Glioblastoma multiforme (GBM) is the most prevalent
type of malignant brain tumor in adults. It is considered the most
aggressive form of primary intracranial tumor and is associated
with a dismal prognosis (1,2).
Survival of patients with GBM remains poor: The overall 5-year
relative survival rate is one of the lowest among all cancer types
(4–5%). Despite aggressive treatment, the median overall survival
(OS) is ~15 months (2). There
have been only modest improvements in survival rates for patients
with GBM in the last 30 years (3).
According to the 2021 World Health Organization
classification of central nervous system (CNS) tumors,
glioblastomas are defined as isocitrate dehydrogenase
(IDH)-wild-type (wt) diffuse astrocytic tumors. Astrocytoma,
IDH-mutant (mut) grade 2, 3 or 4 tumors, are now considered
separate entities (4). The
histopathological features of GBM include diffuse neoplastic
infiltration of the nervous tissue with a necrotic core and cells
resembling astroglia ('angular' nucleus, euchromatin) and vascular
proliferation and/or pseudopalisading necrosis with mitoses
(5). The structure of the
blood-brain barrier (BBB), the suppressive tumor microenvironment
and tumor heterogeneity provide an advantage to glioblastoma cells,
leading to decreased efficacy of chemotherapeutic agents, targeted
therapy and immunotherapy (6,7).
Despite multimodal approaches to treatment, most
patients with GBM have an aggressive course of the disease.
Glioblastomas frequently recur (in 75–90%) within 2–3 cm from the
borders of the initial lesion and with multiple lesions observed in
5% of cases after treatment (2).
Treatment has historically consisted of maximal surgical resection
with adjuvant radiation therapy (RT) or primary RT for inoperable
tumors. Within the last two decades, temozolomide (TMZ) and a
non-invasive device called the tumor-treating field (TTF;
Optune®; Novocure GmbH) have demonstrated clinical
efficacy and achieved improved outcomes (8–10).
Additional treatment options that demonstrated activity include
beva-cizumab, lomustine, carmustine, PCV (combination of
procarbazine, lomustine and vincristine), and, more recently,
multikinase inhibitor regorafenib, which demonstrated superior
outcomes over lomustine in a recent phase 2 trial (11–14).
According to the 2020 National Comprehensive Cancer
Network (NCCN) clinical practice guidelines in oncology (12), GBM treatment options depend on
patient age, performance status (PS) and
O6-methylguanine DNA methyltransferase (MGMT) promoter
methylation status (methylated vs. unmethylated). Patients aged 70
years or younger with a good PS, regardless of the tumor's MGMT
methylation status, should receive standard brain RT plus
concurrent and adjuvant TMZ with alternating electric field therapy
(9,10). Patients older than 70 years with
good PS should receive hypofractionated or standard brain RT plus
concurrent and adjuvant TMZ and alternating electric field
therapy.
According to The Central Brain Tumor Registry of the
United States statistical report, the annual average age-adjusted
incidence rate of GBM between 2012 and 2016 was 3.22 per 100,000
individuals in the US (15). GBM
accounts for ~15% of all brain tumors and is mainly found in adults
aged 45–70 years (16). Seminal
clinical study results indicated that the median survival was 14.6
months for RT plus TMZ and 12.1 months for RT alone (9). The 5-year OS outcome with RT plus
TNZ was 9.8 and 1.9% with RT alone (17). To date, there are no identified
risk factors or underlying carcinogenetic causes for the
development of GBM and the only confirmed risk factor is exposure
to high levels of ionizing radiation (18). Of note, patients with asthma and
other allergic conditions have been described to have a lower risk
to develop GBM. In addition, genotypes that increase the risk of
asthma are associated with a decreased GBM risk (3). Several studies have suggested a
possible inverse relationship between GBM development and
non-steroidal anti-inflammatory drug (NSAID) use. The Glioma
International Case-Control Study reported that daily aspirin use
for ≥6 months was associated with a 38% lower glioma risk (19). Another study assessed the risk of
glioma among 325 glioma cases and 600 frequency-matched controls in
the Houston metropolitan region (2001–2006) and it indicated that
regular use of NSAIDs was related to a 33% reduction in the risk of
glioma (20).
It has been proposed that human cytomegalovirus
modulates the malignant phenotype in glioblastomas (21). In a limited study at Karolinska
University Hospital, 50 patients with GBM received valganciclovir
as adjuvant treatment. The rate of survival at 2 years was 62%
compared with 18% of contemporary controls with a similar disease
stage, surgical resection grade and baseline treatment (P<0.001)
(22). While these results sound
promising, they should be validated in larger randomized studies in
the future.
The mechanisms of action of effective systemic
standard of care treatments for GBM are summarized in Fig. 1. The most frequently used drug in
the first-line setting is TMZ. TMZ is a prodrug that works by being
converted to the monomethyl triazene 5-(3-methyl-1-triazeno)
imidazole-4-carboxamide (23).
The biological actions of TMZ appear to be mediated by methylation
at the O6 position of guanine (24), leading to mutations that
ultimately escape the mismatch repair system (MMR). The MMR
promotes a signaling cascade that activates cell cycle checkpoints
and causes G2-M cell cycle arrest and apoptosis through single-and
double-strand breaks in DNA (25). Tumor cells with methylated MGMT
are more susceptible to the cytotoxic effects of TMZ than cells
with functioning MGMT (26).
Carmustine (also known as BCNU) is a small
alkylating agent and nitrogen mustard compound. It causes guanine
and cytosine bases in DNA to form interstrand crosslinks. Lomustine
is an oral alkylating anti-tumor therapy, which has anti-GBM
efficacy due to its high lipophilicity and small size, which
facilitates BBB crossover (27).
A high degree of tumor vascularization as a result
of increased production of proangiogenic growth factors, including
VEGF, is observed in GBM, which has led to the development of
treatment methods aimed at targeting proangiogenic signaling
pathways. The randomized, multicenter, open-label phase II BRAIN
trial comparing the efficacy of bevacizumab in combination with
irinotecan vs. bevacizumab alone contributed to the accelerated
Food and Drug Administration (FDA) approval of bevacizumab for the
treatment of recurrent glioblastoma in 2009 (28). The primary endpoints in this trial
were 6-month progression-free survival (PFS) and objective response
rate (ORR). The rate of PFS at 6 months was 42.6 and 50.3% for the
bevacizumab-alone and the bevacizumab-plus-irinotecan groups,
respectively. The ORR was 28.2 and 37.8%, respectively. The median
OS was 9.2 and 8.7 months, respectively. Grade 3 adverse events
occurred in 46.4% of individuals treated with bevacizumab alone,
with hypertension (8.3%) and convulsions (6.0%) being the most
common. In comparison, 65.8% of patients in the
bevacizumab-plus-irinotecan group experienced grade 3 adverse
events, including convulsions (13.9%), neutropenia (8.9%) and
fatigue (8.9%) (29).
In patients treated with bevacizumab and standard
therapy, PFS was better than with standard therapy in two studies
at 4.4 months (P<0.0001) in the AVAglio and 3.4 months (P=0.004)
in the RTOG0825 trial (30,31). However, the median OS was not
different between treatment groups in both trials. In recurrent
GBM, studies failed to achieve an improvement in OS and PFS in
patients treated with combination therapy with bevacizumab compared
to the use of bevacizumab alone (32). The combination of bevacizumab and
lomustine was also studied. OS at 9 months was chosen as the
primary endpoint. This combination increased the median OS to 12
months compared to bevacizumab or lomustine alone with 8 months
each. The combination also increased 6-month PFS to 42%. Several
patients in the study had grade 4 or grade 3 thrombocy-topenia
(33). However, the increase in
PFS may be related to the phenomenon of 'pseudo-response'. This
phenomenon consists of improved contrast enhancement due to the
normalization of vascular permeability (34). However, the usage of FLAIR or
T2-weighted images revealed an increase in the nonenhacing part of
the tumor. The Response Assessment in Neuro-Oncology criteria
consider FLAIR/T2 hyperintensity as a surrogate for the
nonenhancing component of the tumor (35).
Cediranib, a multi-kinase inhibitor targeting VEGFR,
failed to achieve a significant improvement in PFS in a phase III
randomized study of either cediranib alone or cediranib in
combination with lomustine vs. lomustine based on independent or
local review of postcontrast T1-weighted MRI (36).
Aflibercept is a recombinantly generated fusion
protein that scavenges both VEGF and placental growth factor. In a
phase II study, it had minimal evidence of single-agent activity in
unselected patients with recurrent malignant glioma (37).
Finally, regorafenib is a recently approved oral
multikinase agent that is now endorsed by the NCCN for the
treatment of relapsed GBM post-radiation and TMZ (12). This drug demonstrated significant
clinical activity and superiority to lomustine in a recent phase 2
trial (13).
TTF is a non-invasive antimitotic therapy, delivered
by an alternating electric field by the Optune® system.
Preclinical studies have indicated that TTF is able to alter
microtubule formation, causing mitotic arrest and death. During
cyto-kinesis, it also induces the dielectrophoretic migration of
polar molecules. The phase III EF-14 study demonstrated a
significant improvement in PFS and OS of patients with newly
diagnosed GBM when Optune® was used with TMZ compared to
chemotherapy alone. Compliance was associated with a better
clinical outcome. The use of TTF with TMZ significantly improved
median OS compared to chemotherapy alone (20.9 vs. 16.0 months,
respectively; P<0.001) (38).
The clinical efficacy and tolerability of TTF in glioblastoma have
been established in 2 large phase III studies and have been
validated in real-world settings. TTF is associated with minimal
adverse events (local or systemic). A limitation of TTF is that it
must be worn continuously with minimal interruption. This
inevitably leads to major lifestyle modifications. Furthermore, the
total monthly therapy cost is ~$21,000 (39). TTF has proven its clinical
efficacy, but its usage is associated with certain disadvantages
and side effects in patients with GBM, including significantly
higher rates of localized skin toxicity (38). Following regulatory approval, NCCN
guide-lines included TTF in conjunction with TMZ for the treatment
of patients with both newly diagnosed (Category 1) and recurrent
glioblastoma (Category 2B) (12).
Treatment strategies in conjunction with TTF provide important
clinical benefits in limiting additional toxicity in patients with
brain cancer and may be extended to patients with other types of
solid tumor.
Despite the absence of randomized trials, surgery
appears effective and the main principle of glioblas-toma surgery
is currently gross-total resection (40). Maximal resection improves survival
irrespective of the age of the patient or the molecular status of
the tumor (41). When resection
is contraindicated, stereotactic biopsy is the method of choice for
both histological verification and molecular evaluation of the
tumor (42). The use of
fluorescence-guided surgery with 5-aminolevulinic acid assessed in
a prospective study provided an improvement (46 vs. 28.3%) of
6-month PFS (43). To perform
safer interventions and reduce postoperative complications,
functional MRI and assessment of diffusion-tensor fibers should be
routinely used. As a mandatory requirement, postoperative
contrast-enhanced MRI should be performed within 48 h of resection
to determine the extent of the intervention (44). In case of recurrence, the most
radical resection of the focus is performed, particularly if >6
months have passed since the intervention or in patients with a
good functional status (high Karnofsky performance score) or a
young age (45). Currently, there
are no data from ongoing randomized clinical trials regarding the
surgical treatment of recurrent glioblastomas.
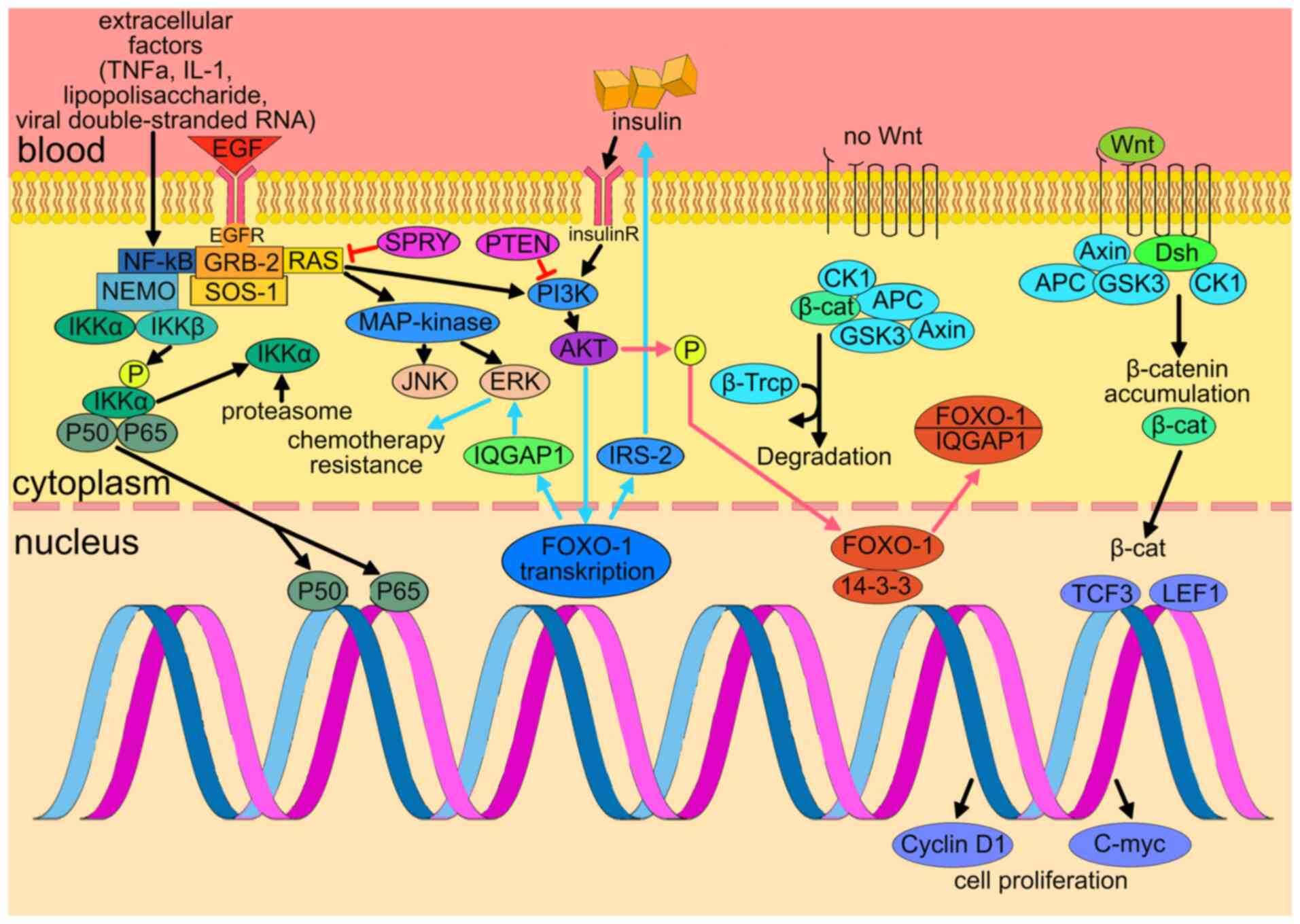
In GBM, alteration and/or upregulation of the Wnt,
transforming growth factor β (TGF-β), VEGF, epidermal growth factor
receptor (EGFR), cyclin-dependent kinase 2A (CDKN2A), nuclear
factor-κB (NF-κB), phosphatidylinositol-3-kinase
(PI3K)/AKT/mammalian target of rapamycin (mTOR) may be associated
with pathogenesis of the disease and aggressive tumor behavior.
Wnt is responsible for the development, regeneration
and homeostasis, where it mediates cellular proliferation,
polarity, differentiation, motility and activity of stem cells
(46). The increased activity of
the canonical Wnt pathway may be responsible for the resistance to
chemotherapy and RT, as well as growth, aggressiveness and invasive
potential of GBM (46). EGFR
(also known as ErbB1/HER1) is a type of receptor tyrosine kinase
(RTK) that has an important role in the division, migration,
adhesion, differentiation and apoptosis of cells (47). Under normal conditions, TGF-β is
an inflammatory pathway responsible for the expression of p21 and
other tumor suppressors. In cancer cells, however, TGF-β disrupts
the cell cycle and mediates malignant characteristics (48). VEGF is a potent stimulator of
endothelial cell growth and a key regulator of normal and
pathologic growth of blood vessels and angiogenesis (49).
Genomic analysis of glioblastoma revealed several
signaling pathways and gene alterations that are critical for its
development: Cell cycle checkpoints, apoptosis, TGF-β, NF-κB, the
notch signaling pathway, also signaling pathways associated with
growth factors and RAS, such as the PI3K/AKT/mTOR pathway, EGFR,
tensin/AKT homologs and the CDKN2A pathway (56,57).
It is worth noting that human malignant gliomas are
rarely dependent on a single oncogene or tumor suppressor gene,
which may explain the lack of efficacy of drugs targeting only one
molecular alteration in clinical trials (58). Furthermore, the BBB restricts the
entry of chemotherapeutic agents into the tumor. Intratumoral
presence of cancer stem cells (CSCs) that are characterized by
chemo-and radioresistance may also contribute to the aggressiveness
and high recurrence rate of GBM (6). Another problem is the infiltrative
nature of GBM cells, which limits the feasibility of complete
surgical resection, despite the advances in neurosurgery techniques
(8).
The Cancer Genome Atlas (TCGA) published a study
analyzing main mutational events in GBM, according to which three
main genetic events occur in human glioblastomas: i) Amplification
and mutational activation of RTK genes; ii) activation of the PI3K
pathway; and iii) inactivation of the p53 and retinoblastoma tumor
suppressor pathways (59). In
terms of epigenetic events, the MGMT promoter is methylated in ~50%
of newly diagnosed GBM cases. MGMT encodes a DNA repair protein
that disrupts the therapeutic process by removing alkyl groups from
guanine-rich areas in DNA, a target for alkylating agents such as
TMZ. The DNA methylation status of this gene may be a useful
biomarker of the chemotherapy response and explain in part why
patients with a methylated MGMT gene promoter may have longer OS
(60).
Glioblastoma may harbor the codeletion 1p/19q. This
codeletion has an association with chemosensitivity and favorable
prognosis in oligodendroglioma (61). Therefore, it was proposed that in
GBMs with oligodendroglioma component 1p/19q codeletion may also
have a prognostic value. However, this still remains to be fully
demonstrated (62). In one study,
the frequency of 1p/19q codeletion in GBM was 3%, but neither
codeletion, nor isolated mutations were associated with increased
survival and had no prognostic value (63). The tumor suppressor gene TP53,
encoding the transcription factor p53, is the most frequently
mutated gene in various types of malignancies, and in GBM, it is
the second-most commonly mutated gene (28,3%) after phosphatase and
tensin homolog (PTEN) (30.7%) (64). In accordance with its primary role
in suppressing oncogenesis, mutations that disrupt the function of
wt-p53 are common in human malignant tumors (65). TCGA project data indicated that
the p53 signaling pathway (including CDKN2A, MDM2 and TP53) is
disrupted in ~85% of glioblastoma cases (66).
Within the glioblastoma tumors reside ontogenically
distinct immunoregulatory macrophages [spalt-like 1-positive
(Sall1+) tumor microglia,
Sall1-monocyte-derived macro-phages), immunosuppressive
T-regulatory cells (e.g., C-C chemokine receptor 8-positive)] and
dysfunctional T-cell populations [high levels of cytotoxic
T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell
death protein 1] (67).
Computational analysis grouped glioblastoma tumors into three
immune response-related subgroups: i) Negative (defined by a
relative paucity of immune cells; enriched in TCGA-proneural cells
and cyclin-dependent kinase 4-membrane associated ring-CH-type
finger 9 amplification); ii) humoral (defined by a high B-cell and
CD4+ T-cell compartment; enriched in TCGA-mesenchymal cells); and
iii) cellular-like (defined by a higher 'negative regulation of
T-cell activation' and 'gamma delta T-cell' cluster; enriched by
classical TCGA-CL cells and samples with a high macrophage content)
(7). For further information on
additional signaling pathways please refer to Garofano et al
(68).
CDKN2A is a gene located on chromosome 9, band
p21.3. It is ubiquitously expressed in numerous tissues and cell
types (50). Germinal CDKN2A
mutations have been described to be associated with familial
glioblastoma (69). The presence
of multiple altered signaling pathways in GBM emphasizes the notion
of tumor dependency on dysregulation of multiple molecular targets
that may alter tumor biology, as illustrated in Fig. 2.
EGFR has an important role in the division,
migration, adhesion, differentiation and apoptosis of cells. EGFR
consists of the extracellular domain, which binds ligands, a
transmembrane domain and the intracellular tyrosine kinase domain.
Activation of EGFR leads to the activation of numerous downstream
signaling pathways, such as PI3K-AKT-mTOR, leading to cancer
proliferation and therapy resistance (47).
Amplification of EGFR and/or its overexpression at
the protein level are common alterations that occur in 35–45% of
cases of GBM (70). Amplification
of its active mutant EGFRvIII in GBM (characterized by an in-frame
deletion of exons 2–7) is also distinctive for GBM and is found in
50% of cases (71). While EGFR
and EGFRvIII have a crucial role in the pathogenesis of GBM,
inhibitors of EGFR tyrosine kinase and antibodies demonstrated low
efficacy in clinical trials (72). Overexpression of EGFR results in
an abnormal activity of downstream signaling pathways, including
son of sevenless 1 (SOS1), growth factor receptor bound protein 2
(GRB2), RAS protein and AKT (73). GRB2 is a molecular adapter protein
that coordinates other signaling molecules, including SOS1, NF-κB,
Ras and Akt. Thus, GRB2 enhances other types of cell activity, such
as cell proliferation, epithelial to mesenchymal transition and
tumor development (74).
Overexpression of EGFR/mutant EGFRvIII is associated
with increases in proliferation and migration of GBM cells. These
properties affect the malignant phenotype of these tumor cells
(75). Expression of EGFRvIII
also stimulated and accelerated angiogenesis in preclinical models
of GBM in vivo (76).
Potential therapies that target EGFR or EGFRvIII, including
tyrosine kinase inhibitors (TKIs), such as erlotinib, gefitinib and
lapatinib, as well as monoclonal antibodies, vaccines and RNA-based
agents, are currently in development or in clinical trials for the
treatment of GBM (72).
Erlotinib exhibited minimal activity only against
tumors that overexpressed EGFR and PTEN (77). Given that PTEN mutations are found
in 41% of patients with GBM (78), erlo-tinib may not be a good
therapeutic option for the majority of GBM cases with
overexpression of EGFR. Erlotinib was indicated to be ineffective
as a monotherapy in patients with recurrent GBM and only slightly
beneficial as a follow-up to RT in patients with non-progressive
GBM (79). Unlike erlo-tinib,
gefitinib has anti-tumor activity regardless of the level of EGFR
expression (80); however, only
minor clinical effects were observed in phase II trials. Several
phase I/II trials have indicated that while adding gefitinib to RT
improves tolerability, it only has a minor effect on the survival
rate (81,82).
One of the difficulties of the analysis of the
impact of EGFR amplification on targeted therapy is that
amplification may be lost when cells from EGFR-amplified GBM are
placed in the cell culture (70).
As a result of this constraint, preclinical models studying the
biology of EGFR in GBM substantially relied on the ectopic
overexpression of EGFR and/or EGFRvIII in non-amplified cell lines
of GBM and subsequent blockage of overexpressed proteins (83). Another reason for the failure of
targeting EGFR is the heterogeneous distribution of EGFR in the
tumor. This may lead to differential EGFR sensitivity, which may
eventually result in treatment failure (84).
Depatuxizumab mafodotin (Depatux-M) is a
tumor-specific combination made up of an antibody directed against
EGFR antibody (ABT-806), conjugated to the toxin
monomethylau-ristatin-F. In a phase II trial, this drug was tested
in patients with centrally confirmed EGFR-amplified glioblastoma at
first recurrence after concurrent chemoradiation with TMZ. The
efficacy of Depatux-M monotherapy was comparable to that of the
control group [hazard ratio (HR)=1.04, 95% CI=0.73–1.48; P=0.83],
thus failing to meet the primary endpoint (85).
Aberrant constitutive activation of the NF-κB
signaling pathway is common in GBM. The constitutive NF-κB
hyperactivation is oncogenic due to the stimulation of tumor growth
and invasion, apoptosis suppression and development of resistance
to therapy (53). The most common
form of NF-κB protein dimer is the heterodimer of p65-p50. This
dimer is able to bind to a specific sequence (i.e., NF-κB sites) of
the target gene to regulate gene transcription. NF-κB regulates
cell activity through slight differences in the binding of these
NF-κB dimers to target sequences (52). In non-stimulated cells, NF-κB
dimers are inactive due to binding to three inhibitory factors
(IκBα, IκBβ and IκBε) of NF-κB in the cytoplasm, which blocks the
nuclear localization sequence and prevents the transfer of NF-κB
into the nucleus. In the nucleus, NF-κB dimers bind to κB-sites in
the regulatory regions of genes participating in a wide range of
cellular processes (86). NF-κB
in neurons maintains neuronal health, synapse growth and
plasticity-related functions (87).
Activation of the NF-κB signaling pathway is common
in various cancer types. Numerous mechanisms have been proposed
that may lead to the disruption of NF-κB signal regulation in
gliomas. For instance, RTK, primarily EGFR and PDGFR that are
frequently activated in glioblastomas, may be triggered secondary
to the activation of NF-κB through several mechanisms such as
AKT-related and -unrelated signaling pathways. In murine models, it
has been demonstrated that inhibition of NF-κB by depletion of IκB
kinase 2, expression of an IκBaM super repressor or using an NF-κB
essential modifier-binding domain attenuated tumor proliferation
and prolonged survival (88). In
one of the studies, NF-κB p65 subunit was indicated to be
overexpressed in 81% of cases of GBM (89).
Oncogenic mechanisms of EGFR and PDGFR signal
transduction make a major contribution to the growth and invasion
of GBM. The NF-κB pathway interplays with these receptors, which
may lead to the development of GBM (90). Loss of the tumor suppressors, such
as neurofibromin 1 is also associated with the disruption of
activation of NF-κB in GBM due to the upregulated activity of PI3K
(91). Disruption of the tumor
suppressor Krüppel-like factor 6, which serves as a negative
regulator of NF-κB, also contributes to the activation of NF-κB
(92).
Exposure of GBM cell lines to NF-κB-p65 small
interfering (si)RNA and NF-κB inhibitors resulted in a significant
decrease in GBM-cell viability. Treatment of GBM with NF-κB
inhibitors overcame cisplatin resistance and led to an increase in
the effects of cisplatin and doxorubicin. Importantly, normal
astrocytes were less sensitive to NF-κB inhibition, implying tumor
cell selectivity (93).
It has been proposed that numerous other mechanisms
have an important role in the disruption of signal transduction of
NF-κB, including facilitation of NF-κB by peptidyl-prolyl cis-trans
isomerase NIMA-interacting 1, mixed lineage kinase 4 and
heterozygous deletion of the NFKBIA gene, which encodes IκBα
(94). These observations
emphasize the potential role of abnormal NF-κB signaling pathways
in various mechanisms of GBM pathogenesis.
Amentofavone is a flavonoid that is able to cross
the BBB and inhibit the NF-κB pathway by inhibiting IκB kinase
degradation. Treatment with this compound reduces the viability and
proliferation of GBM cells, resulting in the emergence of sub-G1
population, which indicates apoptosis. To date, amen-tofavone has
not been evaluated in any randomized clinical trial (95).
Wnt/β-catenin pathway activity has been linked to
neural progenitor cell proliferation in the early stages of brain
development, while it reduces the self-renewal capacity and
promotes neuronal differentiation in later stages (97). Recent reports from a small cohort
have reported mutations of adenomatous polyposis coli (which is
responsible for Wnt activation) in ~13% of GBM cases with a
mutation frequency of close to 14.5% (98). It has been reported that the
increased activity of the canonical Wnt pathway is responsible for
GBM resistance to chemotherapy and RT (99), contributing to the growth,
aggressiveness and invasive potential of GBM (100). It also generates CSCs from
differentiated cells (97).
It should be noted that the hypoxic environment
influences Wnt-induced differentiation. Furthermore,
hypoxia-inducible factor 1α is required to sustain the expression
of transcriptional associates of β-catenin the transcription factor
T cell factor and lymphoid enhancer binding factor) (101). In addition, Wnt-induced
differentiation inhibits Notch signal transduction and thus,
enhancement of Wnt and Notch suppression lead to the activation of
pre-neuronal differentiation (102).
Although the Wnt pathway has been thoroughly studied
and numerous molecular targeted drugs have entered the clinical
trial stage, insight into the efficacy of these medications is
lacking (40). In the canonical
Wnt signaling pathway, the Wnt protein interacts with the Frizzled
and low-density lipoprotein receptor-related protein 5/6 receptor.
This binding was inhibited by monoclonal antibodies, including
vantic-tumab (OMP18R5) and ipafricept (OMP54F28), thus blocking the
Wnt signaling pathway (103).
Vantictumab has been indicated to be well tolerated in a phase I
trial with nearly no gastrointestinal adverse effects at the
effective dose (104). While
clinical trials are ongoing, it remains elusive at this time
whether these drugs have brain efficacy or whether any GBM trials
are planned with these agents.
TERT encodes the catalytic subunit of the telomerase
complex. Since telomerase activity is a function of this catalytic
subunit, mutations of the TERT gene promoter are frequently
associated with cancer. These mutations are most often represented
by nucleotide substitutions in the two most common 'hot spots': At
position-124 up from the transcription initiation site (nucleotide
polymorphism chr5: 1,295,228 G>A, also called C228T) and at
region-146 (nucleotide polymor-phism chr5: 1,295,250 G>A, known
as C250T (105).
The TERT promoter (TERTp) mutation (pTERTmut) was
originally detected in melanoma. Follow-up studies also revealed a
high frequency of pTERTmut in IDH-wt GBM, as well as in IDHmut
oligodendroglioma and oligodendroglioma with 1p/19q co-deletion,
and demonstrated its potential use for glioma classification
(106).
Most glioblastomas may be divided into molecular
subgroups based on mutations in TERTp and IDH 1/2. These molecular
subgroups use different genetic mechanisms to maintain telomeres:
This is either a TERTp mutation leading to telomerase activation or
an a α-thalassemia/mental retardation, X-linked mutation leading to
an alternative extension of the telomere phenotype. TERTp-mutant
GBM demonstrates telomerase activation due to the de novo
generation of transcription factor binding sites, leading to
increased TERT expression. These tumors, designated glioblastomas
TERT pWT-IDH WT, do not have any well-established genetic
biomarkers or specific mechanisms for maintaining telo-meres
(107).
In one of the studies, overexpression of the
C-terminal fragment of human TERT (hTERTC27) was demonstrated to
inhibit the growth and oncogenicity of HeLa cells (108). The therapeutic effect and
molecular mechanisms of gene therapy for hTERTC27-mediated
malignant tumors were further studied in vivo on human
glioblastoma xenografts in thymus-free mice. Intra-tumor injection
of the hTERTC27-carrying adeno-associated virus (rAAV-hTERTC27) has
been demonstrated to be effective in slowing the growth of
subcutaneously transplanted glioblastoma tumors. Histological
analysis suggested that treatment with rAAV-hTERTC27 led to deep
necrosis, apoptosis, infiltration of polymorphonuclear neutro-phils
and a decrease in the density of microvessels in tumor samples
(109). Another pre-clinical
study reported increased expression of the hTERT gene in patients
with high-grade glioma that may be associated with the
aggressiveness of the tumor (patients with low hTERT mRNA levels in
the tumors had a median PFS of 24 months and patients with low
hTERT levels had a PFS of 11 months). It was indicated that when
hTERT mRNA expression was reduced by siRNA, this led to a decrease
in the cell viability. Therefore, targeting TERT using small
molecules or other approaches may lead to the development of novel
therapeutic agents in the future (110).
The PI3K/AKT/mTOR intracellular signaling pathway is
responsible for growth, cell proliferation and metabolism (54). There are three classes (I, II and
III) of PI3K, which differ in substrate specificity and products.
Class I kinases are the most well-studied; they are heterodimers of
regulatory and catalytic subunits. These enzymes may be activated
by G-protein-associated receptors and RTK. After ligand binding,
RTK autophosphorylate tyrosine residues in their cytoplasmic
domains. The regulatory subunits of PI3K contain an SH2 domain that
allows them to recognize and bind phosphotyrosine residues of RTK.
As a result, kinases are in close proximity to the membrane, and
hence their substrates, so the synthesis of PI3K begins (112). PI3K inhibitors may be
clas-sified into pan-PI3K, isoform-selective and dual PI3K/mTOR
types (113). The frequency of
mutations of PIK3CA in GBM (encodes p110α, which is part of a
catalytic subunit of class IA PI3K) ranges from 4 to 27% (113). Through decreased AKT and FAK
activation, PIK3CA knockdown significantly decreases cell survival,
migration and invasion of GBM cells (114). The p110α isoform-selective
inhibitors A66 or PIK-75 effectively suppressed GBM cell growth,
survival and migration in vitro (115). In the absence of PTEN, p110β has
a critical role in GBM cell proliferation, survival and migration.
In vitro and in vivo, knockdown of PIK3CB (encodes
p110β) suppresses cell proliferation and triggers caspase-dependent
apoptosis in GBM, and it works in tandem with PTEN restoration
(116). The selective inhibitor
of p110β TGX-221 significantly reduces cell migration in GBM cells
while having a minimal effect on survival and invasion (115). Thus, AKT-phosphorylated forkhead
box O proteins may have tumor suppressor functions unless they are
degraded by E3 ubiquitin ligases (117).
The deregulation of the mTOR pathway was also
correlated with radioresistance and in pre-clinical studies, it has
also been proposed that PI3K/mTOR inhibition rendered GBM tumors
radiosensitive (124).
mTORC1 inhibitors include rapamycin (sirolimus) and
its analogues, such as RAD001 (everolimus), CCL-779 (temsiro-limus)
and AP23573 (ridaforolimus) (55). These medications are
first-generation mTOR inhibitors. A total of 171 patients with
newly diagnosed GBM took part in the everolimus phase II study. RT
with concurrent and adjuvant TMZ, with or without daily everolimus
(10 mg), was provided to patients. When comparing patients in the
everolimus group to those in the control group, there was no
significant difference in PFS. Patients who received everolimus, on
the other hand, had a considerable increase in toxicities (125). The only known peri-surgical
phase 1 study of ridaforolimus in grade IV malignant glioma was
suspended due to slower than expected patient accrual and
postsurgical drug administration challenges (126). In the phase II study of RT with
temsirolimus vs. radiochemotherapy with TMZ, a total of 257
patients were enrolled. In the temsirolimus arm, the median OS was
14.8 months, while in the control arm, it was 16.0 months. The
temsirolimus arm had a median PFS of 5.4 months, while the control
arm had a median PFS of 6.0 months (127). The combination of RT and
temsirolimus is currently being further studied in a phase I/IIa
trial that seeks to selectively match patients with targeted
therapies based on known alterations (N2M2 trial) (128). Over the past several years,
great effort has been put into developing second-generation
ATP-competitive mTOR kinase inhibitors (TORKi), including INK128,
Torin 1 and AZD8055, and third-generation bivalent mTOR inhibitors
that specifically target mTOR resistance mutations (129). However, TORKi have not yet
demonstrated clinical effectiveness in GBM, likely due to the
limited capacity of mTOR inhibitors to cross the BBB and
compensatory AKT activation (130).
c-Met is an RTK, expressed on the surfaces of
various cells. Hepatocyte growth factor (HGF) is the ligand for
this receptor. HGF binding leads to a sequence of intracellular
signals that mediate embryogenesis and wound healing in normal
cells. In cancer cells, aberrant HGF/c-Met axis activation, which
is closely related to c-Met gene mutations, overexpression and
amplification, promotes tumor development and progression by
stimulating several signaling pathways (131). Approximately 37% of patients
with GBM have c-Met overexpression (132). c-Met also has a role in the
resistance mechanism that drives GBM invasion in xenografts.
Resistance to anti-angiogenic drugs may be mediated by upregulation
of c-Met gene expression (acquired resistance) or due to selective
survival (intrinsic resistance) of tumor cell subpopulations
overexpressing c-Met (133). It
is worth noting that the shortest time to progression and OS was
observed in GBM accompanied by overexpression of c-Met and VEGFR2,
which indicates the primary/innate activation of the two pathways
(134).
Several drugs targeting c-Met have been studied in
clinical trials. Onartuzumab, which is an anti-c-met monoclonal
anti-body, is highly specific for binding c-Met. This antibody is
able to block the binding of c-Met-HGF by blocking the HGF α-chain
and forming a complex with the Sema-PSI domain of c-Met (135). This process does not lead to any
agonistic activity or trigger c-Met dimerization. Recent clinical
trials did not indicate any clinical benefit with onartuzumab in
GBM (136). Other types of drugs
that may affect c-Met are small molecule inhibitors. Crizotinib is
an effective small molecule inhibitor of c-Met, derived from the
first-generation series c-Met inhibitor, PHA-66752. Crizotinib
targets the TK domain of c-Met and is approved by the FDA for the
treatment of advanced non-small-cell lung carcinoma (131). In the context of GBM, a recent
phase Ib dose-escalation study followed by an extension phase with
crizotinib was added to standard RT and TMZ. This study indicated
highly promising efficacy for newly diagnosed GBM, warranting
further investigation (137).
Other small molecule inhibitors of c-Met include cabozantinib,
foretinib, LY280163, MK2461, capmatinib, tivantinib, which may be
tested in future GBM trials (131).
FGFRs control numerous biological functions,
including cell proliferation, survival and cytoskeletal regulation.
The FGFR signal is important in the embryonic development of the
CNS and serves as the survival mechanism of adult neurons and
astrocytes (138). In addition,
it was indicated that FGFR signaling may promote the self-renewal
and fate specification of neural stem cells (139). FGFR expression changes in
astrocytes may prompt malignant transformation and GBM progression
due to the activation of mitogenic, migratory and antiapoptotic
reactions (140). Whole-genome
analyses of patient samples have uncovered that the rate of FGFR
mutations and amplifications are exceptionally low in GBM (<2%)
(141). Several FGFR inhibitors
have been developed over the past years. Fisogatinib is an
inhibitor of the FGFR4 gene. Clinical trials suggested that
fisogatinib has high activity and selectivity, resulting in
considerable anti-tumor efficacy (142). Fisogatinib is able to covalently
bind to a specific cysteine residue identified in FGFR4 (Cys 552),
giving it a high degree of selectivity over other FGFR family
members (133). Currently, this
drug has only been studied in phase I clinical trials for the
treatment of hepatocellular carcinoma (143). However, the study in mice
indicated that brain accumulation of fisogatinib is significantly
limited by ATP binding cassette subfamily B member 1 P-glycoprotein
in the BBB, while oral availability of fisogatinib is markedly
constrained by CYP3A activity (144). Futibatinib is also an
irreversible inhibitor of FGFR. Several tumor cell lines with
distinct genetic abnormalities of FGFR exhibited effective and
specific growth suppression with futibatinib (145). However, to date, it has not been
studied in the settings of GBM or CNS tumors. Another drug is
AZD4547, which is a selective FGFR1–3 inhibitor. In an
FGFR3-transforming acidic coiled-coil containing protein 3 (TACC3)
glioma xenograft model, oral administration of AZD4547 resulted in
longer survival compared with that of mice that were given the
vehicle control (146). AZD4547
has been studied in patients with recurrent IDH-wt gliomas with
FGFR1-TACC1 or FGFR3-TACC3 fusions (147).
The BRAF proto-oncogene serine/threonine kinase
(BRAF) is a member of the Raf kinase family (148), which consists of three kinases:
ARAF, CRAF (RAF-1) and BRAF. BRAF has an important role in
regulating of the MAPK/ERK pathway. Hyperactivation of this pathway
may cause cell cycle arrest, while aberrant regulation of the
pathway may lead to carcinogenesis (149). BRAF activation in human neural
stem and progenitor cells not only triggers tumor growth, but also
subsequently leads to oncogene-induced senescence in certain
low-grade brain tumors (150).
This may explain the relatively high frequency of BRAF mutant brain
tumors associated with good prognosis. BRAF gene alterations, on
the other hand, are also seen in diffusely developing malignancies
in adults, which are associated with poor prognosis (151).
In total, >40 mutations have been discovered in
the BRAF gene, with a single thymine-to-adenine nucleotide base
change at position 1,799 accounting for 90% of them. This missense
mutation causes a substitution of glutamine for valine at position
600 (V600E). BRAFV600E leads to a ~500-fold increase in
gene activity. It allows signaling cascade activation in the
absence of external stimuli such as growth signals, allowing cells
to become self-sufficient in this pathway (152). The BRAFV600E mutation
is rare in primary and metastatic CNS neoplasms, found in 4% of
cases (151).
Another study reported a marked radiological
response and a stable clinical outcome in patients with malignant
BRAF V600E-mutated glioma with leptomeningeal tumor appearance who
received dabrafenib alone for up to 27 months (155). In one of the three instances in
this case series, histology revealed glioblastoma, whereas the
other diagnoses were compatible with anaplastic pleomorphic
xanthoastrocytomas. Primary treatment with BRAF and MEK inhibitors
has been indicated to lead to tumor regression in patients with
BRAF V600E mutant glioblastoma. As a result, it was proposed that
all young patients with GBM, particularly those with an unusually
aggressive tumor behaviour, should be tested for
BRAFV600E mutation (156).
Preclinical and human tumor studies support a
potentially important role for Src in human glioblastoma. In
transgenic mice expressing v-Src, glioblastomas may potentially
develop due to this alteration (157). Dasatinib is a TKI that reduces
Src autophosphorylation and downstream signaling to AKT and
phosphor-S6 in GBM cell lines, also reducing the growth and
invasion of glioblastoma cells. Inhibition of src-family kinases by
dasatinib also causes death of autophagic glioblastoma cells in
vitro (158). Src signaling
is also markedly enhanced in patients with invasive glioblastoma
after administration of bevacizumab. The Src family kinase
inhibitor dasatinib effectively blocked bevacizumab-induced
invasion of glioma in preclinical models, leading to the hypothesis
that combining bevacizumab with dasatinib may increase the efficacy
of bevacizumab in patients with recurrent GBM (159).
Several experimental therapeutic vaccines have been
developed over the past decades to treat GBM. Dendritic cell
vaccines have been able to modify the immune response in patients
with malignant neoplasms and induce anti-tumor immunity (160). Recent advances in vaccination
with dendritic cells have achieved encouraging results in clinical
trials and may improve the survival of patients with GBM (160). A recent double-blind, randomized
phase II trial (161) evaluated
the efficacy of the ICT-107 vaccine based on autologous dendritic
cells loaded with six epitopes targeting GBM-associated antigens:
melanoma antigen gene 1, human epidermal growth factor receptor 2
(HER-2), absent in melanoma 2, tyrosi-nase-related protein-2, gp100
(also known as premelanosome protein) and interleukin-13 receptor
subunit α-2 in patients with newly diagnosed GBM, whose major
histocompatibility complex serotype was human leukocyte antigen
(HLA)-A1+ and/or HLA-A2+. The vaccine increased PFS by 2.2 months
(P=0.011) but did not increase OS (17 months for the treatment
group and 15 for the control group).
The frequency of expression of the primary tumor
antigen HLA-A2 in patients (>90%) was higher than that of HLA-A1
(37.8%). Patients with HLA-A2 exhibited a more pronounced immune
response to the vaccine (assessed using Elispot) and a significant
therapeutic effect was observed in patients with a methylated MGMT
gene promoter (PFS, 24.1 vs. 8.5 months in the control group) and
with an unmethylated one (PFS, 10.5 vs. 6 months in the control
group). This study demonstrated the possible clinical efficacy of
ICT-107 in patients with the HLA-A2 serotype (152).
KHS101 exerts its cytotoxic effects by disrupting
the mito-chondrial chaperone heat shock protein family D member 1
(HSPD1). Research identified that KHS101 exerts cytotoxic activity
in several GBM cell lines obtained from patients, disrupting
cellular metabolism and promoting GBM cell autophagy. The mechanism
of action of KHS101 is to affect HSPD1, which also influences the
mitochondrial protein, leading to the disruption of mitochondrial
metabolism and the activation of autophagy mechanisms. In
vivo injection of KHS101 reduced tumor growth and increased
survival in patient-derived xenograft tumor GBM models in mice
(162).
Another novel approach in GBM research is VB-111,
an anti-cancer gene therapy. The mechanism of action of VB-111 is
determined by two main mechanisms: An anti-angiogenetic action
leading to oxygen starvation of the tumor and the induction of a
tumor-directed immune response. VB-111 is based on a
non-integrating adenovirus type 5 vector carrying a chimeric Fas
receptor transgene that binds to the human TNF-1 receptor (163). Based on preclinical results in
combination with dose escalation, VB-111 demonstrated efficacy as
an anti-angiogenic agent in the treatment of GBM. According to the
results of phase I/II clinical trials, VB-111 monotherapy,
continued after tumor progression with the addition of
beva-cizumab, was associated with promising improvements in OS and
PFS, a favorable safety profile and typical radiological responses.
The observed radiological response and the survival advantage of
the combined regimen with primer VB-111 prompted further study in
the randomized controlled GLOBE trial (164). However, the GLOBE study did not
reproduce the promising results seen in a phase II study (164).
Rindopepimut (also known as CDX-110) is a vaccine
targeting EGFRvIII deletion mutation and consists of an
EGFRvIII-specific peptide conjugated to snail lymph hemocyanin
(165). Preclinical studies
demonstrated that intradermal administration of an
EGFRvIII-specific vaccine-induced humoral immunity and prolonged
survival of mice with intracerebral tumors (166). A randomized phase II trial
confirmed the possibility of its usage in patients with GBM. A
total of 73 patients were randomized (36 rindopepimut, 37
controls). PFS was 28% (10/36) for rindopepimut vs. 16% (6/37) for
the control group (P=0.12). In the experimental group, tumor
response to treatment based on objective assessment was 30% (9/30)
vs. 18% in the control group (6/34; P=0.38) and the median response
duration was 7.8 months (95% CI, 3.5–22.2) compared to 5.6 for the
control group (95% CI, 3.7–7.4); at 6 months, steroid intake was
discontinued in 33% (6/18) vs. 0% (0/19) in the control group
(167).
Immunotherapy with checkpoint inhibitors of
programmed death-ligand 1 and CTLA-4 has improved outcomes in
numerous tumor types. However, in GBM, the exploratory phase I
Checkmate 143 study with nivolumab ± ipilimumab enrolled 40
patients with GBM and demonstrated limited efficacy (3/40
responders) (168,169).
A total of 369 patients with recurrent glioblastoma
were randomized to receive nivolumab or bevacizumab in the
open-label phase 3 CheckMate 143 clinical study. Nivolumab is an
immune checkpoint inhibitor, a fully human IgG4 monoclonal antibody
against programmed cell death-1. The primary endpoint of median OS
did not differ substantially between the two medications at the
study's conclusion: 9.8 months for nivolumab and 10.0 months for
bevacizumab with a considerably lower PFS in the nivolumab group
(1.5 vs. 3.5 months) (170).
In the CheckMate-498 trial, 560 patients were
randomized into either the RT + TMZ + nivolumab or RT + TMZ +
placebo group. The median OS in the treatment group was 13.4
months, while in the control group, the median OS totalled 14.9
months. PFS was also longer in the control group (6.2 vs. 6 months)
(171).
In another study, 80 bevacizumab-naïve patients at
the 1st/2nd recurrence were randomized to receive pembrolizumab
with or without bevacizumab. The group that received bevaci-zumab
had a median OS of 8.8 months, while the other group had a median
OS of 10.3 months. Pembrolizumab was well tolerated, although its
efficacy as a monotherapy for recurrent GBM was limited (172). All these trials failed to
support the utility of checkpoint inhibitors in glioblastoma.
Checkpoint inhibitors were also tested in the neoadjuvant approach.
In one of those trials, a pre-surgical dose of nivolumab was
followed by postsurgical nivolumab until disease progression or
unacceptable toxicity in 30 patients. Neoadjuvant nivolumab
increased chemokine transcript expression, immune cell infiltration
and TCR clonal diversity among tumor-infiltrating T cells,
indicating that the treatment had a local immunomodulatory effect.
However, there was no evidence of a therapeutic benefit. In
evaluated patients, median PFS was 4.1 months and median OS was 7.3
months (173). Another study was
aimed at evaluating immune responses and survival following
neoadjuvant and/or adjuvant therapy with pembrolizumab in 35
patients. In this study, neoadjuvant use was also associated with
an immunomodulatory effect. Patients in the adjuvantonly group had
a median OS of 7.5 months, whereas those in the neoadjuvant arm had
a median OS of 13.7 months (174).
Insulin-like growth factors (IGFs) are associated
with aberrant signaling, cell growth and resistance to therapy, and
are widely expressed in glioblastoma (175), including both a ligand and a
receptor (IGF1R). IGF1R expression also correlates with a worse
response to therapy and the expression of the receptor is
associated with a decrease in the effectiveness of therapy aimed at
EGFR (176), mTOR and HER-2.
Preclinical studies have provided encouraging results for
anti-IGF1R therapy (177), but
clinical trials have not confirmed the benefits of the therapy
(178).
There are 14 types of Ephrin family receptors
(Eph), divided into subcategories EphA and EphB (179). EphA receptors are expressed
largely in stem cells and de-differentiated phenotypes and are
absent in differentiated cell populations (180). EphA2 and EphA3 in glioblastomas
are associated with self-renewal of tumor stem cells (181,182). Ifabotuzumab (KB004) the
anti-EphA3 monoclonal antibody achieved stable disease for 23 weeks
in GBM (NCT03374943) (183).
Additional studies targeting EphA are warranted in GBM.
Despite a large amount of research regarding the
biology and associated disrupted signaling pathways, glioblastoma
remains one of the most difficult tumors to treat, leading to
dismal prognosis. Existing treatment options may only modestly
prolong survival and only a small proportion of patients are cured
with current standard of care therapy. Therefore, there is an unmet
requirement for further research to investigate the mechanisms of
glioblastoma pathogenesis and to look for new treatment targets in
novel signaling pathways that are implicated in glioblastoma
progression. Recent data suggest the use of a personalized approach
for the treatment of GBM with targeted drugs may be promising, but
this will require further research into the safety and efficacy of
novel compounds in selected GBM populations with distinct molecular
targets. Finally, deeper biological studies of CNS development, as
well as GBM cancer biology, are important to discover novel
approaches for the treatment of glioblastomas.
Not applicable.
MK: Conceptualization and ideas, writing of the
original draft, review and editing, project administration,
visualization preparation. AG: Conceptualization and ideas, writing
of the original draft, review and editing, project administration,
visualization preparation, critical revision. YB: Review and
editing, project administration, conceptualization and ideas,
funding acquisition, visualization preparation, critical revision.
KK: Review and editing, project administration, conceptualization
and ideas, visualization preparation, critical revision. MF: Review
and editing. FK: Writing of the original draft, funding
acquisition. LaK, NK, OK: Reviewing and editing, funding
acquisition. LeK: Review and editing, funding acquisition, critical
revision. Data authentication is not applicable. All authors read
and approved the final manuscript.
Not applicable.
Not applicable.
YB has been a speaker for Amgen and Regeneron and
has been an Advisor/Board Member for Alphageneron, G1 Therapeutics
and Jazz Pharmaceuticals. The remaining authors have no competing
interests to declare.
Not applicable.
YB was supported in part by the NIH R01 CA218802 grant, Jimmy V
Foundation T2018-013 Translational Award grant, Northwestern
University 2022 Translational Bridge Award and by the NCI NIH Core
Grant P30 CA060553 to the Robert H. Lurie Comprehensive Cancer
Center at Northwestern University (Chicago, USA). This study was
also partially supported by the Russian government program for
Competitive Growth of the Kazan Federal University (Kazan, Russia)
and Northwestern University (Chicago, USA) start-up funds to
YB.
|
1
|
Darlix A, Zouaoui S, Rigau V, Bessaoud F,
Figarella-Branger D, Mathieu-Daudé H, Trétarre B, Bauchet F, Duffau
H, Taillandier L and Bauchet L: Epidemiology for primary brain
tumors: A nationwide population-based study. J Neurooncol.
131:525–546. 2017. View Article : Google Scholar
|
|
2
|
Tykocki T and Eltayeb M: Ten-year survival
in glioblastoma. A systematic review. J Clin Neurosci. 54:7–13.
2018. View Article : Google Scholar
|
|
3
|
Faleh TA and Juweid M: Epidemiology and
outcome of glioblastoma. Exon Publications. 143–153. 2017.
|
|
4
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar
|
|
5
|
World Health Organization: Histological
classification of tumors of the central nervous system. Lyon,
France: IARC; 2016
|
|
6
|
Zong H, Parada LF and Baker SJ: Cell of
origin for malignant gliomas and its implication in therapeutic
development. Cold Spring Harb Perspect Biol. 7:a0206102015.
View Article : Google Scholar
|
|
7
|
Pombo Antunes AR, Scheyltjens I, Duerinck
J, Neyns B, Movahedi K and Van Ginderachter JA: Understanding the
glioblastoma immune microenvironment as basis for the development
of new immunotherapeutic strategies. Elife. 9:e521762020.
View Article : Google Scholar
|
|
8
|
Khaddour K, Johanns TM and Ansstas G: The
landscape of novel therapeutics and challenges in glioblastoma
multiforme: Contemporary state and future directions.
Pharmaceuticals (Basel). 13:3892020. View Article : Google Scholar
|
|
9
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar
|
|
10
|
Stupp R, Taillibert S, Kanner A, Read W,
Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K,
et al: Effect of tumor-treating fields plus maintenance
temozolomide vs maintenance temozolomide alone on survival in
patients with glioblastoma: A randomized clinical trial. JAMA.
318:2306–2316. 2017. View Article : Google Scholar
|
|
11
|
Cloughesy TF, Brenner A, de Groot JF,
Butowski NA, Zach L, Campian JL, Ellingson BM, Freedman LS, Cohen
YC, Lowenton-Spier N, et al: A randomized controlled phase III
study of VB-111 combined with bevacizumab vs bevacizumab
monotherapy in patients with recurrent glioblastoma (GLOBE). Neuro
Oncol. 22:705–717. 2020. View Article : Google Scholar
|
|
12
|
Nabors LB, Portnow J, Ahluwalia M,
Baehring J, Brem H, Brem S, Butowski N, Campian JL, Clark SW,
Fabiano AJ, et al: Central nervous system cancers, Version 3.2020,
NCCN clinical practice guidelines in oncology. J Natl Compr Canc
Netw. 18:1537–1570. 2020. View Article : Google Scholar
|
|
13
|
Lombardi G, De Salvo GL, Brandes AA, Eoli
M, Rudà R, Faedi M, Lolli I, Pace A, Daniele B, Pasqualetti F, et
al: Regorafenib compared with lomustine in patients with relapsed
glioblastoma (REGOMA): A multicentre, open-label, randomised,
controlled, phase 2 trial. Lancet Oncol. 20:110–119. 2019.
View Article : Google Scholar
|
|
14
|
Grothey A, Blay JY, Pavlakis N, Yoshino T
and Bruix J: Evolving role of regorafenib for the treatment of
advanced cancers. Cancer Treat Rev. 86:1019932020. View Article : Google Scholar
|
|
15
|
Ostrom QT, Cioffi G, Gittleman H, Patil N,
Waite K, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: Primary brain and other central nervous system tumors
diagnosed in the United States in 2012-2016. Neuro Oncol. 21(Suppl
5): v1–v100. 2019. View Article : Google Scholar
|
|
16
|
Levin VA, Leibel SA and Gutin PH:
Neoplasms of the central nervous system In: Cancer: Principles and
Practice of Oncology. 6th edition. Lippincott Williams and Wilkins;
Philadelphia, PA: pp. 2100–2160. 2001
|
|
17
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolo-mide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar
|
|
18
|
Hanif F, Muzaffar K, Perveen K, Malhi SM
and Simjee ShU: Glioblastoma multiforme: A review of its
epidemiology and pathogenesis through clinical presentation and
treatment. Asian Pac J Cancer Prev. 18:3–9. 2017.
|
|
19
|
Amirian ES, Ostrom QT, Armstrong GN, Lai
RK, Gu X, Jacobs DI, Jalali A, Claus EB, Barnholtz-Sloan JS,
Il'yasova D, et al: Aspirin, NSAIDs, and Glioma Risk: Original data
from the glioma international Case-Control study and a
meta-analysis. Cancer Epidemiol Biomarkers Prev. 28:555–562.
2019.
|
|
20
|
Scheurer ME, El-Zein R, Thompson PA,
Aldape KD, Levin VA, Gilbert MR, Weinberg JS and Bondy ML:
Long-term anti-inflam-matory and antihistamine medication use and
adult glioma risk. Cancer Epidemiol Biomarkers Prev. 17:1277–1281.
2008. View Article : Google Scholar
|
|
21
|
Dziurzynski K, Chang SM, Heimberger AB,
Kalejta RF, McGregor Dallas SR, Smit M, Soroceanu L and Cobbs CS;
HCMV and Gliomas Symposium: Consensus on the role of human
cytomegalovirus in glioblastoma. Neuro Oncol. 14:246–255. 2012.
View Article : Google Scholar
|
|
22
|
Söderberg-Nauclér C, Rahbar A and
Stragliotto G: Survival in patients with glioblastoma receiving
valganciclovir. N Engl J Med. 369:985–986. 2013. View Article : Google Scholar
|
|
23
|
Bei R, Marzocchella L and Turriziani M:
The use of temozolo-mide for the treatment of malignant tumors:
Clinical evidence and molecular mechanisms of action. Recent Pat
Anticancer Drug Discov. 5:172–187. 2010. View Article : Google Scholar
|
|
24
|
Lacal PM, D'Atri S, Orlando L, Bonmassar E
and Graziani G: In vitro inactivation of human O6-alkylguanine DNA
alkyl-transferase by antitumor triazene compounds. J Pharmacol Exp
Ther. 279:416–422. 1996.
|
|
25
|
D'Atri S, Tentori L, Lacal PM, Graziani G,
Pagani E, Benincasa E, Zambruno G, Bonmassar E and Jiricny J:
Involvement of the mismatch repair system in temozolomide-induced
apoptosis. Mol Pharmacol. 54:334–341. 1998. View Article : Google Scholar
|
|
26
|
Baer JC, Freeman AA, Newlands ES, Watson
AJ, Rafferty JA and Margison GP: Depletion of O 6-alkylguanine-DNA
alkyltrans-ferase correlates with potentiation of temozolomide and
CCNU toxicity in human tumour cells. Br J Cancer. 67:1299–1302.
1993. View Article : Google Scholar
|
|
27
|
Wu W, Klockow JL, Zhang M, Lafortune F,
Chang E, Jin L, Wu Y and Daldrup-Link HE: Glioblastoma Multiforme
(GBM): An overview of current therapies and mechanisms of
resistance. Pharmacol Res. 17:1057802021. View Article : Google Scholar
|
|
28
|
Grossmann P, Narayan V, Chang K, Rahman R,
Abrey L, Reardon DA, Schwartz LH, Wen PY, Alexander BM, Huang R and
Aerts HJWL: Quantitative imaging biomarkers for risk stratification
of patients with recurrent glioblastoma treated with bevacizumab.
Neuro Oncol. 19:1688–1697. 2017. View Article : Google Scholar
|
|
29
|
Friedman HS, Prados MD, Wen PY, Mikkelsen
T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen
R, et al: Bevacizumab alone and in combination with irinotecan in
recurrent glioblastoma. J Clin Oncol. 27:4733–4740. 2009.
View Article : Google Scholar
|
|
30
|
Chinot OL, de La Motte Rouge T, Moore N,
Zeaiter A, Das A, Phillips H, Modrusan Z and Cloughesy T: AVAglio:
Phase 3 trial of bevacizumab plus temozolomide and radiotherapy in
newly diagnosed glioblastoma multiforme. Adv Ther. 28:334–340.
2011. View Article : Google Scholar
|
|
31
|
Gilbert MR, Dignam J, Won M, Blumenthal
DT, Vogelbaum MA, AldapeHoward Colman KD, Chakravarti A, Jeraj R,
Armstrong TS, Scott Wefel J, et al: RTOG 0825: Phase III
double-blind placebo-controlled trial evaluating bevacizumab (Bev)
in patients (Pts) with newly diagnosed glioblastoma (GBM). J Clin
Oncol. 31:12013. View Article : Google Scholar
|
|
32
|
Diaz RJ, Ali S, Qadir MG, De La Fuente MI,
Ivan ME and Komotar RJ: The role of bevacizumab in the treatment of
glio-blastoma. J Neurooncol. 133:455–467. 2017. View Article : Google Scholar
|
|
33
|
Taal W, Oosterkamp HM, Walenkamp AM,
Dubbink HJ, Beerepoot LV, Hanse MC, Buter J, Honkoop AH, Boerman D,
de Vos FY, et al: Single-agent bevacizumab or lomustine versus a
combination of bevacizumab plus lomustine in patients with
recurrent glioblastoma (BELOB trial): A randomised controlled phase
2 Trial. Lancet Oncol. 15:943–953. 2014. View Article : Google Scholar
|
|
34
|
Brandsma D and van den Bent MJ:
Pseudoprogression and pseu-doresponse in the treatment of gliomas.
Curr Opin Neurol. 22:633–638. 2009. View Article : Google Scholar
|
|
35
|
Wen PY, Macdonald DR, Reardon DA,
Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert
MR, Lassman AB, et al: Updated response assessment criteria for
high-grade gliomas: Response assessment in Neuro-Oncology working
group. J Clin Oncol. 28:1963–1972. 2010. View Article : Google Scholar
|
|
36
|
Batchelor TT, Mulholland P, Neyns B,
Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S,
Ashby LS, et al: Phase III randomized trial comparing the efficacy
of cediranib as monotherapy, and in combination with lomustine,
versus lomustine alone in patients with recurrent glioblastoma. J
Clin Oncol. 31:32122013. View Article : Google Scholar
|
|
37
|
de Groot JF, Lamborn KR, Chang SM, Gilbert
MR, Cloughesy TF, Aldape K, Yao J, Jackson EF, Lieberman F, Robins
HI, et al: Phase II study of aflibercept in recurrent malignant
glioma: A North American Brain Tumor Consortium study. J Clin
Oncol. 29:2689–2995. 2011. View Article : Google Scholar
|
|
38
|
Stupp R, Taillibert S, Kanner AA, Kesari
S, Steinberg DM, Toms SA, Taylor LP, Lieberman F, Silvani A, Fink
KL, et al: Maintenance therapy with tumor-treating fields plus
temozolomide vs temozolomide alone for glioblastoma: A randomized
clinical trial. JAMA. 314:2535–2543. 2015. View Article : Google Scholar
|
|
39
|
Fabian D, Guillermo Prieto Eibl MDP,
Alnahhas I, Sebastian N, Giglio P, Puduvalli V, Gonzalez J and
Palmer JD: Treatment of glioblastoma (GBM) with the addition of
tumor-treating fields (TTF): A review. Cancers (Basel). 11:1742019.
View Article : Google Scholar
|
|
40
|
Brown TJ, Brennan MC, Li M, Church EW,
Brandmeir NJ, Rakszawski KL, Patel AS, Rizk EB, Suki D, Sawaya R
and Glantz M: Association of the extent of resection with survival
in glioblastoma: A systematic review and meta-analysis. JAMA Oncol.
2:1460–1469. 2016. View Article : Google Scholar
|
|
41
|
Noorbakhsh A, Tang JA, Marcus LP,
McCutcheon B, Gonda DD, Schallhorn CS, Talamini MA, Chang DC,
Carter BS and Chen CC: Gross-total resection outcomes in an elderly
population with glioblastoma: A SEER-based analysis. J Neurosurg.
120:31–39. 2014. View Article : Google Scholar
|
|
42
|
Eigenbrod S, Trabold R, Brucker D, Erös C,
Egensperger R, La Fougere C, Göbel W, Rühm A, Kretzschmar HA, Tonn
JC, et al: Molecular stereotactic biopsy technique improves
diagnostic accuracy and enables personalized treatment strategies
in glioma patients. Acta Neurochir (Wien). 156:1427–1440. 2014.
View Article : Google Scholar
|
|
43
|
Stummer W, Tonn JC, Mehdorn HM, Nestler U,
Franz K, Goetz C, Bink A and Pichlmeier U; ALA-Glioma Study Group:
Counterbalancing risks and gains from extended resections in
malignant glioma surgery: A supplemental analysis from the
randomized 5-aminolevulinic acid glioma resection study. J
Neurosurg. 114:613–623. 2011. View Article : Google Scholar
|
|
44
|
Berntsen EM, Gulati S, Solheim O, Kvistad
KA, Torp SH, Selbekk T, Unsgård G and Håberg AK: Functional
magnetic resonance imaging and diffusion tensor tractography
incorporated into an intraoperative 3-dimensional ultrasound-based
neuronavigation system: Impact on therapeutic strategies, extent of
resection, and clinical outcome. Neurosurgery. 67:251–264. 2010.
View Article : Google Scholar
|
|
45
|
Ringel F, Pape H, Sabel M, Krex D, Bock
HC, Misch M, Weyerbrock A, Westermaier T, Senft C, Schucht P, et
al: Clinical benefit from resection of recurrent glioblastomas:
Results of a multicenter study including 503 patients with
recurrent glioblastomas undergoing surgical resection. Neuro Oncol.
18:96–104. 2016. View Article : Google Scholar
|
|
46
|
Zuccarini M, Giuliani P, Ziberi S,
Carluccio M, Iorio PD, Caciagli F and Ciccarelli R: The role of Wnt
signal in glioblastoma development and progression: A possible new
pharmacological target for the therapy of this tumor. Genes
(Basel). 9:1052018. View Article : Google Scholar
|
|
47
|
Sigismund S, Avanzato D and Lanzetti L:
Emerging functions of the EGFR in cancer. Mol Oncol. 12:3–20. 2018.
View Article : Google Scholar
|
|
48
|
Xiao A, Brenneman B, Floyd D, Comeau L,
Spurio K, Olmez I, Lee J, Nakano I, Godlewski J, Bronisz A, et al:
Statins affect human glioblastoma and other cancers through TGF-β
inhibition. Oncotarget. 10:1716–1728. 2019. View Article : Google Scholar
|
|
49
|
Keunen O, Johansson M, Oudin A, Sanzey M,
Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R, et al:
Anti-VEGF treatment reduces blood supply and increases tumor cell
invasion in glioblastoma. Proc Natl Acad Sci USA. 108:3749–3754.
2011. View Article : Google Scholar
|
|
50
|
Cyclin-dependent kinase inhibitor 2A.
GeneCards. Weizmann institute of science. Retrieved December 15,
2021.
|
|
51
|
Albensi BC: What is nuclear factor kappa B
(NF-κB) doing in and to the mitochondrion? Front Cell Dev Biol.
7:1542019. View Article : Google Scholar
|
|
52
|
Xia L, Tan S, Zhou Y, Lin J, Wang H, Oyang
L, Tian Y, Liu L, Su M, Wang H, et al: Role of the NFκB-signaling
pathway in cancer. Onco Targets Ther. 11:2063–2073. 2018.
View Article : Google Scholar
|
|
53
|
Xia Y, Shen S and Verma IM: NF-κB, an
active player in human cancers. Cancer Immunol Res. 2:823–830.
2014. View Article : Google Scholar
|
|
54
|
Li X, Wu C, Chen N, Gu H, Yen A, Cao L,
Wang E and Wang L: PI3K/Akt/mTOR signaling pathway and targeted
therapy for glioblastoma. Oncotarget. 7:33440–33450. 2016.
View Article : Google Scholar
|
|
55
|
Markman B, Dienstmann R and Tabernero J:
Targeting the PI3K/Akt/mTOR pathway-beyond rapalogs. Oncotarget.
1:5302010. View Article : Google Scholar
|
|
56
|
Crespo I, Vital AL, Gonzalez-Tablas M,
Patino Mdel C, Otero A, Lopes MC, de Oliveira C, Domingues P, Orfao
A and Tabernero MD: Molecular and genomic alterations in
glioblastoma multiforme. Am J Pathol. 185:1820–1833. 2015.
View Article : Google Scholar
|
|
57
|
Balça-Silva J, Matias D, Carmo AD,
Sarmento-Ribeiro AB, Lopes MC and Moura-Neto V: Cellular and
molecular mechanisms of glioblastoma malignancy: Implications in
resistance and therapeutic strategies. Semin Cancer Biol.
58:130–141. 2019. View Article : Google Scholar
|
|
58
|
Rajesh Y, Pal I, Banik P, Chakraborty S,
Borkar SA, Dey G, Mukherjee A and Mandal M: Insights into molecular
therapy of glioma: Current challenges and next generation
blueprint. Acta Pharmacol Sin. 38:591–613. 2017. View Article : Google Scholar
|
|
59
|
Cancer Genome Atlas and Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar
|
|
60
|
Hegi ME, Genbrugge E, Gorlia T, Stupp R,
Gilbert MR, Chinot OL, Nabors LB, Jones G, Van Criekinge W, Straub
J and Weller M: MGMT promoter methylation cutoff with safety margin
for selecting glioblastoma patients into trials omitting
temozolomide: A pooled analysis of four clinical trials. Clin
Cancer Res. 25:1809–1816. 2019. View Article : Google Scholar
|
|
61
|
Chai RC, Zhang KN, Chang YZ, Wu F, Liu YQ,
Zhao Z, Wang KY, Chang YH, Jiang T and Wang YZ: Systematically
characterize the clinical and biological significances of 1p19q
genes in 1p/19q non-codeletion glioma. Carcinogenesis.
40:1229–1239. 2019. View Article : Google Scholar
|
|
62
|
Wang Y, Li S, Chen L, You G, Bao Z, Yan W,
Shi Z, Chen Y, Yao K, Zhang W, et al: Glioblastoma with an
oligodendroglioma component: Distinct clinical behavior, genetic
alterations, and outcome. Neuro Oncol. 14:518–525. 2012. View Article : Google Scholar
|
|
63
|
Clark KH, Villano JL, Nikiforova MN,
Hamilton RL and Horbinski C: 1p/19q testing has no significance in
the workup of glioblastomas. Neuropathol Appl Neurobiol.
39:706–717. 2013. View Article : Google Scholar
|
|
64
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar
|
|
65
|
Brosh R and Rotter V: When mutants gain
new powers: News from the mutant p53 field. Nat Rev Cancer.
9:701–713. 2009. View Article : Google Scholar
|
|
66
|
Brennan CW, Verhaak RG, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: Erratum: The somatic genomic landscape of
glioblastoma. Cell. 155:462–477. 2013. View Article : Google Scholar
|
|
67
|
Liu F, Huang J, Liu X, Cheng Q, Luo C and
Liu Z: CTLA-4 correlates with immune and clinical characteristics
of glioma. Cancer Cell Int. 20:72020. View Article : Google Scholar
|
|
68
|
Garofano L, Migliozzi S, Oh YT, D'Angelo
F, Najac RD, Ko A, Frangaj B, Caruso FP, Yu K, Yuan J, et al:
Pathway-based clas-sification of glioblastoma uncovers a
mitochondrial subtype with therapeutic vulnerabilities. Nat Cancer.
2:141–156. 2021. View Article : Google Scholar
|
|
69
|
Lu VM, O'Connor KP, Shah AH, Eichberg DG,
Luther EM, Komotar RJ and Ivan ME: The prognostic significance of
CDKN2A homozygous deletion in IDH-mutant lower-grade glioma and
glioblastoma: A systematic review of the contemporary literature. J
Neurooncol. 148:221–229. 2020. View Article : Google Scholar
|
|
70
|
William D, Mokri P, Lamp N, Linnebacher M,
Classen CF, Erbersdobler A and Schneider B: Amplification of the
EGFR gene can be maintained and modulated by variation of EGF
concentrations in in vitro models of glioblastoma multiforme. PLoS
One. 12:e01852082017. View Article : Google Scholar
|
|
71
|
Felsberg J, Hentschel B, Kaulich K,
Gramatzki D, Zacher A, Malzkorn B, Kamp M, Sabel M, Simon M,
Westphal M, et al: Epidermal growth factor receptor variant III
(EGFRvIII) positivity in EGFR-amplified glioblastomas: Prognostic
role and comparison between primary and recurrent tumors. Clin
Cancer Res. 23:6846–6855. 2017. View Article : Google Scholar
|
|
72
|
An Z, Aksoy O, Zheng T, Fan QW and Weiss
WA: Epidermal growth factor receptor and EGFRvIII in glioblastoma:
Signaling pathways and targeted therapies. Oncogene. 37:1561–1575.
2018. View Article : Google Scholar
|
|
73
|
Xu H, Zong H, Ma C, Ming X, Shang M, Li K,
He X, Du H and Cao L: Epidermal growth factor receptor in
glioblastoma. Oncol Lett. 14:512–516. 2017. View Article : Google Scholar
|
|
74
|
De S, Dermawan JK and Stark GR: EGF
receptor uses SOS1 to drive constitutive activation of NFκB in
cancer cells. Proc Natl Acad Sci USA. 111:11721–11726. 2014.
View Article : Google Scholar
|
|
75
|
Talasila KM, Soentgerath A, Euskirchen P,
Rosland GV, Wang J, Huszthy PC, Prestegarden L, Skaftnesmo KO,
Sakariassen PØ, Eskilsson E, et al: EGFR wild-type amplification
and activation promote invasion and development of glioblastoma
independent of angiogenesis. Acta Neuropathol. 125:683–698. 2013.
View Article : Google Scholar
|
|
76
|
Katanasaka Y, Kodera Y, Kitamura Y,
Morimoto T, Tamura T and Koizumi F: Epidermal growth factor
receptor variant type III markedly accelerates angiogenesis and
tumor growth via inducing c-myc mediated angiopoietin-like 4
expression in malignant glioma. Mol Cancer. 12:312013. View Article : Google Scholar
|
|
77
|
Sarkaria JN, Yang L, Grogan PT, Kitange
GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB
and James CD: Identification of molecular characteristics
correlated with glioblastoma sensitivity to EGFR kinase inhibition
through use of an intracranial xenograft test panel. Mol Cancer
Ther. 6:1167–1174. 2007. View Article : Google Scholar
|
|
78
|
Cetintas VB and Batada NN: Is there a
causal link between PTEN deficient tumors and immunosuppressive
tumor microenvironment? J Transl Med. 18:452020. View Article : Google Scholar
|
|
79
|
Raizer JJ, Abrey LE, Lassman AB, Chang SM,
Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KA, Wen PY, et al:
A phase II trial of erlotinib in patients with recurrent malignant
gliomas and nonprogressive glioblastoma multiforme postradiation
therapy. Neuro Oncol. 12:95–103. 2010. View Article : Google Scholar
|
|
80
|
Mellinghoff IK, Wang MY, Vivanco I,
Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute
DJ, et al: Molecular determinants of the response of glioblastomas
to EGFR kinase inhibitors. N Engl J Med. 353:2012–2024. 2005.
View Article : Google Scholar
|
|
81
|
Chakravarti A, Wang M, Robins HI,
Lautenschlaeger T, Curran WJ, Brachman DG, Schultz CJ, Choucair A,
Dolled-Filhart M, Christiansen J, et al: RTOG 0211: A phase 1/2
study of radiation therapy with concurrent gefitinib for newly
diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phy.
85:1206–1211. 2013. View Article : Google Scholar
|
|
82
|
Uhm JH, Ballman KV, Wu W, Giannini C,
Krauss JC, Buckner JC, James CD, Scheithauer BW, Behrens RJ, Flynn
PJ, et al: Phase II evaluation of gefitinib in patients with newly
diagnosed Grade 4 astrocytoma: Mayo/North central cancer treatment
group study N0074. Int J Radiat Oncol Biol Phys. 80:347–353. 2011.
View Article : Google Scholar
|
|
83
|
Inda MM, Bonavia R, Mukasa A, Narita Y,
Sah DW, Vandenberg S, Brennan C, Johns TG, Bachoo R, Hadwiger P, et
al: Tumor heterogeneity is an active process maintained by a mutant
EGFR-induced cytokine circuit in glioblastoma. Genes Dev.
24:1731–1745. 2010. View Article : Google Scholar
|
|
84
|
Mazzoleni S, Politi LS, Pala M, Cominelli
M, Franzin A, Sergi Sergi L, Falini A, De Palma M, Bulfone A,
Poliani PL and Galli R: Epidermal growth factor receptor expression
identifies functionally and molecularly distinct tumor-initiating
cells in human glioblastoma multiforme and is required for
gliomagenesis. Cancer Res. 70:7500–7513. 2010. View Article : Google Scholar
|
|
85
|
Van Den Bent M, Eoli M, Sepulveda JM,
Smits M, Walenkamp A, Frenel JS, Franceschi E, Clement PM, Chinot
O, De Vos F, et al: INTELLANCE 2/EORTC 1410 randomized phase II
study of Depatux-M alone and with temozolomide vs temozolomide or
lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol.
22:684–693. 2020. View Article : Google Scholar
|
|
86
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar
|
|
87
|
Dresselhaus EC and Meffert MK: Cellular
specificity of NF-κB function in the nervous system. Front Immunol.
10:10432019. View Article : Google Scholar
|
|
88
|
Friedmann-Morvinski D, Narasimamurthy R,
Xia Y, Myskiw C, Soda Y and Verma IM: Targeting NF-κB in
glioblastoma: A therapeutic approach. Sci Adv. 2:e15012922016.
View Article : Google Scholar
|
|
89
|
Wang H, Wang H, Zhang W, Huang HJ, Liao WS
and Fuller GN: Analysis of the activation status of Akt, NFkappaB,
and Stat3 in human diffuse gliomas. Lab Invest. 84:941–951. 2004.
View Article : Google Scholar
|
|
90
|
Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang
L, You MJ, Koh MY, Cote G, Aldape K, et al: EGFR-induced and PKCε
monoubiquitylation-dependent NF-κB activation upregulates PKM2
expression and promotes tumorigenesis. Mol Cell. 48:771–784. 2012.
View Article : Google Scholar
|
|
91
|
Yap YS, McPherson JR, Ong CK, Rozen SG,
The BT, Lee AS and Callen DF: The NF1 gene revisited-from bench to
bedside. Oncotarget. 5:5873–5892. 2014. View Article : Google Scholar
|
|
92
|
Schäfer C, Göder A, Beyer M, Kiweler N,
Mahendrarajah N, Rauch A, Nikolova T, Stojanovic N, Wieczorek M,
Reich TR, et al: Class I histone deacetylases regulate p53/NF-κB
crosstalk in cancer cells. Cell Signal. 29:218–225. 2018.
View Article : Google Scholar
|
|
93
|
Zanotto-Filho A, Braganhol E, Schröder R,
de Souza LH, Dalmolin RJ, Pasquali MA, Gelain DP, Battastini AM and
Moreira JC: NFκB inhibitors induce cell death in glioblastomas.
Biochem Pharmacol. 81:412–424. 2011. View Article : Google Scholar
|
|
94
|
Shinoda K, Kuboki S, Shimizu H, Ohtsuka M,
Kato A, Yoshitomi H, Furukawa K and Miyazaki M: Pin1 facilitates
NF-κ B activation and promotes tumour progression in human
hepatocellular carcinoma. Br J Cancer. 113:1323–1331. 2015.
View Article : Google Scholar
|
|
95
|
Medeiros M, Candido MF, Valera ET and
Brassesco MS: The multifaceted NF-kB: Are there still prospects of
its inhibition for clinical intervention in pediatric central
nervous system tumors? Cell Mol Life Sci. 78:6161–6200. 2021.
View Article : Google Scholar
|
|
96
|
Phesse T, Flanagan D and Vincan E:
Frizzled7: A promising Achilles' Heel for targeting the Wnt
receptor complex to treat cancer. Cancers (Basel). 8:502016.
View Article : Google Scholar
|
|
97
|
Gao J, Liao Y, Qiu M and Shen W:
Wnt/β-catenin signaling in neural stem cell homeostasis and
neurological diseases. Neuroscientist. 27:58–72. 2021. View Article : Google Scholar
|
|
98
|
Tang C, Guo J, Chen H, Yao CJ, Zhuang DX,
Wang Y, Tang WJ, Ren G, Yao Y, Wu JS, et al: Gene mutation
profiling of primary glioblastoma through multiple tumor biopsy
guided by 1H-magnetic resonance spectroscopy. Int J Clin Exp
Pathol. 8:5327–5335. 2015.
|
|
99
|
Yun EJ, Kim S, Hsieh JT and Baek ST:
Wnt/β-catenin signaling pathway induces autophagy-mediated
temozolomide-resistance in human glioblastoma. Cell Death Dis.
11:7712020. View Article : Google Scholar
|
|
100
|
Tompa M, Kalovits F, Nagy A and Kalman B:
Contribution of the Wnt pathway to defining biology of
glioblastoma. Neuromolecular Med. 20:437–451. 2018. View Article : Google Scholar
|
|
101
|
Mori H, Yao Y, Learman BS, Kurozumi K,
Ishida J, Ramakrishnan SK, Overmyer KA, Xue X, Cawthorn WP, Reid
MA, et al: Induction of WNT11 by hypoxia and hypoxia-inducible
factor-1α regulates cell proliferation, migration and invasion. Sci
Rep. 6:215202016. View Article : Google Scholar
|
|
102
|
Rampazzo E, Persano L, Pistollato F, Moro
E, Frasson C, Porazzi P, Della Puppa A, Bresolin S, Battilana G,
Indraccolo S, et al: Wnt activation promotes neuronal
differentiation of glioblastoma. Cell Death Dis. 4:e5002013.
View Article : Google Scholar
|
|
103
|
Liu C, Takada K and Di Z: Targeting
Wnt/β-catenin pathway for drug therapy. Med Drug Discovery.
8:1000662020. View Article : Google Scholar
|
|
104
|
Diamond JR, Becerra C, Richards D, Mita A,
Osborne C, O'Shaughnessy J, Zhang C, Henner R, Kapoun AM, Xu L, et
al: Phase Ib clinical trial of the anti-frizzled antibody
vantictumab (OMP-18R5) plus paclitaxel in patients with locally
advanced or metastatic HER2-negative breast cancer. Breast Cancer
Res Treat. 184:53–62. 2020. View Article : Google Scholar
|
|
105
|
Selivanova LS, Volganova KS and Abrosimov
AY: Telomerase reverse transcriptase (TERT) promoter mutations in
the tumors of human endocrine organs: Biological and prognostic
value. Arkh Patol. 78:62–69. 2016.In Russian. View Article : Google Scholar
|
|
106
|
Pekmezci M, Rice T, Molinaro AM, Walsh KM,
Decker PA, Hansen H, Sicotte H, Kollmeyer TM, McCoy LS, Sarkar G,
et al: Adult infiltrating gliomas with WHO 2016 integrated
diagnosis: Additional prognostic roles of ATRX and TERT. Acta
Neuropathol. 133:1001–1016. 2017. View Article : Google Scholar
|
|
107
|
Bell RJ, Rube HT, Xavier-Magalhães A,
Costa BM, Mancini A, Song JS and Costello JF: Understanding TERT
promoter mutations: A common path to immortality. Mol Cancer Res.
14:315–323. 2016. View Article : Google Scholar
|
|
108
|
Huang JJ, Lin MC, Bai YX, Jing DD, Wong
BC, Han SW, Lin J, Xu B, Huang CF and Kung HF: Ectopic expression
of a COOH-terminal fragment of the human telomerase reverse
transcriptase leads to telomere dysfunction and reduction of growth
and tumorigenicity in HeLa cells. Cancer Res. 62:3226–3232.
2002.
|
|
109
|
Ng SS, Gao Y, Chau DH, Li GH, Lai LH,
Huang PT, Huang CF, Huang JJ, Chen YC, Kung HF and Lin MC: A novel
glioblastoma cancer gene therapy using AAV-mediated long-term
expression of human TERT C-terminal polypeptide. Cancer Gene Ther.
14:561–572. 2007. View Article : Google Scholar
|
|
110
|
Lavanya C, Sibin MK, Srinivas Bharath MM,
Manoj MJ, Venkataswamy MM, Bhat DI, Narasinga Rao KV and Chetan GK:
RNA interference mediated downregulation of human telomerase
reverse transcriptase (hTERT) in LN18 cells. Cytotechnology.
68:2311–2321. 2016. View Article : Google Scholar
|
|
111
|
Li X, Qian X, Wang B, Xia Y, Zheng Y, Du
L, Xu D, Xing D, DePinho RA and Lu Z: Programmable base editing of
mutated TERT promoter inhibits brain tumour growth. Nat Cell Biol.
22:282–288. 2020. View Article : Google Scholar
|
|
112
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte
G, Mazzarino MC, et al: Mutations and deregulation of
Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy
response. Oncotarget. 3:954–987. 2012. View Article : Google Scholar
|
|
113
|
Zhao HF, Wang J, Shao W, Wu CP, Chen ZP,
To ST and Li WP: Recent advances in the use of PI3K inhibitors for
glioblastoma multiforme: Current preclinical and clinical
development. Mol Cancer. 16:1002017. View Article : Google Scholar
|
|
114
|
Gymnopoulos M, Elsliger MA and Vogt PK:
Rare cancer-specific mutations in PIK3CA show gain of function.
Proc Natl Acad Sci USA. 104:5569–5574. 2007. View Article : Google Scholar
|
|
115
|
Höland K, Boller D, Hagel C, Dolski S,
Treszl A, Pardo OE, Cwiek P, Salm F, Leni Z, Shepherd PR, et al:
Targeting class IA PI3K isoforms selectively impairs cell growth,
survival, and migration in glioblastoma. PLoS One. 9:e941322014.
View Article : Google Scholar
|
|
116
|
Chen H, Mei L, Zhou L, Shen X, Guo C,
Zheng Y, Zhu H, Zhu Y and Huang L: PTEN restoration and PIK3CB
knockdown synergistically suppress glioblastoma growth in vitro and
in xenografts. J Neurooncol. 104:155–167. 2011. View Article : Google Scholar
|
|
117
|
Huang H, Regan KM, Wang F, Wang D, Smith
DI, van Deursen JM and Tindall DJ: Skp2 inhibits FOXO1 in tumor
suppression through ubiquitin-mediated degradation. Proc Natl Acad
Sci USA. 102:1649–1654. 2005. View Article : Google Scholar
|
|
118
|
Baretić D and Williams RL: PIKKs-the
solenoid nest where partners and kinases meet. Curr Opin Struct
Biol. 29:134–142. 2014. View Article : Google Scholar
|
|
119
|
Sarbassov DD, Ali SM, Kim DH, Guertin DA,
Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM: Rictor, a
novel binding partner of mTOR, defines a rapamycin-insensitive and
raptor-independent pathway that regulates the cytoskeleton. Curr
Biol. 14:1296–1302. 2004. View Article : Google Scholar
|
|
120
|
Cornu M, Albert V and Hall MN: mTOR in
aging, metabolism, and cancer. Curr Opin Genet Dev. 23:53–62. 2013.
View Article : Google Scholar
|
|
121
|
Lawlor MA and Alessi DR: PKB/Akt: A key
mediator of cell proliferation, survival and insulin responses? J
Cell Sci. 114:2903–2910. 2001. View Article : Google Scholar
|
|
122
|
Gini B, Zanca C, Guo D, Matsutani T, Masui
K, Ikegami S, Yang H, Nathanson D, Villa GR, Shackelford D, et al:
The mTOR kinase inhibitors, CC214-1 and CC214-2, preferentially
block the growth of EGFRvIII-activated glioblastomas. Clin Cancer
Res. 19:5722–5732. 2013. View Article : Google Scholar
|
|
123
|
Masri J, Bernath A, Martin J, Jo OD,
Vartanian R, Funk A and Gera J: mTORC2 activity is elevated in
gliomas and promotes growth and cell motility via overexpression of
rictor. Cancer Res. 67:11712–11720. 2007. View Article : Google Scholar
|
|
124
|
Agliano A, Balarajah G, Ciobota DM, Sidhu
J, Clarke PA, Jones C, Workman P, Leach MO and Al-Saffar NMS:
Pediatric and adult glioblastoma radiosensitization induced by
PI3K/mTOR inhibition causes early metabolic alterations detected by
nuclear magnetic resonance spectroscopy. Oncotarget. 8:47969–47983.
2017. View Article : Google Scholar
|
|
125
|
Chinnaiyan P, Won M, Wen PY, Rojiani AM,
Werner-Wasik M, Shih HA, Ashby LS, Michael Yu HH, Stieber VW,
Malone SC, et al: A randomized phase II study of everolimus in
combination with chemoradiation in newly diagnosed glioblas-toma:
Results of NRG Oncology RTOG 0913. Neuro Oncol. 20:666–673. 2018.
View Article : Google Scholar
|
|
126
|
Reardon DA, Wen PY, Alfred Yung WK, Berk
L, Narasimhan N, Turner CD, Clackson T, Rivera VM and Vogelbaum MA:
Ridaforolimus for patients with progressive or recurrent malignant
glioma: A perisurgical, sequential, ascending-dose trial. Cancer
Chemother Pharmacol. 69:849–860. 2012. View Article : Google Scholar
|
|
127
|
Wick W, Gorlia T, Bady P, Platten M, van
den Bent MJ, Taphoorn MJ, Steuve J, Brandes AA, Hamou MF, Wick A,
et al: Phase II study of radiotherapy and temsirolimus versus
radiochemotherapy with temozolomide in patients with newly
diagnosed glioblastoma without MGMT promoter hypermethylation
(EORTC 26082). Clin Cancer Res. 22:4797–4806. 2016. View Article : Google Scholar
|
|
128
|
U.S National Library of Medincine (NIH):
NCT Neuro Master Match-N2M2 (NOA-20).
ClinicalTrials.gov Identifier: NCT03158389.
https://clinicaltrials.gov/ct2/show/NCT03158389.
Accessed May 18, 2017.
|
|
129
|
Rodrik-Outmezguine VS, Okaniwa M, Yao Z,
Novotny CJ, McWhirter C, Banaji A, Won H, Wong W, Berger M, de
Stanchina E, et al: Overcoming mTOR resistance mutations with a
new-generation mTOR inhibitor. Nature. 534:272–276. 2016.
View Article : Google Scholar
|
|
130
|
Babak S and Mason WP: mTOR inhibition in
glioblastoma: Requiem for a dream? Neuro Oncol. 20:584–585. 2018.
View Article : Google Scholar
|
|
131
|
Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan
C, Wu Y, Li X, Li X, Li G, et al: Function of the c-Met receptor
tyrosine kinase in carcinogenesis and associated therapeutic
opportunities. Mol Cancer. 17:752018. View Article : Google Scholar
|
|
132
|
Carvalho B, Lopes JM, Silva R, Peixoto J,
Leitão D, Soares P, Fernandes AC, Linhares P, Vaz R and Lima J: The
role of c-Met and VEGFR2 in glioblastoma resistance to bevacizumab.
Sci Rep. 11:60672021. View Article : Google Scholar
|
|
133
|
McCarty JH: Glioblastoma resistance to
anti-VEGF therapy: Has the challenge been MET? Clin Cancer Res.
19:1631–1633. 2013. View Article : Google Scholar
|
|
134
|
Manneh Kopp RA, Sepúlveda-Sánchez JM,
Ruano Y, Toldos O, Pérez Núñez A, Cantero D, Hilario A, Ramos A, de
Velasco G, Sánchez-Gómez P and Hernández-Laín A: Correlation of
radiological and immunochemical parameters with clinical outcome in
patients with recurrent glioblastoma treated with Bevacizumab. Clin
Transl Oncol. 21:1413–1423. 2019. View Article : Google Scholar
|
|
135
|
Merchant M, Ma X, Maun HR, Zheng Z, Peng
J, Romero M, Huang A, Yang NY, Nishimura M, Greve J, et al:
Monovalent antibody design and mechanism of action of onartuzumab,
a MET antagonist with anti-tumor activity as a therapeutic agent.
Proc Natl Acad Sci USA. 110:E2987–E2996. 2013. View Article : Google Scholar
|
|
136
|
Cloughesy T, Finocchiaro G, Belda-Iniesta
C, Recht L, Brandes AA, Pineda E, Mikkelsen T, Chinot OL, Balana C,
Macdonald DR, et al: Randomized, double-blind, placebo-controlled,
multicenter phase II study of onartuzumab plus bevacizumab versus
placebo plus bevacizumab in patients with recurrent glioblastoma:
Efficacy, safety, and hepatocyte growth factor and
O6-methylguanine-DNA methyltransferase biomarker
analyses. J Clin Oncol. 35:343–351. 2017. View Article : Google Scholar
|
|
137
|
Garcia MM, Gil MJ, Losada E, Martin
Soberón MC, Mesia Barroso C, Foro P, Capellades J, Sarmiento B,
Bruna J, Verger E, et al: GEINO 1402: A phase Ib dose-escalation
study followed by an extension phase to evaluate safety and
efficacy of crizotinib in combination with temozolomide (TMZ) and
radiotherapy (RT) in patients with newly diagnosed glioblastoma
(GB. Ann Oncol. 30:v1472019. View Article : Google Scholar
|
|
138
|
Guillemot F and Zimmer C: From cradle to
grave: The multiple roles of fibroblast growth factors in neural
development. Neuron. 71:574–588. 2011. View Article : Google Scholar
|
|
139
|
Frinchi M, Bonomo A, Trovato-Salinaro A,
Condorelli DF, Fuxe K, Spampinato MG and Mudò G: Fibroblast growth
factor-2 and its receptor expression in proliferating precursor
cells of the subventricular zone in the adult rat brain. Neurosci
Lett. 447:20–25. 2008. View Article : Google Scholar
|
|
140
|
Dienstmann R, Rodon J, Prat A,
Perez-Garcia J, Adamo B, Felip E, Cortes J, Iafrate AJ, Nuciforo P
and Tabernero J: Genomic aberrations in the FGFR pathway:
Opportunities for targeted therapies in solid tumors. Ann Oncol.
25:552–563. 2014. View Article : Google Scholar
|
|
141
|
Forbes SA, Beare D, Boutselakis H, Bamford
S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al:
COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids
Res. 45:D777–D783. 2017. View Article : Google Scholar
|
|
142
|
Hatlen MA, Schmidt-Kittler O, Sherwin CA,
Rozsahegyi E, Rubin N, Sheets MP, Kim JL, Miduturu C, Bifulco N,
Brooijmans N, et al: Acquired on-target clinical resistance
validates FGFR4 as a driver of hepatocellular carcinoma. Cancer
Discov. 9:1686–1695. 2019.
|
|
143
|
Kim RD, Sarker D, Meyer T, Yau T,
Macarulla T, Park JW, Choo SP, Hollebecque A, Sung MW, Lim HY, et
al: First-in-human phase I study of fisogatinib (BLU-554) validates
aberrant FGF19 signaling as a driver event in hepatocellular
carcinoma. Cancer Discov. 9:1696–1707. 2019. View Article : Google Scholar
|
|
144
|
Li W, Sparidans R, El-Lari M, Wang Y,
Lebre MC, Beijnen JH and Schinkel AH: P-glycoprotein (ABCB1/MDR1)
limits brain accumulation and Cytochrome P450-3A (CYP3A) restricts
oral availability of the novel FGFR4 inhibitor fisogatinib
(BLU-554). Int J Pharm. 573:1188422020. View Article : Google Scholar
|
|
145
|
Sootome H, Fujita H, Ito K, Ochiiwa H,
Fujioka Y, Ito K, Miura A, Sagara T, Ito S, Ohsawa H, et al:
Futibatinib is a novel irreversible FGFR 1-4 inhibitor that shows
selective antitumor activity against FGFR-deregulated tumors.
Cancer Res. 80:4986–4997. 2020. View Article : Google Scholar
|
|
146
|
Singh D, Chan JM, Zoppoli P, Niola F,
Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S,
et al: Transforming fusions of FGFR and TACC genes in human
glioblastoma. Science. 337:1231–1235. 2012. View Article : Google Scholar
|
|
147
|
Andre F, Ranson M, Dean E, Varga A, van
der Noll R, Stockman PK, Ghiorghiu D, Kilgour E, Smith PD,
Macpherson M, et al: Abstract LB-145: Results of a phase I study of
AZD4547, an inhibitor of fibroblast growth factor receptor (FGFR),
in patients with advanced solid tumors. Cancer Res. 73:LB–145.
2013.
|
|
148
|
Takahashi Y, Akahane T, Sawada T, Ikeda H,
Tempaku A, Yamauchi S, Nishihara H, Tanaka S, Nitta K, Ide W, et
al: Adult classical glioblastoma with a BRAF V600E mutation. World
J Surg Oncol. 13:1002015. View Article : Google Scholar
|
|
149
|
Tosuner Z, Geçer MÖ, Hatiboğlu MA,
Abdallah A and Turna S: BRAF V600E mutation and BRAF VE1
immunoexpression profiles in different types of glioblastoma. Oncol
Lett. 16:2402–2408. 2018.
|
|
150
|
Raabe EH, Lim KS, Kim JM, Meeker A, Mao
XG, Nikkhah G, Maciaczyk J, Kahlert U, Jain D, Bar E, et al: BRAF
activation induces transformation and then senescence in human
neural stem cells: A pilocytic astrocytoma model. Clin Cancer Res.
17:3590–3599. 2011. View Article : Google Scholar
|
|
151
|
Behling F and Schittenhelm J: Oncogenic
BRAF alterations and their role in brain tumors. Cancers (Basel).
11:7942019. View Article : Google Scholar
|
|
152
|
Cantwell-Dorris ER, O'Leary JJ and Sheils
OM: BRAFV600E: Implications for carcinogenesis and molecular
therapy. Mol Cancer Ther. 10:385–394. 2011. View Article : Google Scholar
|
|
153
|
Nakajima N, Nobusawa S, Nakata S, Nakada
M, Yamazaki T, Matsumura N, Harada K, Matsuda H, Funata N, Nagai S,
et al: BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous
deletions are frequent in epithelioid glioblastomas: A histological
and molecular analysis focusing on intratumoral heterogeneity.
Brain Pathol. 28:663–673. 2018. View Article : Google Scholar
|
|
154
|
Chapman PB, Robert C, Larkin J, Haanen JB,
Ribas A, Hogg D, Hamid O, Ascierto PA, Testori A, Lorigan PC, et
al: Vemurafenib in patients with BRAFV600 mutation-positive
metastatic melanoma: Final overall survival results of the
randomized BRIM-3 study. Ann Oncol. 28:2581–2587. 2017. View Article : Google Scholar
|
|
155
|
Burger MC, Ronellenfitsch MW, Lorenz NI,
Wagner M, Voss M, Capper D, Tzaridis T, Herrlinger U, Steinbach JP,
Stoffels G, et al: Dabrafenib in patients with recurrent, BRAF
V600E mutated malignant glioma and leptomeningeal disease. Oncol
Rep. 38:3291–3296. 2017.
|
|
156
|
Woo PYM, Lam TC, Pu JKS, Li LF, Leung RCY,
Ho JMK, Zhung JTF, Wong B, Chan TSK, Loong HHF and Ng HK:
Regression of BRAFV600E mutant adult glioblastoma after primary
combined BRAF-MEK inhibitor targeted therapy: A report of two
cases. Oncotarget. 10:3818–3826. 2019. View Article : Google Scholar
|
|
157
|
Schiff D and Sarkaria J: Dasatinib in
recurrent glioblastoma: Failure as a teacher. Neuro Oncol.
17:910–911. 2015. View Article : Google Scholar
|
|
158
|
Dumont RA, Hildebrandt I, Su H, Haubner R,
Reischl G, Czernin JG, Mischel PS and Weber WA: Noninvasive imaging
of alphaVbeta3 function as a predictor of the antimigratory and
anti-proliferative effects of dasatinib. Cancer Res. 69:3173–3179.
2009. View Article : Google Scholar
|
|
159
|
Galanis E, Anderson SK, Twohy EL, Carrero
XW, Dixon JG, Tran DD, Jeyapalan SA, Anderson DM, Kaufmann TJ,
Feathers RW, et al: A phase 1 and randomized, placebo-controlled
phase 2 trial of bevacizumab plus dasatinib in patients with
recurrent glioblastoma: Alliance/North Central Cancer Treatment
Group N0872. Cancer. 125:3790–3800. 2019. View Article : Google Scholar
|
|
160
|
Srivastava S, Jackson C, Kim T, Choi J and
Lim M: A characterization of dendritic cells and their role in
immunotherapy in glioblastoma: From preclinical studies to clinical
trials. Cancers. 11:5372019. View Article : Google Scholar
|
|
161
|
Wen PY, Reardon DA, Armstrong TS,
Phuphanich S, Aiken RD, Landolfi JC, Curry WT, Zhu JJ, Glantz M,
Peereboom DM, et al: A randomized double-blind placebo-controlled
phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed
patients with glioblastoma. Clin Cancer Res. 25:5799–5807. 2019.
View Article : Google Scholar
|
|
162
|
Polson ES, Kuchler VB, Abbosh C, Ross EM,
Mathew RK, Beard HA, da Silva B, Holding AN, Ballereau S,
Chuntharpursat-Bon E, et al: KHS101 disrupts energy metabolism in
human glioblastoma cells and reduces tumor growth in mice. Sci
Transl Med. 10:eaar27182018. View Article : Google Scholar
|
|
163
|
Gruslova A, Cavazos DA, Miller JR,
Breitbart E, Cohen YC, Bangio L, Yakov N, Soundararajan A, Floyd JR
and Brenner AJ: VB-111: A novel anti-vascular therapeutic for
glioblastoma multiforme. J Neurooncol. 124:365–372. 2015.
View Article : Google Scholar
|
|
164
|
Brenner AJ, Peters KB, Vredenburgh J,
Bokstein F, Blumenthal DT, Yust-Katz S, Peretz I, Oberman B,
Freedman LS, Ellingson BM, et al: Safety and efficacy of VB-111, an
anticancer gene therapy, in patients with recurrent glioblastoma:
Results of a phase I/II study. Neuro Oncol. 22:694–704. 2020.
View Article : Google Scholar
|
|
165
|
Weller M, Butowski N, Tran DD, Recht LD,
Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al:
Rindopepimut with temozolomide for patients with newly diagnosed,
EGFRvIII-expressing glioblastoma (ACT IV): A randomized,
double-blind, international phase 3 trial. Lancet Oncol.
18:1373–1385. 2017. View Article : Google Scholar
|
|
166
|
Reardon DA, Desjardins A, Vredenburgh JJ,
O'Rourke DM, Tran DD, Fink KL, Nabors LB, Li G, Bota DA, Lukas RV,
et al: Rindopepimut with bevacizumab for patients with relapsed
EGFRvIII-expressing glioblastoma (ReACT): Results of a double-blind
randomized phase II trial. Clin Cancer Res. 26:1586–1594. 2020.
View Article : Google Scholar
|
|
167
|
Heimberger AB, Archer GE, Crotty LE,
McLendon RE, Friedman AH, Friedman HS, Bigner DD and Sampson JH:
Dendritic cells pulsed with a tumor-specific peptide induce
long-lasting immunity and are effective against murine
intrace-rebral melanoma. Neurosurgery. 50:158–166. 2002.
|
|
168
|
Filley AC, Henriquez M and Dey M:
Recurrent glioma clinical trial, CheckMate-143: The game is not
over yet. Oncotarget. 8:91779–91794. 2017. View Article : Google Scholar
|
|
169
|
Ferris RL, Blumenschein G Jr, Fayette J,
Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE,
Even C, et al: Nivolumab for recurrent squamous-cell carcinoma of
the head and neck. N Engl J Med. 375:1856–1867. 2016. View Article : Google Scholar
|
|
170
|
Reardon DA, Brandes AA, Omuro A,
Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bähr
O, et al: Effect of nivolumab vs bevaci-zumab in patients with
recurrent glioblastoma: The CheckMate 143 phase 3 randomized
clinical trial. JAMA Oncol. 6:1003–1010. 2020. View Article : Google Scholar
|
|
171
|
Sampson JH, Padula Omuro AM, Preusser M,
Lim M, Butowski NA, Cloughesy TF, Strauss LC, Latek RR, Paliwal P,
Weller M and Reardon DA: A randomized, phase 3, open-label study of
nivolumab versus temozolomide (TMZ) in combination with
radiotherapy (RT) in adult patients (pts) with newly diagnosed,
O-6-methylguanine DNA methyltransferase (MGMT)-unmethylated
glioblastoma (GBM): CheckMate-498. J Clin Oncol. 34:TPS20792016.
View Article : Google Scholar
|
|
172
|
Reardon DA, Nayak L, Peters KB, Clarke JL,
Jordan JT, De Groot JF, Nghiemphu PL, Kaley TJ, Colman H, Gaffey
SC, et al: Phase II study of pembrolizumab or pembrolizumab plus
bevacizumab for recurrent glioblastoma (rGBM) patients. J Clin
Oncol. 36:20062018. View Article : Google Scholar
|
|
173
|
Schalper KA, Rodriguez-Ruiz ME, Diez-Valle
R, López-Janeiro A, Porciuncula A, Idoate MA, Inogés S, de Andrea
C, López-Diaz de Cerio A, Tejada S, et al: Neoadjuvant nivolumab
modifies the tumor immune microenvironment in resectable
glioblastoma. Nat Med. 25:470–476. 2019. View Article : Google Scholar
|
|
174
|
Cloughesy TF, Mochizuki AY, Orpilla JR,
Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA,
Sanders CM, et al: Neoadjuvant anti-PD-1 immunotherapy promotes a
survival benefit with intratumoral and systemic immune responses in
recurrent glioblastoma. Nat Med. 25:477–486. 2019. View Article : Google Scholar
|
|
175
|
Li R, Pourpak A and Morris SW: Inhibition
of the insulin-like growth factor-1 receptor (IGF1R) tyrosine
kinase as a novel cancer therapy approach. J Med Chem.
52:4981–5004. 2009. View Article : Google Scholar
|
|
176
|
Chakravarti A, Loeffler JS and Dyson NJ:
Insulin-like growth factor receptor I mediates resistance to
anti-epidermal growth factor receptor therapy in primary human
glioblastoma cells through continued activation of phosphoinositide
3-kinase signaling. Cancer Res. 62:200–207. 2002.
|
|
177
|
Zhou X, Shen F, Ma P, Hui H, Pei S, Chen
M, Wang Z, Zhou W and Jin B: GSK1838705A, an IGF-1R inhibitor,
inhibits glioma cell proliferation and suppresses tumor growth in
vivo. Mol Med Rep. 12:5641–5646. 2015. View Article : Google Scholar
|
|
178
|
Osher E and Macaulay VM: Therapeutic
targeting of the IGF axis. Cells. 8:8952019. View Article : Google Scholar
|
|
179
|
Janes PW, Vail ME, Gan HK and Scott AM:
Antibody targeting of eph receptors in cancer. Pharmaceuticals.
13:882020. View Article : Google Scholar
|
|
180
|
Anderton M, van der Meulen E, Blumenthal
MJ and Schäfer G: The role of the Eph receptor family in
tumorigenesis. Cancers (Basel). 13:2062021. View Article : Google Scholar
|
|
181
|
Binda E, Visioli A, Giani F, Lamorte G,
Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, DiMeco F, et
al: The EphA2 receptor drives self-renewal and tumorigenicity in
stem-like tumor-propagating cells from human glioblastomas. Cancer
Cell. 22:765–780. 2012. View Article : Google Scholar
|
|
182
|
Wykosky J, Gibo DM, Stanton C and Debinski
W: EphA2 as a novel molecular marker and target in glioblastoma
multiforme. Mol Cancer Res. 3:541–551. 2005. View Article : Google Scholar
|
|
183
|
Swords RT, Greenberg PL, Wei AH, Durrant
S, Advani AS, Hertzberg MS, Jonas BA, Lewis ID, Rivera G,
Gratzinger D, et al: KB004, a first in class monoclonal antibody
targeting the receptor tyrosine kinase EphA3, in patients with
advanced hematologic malignancies: Results from a phase 1 study.
Leuk Res. 50:123–131. 2016. View Article : Google Scholar
|
|
184
|
Wade M, Li YC and Wahl GM: MDM2, MDMX and
p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 13:83–96.
2013. View Article : Google Scholar
|
|
185
|
Avci NG, Ebrahimzadeh-Pustchi S, Akay YM,
Esquenazi Y, Tandon N, Zhu JJ and Akay M: NF-κB inhibitor with
Temozolomide results in significant apoptosis in glioblastoma via
the NF-κB(p65) and actin cytoskeleton regulatory pathways. Sci Rep.
10:133522020. View Article : Google Scholar
|
|
186
|
Beck S, Jin X, Sohn YW, Kim JK, Kim SH,
Yin J, Pian X, Kim SC, Nam DH, Choi YJ and Kim H: Telomerase
activity-independent function of TERT allows glioma cells to attain
cancer stem cell characteristics by inducing EGFR expression. Mol
Cells. 31:9–15. 2011. View Article : Google Scholar
|
|
187
|
Olympios N, Gilard V, Marguet F, Clatot F,
Di Fiore F and Fontanilles M: TERT promoter alterations in
glioblastoma: A systematic review. Cancers (Basel). 13:11472021.
View Article : Google Scholar
|
|
188
|
Metro G, Pierini T and La Starza R: TERT
Mutations in Glioma: ESMO Biomarker Factsheet. European Society for
Medical Oncology; Lugano: 2019, https://oncologypro.esmo.org/education-library/factsheets-on-biomarkers/tert-mutations-in-glioma.
Accessed January 25, 2019.
|