E2 conjugating enzyme involved
in selecting and binding directly to the specific SUMO targets that
possess SUMOylation consensus sites. There are two prerequisites
for a protein to be SUMOylated: A direct interaction with the
Ubc9-SUMO thioester and the recognition of a specific SUMO ligase
in proximity with Ubc9 (2). Ubc9
acts as a hub protein of SUMOylation, and has been found to be
upregulated in various types of cancer cells (48-50). Functionally, Ubc9 serves an
important role in cell cycle regulation, DNA repair, transcription
and nuclear transport (51,52). Previous studies have reported that
Ubc9 predominantly exerts its effects via SUMOylation (53,54). In addition, the deregulation of
Ubc9 may lead to alterations in SUMOylation and deSUMOylation of
proteins, and this may affect cancer development and tumor drug
resistance (55). Therefore,
targeting Ubc9 may be a novel therapy for SUMOylation-induced
tumors.
SUMO E3 ligases enhance the transfer of SUMO from
the charged E2 enzyme to the substrate. In contrast to the
ubiquitination system, where hundreds of distinct E3 ligases
mediate the recognition of specific substrates, only the PIAS
family and a few other SUMO E3 ligases have been described, to the
best of our knowledge (36).
Previous findings indicate that SUMO E3 ligases possess a role in
regulating protein stability and signaling transduction pathways.
PIAS proteins were initially identified as inhibitors of STAT
transcription factors. For example, PIAS1 and PIAS3 block the DNA
binding activity of activated STAT1 and STAT3 to inhibit
STAT-mediated transcription, respectively (56). It is becoming increasingly clear
that PIAS proteins regulate nuclear trafficking, DNA damage repair,
NF-κB signaling, several transcription factors and numerous nuclear
receptors (57-59). PIAS1 is upregulated in various
human malignancies, including prostate cancer, MM and B-cell
lymphomas (60,61). Previously, PIAS1 was also reported
as a mediator in lymphomagenesis via SUMOylation of Myc, which
further increases the half-life of Myc and increased oncogenic
activity (61). PIAS1 can also
SUMOylate focal adhesion kinase (FAK) at K152, a modification that
enhances its ability to autophosphorylate threonine (Thr/T)397,
activate FAK and recruit several Src family kinases (62). Furthermore, PIAS1 can regulate
oncogenic signaling via the SUMOylation of promyelocytic leukemia
(PML), and has been shown to be involved in the cancer therapeutic
mechanism of arsenic trioxide (ATO/As2O3) by
promoting the ubiquitin-dependent proteasomal degradation of
PML/retinoic acid receptor α (RARα) (63); this is further described in the
next section. Intriguingly, the PIAS protein can mediate the
SUMOylation of p53 and c-Jun (64). Moreover, PIASγ inhibits
p53-mediated transactivation by inhibiting the DNA binding activity
of p53 in nuclear extracts (64).
The SUMO E3 ligase RanBP2 is a large nucleoporin
possessing SUMO-stabilizing activity, which is involved in several
types of cancer (65,66). RanBP2 is part of a larger complex,
consisting of SUMO-modified Ran GTPase activating protein 1
(RanGAP1), the GTP-hydrolysis activating factor for the GTPase RAN
(67). It has been shown that
RanBP2 mediates the SUMOylation of Polλ at K27, which promotes the
incorporation of Polλ into the nucleus DNA damage repair group with
appropriate polymerase activity (65). RanBP2 also promotes the
SUMOylation of endogenous tripartite motif-containing protein 5
(TRIM5α) on K84 within the predicted phosphorylated SUMOylation
motif in the cytoplasm, thereby forming a complex of
RanGAP1/Ubc9/TRIM5α at the nuclear pore (68).
SUMO-specific proteases are primarily composed of
SENP family members. These SENPs enzymes possessing hydrolase
activity not only catalyze the removal of a C-terminus short
fragment of inactive SUMO to expose two Gly residues during SUMO
maturation, but also cleave and release free SUMO conjugates to
maintain the level of deSUMOylated proteins required for cellular
physiology (69,70). The abnormality of SUMO specific
kinases has been reported in several types of cancer. A total of 6
SENP isoforms (SENP1, SENP2, SENP3, SENP5, SENP6 and SENP7) in
mammals are divided into three subfamilies based on their sequence
homology, substrate specificity and subcellular localization
(27). The SUMO balance dictates
the normal distribution of PTEN between the nucleus and cytoplasm.
Following induction by SENP1, the interaction between PTEN and
NEDD4-like E3 ubiquitin-protein ligase WWP2 is significantly
reduced by ubiquitin-mediated PTEN degradation. Loss of PTEN
expression allows for the elevated SENP1 levels to drive
microinvasive carcinoma (71,72). SENP1 has recently been shown to
possess an oncogenic role in cancer by deSUMOylating c-Myc in cells
and in vitro, which can be co-modified by ubiquitination and
SUMOylation. DeSUMOylation of c-Myc by SENP1 suppresses c-Myc
proteasome degradation and increases the expression levels of
transcriptionally active c-Myc (73). The SENP1-mediated regulation of
myeloid-derived suppressor cells is dependent on STAT3 signaling.
Increased SUMOylation of CD45, a specific STAT3 phosphatase, due to
loss of SENP1 suppresses CD45-mediated dephosphorylation of STAT3,
thereby promoting tumorigenesis (74). Therefore, SENP family members not
only participate in the SUMOylation process, but also actively
regulate tumorigenesis and development mediated by the crosstalk
between SUMOylation and other PTMs.
As mentioned above, the activity of SUMOylation
signaling components not only affects the SUMO process, but also
regulates thousands of downstream targets. Moreover, it may
contribute to the occurrence and development of tumors together
with numerous oncogenic or tumor-suppressive factors, including,
but not limited to, the factors that are discussed further
below.
p53 is the most frequently mutated tumor suppressor
protein and is a well-known transcription factor that can govern
cellular proliferation, apoptosis and senescence (75). Furthermore, p53 is modified by
ubiquitin to directly facilitate its interactions with SUMO family
members (76). MDM2
proto-oncogene (MDM2), as a critical E3 ligase to ubiquitinate p53,
can promote the interaction of p53 with SUMO E3 PIAS. SUMOylation
at the K386 residue of p53 further enhances the p53/MDM2
interaction, thus degrading p53, and the resultant reduced activity
of p53 by SUMOylation may be associated with the acetylation PTM
(77). Furthermore, the oncogenic
SKI proto-oncogene, which is upregulated in numerous types of
cancer, including leukemia, negatively regulates p53 by enhancing
MDM2 SUMOylation to stabilize MDM2 (78).
It has been reported that c-Myc activation
abnormally induces a variety of hematological malignancies in
single Myc transgenic mice (79).
In addition, Myc-driven B cell lymphomas were shown to exhibit
increased expression of core SUMO pathway components, including
SUMO proteins, SAE1, UBA2 and Ubc9 (45). Inhibition of the SUMO pathway
triggers G2/M phase arrest of the cell cycle, polyploidy
and apoptosis in a Myc-specific manner (45). Moreover, SAE1 is activated by
direct binding and transcription of Myc, and then the induced
SUMOylation initiates the Myc oncogenic program (80). The loss of SAE1/UBA2 function
drives the lethal rate of Myc synthesis, and the abnormality of
UBA2 can affect the Myc transcription program, leading to the
abnormality of mitosis (81). The
SUMO E3 ligase PIAS1 can modify Myc to increase the stability and
transactivation activity of Myc, and thus promote Myc-driven
tumorigenesis. The resultant SUMOylation, mainly at residues K51
and K52 of Myc, provides docking sites for JNK1 to phosphorylate
the protein on S62, and prevents the recruitment of GSK3β to
phosphorylate T58 of Myc. S62 phosphorylation activates Myc, whilst
T58 phosphorylation promotes its degradation via the
ubiquitin-proteasome system (61). In addition, SENP1 directly
interacts with Myc, as it deconjugates Myc SUMOylation with either
SUMO1 or SUMO2, and stimulates Myc transactivation activity
(73). Thus, SUMOylation could
also further change the interaction partner and PTM profile of
Myc.
Ras GTPase is a classic membrane-bound signal factor
found in either the active GTP-bound state or the inactive GDP
binding form. Genetic mutations affecting the activity of Ras
GTPase are common in all three subtypes of the Ras family (HRas,
KRas and NRas), with KRas being the most frequently mutated
(82,83). Using gene library screening, SAE1
and UBA2 were found to be closely associated with the lethal effect
of KRas synthesis (84). Further
studies reported that KRas could be SUMOylated both endogenously
and exogenously, and all three Ras protein members were modified by
SUMO3. Moreover, SUMO1/SENP1 and SENP2 can remove the SUMO3
modification from the KRas protein, indicating that the
Ras/SUMOylation process is highly dynamic and reversible (85).
PML and its fused form of PML/RARα, generated as a
result of the specific t(15:17) translocation, established the
initial connection between SUMOylation and cancer, which was first
identified along with discovery of SUMO in the 1990's (86). Acute promyelocytic leukemia (APL)
as a rare, yet highly aggressive and fatal subtype of acute myeloid
leukemia (AML), is currently the most curable leukemia through the
combination of ATO and all-trans retinoic acid (ATRA) treatment
(87). As a result of the
pioneering therapeutic groundwork of Rao et al (88) in 1973 using ATO, the curative
effect of ATO in APL and the subsequent mechanistic study of this
therapy set APL as the paradigmatic example of SUMOylation on tumor
suppressive (PML) or oncogenic proteins (PML/RARα).
The PML gene, encoding the N-terminal region of the
PML/RARα fusion oncoprotein, was first discovered in a patient with
APL and was found to contribute to the formation of the PML nuclear
body (PML-NB) (89). A key
feature of SUMOylation is its association with PML-NB, which is a
membraneless super-assembled sub-nuclear organelle organized mainly
by the PML protein, with specific enrichment of numerous
SUMO-modified client proteins, including p53 and its regulators
(90,91). PML constitutes the outer shell of
the NB spheres, and SUMOylation is dispensable for both PML and the
recruitment of multiple proteins (92). Of note, both the pathogeny and the
treatment of APL rely on SUMOylation. On the one hand, under a
normal state, the PML and SUMO proteins crosstalk reciprocally,
with SUMO heavily modifying PML, and in turn, PML facilitating the
SUMOylation of numerous client proteins by concentrating them into
the PML-NB, where it serves as a scaffold via SUMO-SIM
interactions. On the other hand, high SUMOylation of PML/RARα
precipitates its targeted for degradation at the
ubiquitin-proteasome system, which is engaged by the curative
therapy of As2O3 and ATRA (93). However, the relationship between
PML and SUMO requires further study (90).
pRb, as a negative regulator of cell proliferation,
interacts with >100 cellular proteins and co-regulates cell
senescence together with the SUMO cascade (94,95). The formation of heterochromatin
during aging requires SUMOylation, which can be used as a scaffold
to stabilize pRb repressor complexes. SUMO also conjugates to pRb
and preferentially targets K720 of active, hypophosphorylated pRb
(96). The E2F transcription
factor (E2F) is a key partner of pRb, and the highly regulated cell
cycle depends on the association between pRb and E2F. Moreover,
co-expression of PIASγ and SUMO potently represses E2F-regulated
promoters in a pRb-dependent manner (97,98).
Since the pathological and potentially therapeutic
roles of SUMO extend beyond APL, this review will discuss SUMO's
participation in a range of cellular mechanisms, such as
SUMO-mediated cellular proliferation and stress responsive drug
resistance in hematological malignancies.
Aberrant expression levels of SUMOylation family
members and substrates have been shown to be critical in the
development of various solid tumors and contribute to the stress
response to intrinsic or extrinsic stimuli (47,99,100). Hematologic malignancies arise
generally due to dysregulation of cellular differentiation and
functional blood production throughout the maturation of the human
blood system (101,102). In hematologic tumors, alteration
of SUMO pathway activity can sensitize cells to various stressors
(9,103). MM is a plasma cell malignancy,
which is usually characterized by abnormal elevation of monoclonal
immunoglobulin and a high degree of inter- or intra-individual
genetic heterogeneity, with numerous cytogenetic and epigenetic
changes (104,105). Despite the significant
improvement in the survival of patients over the past decades as a
result of the development of proteasome inhibitors,
immunomodulatory drugs, autologous stem cell transplantation,
monoclonal antibody treatment and chimeric antigen receptor T-cell
immunotherapy (106-109), MM remains incurable as most
patients are either refractory to treatment or relapse due to
drug-resistance (110).
Previous evidence has suggested that targeting the
SUMOylation pathway may serve as a novel therapeutic means to treat
MM. DeSUMOylase SENP1 is upregulated in certain MM cell lines and
patient samples. Knocking down SENP1 in MM cell lines inhibits
proliferation and increases apoptosis (111). Additionally, loss of SENP2
expression in MM leads to an increase in IκBα SUMOylation and the
activation of NF-κB, which induces bortezomib resistance in MM
(112). Therefore,
therapeutically targeting the SUMOylation pathways may become a
promising anticancer strategy to control a variety of events,
including mitotic chromosome separation, cellular proliferation,
metastasis and chemoresistance. This section will summarize several
detailed possible adaptive mechanisms that may contribute to the
progression of MM.
CIN is an intricate phenomenon common in human
cancers, especially in hematological malignancies, and is
characterized by persisting structural and/or numerical chromosome
segregation errors across all stages of tumorigenesis (113-115). Compared with most normal cells,
cancer cells are subject to the stressors of compromised DNA damage
sensing and repair (genotoxic stress), hypoxia and a poor provision
of nutrients. Accumulating evidence has suggested that SUMO
modification serves a pivotal role in almost all types of DNA
repair mechanisms, including base excision repair, non-homologous
end-joining and homologous recombination (116). Studies using microscopy and
laser microablation showed the orderly recruitment of SUMO
components and downstream effectors, including STUbLs, to sites of
double-strand DNA breaks, thus confirming the direct in situ
roles of SUMOylation in DNA repair (117). Early genetic studies reported
that SUMO conjugation and deconjugation are indispensable for
sister chromatid aggregation, chromosomal aggregation, centromere
and motility function, and chromosomal separation (118). Furthermore, multiple centromeres
and kinetogranular proteins have been identified as SUMO targets
(119-121). It has been shown that disruption
of the SUMO bonding mechanism leads to delayed mitosis and mitotic
chromosome separation defects (122). The SUMO E3 ligase PIASγ is
specifically targeted to mitotic chromatin (123). Furthermore, downregulation of
RanBP2 expression in mice results in severe defects of chromosomal
segregation and increased aneuploidy (124). The stable complex of RanBP2 and
Ubc9 may appear on the nuclear membrane during interphase, and can
translocate to the centromere and the spinneret during mitosis
(125). Interestingly, as a
maintenance factor for heterochromatin 1 (HP1) accumulation in
peri-central heterochromatin, SENP7 is essential for centromere
organization and accurate chromosomal segregation by directly
binding and stabilizing HP1 in the central region of mouse cells.
Accordingly, SENP7 can deSUMOylate HP1 in vivo, and
depletion of SENP7 results in a prolonged period of time spent in
mitosis (126). These examples
highlight the significance of further studying the dynamic and
precise SUMO mechanism in the process of CIN.
Flap structure-specific endonuclease 1 (FEN1)
nuclease activity is also essential for cell-cycle progression, as
well as the maintenance of genomic stability. SUMOylation of FEN1
mediated by SUMO3 is stimulated by phosphorylation, which enhances
its ubiquitination and degradation via the SUMOylation-dependent
ubiquitin proteasome pathway. Conversely, blocking FEN1 degradation
by mutating the SUMOylation or ubiquitination sites leads to the
accumulation of cyclins B/E and a delayed cell cycle (132). The overexpression of SENP1 can
increase cyclin D1 expression and reduce the sensitivity of cancer
cells to radiotherapy. Additionally, SENP1 mutations in its
catalytic domain affect its ability to regulate cyclin D1
transcription (133,134). It has also been reported that
SENP1 can increase IL-6-induced phosphorylation of p65 and IκBα to
activate NF-κB signaling, thereby regulating cell cycle progression
and proliferation of MM cells (111). These examples demonstrate that
numerous important cell cycle regulators are functionally regulated
by SUMOylation. Moreover, SUMOylation regulates tumor progression
by modifying the proteome at various stages of the cell cycle.
Cellular senescence, recognized as a critical tumor
suppressive mechanism, refers to the usually irreversible cell
cycle arrest, which is a common cellular response to stressors,
such as DNA damage, oxidative stress or the aberrant expression of
critical regulators (134). p53,
as a tumor suppressor, promotes cellular senescence, as well as
programmed cell death (135).
Moreover, SUMO1-modified p53 can cause p53 stabilization and the
induction of senescence (136).
The repression of SENP1 can mediate premature senescence of normal
human fibroblasts by increasing the transcriptional activity of p53
(137), and knockdown of Ubc9 in
primary human fibroblasts can result in senescence-like growth
arrest (129). Therefore,
cellular senescence is induced via dysregulated SUMOylation and can
further affect the SUMOylation of several genes involved in
regulating cell proliferation.
Apoptosis is a mechanism that negatively controls
cell proliferation, and is also referred to as programmed cell
death. If DNA damage is not repaired in time for maintaining the
genomic stability and cellular integrity, cells will die via this
mechanism (138). Certain
malignancies possess defects in regulatory apoptotic pathways, such
as NF-κB, p53 and PI3K/Akt, which lead to apoptosis defects and
permanent proliferation of tumor cells (139). Amongst these, the first pathway,
known as the cytoplasmic pathway, is triggered by Fas death
receptors (140), and the second
is the mitochondrial pathway, which releases the cytochrome c death
signal from the mitochondria (141). Recent research has indicated
that SUMOylation may indirectly regulate cell apoptosis by
affecting the expression and activity of related signaling factors.
The interaction of SUMO1 with the caspase activation and
recruitment domain of caspase-2 relies on the K60 site, and the
K60R mutation abolishes this modification, delays enzyme maturation
and reduces caspase-2 activity (142). It has been reported that DCB1 is
a major inhibitor of sirtuin 1 (SIRT1) and could be phosphorylated
at T454 by the ATM/ATR kinases to switch the binding partner of
DBC1 from SENP1 to PIAS3. Subsequently, SUMOylation modification of
DBC1 by SUMO2/3 increases the interaction between DBC1 and SIRT1,
leading to the release of p53 from SIRT1 for transcriptional
activation-mediated apoptosis (143,144). Moreover, SENP1 deficiency
significantly increases ER stress-induced apoptosis by accumulating
X-box binding protein 1 SUMOylation (145).
Myeloid cell leukemia 1 (MCL1), an anti-apoptotic
protein that belongs to the Bcl-2 family, maintains its stability
via its SUMOylation at sites K234 and K238, and inhibits
TRIM11-mediated ubiquitination of MCL1 and cancer cell apoptosis
(149). Moreover, death domain
associated protein (DAXX) has been shown to contain SIMs, and
phosphorylation of DAXX-SIM promotes SUMOylation by binding to
SUMO1 and SUMO2/3, thereby enhancing stress-induced apoptosis via
inhibiting anti-apoptotic genes (150,151). In addition, when p53 is
transferred to the nucleus and is modified by SUMO, the
transcriptional expression of the pro-apoptotic gene Bax can be
upregulated (152). Recently, it
has been found that SUMOylation could serve a dual role in the
mechanism of apoptosis. For instance, in addition to its
traditional anti-apoptotic role, it can also stimulate the
activation of other cytokines to promote apoptosis (153).
Autophagy signaling events, including induction,
regulation and fine-tuning under various stresses, are also
dependent on PTM of proteins, such as phosphorylation, acetylation,
ubiquitination and SUMOylation. Phosphorylation during autophagy
regulates the activity of autophagy-related proteins, as well as
the initiation and progression of autophagy by regulating signaling
pathways (154). Both
acetylation and deacetylation are involved in autophagy initiation
and selective autophagy regulation by controlling the levels of
acetylation of important proteins (155,156). The crosstalk between
ubiquitylation and SUMOylation may be involved in mediating
autophagy (157). For example,
SUMOylated 3-phosphoinositide dependent protein kinase 1 modulates
the activation of its own phosphorylation to initiate
macroautophagy/autophagy (158).
Furthermore, overexpression of SUMO1 or the SUMO E2 enzyme Ubc9 can
accelerate the accumulation of autophagosomes. It has also been
shown that the increase of beclin 1 (BECN1) base SUMOylation
promotes the interaction of BECN1 with UV radiation resistance
associated PIK3C3 and autophagy related 14 to form a complex,
thereby enhancing autophagic protein localization and autophagosome
formation (159).
Metastasis, the cause of the majority of
cancer-related deaths, involves the metastasis of cells from a
tumor to a distant organ. TGF-β signaling is essential for cellular
proliferation, metastasis and angiogenesis (160). TGF-β may contribute to the
inhibition of tumor growth during the early stages of cancer
development, whereas it can promote epithelial-mesenchymal
transition and metastasis during the later stages. Furthermore,
TGF-β can be modified by SUMO and this amplifies TGF-β signaling at
multiple levels under various conditions (161), such as hypoxia and chemical drug
treatment (162).
Chemoresistance involves multiple complicated
cellular factors, and amongst them, SUMOylation was firstly shown
to be involved in AML, which is a heterogeneous group of severe
hematological leukemias resulting from the oncogenic transformation
of hematopoietic stem cells and myeloid progenitors (163). In AMLs, the standard
genotoxic-based chemotherapies, combining anthracyclines
(daunorubicine or idarubicine) and a nucleoside analogue
(cytarabine) (161), produce
reactive oxygen species (ROS) and inhibit SUMOylation via oxidative
crosslinking of E1 and E2 enzymes, thereby causing death of
chemosensitive cells. By contrast, the chemoresistant AML cells
that produce fewer ROS with active SUMOylation can be re-sensitized
either by inhibiting SUMOylation or restoring a pro-oxidative
condition (164).
The most frequent chromosomal translocation is
t(12;21) in acute lymphoblastic leukemia (ALL), which is also the
most common genetic rearrangement found in pre-B ALL in children,
accounting for ~25% of ALL cases (165). Translocation-ETS-leukemia (TEL)
is the main target of chromosomal translocation in lymphoid and
myeloid leukemias (166). A
previous study has reported that TEL can be covalently modified by
SUMO1, and SUMOylation changes the nuclear localization of TEL,
whereas mutation of the lysine residue at position 99 of TEL leads
to its nuclear accumulation, which is critical in the regulation of
intercellular adhesion molecules, and may be associated with ALL
(166,167). As mentioned above, the exception
concerns one rare (~10%) type of AML, known as APL, which is
associated with the common t(15;17) chromosomal translocations,
producing a PML/RARα chimeric protein (168). APL is efficiently treated using
the differentiation therapy of ATRA and ATO. A previous mechanistic
study demonstrated that an active SUMOylation pathway was crucial
for the successful treatment of APLs, as ATO directly bound to
PML/RARα and PML and triggered their polymerization via the
oxidation of specific cysteines and the formation of disulfide
bonds (169). Then, PML was
concentrated and highly SUMOylated to form the PML-NBs, whereas the
highly SUMOylated PML/RARα could be degraded by the proteasome via
its recruitment of the SUMO-dependent ubiquitin ligase ring finger
protein 4. The degradation of whole oncogenic fusion protein
reactivates the RARα signaling pathway, as well as restoring
PML-NBs and the p53 pathway (170,171). In addition, B-cell lymphomas are
a group of blood cell malignancies that affect B cells, and
aberrantly high SUMO1 expression has been found in all lymphomas
(42). Therefore, therapeutically
targeting the SUMOylation pathways may become a novel anticancer
strategy for treatment of MM and other hematological tumors.
Given the aforementioned findings, there is no doubt
that SUMOylation contributes to tumorigenesis and progression in
various types of cancer, by allowing survival from internal or
external stressors, and it may be a promising target for cancer
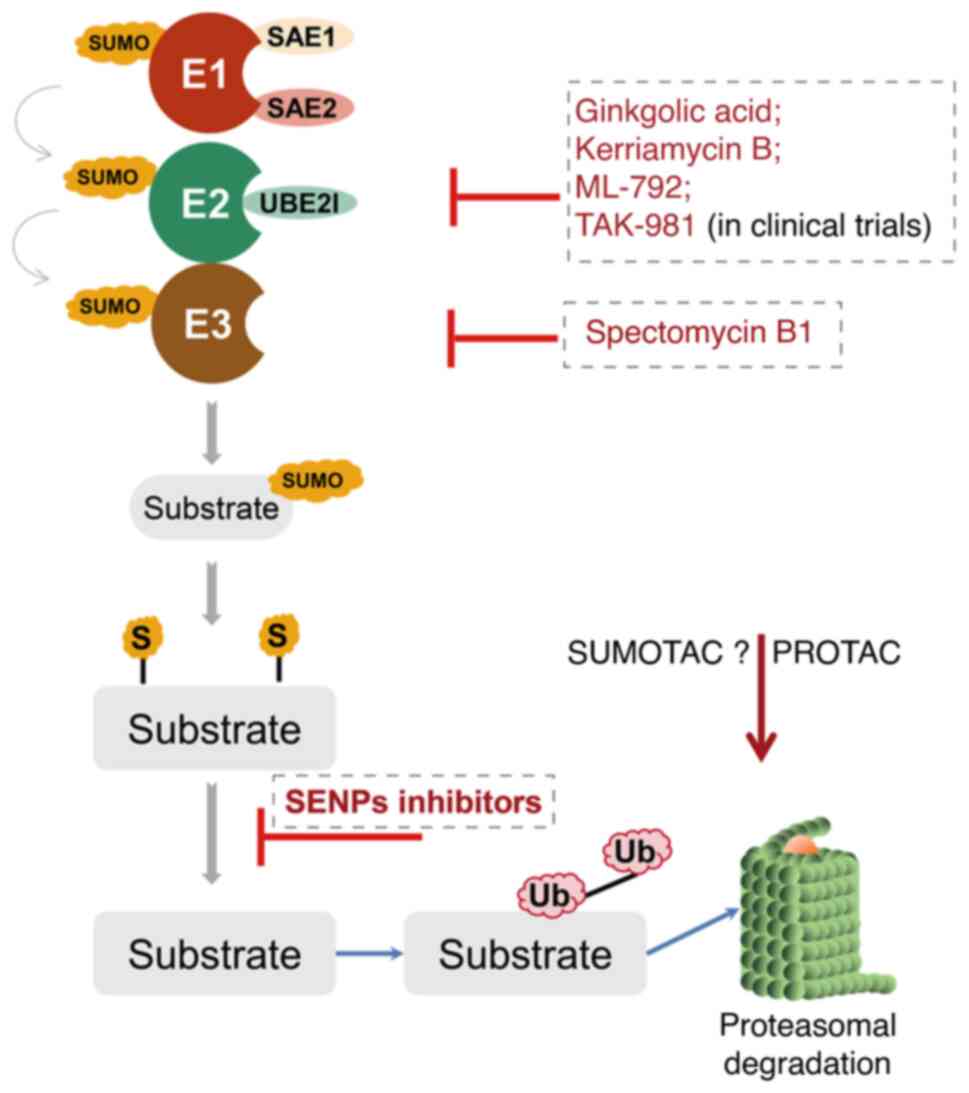
therapy. At present, there are a few types of SUMO inhibitors
reported in the literature (172). However, these molecules mostly
lack sufficient specificity and efficiency, as well as clinically
relevant pharmacological properties (173). Most of them can be categorized
into either natural or synthetic small molecular inhibitors
(Fig. 4).
Ginkgolic acid and anacardic acid were identified
via high-throughput screening of plant extracts and have been shown
to inhibit protein synthesis in vivo and in vitro by
directly binding to SUMO E1 (174,175). Spectomycin B1 can directly
interact with Ubc9 and selectively block the formation of the
E2-SUMO intermediate (176).
Another particular compound is the aforementioned traditional
Chinese medicine ATO (As2O3), which has been
approved for the treatment of relapsed or refractory APL in
combination with ATRA by the Food and Drug Administration of the
United States in 2000 (177). An
important advance in the field of SUMOylation was the synthesis of
mechanism-based inhibitors targeting the SUMO pathway. For example,
ML-792 forms an adduct with SUMO and blocks the SUMO E1 (nM range),
as well as exerting anti-tumor activity, with only minor effects on
gene expression without affecting DNA-repair. However, ML-792 also
leads to chromosome-segregation defects during mitosis,
proliferation arrest and even cell death (178). TAK-981, a derivative of ML-792,
is currently being tested in a phase I clinical trial in patients
with metastatic solid tumors and lymphomas (ClinicalTrials.gov Identifier: NCT03648372) (179). The recently discovered COH000,
although less potent than ML-792 (µM range), can decrease
Myc expression in lymphoma cell lines in vitro and inhibit
SUMO E1 by binding to a cryptic allosteric site (180,181). Other inhibitors targeting the
SUMOylation cascade, including SAE1/UBA2 (182-188), Ubc9 (189-192), SENP1 (193-201) and SENP2 (202,203), are summarized in Table I.
In addition, protein- or peptide-derived drugs may
also be designed to target the SUMO pathway. The adenovirus
protein, Gam1, was the first reported SUMO E1 inhibitor that could
directly block the activity of the E1 enzyme in vitro,
thereby blocking the formation of intermediate products of the SUMO
E1 thioester reaction, and thus inhibiting the SUMO E1 of the
substrate. In vivo, the expression of Gam1 can lead to the
inactivation of SAE1/UBA2, the disappearance of SAE1/UBA2 and Ubc9
can inhibit the SUMOylation of proteins (204). The protein inhibitors based on a
semisynthetic mechanism can mimic the tetrahedral intermediates
produced during the formation of adenylate intermediates or
thioester bonds (184).
Recently, a study using a SUMO1-derived peptide inhibitor found
that it targeted two SIMs within α-synuclein aggregation and
reduced α-synuclein-induced cytotoxicity in cell-based and
Drosophila disease models (205).
In summary, clinical trials have recently begun and
therapeutic utilization of SUMOylation inhibitors will face the
classic challenges of drug discovery and development before
benefiting patients. SUMOylation serves an important role in the
development of tumors and targeting SUMOylation is a novel
therapeutic strategy, which could be used in the diagnosis and
treatment of hematologic malignancies and numerous other cancer
types.
In recent years, the key roles of PTMs in the
physiological and pathological development of cells has been
extensively studied. Increasing evidence has shown that SUMO
enzymes participate in cell proliferation, cell division,
chromosomal instability, apoptosis and stress-response in numerous
types of cancer, including hematological malignancies. Based on the
influence of SUMOylation on the development of hematologic
malignancies, the further development of small molecules that
specifically modulate these PTMs is a highly promising approach of
novel targeted therapies for cancer. However, additional research
is required to further understand the function and the relevance of
SUMOylation targeting anti-cancer therapy from bench to
bedside.
Given that deregulation of SUMO pathways
contributes to increased cell proliferation with reduced apoptosis
in tumors, further investigation into the regulatory mechanism of
the dynamic redistribution/activity of SUMO signaling is necessary.
At present, thousands of novel SUMO target proteins have been
identified in a site-specific manner using proteomics approaches.
However, a more global examination of overall SUMOylation levels in
a broad range of tumors should be performed. Examination of the
functional relevance of SUMOylation for all novel SUMO substrates
should also be conducted. Identification of the effects of
SUMOylation during cell cycle progression will provide functional
insights into the role of SUMOylation in tumorigenesis.
Furthermore, it will be interesting to investigate how different
PTMs cooperate in a cell-wide manner to drive cell cycle
progression. For example, the SUMOylation pathway may crosstalk
with ubiquitylation, phosphorylation and other PTMs during cell
proliferation.
Current evidence has shown that in certain tumors,
hyper-SUMOylation is required for tumor cells to survive,
especially under conditions of stress. Different types of tumors
depend on a functioning SUMOylation system. A potential challenge
may be the specificity of SUMOylation modification during
therapeutic development of SUMO-targeted interference. Whether it
is possible to develop SUMOylation or deSUMOylation inhibitors that
can only kill tumor cells, whilst balancing toxicity or tolerance
in healthy tissues should be determined. Since hematological cells
proliferating in bone marrow and elsewhere may be similarly
dependent on the SUMOylation system, it would be interesting to
test whether inhibiting SUMOylation is a valid anticancer
strategy.
As discussed extensively, PML-NBs are druggable
SUMOylation targets that provide scaffolds for leukemia
intervention. Whilst at present, there is a lack of an effective
method to target certain proteins to PML-NBs, a robust and
generalized induction of PML-NBs biogenesis using ATO treatment can
benefit patients. The enhanced PML-NB formation can enrich the
hyper-SUMOylated pathogenic proteins, leading to distinct
processes, such as degradation, sequestration into the PML-NBs,
alteration in activity and modification via other PTMs. Therefore,
targeting PML/SUMO mediated oncoprotein oligomerization may
represent a promising strategy in the treatment of hematological
malignancies and other solid tumors (93).
Considering the critical role of the
ubiquitin-proteasome system in maintaining protein homeostasis for
MM and the deregulated SUMOylation in various hematological
malignancies, these add the complicated interactions between
ubiquitination and SUMOylation. On the one hand, SUMO may act
together with ubiquitin to degrade proteins via STUbL. On the other
hand, SUMOylation antagonizes the ubiquitination degradation of
SUMOylated proteins by competing for ubiquitination sites. Thus,
clarifying the crosstalk between SUMOylation and ubiquitination in
hematological tumors is also crucial for the development of further
targeted therapies. Targeting the SUMO pathway, alone or in
combination with other drugs, is therefore a promising approach in
the treatment of hematological malignancies.
Finally, similar to the emerging ubiquitylation
based technology of 'proteolysis targeting chimera' to degrade a
given substrate via the ubiquitin proteasomal system (206), the SUMO pathway may be precisely
targeted for a given substrate to fine-tune a substrate protein's
stability, localization, solubility and interaction network. Whilst
this system is yet to be developed, a chimeric construct where Ubc9
is directly attached to a substrate to specifically induce its
SUMOylation has been developed (207). A similar effect could be
achieved via the use of a chimeric construct that consists of a
protein domain specifically binding the target substrate and a SUMO
machinery component attaching to this protein domain via a linker
peptide (208).
Taken together, over the past two decades,
significant advances have been achieved in the field of SUMOylation
in cancer. The identification of novel combination therapies using
PTM-based depressant agents is of great significance for clinical
practice. Collectively, developing and investigating inhibitors of
SUMO conjugation in the coming years are of promising potential for
novel therapeutic strategies.
Not applicable.
LW wrote the first draft of the manuscript. JJQ and
LW drew the figures and revised the manuscript. YY and CYG reviewed
and edited the manuscript. All authors have read and approved the
final manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
|
1
|
Deribe YL, Pawson T and Dikic I:
Post-translational modifications in signal integration. Nat Struct
Mol Biol. 17:666–672. 2010. View Article : Google Scholar
|
|
2
|
Flotho A and Melchior F: Sumoylation: A
regulatory protein modification in health and disease. Ann Rev
Biochem. 82:357–385. 2013. View Article : Google Scholar
|
|
3
|
Bergink S and Jentsch S: Principles of
ubiquitin and SUMO modifications in DNA repair. Nature.
458:461–467. 2009. View Article : Google Scholar
|
|
4
|
Iribarren PA, Di Marzio LA, Berazategui
MA, De Gaudenzi JG and Alvarez VE: SUMO polymeric chains are
involved in nuclear foci formation and chromatin organization in
Trypanosoma brucei procyclic forms. PLoS One. 13:e01935282018.
View Article : Google Scholar
|
|
5
|
Hendriks I, Lyon D, Young C, Jensen L,
Vertegaal A and Nielsen M: Site-specific mapping of the human SUMO
proteome reveals co-modification with phosphorylation. Nat Struct
Mol Biol. 24:325–336. 2017. View Article : Google Scholar
|
|
6
|
Hendriks I and Vertegaal A: A
comprehensive compilation of SUMO proteomics. Nat Rev Mol Cell
Biol. 17:581–595. 2016. View Article : Google Scholar
|
|
7
|
Beltrao P, Bork P, Krogan N and van Noort
V: Evolution and functional cross-talk of protein
post-translational modifications. Mol Syst Biol. 9:7142013.
View Article : Google Scholar
|
|
8
|
Zhao X: SUMO-mediated regulation of
nuclear functions and signaling processes. Mol Cell. 71:409–418.
2018. View Article : Google Scholar
|
|
9
|
Enserink JM: Sumo and the cellular stress
response. Cell Div. 10:42015. View Article : Google Scholar
|
|
10
|
Minguez P, Parca L, Diella F, Mende DR,
Kumar R, Helmer-Citterich M, Gavin AC, van Noort V and Bork P:
Deciphering a global network of functionally associated
post-translational modifications. Mol Syst Biol. 8:5992012.
View Article : Google Scholar
|
|
11
|
Han ZJ, Feng YH, Gu BH, Li YM and Chen H:
The post-translational modification, SUMOylation, and cancer
(Review). Int J Oncol. 52:1081–1094. 2018.
|
|
12
|
Mattoscio D and Chiocca S: SUMO pathway
components as possible cancer biomarkers. Future Oncol.
11:1599–1610. 2015. View Article : Google Scholar
|
|
13
|
Xie M, Yu J, Ge S, Huang J and Fan X:
SUMOylation homeostasis in tumorigenesis. Cancer Lett. 469:301–309.
2020. View Article : Google Scholar
|
|
14
|
Neuse CJ, Lomas OC, Schliemann C, Shen YJ,
Manier S, Bustoros M and Ghobrial IM: Genome instability in
multiple myeloma. Leukemia. 34:2887–2897. 2020. View Article : Google Scholar
|
|
15
|
Rajkumar SV and Kumar S: Multiple myeloma
current treatment algorithms. Blood Cancer J. 10:942020. View Article : Google Scholar
|
|
16
|
Sha Z and Goldberg AL: Multiple myeloma
cells are exceptionally sensitive to heat shock, which overwhelms
their proteostasis network and induces apoptosis. Proc Natl Acad
Sci USA. 117:21588–21597. 2020. View Article : Google Scholar
|
|
17
|
Zhao Q, Ma Y, Li Z, Zhang K, Zheng M and
Zhang S: The function of SUMOylation and its role in the
development of cancer cells under stress conditions: A systematic
review. Stem Cells Int. 2020:88357142020. View Article : Google Scholar
|
|
18
|
Li YY, Wang H, Yang XX, Geng HY, Gong G,
Kim HJ, Zhou YH and Wu JJ: Small Ubiquitin-Like Modifier 4 (SUMO4)
Gene M55V polymorphism and type 2 diabetes mellitus: A
Meta-analysis including 6,823 subjects. Front Endocrinol.
8:3032017. View Article : Google Scholar
|
|
19
|
Bohren KM, Nadkarni V, Song JH, Gabbay KH
and Owerbach D: A M55V polymorphism in a novel SUMO gene (SUMO-4)
differentially activates heat shock transcription factors and is
associated with susceptibility to type I diabetes mellitus. J Biol
Chem. 279:27233–27238. 2004. View Article : Google Scholar
|
|
20
|
Liang YC, Lee CC, Yao YL, Lai CC, Schmitz
ML and Yang WM: SUMO5, a Novel Poly-SUMO isoform, regulates PML
nuclear bodies. Sci Rep. 6:265092016. View Article : Google Scholar
|
|
21
|
Gong L, Li B, Millas S and Yeh ET:
Molecular cloning and characterization of human AOS1 and UBA2,
components of the sentrin-activating enzyme complex. FEBS Lett.
448:185–189. 1999. View Article : Google Scholar
|
|
22
|
Desterro JM, Rodriguez MS, Kemp GD and Hay
RT: Identification of the enzyme required for activation of the
small ubiquitin-like protein SUMO-1. J Biol Chem. 274:10618–10624.
1999. View Article : Google Scholar
|
|
23
|
Müller S, Hoege C, Pyrowolakis G and
Jentsch S: SUMO, ubiquitin's mysterious cousin. Nat Rev Mol Cell
Biol. 2:202–210. 2001. View Article : Google Scholar
|
|
24
|
Johnson ES: Protein modification by SUMO.
Ann Rev Biochem. 73:355–382. 2004. View Article : Google Scholar
|
|
25
|
Tatham MH, Jaffray E, Vaughan OA, Desterro
JM, Botting CH, Naismith JH and Hay RT: Polymeric chains of SUMO-2
and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and
Ubc9. J Biol Chem. 276:35368–35374. 2001. View Article : Google Scholar
|
|
26
|
Sriramachandran AM, Meyer-Teschendorf K,
Pabst S, Ulrich HD, Gehring NH, Hofmann K, Praefcke GJ and Dohmen
RJ: Arkadia/RNF111 is a SUMO-targeted ubiquitin ligase with
preference for substrates marked with SUMO1-capped SUMO2/3 chain.
Nat Commun. 10:36782019. View Article : Google Scholar
|
|
27
|
Drag M and Salvesen GS: DeSUMOylating
enzymes-SENPs. IUBMB Life. 60:734–742. 2008. View Article : Google Scholar
|
|
28
|
Hecker CM, Rabiller M, Haglund K, Bayer P
and Dikic I: Specification of SUMO1- and SUMO2-interacting motifs.
J Biol Chem. 281:16117–16127. 2006. View Article : Google Scholar
|
|
29
|
Chen Y, Sun XX, Sears RC and Dai MS:
Writing and erasing MYC ubiquitination and SUMOylation. Genes Dis.
6:359–371. 2019. View Article : Google Scholar
|
|
30
|
Fan L, Bi T, Wang L and Xiao W: DNA-damage
tolerance through PCNA ubiquitination and sumoylation. Biochem J.
477:2655–2677. 2020. View Article : Google Scholar
|
|
31
|
Sriramachandran AM and Dohmen RJ:
SUMO-targeted ubiquitin ligases. Biochim Biophys Acta. 1843:75–85.
2014. View Article : Google Scholar
|
|
32
|
Johnson ES, Schwienhorst I, Dohmen RJ and
Blobel G: The ubiquitin-like protein Smt3p is activated for
conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO
J. 16:5509–5519. 1997. View Article : Google Scholar
|
|
33
|
Lois LM and Lima CD: Structures of the
SUMO E1 provide mechanistic insights into SUMO activation and E2
recruitment to E1. EMBO J. 24:439–451. 2005. View Article : Google Scholar
|
|
34
|
Cappadocia L, Pichler A and Lima CD:
Structural basis for catalytic activation by the human ZNF451 SUMO
E3 ligase. Nat Struct Mol Biol. 22:968–975. 2015. View Article : Google Scholar
|
|
35
|
Eisenhardt N, Chaugule VK, Koidl S,
Droescher M, Dogan E, Rettich J, Sutinen P, Imanishi SY, Hofmann K,
Palvimo JJ and Pichler A: A new vertebrate SUMO enzyme family
reveals insights into SUMO-chain assembly. Nat Struct Mol Biol.
22:959–967. 2015. View Article : Google Scholar
|
|
36
|
Werner A, Flotho A and Melchior F: The
RanBP2/RanGAP1*SUMO1/Ubc9 complex is a multisubunit SUMO E3 ligase.
Mol Cell. 46:287–298. 2012. View Article : Google Scholar
|
|
37
|
Drabikowski K: Ubiquitin and SUMO
Modifications in Caenorhabditis elegans stress response. Curr
Issues Mol Biol. 35:145–158. 2020. View Article : Google Scholar
|
|
38
|
Lu W, Wang Q, Xu C, Yuan H, Fan Q, Chen B,
Cai R, Wu D and Xu M: SUMOylation is essential for Sirt2
tumor-suppressor function in neuroblastoma. Neoplasia. 23:129–139.
2021. View Article : Google Scholar
|
|
39
|
Chanda A, Sarkar A and Bonni S: The SUMO
System and TGFβ signaling interplay in regulation of
epithelial-mesenchymal transition: Implications for cancer
progression. Cancers (Basel). 10:2642018. View Article : Google Scholar
|
|
40
|
Wu G, Xu Y, Ruan N, Li J, Lv Q, Zhang Q,
Chen Y, Wang Q, Xia Q and Li Q: Genetic alteration and clinical
significance of SUMOylation regulators in multiple cancer types. J
Cancer. 11:6823–6833. 2020. View Article : Google Scholar
|
|
41
|
Boulanger M, Paolillo R, Piechaczyk M and
Bossis G: The SUMO pathway in Hematomalignancies and their response
to therapies. Int J Mol Sci. 20:38952019. View Article : Google Scholar
|
|
42
|
Küppers R: Mechanisms of B-cell lymphoma
pathogenesis. Nat Rev Cancer. 5:251–262. 2005. View Article : Google Scholar
|
|
43
|
Driscoll JJ, Pelluru D, Lefkimmiatis K,
Fulciniti M, Prabhala RH, Greipp PR, Barlogie B, Tai YT, Anderson
KC, Shaughnessy JD Jr, et al: The sumoylation pathway is
dysregulated in multiple myeloma and is associated with adverse
patient outcome. Blood. 115:2827–2834. 2010. View Article : Google Scholar
|
|
44
|
Chen YC, Hsu WL, Ma YL, Tai DJ and Lee EH:
CREB SUMOylation by the E3 ligase PIAS1 enhances spatial memory. J
Neurosci. 34:9574–9589. 2014. View Article : Google Scholar
|
|
45
|
Hoellein A, Fallahi M, Schoeffmann S,
Steidle S, Schaub FX, Rudelius M, Laitinen I, Nilsson L, Goga A,
Peschel C, et al: Myc-induced SUMOylation is a therapeutic
vulnerability for B-cell lymphoma. Blood. 124:2081–2090. 2014.
View Article : Google Scholar
|
|
46
|
Jiang B, Fan X, Zhang D, Liu H and Fan C:
Identifying UBA2 as a proliferation and cell cycle regulator in
lung cancer A549 cells. J Cell Biochem. 120:12752–12761. 2019.
View Article : Google Scholar
|
|
47
|
Licciardello MP, Müllner MK, Dürnberger G,
Kerzendorfer C, Boidol B, Trefzer C, Sdelci S, Berg T, Penz T,
Schuster M, et al: NOTCH1 activation in breast cancer confers
sensitivity to inhibition of SUMOylation. Oncogene. 34:3780–3790.
2015. View Article : Google Scholar
|
|
48
|
Yan S, Li A and Liu Y: CacyBP/SIP inhibits
the migration and invasion behaviors of glioblastoma cells through
activating Siah1 mediated ubiquitination and degradation of
cytoplasmic p27. Cell Biol Int. 42:216–226. 2018. View Article : Google Scholar
|
|
49
|
Imamura Y, Wang PL, Masuno K and Sogawa N:
Salivary protein histatin 3 regulates cell proliferation by
enhancing p27(Kip1) and heat shock cognate protein 70
ubiquitination. Biochem Biophys Res Commun. 470:269–274. 2016.
View Article : Google Scholar
|
|
50
|
Wang L, Bai G and Chen F: Human bone
marrow mesenchymal stem cells suppress the proliferation of hepatic
stellate cells by inhibiting the ubiquitination of p27. Biochem
Cell Biol. 95:628–633. 2017. View Article : Google Scholar
|
|
51
|
Huang X, Tao Y, Gao J, Zhou X, Tang S,
Deng C, Lai Z, Lin X, Wang Q and Li T: UBC9 coordinates
inflammation affecting development of bladder cancer. Sci Rep.
10:206702020. View Article : Google Scholar
|
|
52
|
He W, Verhees GF, Bhagwat N, Yang Y,
Kulkarni DS, Lombardo Z, Lahiri S, Roy P, Zhuo J, Dang B, et al:
SUMO fosters assembly and functionality of the MutSγ complex to
facilitate meiotic crossing over. Dev Cell. 56:2073–2088.e3. 2021.
View Article : Google Scholar
|
|
53
|
Kaul S, Blackford JA Jr, Cho S and Simons
SS Jr: Ubc9 is a novel modulator of the induction properties of
glucocorticoid receptors. J Biol Chem. 277:12541–12549. 2002.
View Article : Google Scholar
|
|
54
|
Chakrabarti SR, Sood R, Ganguly S,
Bohlander S, Shen Z and Nucifora G: Modulation of TEL transcription
activity by interaction with the ubiquitin-conjugating enzyme UBC9.
Proc Natl Acad Sci USA. 96:7467–7472. 1999. View Article : Google Scholar
|
|
55
|
Zhu S, Sachdeva M, Wu F, Lu Z and Mo YY:
Ubc9 promotes breast cell invasion and metastasis in a
sumoylation-independent manner. Oncogene. 29:1763–1772. 2010.
View Article : Google Scholar
|
|
56
|
Li C, McManus FP, Plutoni C, Pascariu CM,
Nelson T, Alberici Delsin LE, Emery G and Thibault P: Quantitative
SUMO proteomics identifies PIAS1 substrates involved in cell
migration and motility. Nat Commun. 11:8342020. View Article : Google Scholar
|
|
57
|
Liu B, Tahk S, Yee KM, Fan G and Shuai K:
The ligase PIAS1 restricts natural regulatory T cell
differentiation by epigenetic repression. Science. 330:521–525.
2010. View Article : Google Scholar
|
|
58
|
Kotaja N, Vihinen M, Palvimo JJ and Jänne
OA: Androgen receptor-interacting protein 3 and other PIAS proteins
cooperate with glucocorticoid receptor-interacting protein 1 in
steroid receptor-dependent signaling. J Biol Chem. 277:17781–17788.
2002. View Article : Google Scholar
|
|
59
|
Galanty Y, Belotserkovskaya R, Coates J,
Polo S, Miller KM and Jackson SP: Mammalian SUMO E3-ligases PIAS1
and PIAS4 promote responses to DNA double-strand breaks. Nature.
462:935–939. 2009. View Article : Google Scholar
|
|
60
|
Hoefer J, Schäfer G, Klocker H, Erb HH,
Mills IG, Hengst L, Puhr M and Culig Z: PIAS1 is increased in human
prostate cancer and enhances proliferation through inhibition of
p21. Am J Pathol. 180:2097–2107. 2012. View Article : Google Scholar
|
|
61
|
Rabellino A, Melegari M, Tompkins VS, Chen
W, Van Ness BG, Teruya-Feldstein J, Conacci-Sorrell M, Janz S and
Scaglioni PP: PIAS1 promotes Lymphomagenesis through MYC
upregulation. Cell Rep. 15:2266–2278. 2016. View Article : Google Scholar
|
|
62
|
Kadaré G, Toutant M, Formstecher E, Corvol
JC, Carnaud M, Boutterin MC and Girault JA: PIAS1-mediated
sumoylation of focal adhesion kinase activates its
autophosphorylation. J Biol Chem. 278:47434–47440. 2003. View Article : Google Scholar
|
|
63
|
Rabellino A, Carter B, Konstantinidou G,
Wu SY, Rimessi A, Byers LA, Heymach JV, Girard L, Chiang CM,
Teruya-Feldstein J and Scaglioni PP: The SUMO E3-ligase PIAS1
regulates the tumor suppressor PML and its oncogenic counterpart
PML-RARA. Cancer Res. 72:2275–2284. 2012. View Article : Google Scholar
|
|
64
|
Schmidt D and Müller S: Members of the
PIAS family act as SUMO ligases for c-Jun and p53 and repress p53
activity. Proc Natl Acad Sci USA. 99:2872–2877. 2002. View Article : Google Scholar
|
|
65
|
Moreno-Oñate M, Herrero-Ruiz AM,
García-Dominguez M, Cortés-Ledesma F and Ruiz JF: RanBP2-mediated
SUMOylation promotes human DNA polymerase lambda nuclear
localization and DNA repair. J Mol Biol. 432:3965–3979. 2020.
View Article : Google Scholar
|
|
66
|
Wang H, Luo Q, Kang J, Wei Q, Yang Y, Yang
D, Liu X, Liu T and Yi P: YTHDF1 aggravates the progression of
cervical cancer through m6A-mediated up-regulation of
RANBP2. Front Oncol. 11:6503832021. View Article : Google Scholar
|
|
67
|
Gilistro E, de Turris V, Damizia M,
Verrico A, Moroni S, De Santis R, Rosa A and Lavia P: Importin-β
and CRM1 control a RANBP2 spatiotemporal switch essential for
mitotic kinetochore function. J Cell Sci. 130:2564–2578. 2017.
|
|
68
|
Maarifi G, Fernandez J, Portilho DM,
Boulay A, Dutrieux J, Oddos S, Butler-Browne G, Nisole S and Arhel
NJ: RanBP2 regulates the anti-retroviral activity of TRIM5α by
SUMOylation at a predicted phosphorylated SUMOylation motif. Commun
Biol. 1:1932018. View Article : Google Scholar
|
|
69
|
Kunz K, Piller T and Müller S:
SUMO-specific proteases and isopeptidases of the SENP family at a
glance. J Cell Sci. 131:jcs2119042018. View Article : Google Scholar
|
|
70
|
Chauhan KM, Chen Y, Chen Y, Liu AT, Sun XX
and Dai MS: The SUMO-specific protease SENP1 deSUMOylates p53 and
regulates its activity. J Cell Biochem. 122:189–197. 2021.
View Article : Google Scholar
|
|
71
|
Bawa-Khalfe T, Yang FM, Ritho J, Lin HK,
Cheng J and Yeh ET: SENP1 regulates PTEN stability to dictate
prostate cancer development. Oncotarget. 8:17651–17664. 2017.
View Article : Google Scholar
|
|
72
|
Song MS, Salmena L, Carracedo A, Egia A,
Lo-Coco F, Teruya-Feldstein J and Pandolfi PP: The
deubiquitinylation and localization of PTEN are regulated by a
HAUSP-PML network. Nature. 455:813–817. 2008. View Article : Google Scholar
|
|
73
|
Sun XX, Chen Y, Su Y, Wang X, Chauhan KM,
Liang J, Daniel CJ, Sears RC and Dai MS: SUMO protease SENP1
deSUMOylates and stabilizes c-Myc. Proc Natl Acad Sci USA.
115:10983–10988. 2018. View Article : Google Scholar
|
|
74
|
Huang X, Zuo Y, Wang X, Wu X, Tan H, Fan
Q, Dong B, Xue W, Chen GQ and Cheng J: SUMO-specific protease 1 is
critical for myeloid-derived suppressor cell development and
function. Cancer Res. 79:3891–3902. 2019. View Article : Google Scholar
|
|
75
|
Kanapathipillai M: Treating p53 mutant
aggregation-associated cancer. Cancers (Basel). 10:1542018.
View Article : Google Scholar
|
|
76
|
Carter S, Bischof O, Dejean A and Vousden
KH: C-terminal modifications regulate MDM2 dissociation and nuclear
export of p53. Nat Cell Biol. 9:428–435. 2007. View Article : Google Scholar
|
|
77
|
Wu SY and Chiang CM: Crosstalk between
sumoylation and acetylation regulates p53-dependent chromatin
transcription and DNA binding. EMBO J. 28:1246–1259. 2009.
View Article : Google Scholar
|
|
78
|
Ding B, Sun Y and Huang J: Overexpression
of SKI oncoprotein leads to p53 degradation through regulation of
MDM2 protein sumoylation. J Biol Chem. 287:14621–14630. 2012.
View Article : Google Scholar
|
|
79
|
Deng C, Lipstein MR, Scotto L, Jirau
Serrano XO, Mangone MA, Li S, Vendome J, Hao Y, Xu X, Deng SX, et
al: Silencing c-Myc translation as a therapeutic strategy through
targeting PI3Kδ and CK1ε in hematological malignancies. Blood.
129:88–99. 2017. View Article : Google Scholar
|
|
80
|
Amente S, Lavadera ML, Palo GD and Majello
B: SUMOactivating SAE1 transcription is positively regulated by
Myc. Am J Cancer Res. 2:330–334. 2012.
|
|
81
|
Kessler JD, Kahle KT, Sun T, Meerbrey KL,
Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, et
al: A SUMOylation-dependent transcriptional subprogram is required
for Myc-driven tumorigenesis. Science. 335:348–353. 2012.
View Article : Google Scholar
|
|
82
|
Wang WH, Yuan T, Qian MJ, Yan FJ, Yang L,
He JQ, Yang B, Lu JJ and Zhu H: Post-translational modification of
KRAS: Potential targets for cancer therapy. Acta Pharmacol Sin.
42:1201–1211. 2021. View Article : Google Scholar
|
|
83
|
Uprety D and Adjei AA: KRAS: From
undruggable to a druggable Cancer Target. Cancer Treat Rev.
89:1020702020. View Article : Google Scholar
|
|
84
|
Moutty MC, Sakin V and Melchior F:
Importin α/β mediates nuclear import of individual SUMO E1 subunits
and of the holo-enzyme. Mol Biol Cell. 22:652–660. 2011. View Article : Google Scholar
|
|
85
|
Choi BH, Philips MR, Chen Y, Lu L and Dai
W: K-Ras Lys-42 is crucial for its signaling, cell migration, and
invasion. J Biol Chem. 293:17574–17581. 2018. View Article : Google Scholar
|
|
86
|
Boddy MN, Howe K, Etkin LD, Solomon E and
Freemont PS: PIC 1, a novel ubiquitin-like protein which interacts
with the PML component of a multiprotein complex that is disrupted
in acute promyelocytic leukaemia. Oncogene. 13:971–982. 1996.
|
|
87
|
de Thé H, Pandolfi PP and Chen Z: Acute
promyelocytic leukemia: A paradigm for oncoprotein-targeted cure.
Cancer Cell. 32:552–560. 2017. View Article : Google Scholar
|
|
88
|
Rao Y, Li R and Zhang D: A drug from
poison: How the therapeutic effect of arsenic trioxide on acute
promyelocytic leukemia was discovered. Sci China Life Sci.
56:495–502. 2013. View Article : Google Scholar
|
|
89
|
Zhu J, Zhou J, Peres L, Riaucoux F, Honoré
N, Kogan S and de Thé H: A sumoylation site in PML/RARA is
essential for leukemic transformation. Cancer Cell. 7:143–153.
2005. View Article : Google Scholar
|
|
90
|
Stubbe M, Mai J, Paulus C, Stubbe HC,
Berscheminski J, Karimi M, Hofmann S, Weber E, Hadian K, Hay R, et
al: Viral DNA binding protein SUMOylation promotes PML nuclear body
localization next to viral replication centers. mBio. 11:e00049–20.
2020. View Article : Google Scholar
|
|
91
|
El-Asmi F, El-Mchichi B, Maroui MA,
Dianoux L and Chelbi-Alix MK: TGF-β induces PML SUMOylation,
degradation and PML nuclear body disruption. Cytokine. 120:264–272.
2019. View Article : Google Scholar
|
|
92
|
Sahin U, Ferhi O, Jeanne M, Benhenda S,
Berthier C, Jollivet F, Niwa-Kawakita M, Faklaris O, Setterblad N,
de Thé H and Lallemand-Breitenbach V: Oxidative stress-induced
assembly of PML nuclear bodies controls sumoylation of partner
proteins. J Cell Biol. 204:931–945. 2014. View Article : Google Scholar
|
|
93
|
Li Y, Ma X, Wu W, Chen Z and Meng G: PML
nuclear body biogenesis, carcinogenesis, and targeted therapy.
Trends Cancer. 6:889–906. 2020. View Article : Google Scholar
|
|
94
|
Ben-Porath I and Weinberg RA: The signals
and pathways activating cellular senescence. Int J Biochem Cell
Biol. 37:961–976. 2005. View Article : Google Scholar
|
|
95
|
Morris EJ and Dyson NJ: Retinoblastoma
protein partners. Adv Cancer Res. 82:1–54. 2001. View Article : Google Scholar
|
|
96
|
Lee BY, Han JA, Im JS, Morrone A, Johung
K, Goodwin EC, Kleijer WJ, DiMaio D and Hwang ES:
Senescence-associated beta-galactosidase is lysosomal
beta-galactosidase. Aging Cell. 5:187–195. 2006. View Article : Google Scholar
|
|
97
|
Trimarchi JM and Lees JA: Sibling rivalry
in the E2F family. Nat Rev Mol Cell Biol. 3:11–20. 2002. View Article : Google Scholar
|
|
98
|
Bischof O, Schwamborn K, Martin N, Werner
A, Sustmann C, Grosschedl R and Dejean A: The E3 SUMO ligase PIASy
is a regulator of cellular senescence and apoptosis. Mol Cell.
22:783–794. 2006. View Article : Google Scholar
|
|
99
|
Yu B, Swatkoski S, Holly A, Lee LC, Giroux
V, Lee CS, Hsu D, Smith JL, Yuen G, Yue J, et al: Oncogenesis
driven by the Ras/Raf pathway requires the SUMO E2 ligase Ubc9.
Proc Natl Acad Sci USA. 112:E1724–E1733. 2015. View Article : Google Scholar
|
|
100
|
Luo J, Emanuele MJ, Li D, Creighton CJ,
Schlabach MR, Westbrook TF, Wong KK and Elledge SJ: A genome-wide
RNAi screen identifies multiple synthetic lethal interactions with
the Ras oncogene. Cell. 137:835–848. 2009. View Article : Google Scholar
|
|
101
|
Gallipoli P and Huntly BJP: Novel
epigenetic therapies in hematological malignancies: Current status
and beyond. Semin Cancer Biol. 51:198–210. 2018. View Article : Google Scholar
|
|
102
|
Bryder D, Rossi DJ and Weissman IL:
Hematopoietic stem cells: The paradigmatic tissue-specific stem
cell. Am J Pathol. 169:338–346. 2006. View Article : Google Scholar
|
|
103
|
Tempé D, Piechaczyk M and Bossis G: SUMO
under stress. Biochem Soc Trans. 36:874–878. 2008. View Article : Google Scholar
|
|
104
|
Rajkumar SV: Multiple myeloma. Curr Probl
Cancer. 33:7–64. 2009. View Article : Google Scholar
|
|
105
|
Röllig C, Knop S and Bornhäuser M:
Multiple myeloma. Lancet. 385:2197–2208. 2015. View Article : Google Scholar
|
|
106
|
Susanibar Adaniya SP, Cohen AD and Garfall
AL: Chimeric antigen receptor T cell immunotherapy for multiple
myeloma: A review of current data and potential clinical
applications. Am J Hematol. 94(Suppl 1): S28–S33. 2019. View Article : Google Scholar
|
|
107
|
Atrash S, Bano K, Harrison B and Abdallah
AO: CAR-T treatment for hematological malignancies. J Investig Med.
68:956–964. 2020. View Article : Google Scholar
|
|
108
|
Gagelmann N, Riecken K, Wolschke C, Berger
C, Ayuk FA, Fehse B and Kröger N: Development of CAR-T cell
therapies for multiple myeloma. Leukemia. 34:2317–2332. 2020.
View Article : Google Scholar
|
|
109
|
Feng D and Sun J: Overview of anti-BCMA
CAR-T immunotherapy for multiple myeloma and relapsed/refractory
multiple myeloma. Scand J Immunol. 92:e129102020. View Article : Google Scholar
|
|
110
|
Minnie SA and Hill GR: Immunotherapy of
multiple myeloma. J Clin Invest. 130:1565–1575. 2020. View Article : Google Scholar
|
|
111
|
Xu J, Sun HY, Xiao FJ, Wang H, Yang Y,
Wang L, Gao CJ, Guo ZK, Wu CT and Wang LS: SENP1 inhibition induces
apoptosis and growth arrest of multiple myeloma cells through
modulation of NF-κB signaling. Biochem Biophys Res Commun.
460:409–415. 2015. View Article : Google Scholar
|
|
112
|
Xie H, Gu Y, Wang W, Wang X, Ye X, Xin C,
Lu M, Reddy BA and Shu P: Silencing of SENP2 in multiple myeloma
induces bortezomib resistance by activating NF-κB through the
modulation of IκBα sumoylation. Sci Rep. 10:7662020. View Article : Google Scholar
|
|
113
|
Tanaka K and Hirota T: Chromosomal
instability: A common feature and a therapeutic target of cancer.
Biochim Biophys Acta. 1866:64–75. 2016.
|
|
114
|
McGranahan N, Burrell RA, Endesfelder D,
Novelli MR and Swanton C: Cancer chromosomal instability:
Therapeutic and diagnostic challenges. EMBO Rep. 13:528–538. 2012.
View Article : Google Scholar
|
|
115
|
Giam M and Rancati G: Aneuploidy and
chromosomal instability in cancer: A jackpot to chaos. Cell Div.
10:32015. View Article : Google Scholar
|
|
116
|
Dantuma NP and van Attikum H:
Spatiotemporal regulation of posttranslational modifications in the
DNA damage response. EMBO J. 35:6–23. 2016. View Article : Google Scholar
|
|
117
|
Galanty Y, Belotserkovskaya R, Coates J
and Jackson SP: RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes
DNA double-strand break repair. Genes Dev. 26:1179–1195. 2012.
View Article : Google Scholar
|
|
118
|
Biggins S, Bhalla N, Chang A, Smith DL and
Murray AW: Genes involved in sister chromatid separation and
segregation in the budding yeast Saccharomyces cerevisiae.
Genetics. 159:453–470. 2001. View Article : Google Scholar
|
|
119
|
Ohta S, Bukowski-Wills JC, Sanchez-Pulido
L, Alves Fde L, Wood L, Chen ZA, Platani M, Fischer L, Hudson DF,
Ponting CP, et al: The protein composition of mitotic chromosomes
determined using multiclassifier combinatorial proteomics. Cell.
142:810–821. 2010. View Article : Google Scholar
|
|
120
|
Thomas GE, Renjith MR and Manna TK:
Kinetochore-microtubule interactions in chromosome segregation:
Lessons from yeast and mammalian cells. Biochem J. 474:3559–3577.
2017. View Article : Google Scholar
|
|
121
|
Verdaasdonk JS and Bloom K: Centromeres:
Unique chromatin structures that drive chromosome segregation. Nat
Rev Mol Cell Biol. 12:320–332. 2011. View Article : Google Scholar
|
|
122
|
Eifler K, Cuijpers SAG, Willemstein E,
Raaijmakers JA, El Atmioui D, Ovaa H, Medema RH and Vertegaal AC:
SUMO targets the APC/C to regulate transition from metaphase to
anaphase. Nat Commun. 9:11192018. View Article : Google Scholar
|
|
123
|
Azuma Y, Arnaoutov A, Anan T and Dasso M:
PIASy mediates SUMO-2 conjugation of Topoisomerase-II on mitotic
chromosomes. EMBO J. 24:2172–2182. 2005. View Article : Google Scholar
|
|
124
|
Dawlaty MM, Malureanu L, Jeganathan KB,
Kao E, Sustmann C, Tahk S, Shuai K, Grosschedl R and van Deursen
JM: Resolution of sister centromeres requires RanBP2-mediated
SUMOylation of topoisomerase IIalpha. Cell. 133:103–115. 2008.
View Article : Google Scholar
|
|
125
|
Joseph J, Liu ST, Jablonski SA, Yen TJ and
Dasso M: The RanGAP1-RanBP2 complex is essential for
microtubule-kinetochore interactions in vivo. Curr Biol.
14:611–617. 2004. View Article : Google Scholar
|
|
126
|
Maison C, Romeo K, Bailly D, Dubarry M,
Quivy JP and Almouzni G: The SUMO protease SENP7 is a critical
component to ensure HP1 enrichment at pericentric heterochromatin.
Nat Struct Mol Biol. 19:458–460. 2012. View Article : Google Scholar
|
|
127
|
Eifler K and Vertegaal ACO:
SUMOylation-mediated regulation of cell cycle progression and
cancer. Trends Biochem Sci. 40:779–793. 2015. View Article : Google Scholar
|
|
128
|
Schimmel J, Eifler K, Sigurðsson JO,
Cuijpers SA, Hendriks IA, Verlaan-de Vries M, Kelstrup CD,
Francavilla C, Medema RH, Olsen JV and Vertegaal AC: Uncovering
SUMOylation dynamics during cell-cycle progression reveals FoxM1 as
a key mitotic SUMO target protein. Mol Cell. 53:1053–1066. 2014.
View Article : Google Scholar
|
|
129
|
Neyret-Kahn H, Benhamed M, Ye T, Le Gras
S, Cossec JC, Lapaquette P, Bischof O, Ouspenskaia M, Dasso M,
Seeler J, et al: Sumoylation at chromatin governs coordinated
repression of a transcriptional program essential for cell growth
and proliferation. Genome Res. 23:1563–1579. 2013. View Article : Google Scholar
|
|
130
|
Nacerddine K, Lehembre F, Bhaumik M, Artus
J, Cohen-Tannoudji M, Babinet C, Pandolfi PP and Dejean A: The SUMO
pathway is essential for nuclear integrity and chromosome
segregation in mice. Dev Cell. 9:769–779. 2005. View Article : Google Scholar
|
|
131
|
Bellail AC, Olson JJ and Hao C: SUMO1
modification stabilizes CDK6 protein and drives the cell cycle and
glioblastoma progression. Nat Commun. 5:42342014. View Article : Google Scholar
|
|
132
|
Guo Z, Kanjanapangka J, Liu N, Liu S, Liu
C, Wu Z, Wang Y, Loh T, Kowolik C, Jamsen J, et al: Sequential
posttranslational modifications program FEN1 degradation during
cell-cycle progression. Mol Cell. 47:444–456. 2012. View Article : Google Scholar
|
|
133
|
Cheng J, Wang D, Wang Z and Yeh ET: SENP1
enhances androgen receptor-dependent transcription through
desumoylation of histone deacetylase 1. Mol Cell Biol.
24:6021–6028. 2004. View Article : Google Scholar
|
|
134
|
Childs BG, Baker DJ, Kirkland JL, Campisi
J and van Deursen JM: Senescence and apoptosis: Dueling or
complementary cell fates? EMBO Rep. 15:1139–1153. 2014. View Article : Google Scholar
|
|
135
|
Lujambio A, Akkari L, Simon J, Grace D,
Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA,
Krizhanovsky V and Lowe SW: Non-cell-autonomous tumor suppression
by p53. Cell. 153:449–460. 2013. View Article : Google Scholar
|
|
136
|
Ivanschitz L, Takahashi Y, Jollivet F,
Ayrault O, Le Bras M and de Thé H: PML IV/ARF interaction enhances
p53 SUMO-1 conjugation, activation, and senescence. Proc Natl Acad
Sci USA. 112:14278–14283. 2015. View Article : Google Scholar
|
|
137
|
Yates KE, Korbel GA, Shtutman M, Roninson
IB and DiMaio D: Repression of the SUMO-specific protease Senp1
induces p53-dependent premature senescence in normal human
fibroblasts. Aging Cell. 7:609–621. 2008. View Article : Google Scholar
|
|
138
|
Lowe SW and Lin AW: Apoptosis in cancer.
Carcinogenesis. 21:485–495. 2000. View Article : Google Scholar
|
|
139
|
Kaufmann SH and Hengartner MO: Programmed
cell death: Alive and well in the new millennium. Trends Cell Biol.
11:526–534. 2001. View Article : Google Scholar
|
|
140
|
Zapata JM, Pawlowski K, Haas E, Ware CF,
Godzik A and Reed JC: A diverse family of proteins containing tumor
necrosis factor receptor-associated factor domains. J Biol Chem.
276:24242–24252. 2001. View Article : Google Scholar
|
|
141
|
Hockenbery D, Nuñez G, Milliman C,
Schreiber RD and Korsmeyer SJ: Bcl-2 is an inner mitochondrial
membrane protein that blocks programmed cell death. Nature.
348:334–336. 1990. View Article : Google Scholar
|
|
142
|
Zamaraev AV, Kopeina GS, Prokhorova EA,
Zhivotovsky B and Lavrik IN: Post-translational modification of
Caspases: The other side of apoptosis regulation. Trends Cell Biol.
27:322–339. 2017. View Article : Google Scholar
|
|
143
|
Park JH, Lee SW, Yang SW, Yoo HM, Park JM,
Seong MW, Ka SH, Oh KH, Jeon YJ and Chung CH: Modification of DBC1
by SUMO2/3 is crucial for p53-mediated apoptosis in response to DNA
damage. Nat Commun. 5:54832014. View Article : Google Scholar
|
|
144
|
Xia W, Tian H, Cai X, Kong H, Fu W, Xing
W, Wang Y, Zou M, Hu Y and Xu D: Inhibition of SUMO-specific
protease 1 induces apoptosis of astroglioma cells by regulating
NF-κB/Akt pathways. Gene. 595:175–179. 2016. View Article : Google Scholar
|
|
145
|
Jiang Z, Fan Q, Zhang Z, Zou Y, Cai R,
Wang Q, Zuo Y and Cheng J: SENP1 deficiency promotes ER
stress-induced apoptosis by increasing XBP1 SUMOylation. Cell
Cycle. 11:1118–1122. 2012. View Article : Google Scholar
|
|
146
|
He X, Riceberg J, Pulukuri SM, Grossman S,
Shinde V, Shah P, Brownell JE, Dick L, Newcomb J and Bence N:
Characterization of the loss of SUMO pathway function on cancer
cells and tumor proliferation. PLoS One. 10. pp. e01238822015,
View Article : Google Scholar
|
|
147
|
Zhang M, Jiang D, Xie X, He Y, Lv M and
Jiang X: miR-129-3p inhibits NHEJ pathway by targeting SAE1 and
represses gastric cancer progression. Int J Clin Exp Pathol.
12:1539–1547. 2019.
|
|
148
|
Kalamarz ME, Paddibhatla I, Nadar C and
Govind S: Sumoylation is tumor-suppressive and confers
proliferative quiescence to hematopoietic progenitors in Drosophila
melanogaster larvae. Biol Open. 1:161–172. 2012. View Article : Google Scholar
|
|
149
|
Li S, Wang J, Hu G, Aman S, Li B, Li Y,
Xia K, Yang Y, Ahmad B, Wang M and Wu H: SUMOylation of MCL1
protein enhances its stability by regulating the
ubiquitin-proteasome pathway. Cell Signal. 73:1096862020.
View Article : Google Scholar
|
|
150
|
Chang CC, Naik MT, Huang YS, Jeng JC, Liao
PH, Kuo HY, Ho CC, Hsieh YL, Lin CH, Huang NJ, et al: Structural
and functional roles of Daxx SIM phosphorylation in SUMO
paralog-selective binding and apoptosis modulation. Mol Cell.
42:62–74. 2011. View Article : Google Scholar
|
|
151
|
Santiago A, Godsey AC, Hossain J, Zhao LY
and Liao D: Identification of two independent SUMO-interacting
motifs in Daxx: Evolutionary conservation from Drosophila to humans
and their biochemical functions. Cell Cycle. 8:76–87. 2009.
View Article : Google Scholar
|
|
152
|
Wasiak S, Zunino R and McBride HM: Bax/Bak
promote sumoylation of DRP1 and its stable association with
mitochondria during apoptotic cell death. J Cell Biol. 177:439–450.
2007. View Article : Google Scholar
|
|
153
|
Li P, Jing H, Wang Y, Yuan L, Xiao H and
Zheng Q: SUMO modification in apoptosis. J Mol Histol. 52:1–10.
2021. View Article : Google Scholar
|
|
154
|
Menon MB and Dhamija S: Beclin 1
Phosphorylation-at the center of autophagy regulation. Front Cell
Dev Biol. 6:1372018. View Article : Google Scholar
|
|
155
|
Sun J, Tai S, Tang L, Yang H, Chen M, Xiao
Y, Li X, Zhu Z and Zhou S: Acetylation modification during
autophagy and vascular aging. Front Physiol. 12:5982672021.
View Article : Google Scholar
|
|
156
|
Son SM, Park SJ, Fernandez-Estevez M and
Rubinsztein DC: Autophagy regulation by acetylation-implications
for neurodegenerative diseases. Exp Mol Med. 53:30–41. 2021.
View Article : Google Scholar
|
|
157
|
Wang R and Wang G: Protein modification
and autophagy activation. Adv Exp Med Biol. 1206:237–259. 2019.
View Article : Google Scholar
|
|
158
|
Hu B, Zhang Y, Deng T, Gu J, Liu J, Yang
H, Xu Y, Yan Y, Yang F, Zhang H, et al: PDPK1 regulates
autophagosome biogenesis by binding to PIK3C3. Autophagy. Sep
10–2020.Epub ahead of print. View Article : Google Scholar
|
|
159
|
Liu K, Guo C, Lao Y, Yang J, Chen F, Zhao
Y, Yang Y, Yang J and Yi J: A fine-tuning mechanism underlying
self-control for autophagy: DeSUMOylation of BECN1 by SENP3.
Autophagy. 16:975–990. 2020. View Article : Google Scholar
|
|
160
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar
|
|
161
|
Morrison CD, Parvani JG and Schiemann WP:
The relevance of the TGF-β Paradox to EMT-MET programs. Cancer
Lett. 341:30–40. 2013. View Article : Google Scholar
|
|
162
|
Lin X, Wang Y, Jiang Y, Xu M, Pang Q, Sun
J, Yu Y, Shen Z, Lei R and Xu J: Sumoylation enhances the activity
of the TGF-β/SMAD and HIF-1 signaling pathways in keloids. Life
Sci. 255:1178592020. View Article : Google Scholar
|
|
163
|
Xu R, Yu S, Zhu D, Huang X, Xu Y, Lao Y,
Tian Y, Zhang J, Tang Z, Zhang Z, et al: hCINAP regulates the
DNA-damage response and mediates the resistance of acute myelocytic
leukemia cells to therapy. Nat Commun. 10:38122019. View Article : Google Scholar
|
|
164
|
Bossis G, Sarry JE, Kifagi C, Ristic M,
Saland E, Vergez F, Salem T, Boutzen H, Baik H, Brockly F, et al:
The ROS/SUMO axis contributes to the response of acute myeloid
leukemia cells to chemotherapeutic drugs. Cell Rep. 7:1815–1823.
2014. View Article : Google Scholar
|
|
165
|
Golub TR, Barker GF, Bohlander SK, Hiebert
SW, Ward DC, Bray-Ward P, Morgan E, Raimondi SC, Rowley JD and
Gilliland DG: Fusion of the TEL gene on 12p13 to the AML1 gene on
21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA.
92:4917–4921. 1995. View Article : Google Scholar
|
|
166
|
Chakrabarti SR, Sood R, Nandi S and
Nucifora G: Posttranslational modification of TEL and TEL/AML1 by
SUMO-1 and cell-cycle-dependent assembly into nuclear bodies. Proc
Natl Acad Sci USA. 97:13281–13285. 2000. View Article : Google Scholar
|
|
167
|
Hanson CA, Wood LD and Hiebert SW:
Cellular stress triggers TEL nuclear export via two genetically
separable pathways. J Cell Biochem. 104:488–498. 2008. View Article : Google Scholar
|
|
168
|
de Thé H, Lavau C, Marchio A, Chomienne C,
Degos L and Dejean A: The PML-RAR alpha fusion mRNA generated by
the t(15;17) translocation in acute promyelocytic leukemia encodes
a functionally altered RAR. Cell. 66:675–684. 1991. View Article : Google Scholar
|
|
169
|
Jeanne M, Lallemand-Breitenbach V, Ferhi
O, Koken M, Le Bras M, Duffort S, Peres L, Berthier C, Soilihi H,
Raught B and de Thé H: PML/RARA oxidation and arsenic binding
initiate the antileukemia response of As2O3. Cancer Cell. 18:88–98.
2010. View Article : Google Scholar
|
|
170
|
Ablain J, Rice K, Soilihi H, de Reynies A,
Minucci S and de Thé H: Activation of a promyelocytic
leukemia-tumor protein 53 axis underlies acute promyelocytic
leukemia cure. Nat Med. 20:167–174. 2014. View Article : Google Scholar
|
|
171
|
Tatham MH, Geoffroy MC, Shen L,
Plechanovova A, Hattersley N, Jaffray EG, Palvimo JJ and Hay RT:
RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for
arsenic-induced PML degradation. Nat Cell Biol. 10:538–546. 2008.
View Article : Google Scholar
|
|
172
|
Brackett CM and Blagg BS: Current status
of SUMOylation inhibitors. Curr Med Chem. 28:3892–3912. 2021.
View Article : Google Scholar
|
|
173
|
Yang Y, Xia Z, Wang X, Zhao X, Sheng Z, Ye
Y, He G, Zhou L, Zhu H, Xu N and Liang S: Small-molecule inhibitors
targeting protein SUMOylation as novel anticancer compounds. Mol
Pharmacol. 94:885–894. 2018. View Article : Google Scholar
|
|
174
|
Fukuda I, Ito A, Uramoto M, Saitoh H,
Kawasaki H, Osada H and Yoshida M: Kerriamycin B inhibits protein
SUMOylation. J Antibiot (Tokyo). 62:221–224. 2009. View Article : Google Scholar
|
|
175
|
Fukuda I, Ito A, Hirai G, Nishimura S,
Kawasaki H, Saitoh H, Kimura K, Sodeoka M and Yoshida M: Ginkgolic
acid inhibits protein SUMOylation by blocking formation of the
E1-SUMO intermediate. Chem Biol. 16:133–140. 2009. View Article : Google Scholar
|
|
176
|
Hirohama M, Kumar A, Fukuda I, Matsuoka S,
Igarashi Y, Saitoh H, Takagi M, Shin-ya K, Honda K, Kondoh Y, et
al: Spectomycin B1 as a novel SUMOylation inhibitor that directly
binds to SUMO E2. ACS Chem Biol. 8:2635–2642. 2013. View Article : Google Scholar
|
|
177
|
Kayser S, Schlenk RF and Platzbecker U:
Management of patients with acute promyelocytic leukemia. Leukemia.
32:1277–1294. 2018. View Article : Google Scholar
|
|
178
|
He X, Riceberg J, Soucy T, Koenig E,
Minissale J, Gallery M, Bernard H, Yang X, Liao H, Rabino C, et al:
Probing the roles of SUMOylation in cancer cell biology by using a
selective SAE inhibitor. Nat Chem Biol. 13:1164–1171. 2017.
View Article : Google Scholar
|
|
179
|
Langston SP, Grossman S, England D, Afroze
R, Bence N, Bowman D, Bump N, Chau R, Chuang BC, Claiborne C, et
al: Discovery of TAK-981, a first-in-class inhibitor of
SUMO-activating enzyme for the treatment of cancer. J Med Chem.
64:2501–2520. 2021. View Article : Google Scholar
|
|
180
|
Lv Z, Yuan L, Atkison JH, Williams KM,
Vega R, Sessions EH, Divlianska DB, Davies C, Chen Y and Olsen SK:
Molecular mechanism of a covalent allosteric inhibitor of SUMO E1
activating enzyme. Nat Commun. 9:51452018. View Article : Google Scholar
|
|
181
|
Li YJ, Du L, Wang J, Vega R, Lee TD, Miao
Y, Aldana-Masangkay G, Samuels ER, Li B, Ouyang SX, et al:
Allosteric inhibition of ubiquitin-like modifications by a class of
inhibitor of SUMO-activating enzyme. Cell Chem Biol. 26:278–288.e6.
2019. View Article : Google Scholar
|
|
182
|
Takemoto M, Kawamura Y, Hirohama M,
Yamaguchi Y, Handa H, Saitoh H, Nakao Y, Kawada M, Khalid K,
Koshino H, et al: Inhibition of protein SUMOylation by davidiin, an
ellagitannin from Davidia involucrata. J Antibiot (Tokyo).
67:335–338. 2014. View Article : Google Scholar
|
|
183
|
Suzawa M, Miranda DA, Ramos KA, Ang KK,
Faivre EJ, Wilson CG, Caboni L, Arkin MR, Kim YS, Fletterick RJ, et
al: A gene-expression screen identifies a non-toxic sumoylation
inhibitor that mimics SUMO-less human LRH-1 in liver. Elife.
4:e090032015. View Article : Google Scholar
|
|
184
|
Lu X, Olsen SK, Capili AD, Cisar JS, Lima
CD and Tan DS: Designed semisynthetic protein inhibitors of Ub/Ubl
E1 activating enzymes. J Am Chem Soc. 132:1748–1749. 2010.
View Article : Google Scholar
|
|
185
|
Berger AJ, Friedlander S, Ghasemi O,
Grossman S and Huszar D: Abstract 3079: Pharmacodynamic evaluation
of the novel SUMOylation inhibitor TAK-981 in a mouse tumor model.
In: Proceedings: AACR Annual Meeting 2019; March 29-April 3, 2019;
Atlanta, GA. pp. 792019
|
|
186
|
Kumar A, Ito A, Hirohama M, Yoshida M and
Zhang KY: Identification of sumoylation activating enzyme 1
inhibitors by structure-based virtual screening. J Chem Inf Model.
53:809–820. 2013. View Article : Google Scholar
|
|
187
|
Biederstädt A, Hassan Z, Schneeweis C,
Schick M, Schneider L, Muckenhuber A, Hong Y, Siegers G, Nilsson L,
Wirth M, et al: SUMO pathway inhibition targets an aggressive
pancreatic cancer subtype. Gut. 69:1472–1482. 2020. View Article : Google Scholar
|
|
188
|
Kumar A, Ito A, Hirohama M, Yoshida M and
Zhang KY: Identification of new SUMO activating enzyme 1 inhibitors
using virtual screening and scaffold hopping. Bioorg Med Chem Lett.
26:1218–1223. 2016. View Article : Google Scholar
|
|
189
|
Brandt M, Szewczuk LM, Zhang H, Hong X,
McCormick PM, Lewis TS, Graham TI, Hung ST, Harper-Jones AD,
Kerrigan JJ, et al: Development of a high-throughput screen to
detect inhibitors of TRPS1 sumoylation. Assay Drug Dev Technol.
11:308–325. 2013. View Article : Google Scholar
|
|
190
|
Zlotkowski K, Hewitt WM, Sinniah RS,
Tropea JE, Needle D, Lountos GT, Barchi JJ Jr, Waugh DS and
Schneekloth JS Jr: A Small-molecule microarray approach for the
identification of E2 enzyme inhibitors in ubiquitin-like
conjugation pathways. SLAS Discov. 22:760–766. 2017.
|
|
191
|
Wiechmann S, Gärtner A, Kniss A, Stengl A,
Behrends C, Rogov VV, Rodriguez MS, Dötsch V, Müller S and Ernst A:
Site-specific inhibition of the small ubiquitin-like modifier
(SUMO)-conjugating enzyme Ubc9 selectively impairs SUMO chain
formation. J Biol Chem. 292:15340–15351. 2017. View Article : Google Scholar
|
|
192
|
Kim YS, Nagy K, Keyser S and Schneekloth
JS Jr: An electrophoretic mobility shift assay identifies a
mechanistically unique inhibitor of protein sumoylation. Chem Biol.
20:604–613. 2013. View Article : Google Scholar
|
|
193
|
Ambaye N, Chen CH, Khanna S, Li YJ and
Chen Y: Streptonigrin inhibits SENP1 and reduces the protein level
of hypoxia-inducible factor 1α (HIF1α) in cells. Biochemistry.
57:1807–1813. 2018. View Article : Google Scholar
|
|
194
|
Chen Y, Wen D, Huang Z, Huang M, Luo Y,
Liu B, Lu H, Wu Y, Peng Y and Zhang J:
2-(4-Chlorophenyl)-2-oxoethyl 4-benzamidobenzoate derivatives, a
novel class of SENP1 inhibitors: Virtual screening, synthesis and
biological evaluation. Bioorg Med Chem Lett. 22:6867–6870. 2012.
View Article : Google Scholar
|
|
195
|
Qiao Z, Wang W, Wang L, Wen D, Zhao Y,
Wang Q, Meng Q, Chen G, Wu Y and Zhou H: Design, synthesis, and
biological evaluation of benzodiazepine-based SUMO-specific
protease 1 inhibitors. Bioorg Med Chem Lett. 21:6389–6392. 2011.
View Article : Google Scholar
|
|
196
|
Uno M, Koma Y, Ban HS and Nakamura H:
Discovery of 1-[4-(N-benzylamino)phenyl]-3-phenylurea derivatives
as non-peptidic selective SUMO-sentrin specific protease (SENP)1
inhibitors. Bioorg Med Chem Lett. 22:5169–5173. 2012. View Article : Google Scholar
|
|
197
|
Xie W, Wang Z, Zhang J, Wang L, Zhao Y and
Zhou H: Development and evaluation of a highly reliable assay for
SUMO-specific protease inhibitors. Bioorg Med Chem Lett.
26:2124–2128. 2016. View Article : Google Scholar
|
|
198
|
Lindenmann U, Brand M, Gall F, Frasson D,
Hunziker L, Kroslakova I, Sievers M and Riedl R: Discovery of a
class of potent and selective Non-competitive Sentrin-Specific
protease 1 inhibitors. ChemMedChem. 15:675–679. 2020. View Article : Google Scholar
|
|
199
|
Zhao Y, Wang Z, Zhang J and Zhou H:
Identification of SENP1 inhibitors through in silico screening and
rational drug design. Eur J Med Chem. 122:178–184. 2016. View Article : Google Scholar
|
|
200
|
Wu J, Lei H, Zhang J, Chen X, Tang C, Wang
W, Xu H, Xiao W, Gu W and Wu Y: Momordin Ic, a new natural SENP1
inhibitor, inhibits prostate cancer cell proliferation. Oncotarget.
7:58995–59005. 2016. View Article : Google Scholar
|
|
201
|
Huang W, He T, Chai C, Yang Y, Zheng Y,
Zhou P, Qiao X, Zhang B, Liu Z, Wang J, et al: Triptolide inhibits
the proliferation of prostate cancer cells and down-regulates
SUMO-specific protease 1 expression. PLoS One. 7:e376932012.
View Article : Google Scholar
|
|
202
|
Bernstock JD, Ye D, Smith JA, Lee YJ,
Gessler FA, Yasgar A, Kouznetsova J, Jadhav A, Wang Z, Pluchino S,
et al: Quantitative high-throughput screening identifies
cytoprotective molecules that enhance SUMO conjugation via the
inhibition of SUMO-specific protease (SENP)2. FASEB J.
32:1677–1691. 2018. View Article : Google Scholar
|
|
203
|
Kumar A, Ito A, Takemoto M, Yoshida M and
Zhang KY: Identification of 1,2,5-oxadiazoles as a new class of
SENP2 inhibitors using structure based virtual screening. J Chem
Inf Model. 54:870–880. 2014. View Article : Google Scholar
|
|
204
|
Boggio R, Colombo R, Hay RT, Draetta GF
and Chiocca S: A mechanism for inhibiting the SUMO pathway. Mol
Cell. 16:549–561. 2004. View Article : Google Scholar
|
|
205
|
Liang Z, Chan HYE, Lee MM and Chan MK: A
SUMO1-derived peptide targeting SUMO-interacting motif inhibits
α-Synuclein aggregation. Cell Chem Biol. 28:180–190.e6. 2021.
View Article : Google Scholar
|
|
206
|
Sakamoto KM, Kim KB, Kumagai A, Mercurio
F, Crews CM and Deshaies RJ: Protacs: Chimeric molecules that
target proteins to the Skp1-Cullin-F box complex for ubiquitination
and degradation. Proc Natl Acad Sci USA. 98:8554–8559. 2001.
View Article : Google Scholar
|
|
207
|
Jakobs A, Koehnke J, Himstedt F, Funk M,
Korn B, Gaestel M and Niedenthal R: Ubc9 fusion-directed
SUMOylation (UFDS): A method to analyze function of protein
SUMOylation. Nat Methods. 4:245–250. 2007. View Article : Google Scholar
|
|
208
|
Chen X, Zaro JL and Shen WC: Fusion
protein linkers: Property, design and functionality. Adv Drug Deliv
Rev. 65:1357–1369. 2013. View Article : Google Scholar
|