Introduction
Triple-negative breast cancer (TNBC) is one of the most aggressive molecular subtypes of breast cancer, accounting for 15-20% of all cases worldwide (1). High heterogeneity underlies the poor clinical prognosis of TNBC, which is characterised by early recurrence and a significantly shortened overall survival (~18 months) in patients with distant metastases (2). Another major feature of TNBC is the absence of oestrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 (HER2) expression (3). Cytotoxic agents have been shown to be effective treatment options, and the precision treatment of patients with TNBC relies on the molecular classification. This classification categorizes TNBC into the following subtypes: Basal-like, mesenchymal-like, immunomodulatory and luminal androgen receptor (LAR) subtypes, based on genomic data outlined by Lehman et al (3) and Jiang et al (4).
Notably, 10-35% of patients with TNBC show clinically positive expression of AR (5), whereas the LAR subtype accounts for ~10% (8.7% in The Cancer Genome Atlas cohort and 9% in the Molecular Taxonomy of Breast Cancer International Consortium cohort) of patients with TNBC based on genotypic classification (6,7). AR has been suggested to promote carcinogenesis of TNBC by interacting with circulating androgens or activating the cytoplasmic signalling pathway (1,8). Enzalutamide (Enz) is a second generation AR inhibitor, which has been approved by the US Food and Drug Administration for the treatment of metastatic prostate cancer (9,10). Pre-clinical experiments have shown that Enz is able to suppress the proliferation of AR-positive TNBC cell lines (11). However, two clinical studies demonstrated that anti-AR therapy is not significantly effective for AR-positive TNBC (12,13). Therefore, there is an urgent need to explore more effective treatment strategies for the management of patients with AR-positive TNBC.
Dysregulation of histone deacetylases (HDACs), which is known to cause abnormal cell proliferation, has been observed in numerous types of human cancer, including breast, colon and prostate cancer (14). HDAC inhibitors (HDACis) can mechanistically suppress malignant biological behaviours, by directly interacting with HDAC to prevent the deacetylation of histones, thereby relaxing chromatin and promoting the transcription of tumour suppressors and differentiation-induced genes (15). Additionally, HDACis can inhibit tumour growth by restraining angiogenesis, inhibiting oestrogen receptor α, targeting cancer stem cells and promoting the antitumour effect of T cells (16). Chidamide (Chid) is an innovative drug developed in China (Shenzhen Chipscreen Biosciences Co., Ltd.), which selectively inhibits the activity of class I (HDAC1, 2 and 3) and class II (HDAC10) HDACs (17). Chid has been approved by the China Food and Drug Administration for the treatment of relapsed or refractory peripheral T-cell lymphoma (18,19). Chid combined with exemestane has been shown to be well-tolerated in Chinese patients with advanced hormone receptor-positive breast cancer, highlighting the efficacy and safety of this treatment (20).
Several studies have shown that HDACis enhance antitumour immunity towards TNBC, and inhibit tumour growth and metastasis in prostate cancer (21). Therefore, it was hypothesised that patients with AR-positive TNBC may respond to combined treatment with Enz and Chid. The present study first observed the synergistic effect of the AR inhibitor Enz and the HDACi Chid in a TNBC cell line, and then explored its intrinsic mechanism. it is hypothesized that the combination of Enz and Chid may achieve an effective antitumour effect. This treatment strategy may have the potential for implementation as a novel therapeutic regimen for patients with AR-positive TNBC.
Materials and methods
Cell culture and reagents
Human breast cancer cell lines MDA-MB-231 and CAL-51, and the human prostate cancer cell line LNcap were donated from the Public Laboratory of Tianjin Medical University Cancer Institute and Hospital (Tianjin, China). The human breast cancer cell line MDA-MB-453 was purchased from the BeNa Culture Collection; Beijing Beina Chunglian Institute of Biotechnology. All of the cell lines were identified and characterised by mycoplasma assays and DNA profiling (short tandem repeat) Cells were incubated at 37°C in an incubator containing 95% air and 5% CO2. CAL-51 and LNcap cell lines were maintained in RPMI 1640 medium (cat. no. C11875500BT; Gibco; Thermo Fisher Scientific, Inc.); MDA-MB-231 cells were maintained in Dulbecco's modified Eagle medium (cat. no. C11995500BT; Gibco; Thermo Fisher Scientific, Inc.); and MDA-MB-453 cells were maintained in L15 medium (cat. no. SH30525.01; Hyclone; Cytiva). The aforementioned media were supplemented with 10% foetal bovine serum (FBS; cat. no. 10270-106; Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin streptomycin (cat. no. 15140122; Gibco; Thermo Fisher Scientific, Inc.). Enz was purchased from Beijing Solarbio Science & Technology Co., Ltd. (cat. no. 915087-33-1) and Chid was kindly provided by Chipscreen Biosciences Ltd. Enz and Chid were stored at a concentration of 10 mM in DMSO, the maximum concentration of DMSO was 2%. Various concentrations of AMG510 (KRAS inhibitor; BeiGene) were used to treat the CAL-51 cell line.
Proliferation assay
The MTS assay (cat. no. G3582; Promega Corporation) was used to assess the inhibitory effect of Enz and Chid, both individually and in combination. MDA-MB-231, CAL-51, MDA-MB-453 and LNcap cells in the logarithmic growth phase were collected and inoculated into 96-well plates (5×103 cells/well, six biological replicates) with 200 μl medium. Cells were pretreated with Enz (3 μM for LNcap, 10 μM for MDA-MB-453, 20 μM for CAL-51 and 20 μM for MDA-MB-231 cells) or Chid (0.125 μM for LNcap, 1 μM for MDA-MB-453, 2 μM for CAL-51 and 10 μM for MDA-MB-231 cells) for 48 h at 37°C. Subsequently, 20 μl MTS reagent was added to each well and cultured for 2 h. Absorbance was measured at 490 nm using a microplate reader (Bio-Rad Laboratories, Inc.). The half-maximal inhibitory concentration (IC50) was calculated based on cell viability rate. Statistical analyses were performed using GraphPad Prism 8.0 (Dotmatics).
Combination treatment synergy quantification
LNcap, MDA-MB-231, MDA-MB-453 and CAL-51 cells were treated with Enz and Chid for 48 h at the corresponding concentrations based on IC50. The combination index (CI) was calculated using CompuSyn software (version 1.0; ComboSyn, Inc.) to evaluate the combined effect on cells. CI >1.0 indicates an antagonistic effect, CI <1.0 indicates a synergistic effect, and CI=1.0 indicates an additive effect.
Colony formation assay
Cell proliferation was analysed using a colony formation assay. A total of 800-1,000 MDA-MB-231 and CAL-51 cells were inoculated in 6-well plates were cultured overnight, then treated with Enz (20 μM for CAL-51 and 20 μM for MDA-MB-231 cells) or Chid (2 μM for CAL-51 and 10 μM for MDA-MB-231 cells) alone or in combination for 48 h at 37°C. The plates were cultured for 10-14 days and the medium was replaced every 3 days. When the colonies became visible to the naked eye, the culture was terminated. The colonies were then fixed with 4% paraformaldehyde for 30 min and stained with 0.5% crystal violet for 30 min at room temperature. Subsequently, a light microscope (Olympus Corporation) was used for observation and imaging. Adobe Photoshop software (version 22.5.0; Adobe Systems, Inc.) was used to count the colonies.
Cell migration assay
Migration was assessed using a Transwell assay (pore size, 7 μm; Corning, Inc.). The MDA-MB-231 and CAL-51 cells were pretreated with Enz (20 μM for CAL-51 and 20 μM for MDA-MB-231 cells) or Chid (2 μM for CAL-51 and 10 μM for MDA-MB-231 cells) alone or in combination for 48 h at 37°C, and 1×105 pretreated cells (in 200 μl FBS-free medium) were subsequently inoculated in the upper chamber of the Transwell system, and 500 μl medium supplemented with 20% FBS was added to the lower chamber. The upper chamber was fixed and stained with a Three-Step Stain Set according to the manufacturer's instruction at room temperature (Thermo Fisher Scientific, Inc.) once a single adherent cell was detected. The membrane was then cut, placed on a slide and sealed with paraffin. Images of the migrated cells were captured under an optical microscope at ×200 magnification and the number of cells was counted using Adobe Photoshop.
Cell cycle analysis
Cell cycle profiles were analysed using flow cytometry. The MDA-MB-231, CAL-51 and MDA-MB-453 cells pretreated with Enz (10 μM for MDA-MB-453, 20 μM for CAL-51 and 20 μM for MDA-MB-231 cells) or Chid (1 μM for MDA-MB-453, 2 μM for CAL-51 and 10 μM for MDA-MB-231 cells) at 37°C, individually or in combination, were collected and fixed with 95% pre-cooled ethanol at -20°C overnight, followed by washing with PBS and staining with 3 μM propidium iodide and 10 μg/ml RNase in the dark at 37°C for 30 min. Cell cycle distribution was identified and quantified using a flow cytometer (FACSAria III; BD Biosciences). The cell cycle distribution was analysed using ModFit LT software (v3.3; BD Biosciences).
Western blotting
CAL-51 cells were lysed in RIPA buffer supplemented with 1% phenylmethylsulphonyl fluoride (cat. no. R0020; Beijing Solarbio Science & Technology Co., Ltd.) to extract total proteins. The lysates were centrifuged at 8,000 x g for 10 min at 4°C to collect the supernatants, and protein concentrations were measured using a Pierce™ BCA Protein Assay Kit (cat. no. 23227; Thermo Fisher Scientific, Inc.). The protein lysates were mixed with 5X SDS-PAGE sample loading buffer (cat. no. P0015; Beyotime Institute of Biotechnology) and boiled at 95°C for 5 min. Subsequently, 25 μg proteins were separated by SDS-PAGE on 8-12% gels and were transferred to PVDF membranes (MilliporeSigma). The membranes were blocked with 5% skim milk at room temperature for 2 h and were then incubated with the primary antibodies at 4°C overnight, according to the manufacturer's instructions. Subsequently, the membranes were incubated for 60 min at room temperature with a secondary antibody (HRP-anti-rabbit, cat. no. 15015; HRP-anti-mouse, cat. no. 15014; both 1:5,000; both from ProteinTech Group, Inc.). The protein bands were visualized using ECL luminescence reagent (cat. no. abs920; Absin) and the Western blotting Imaging System (Amersham Imager 600 RGB; Cytiva). The grey value of the target protein band was analysed using ImageJ software (V1.8.0; National Institutes of Health). The primary antibodies used for western blotting were: AR (1:1,000; cat. no. 5153S), cyclin E1 (1:1,000; cat. no. 4129) (both from Cell Signaling Technology, Inc.), p16INK4 (1:1,000; cat. no. ab189034; Abcam), cyclin D1 (1:1,000; cat. no. sc-450; Santa Cruz Biotechnology, Inc.), mTOR (1:1,000; cat. no. 2983S), phosphorylated (p)-mTORSer2448 (1:1,000; cat. no. 5536S), p70S6K (1:1,000; cat. no. 34475S), p-p70S6KThr421/Ser424 (1:1,000; cat. no. 9204S) (all from Cell Signaling Technology, Inc.), insulin receptor substrate 4 (IRS4; 1:500; cat. no. sc-373778; Santa Cruz Biotechnology, Inc.), GAPDH (1:5,000; cat. no. 6004-1-Ig) and β-actin (1:5,000; cat. no. 81115-1-RR) (both from ProteinTech Group, Inc.).
Xenograft in vivo model
All in vivo experiments were performed according to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (22). The animal protocol was approved by the Animal Ethics Committee of Tianjin Medical University Cancer Institute and Hospital (approval no. HSTF-AE-2023023). CAL-51 cells (4×106 cells/mouse) were collected and resuspended in 100 μl pre-cooled PBS supplemented with 5 μl Matrigel® matrix (cat. no. 354248; Corning, Inc.). Cells were injected into the mammary gland fat pad of female NSG mice (age, 4-5 weeks; weight, 18-20 g; n=24; Shanghai Biomodel Organism Science & Technology Development Co., Ltd./). Mice were maintained at a temperature of 18-22°C and humidity of 50-60% under a 12-h light/dark cycle. Each cage contained six mice, and standard rodent diet and water were freely available. Tumour volumes were measured with a Vernier calliper every 3 days and calculated using the following formula: Length x width2/2. Mice were randomly allocated to four groups (n=6 mice/group) when the average tumour volume reached ~100 mm3. The four groups were treated with the control solvent once every 2 days (CMC-Na; cat. no. S6703; Selleck Chemicals), Enz, Chid, or their combination (Enz + Chid) at approximately the same time each day. Enz was dissolved in 0.5% CMC-Na for intragastric administration at 25 mg/kg body weight (BW) every 2 days. Chid was dissolved in 0.5% CMC-Na for intragastric administration and dosed at 12.5 mg/kg BW five times a week, as described in the drug instructions. Mice were sacrificed, and tumours were collected after 2 weeks of treatment. According to the Guidelines for Euthanasia of Rodents Using Carbon Dioxide (NIH 2020) (23), without pre-charging the chamber, the animals were placed in the chamber and 100% CO2 was introduced at a fill rate of 50% chamber volume per minute.
RNA-sequencing (RNA-seq)
Total RNA was extracted from CAL-51 cells treated with 20 μM Enz, 2 μM Chid or Enz + Chid for 48 h at 37°C using TRIzol® reagent (Thermo Fisher Scientific, Inc.). RNA was quantified using the RNA Nano 6000 Assay Kit on the Bioanalyzer 2100 system (Agilent Technologies, Inc.). Each sample (1 μg RNA) was used for cDNA library construction. The effective concentration of the library was accurately quantified by RT-PCR, and the loading concentration of the library was >1.5 nM. The library was prepared using the NEBNext® Ultra RNA Library Prep Kit for Illumina® (New England BioLabs) and sequenced on Illumina Novaseq 6000 platform (Illumina, Inc.) according to the manufacturer's instruction and 150 bp paired-end reads were generated.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to identify enriched signalling pathways from the differentially expressed genes. KEGG pathway enrichment analysis was performed using the R packages clusterProfiler (24), enrichplot, org.Hs.eg.db and ggplot2 (v4.1.2). All packages were downloaded from https://bioconductor.org/, with P<0.05 used as the screening criteria.
Gene Set Enrichment Analysis (GSEA) was used to explore significantly dysregulated pathways between the groups. Gene expression files and phenotype label files were generated and loaded into GSEA software (v4.1.0; Broad Institute, Cambridge, MA, USA) (25). The enrichment analyses were focused on the MsigDB HALLMARKER gene set. The permutation test was carried out 1,000 times, and the criteria were normalized enrichment score absolute value >1.5, nominal P<0.05.
Haematoxylin and eosin (H&E) and immunohistochemical (IHC) staining
Fresh tumour tissues obtained from mice were fixed with 10% neutral buffered formalin immediately for at least 12 h at room temperature. Fixed tissues were embedded in paraffin blocks and cut into 4-μm sections for H&E and IHC staining.
For H&E staining, tissue sections were stained with modified Lillie-Mayer Haematoxylin Solution (cat. no. G4070; Beijing Solarbio Science & Technology Co., Ltd.) at room temperature, and counterstained with Eosin Staining Solution (cat. no. C0109; Beyotime Institute of Biotechnology) according to the manufacturer's instructions.
For IHC staining, the slides were de-paraffinized in dimethylbenzene and hydrated in a series of ethanol solutions. Antigen retrieval was performed in a microwave oven with citric acid buffer (pH 6.0) for 15 min and cooled to room temperature. The endogenous peroxidase activity was quenched using 3% H2O2 at room temperature for 15 min. The tissues were blocked with 5% BSA in PBS 1 h at 37°C (cat. no. A8020; Beijing Solarbio Science & Technology Co., Ltd.) and incubated with primary antibodies overnight at 4°C. Subsequently, Biotinylated Goat anti-Mouse and anti-Rabbit secondary antibodies were obtained from OriGene Technologies, Inc. (cat. nos. TA373082L and TA373083L; 1:500) for 1 h at room temperature according to the manufacturer's instructions. Slides were visualised using a DAB horseradish peroxidase colour development kit (OriGene Technologies, Inc.). The primary antibodies used were as follows: AR (1:500; cat. no. 5153S), mTOR (1:500; cat. no. 2983S) (both from Cell Signaling Technology, Inc.), IRS4 (1:50; cat. no. sc-373778; Santa Cruz Biotechnology, Inc.) and Ki-67 (1:200,cat. no. ZM-0166; OriGene Technologies, Inc.). Finally, chromogenic detection was carried out (×200 magnification) under an optical microscope and the expression of various indicators was observed.
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA of CAL-51 cells was extracted using TRIzol reagent and 1,000 ng was used to generate cDNA using the PrimeScript RT Reagent Kit (cat. no. RR047A; Takara Bio, Inc.) according to the manufacturer's instructions. qPCR was performed using TB Green Premix Ex Taq (cat. no. RR420A; Takara Bio, Inc.) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.) in triplicate, according to the manufacturer's instructions. The thermocycling conditions were as follows: 95°C for 2 min, followed by 40 cycles at 95°C for 10 sec, 60°C for 30 sec and 72°C for 30 sec, and a final step at 72°C for 5 min. All primers were synthesised by Tsingke Biological Technology. The primer sequences are listed in Table I. The results were analysed using 2−ΔΔCq (26) and were normalised to β-actin expression levels.
 |
Table I.
Reverse transcription-quantitative PCR primers used in the present study.
|
Table I.
Reverse transcription-quantitative PCR primers used in the present study.
| Gene |
Forward sequence, 5′-3′ |
Reverse sequence, 5′-3′ |
| IRS4 |
GGCGCTACTTCGTGCTCAA |
GAACTTCCTGGCATTTTCGTAGT |
| CHST2 |
CCTCTCGGAATGAAGGTGTTC |
GTTGAGCATAGTGAGCACCAG |
| ADRA2C |
GCCTCAACGACGAGACCTG |
CCCAGCCCGTTTTCGGTAG |
| CAMK1D |
TGAGATAGCCGTCCTGAGAAA |
GCATGACCAAGTACAGGTGATT |
| TEX15 |
TATCAGACTGGTTGCCAAACG |
TCCAGGTCAGCGAAGTTTTTC |
| KLHDC8A |
TGGACTGCTTCGAGGTCTACT |
CTTTGGCCGTGACAGAAATGC |
| RYR2 |
CATCGAACACTCCTCTACGGA |
GGACACGCTAACTAAGATGAGGT |
| WNT16 |
AGTATGGCATGTGGTTCAGCA |
GCGGCAGTCTACTGACATCAA |
| GDNF |
GGCAGTGCTTCCTAGAAGAGA |
AAGACACAACCCCGGTTTTTG |
| CALCB |
ACCCGGCCACACTCAGTAA |
GGGCACGAAGTTGCTCTTCA |
| BRDT |
CTGTTGACGTTAATGCTTTGGG |
CACAACTTCGTGATCTGGAGG |
| β-actin |
CATGTACGTTGCTATCCAGGC |
CTCCTTAATGTCACGCACGAT |
Statistical analysis
Quantification and statistical analyses were performed using GraphPad Prism 8.0 (Dotmatics) or R software (v4.0.5). For in vitro analyses, the experiments were repeated three times. Data are presented as the mean ± SD. The statistical significance of differences between groups were analysed using one-way analysis of variance followed by Bonferroni's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Synergistic effect of Enz and Chid on AR-positive TNBC cell lines
To evaluate the inhibitory effect of the AR inhibitor Enz and the HDACi Chid, the expression levels of AR were detected in TNBC cell lines by western blotting and comparisons were made between the four cell lines. The prostate cancer cell line LNcap, which was used as a positive control, expressed the highest level of AR, whereas the expression levels of AR in MDA-MB-453, CAL-51 and MDA-MB-231 cells were gradually lower (Fig. 1A). Androgen deprivation therapy is a standard therapy regimen for patients with prostate cancer, and Enz, a second-generation AR antagonist, can substantially increase the survival of patients with metastatic or non-metastatic, castration-resistant prostate cancer (27,28). The AR-positive TNBC cell lines may also respond to Enz, as described previously (11). TNBC cell lines were treated with Enz in a series of concentration gradients to determine the IC50 of Enz for each cell line. The IC50 value of Enz was negatively associated with AR expression. The IC50 of Enz for MDA-MB-453 cells was the lowest (16.66 μM), followed by CAL-51 cells (24.26 μM), whereas MDA-MB-231 cells had the highest IC50 value (26.51 μM) (Fig. 1B and C). The present study then evaluated the IC50 of Chid; the results revealed that the IC50 of Chid was lowest in MDA-MB-453 cells (1.44 μM), followed by CAL-51 (7.75 μM) and MDA-MB-231 cells (18.31 μM) (Fig. 1B and D). In addition, the present study verified that Chid could effectively reduce the protein expression levels of AR (Fig. S1).
 |
Figure 1.
Synergistic effect of Enz and Chid on AR-positive TNBC cell lines. (A) Expression levels of AR protein in different cell lines. The prostate cancer cell line LNcap was used as a positive control. (B) IC50 values were measured using the MTS assay. (C) Effect of Enz on the inhibition of different cell lines. (D) Effect of Chid on the inhibition of different cell lines. Data are presented as the mean ± SEM of three independent experiments. (E) CI of AR-positive TNBC cell lines treated with Enz and Chid; CI <1 indicates a synergistic effect. AR, androgen receptor; Chid, chidamide; CI, combination index; Enz, enzalutamide; IC50, half maximal inhibitory concentration; TNBC, triple-negative breast cancer.
|
To assess the synergistic effect of Enz and Chid treatment on TNBC cell lines, the CI was calculated. The CI ranged between 0.1 and 0.9, suggesting that Enz and Chid had potential synergistic effects at certain concentrations in AR-positive TNBC cell lines, including MDA-MB-453, CAL-51 and MDA-MB-231 (Fig. 1E).
Combination of Chid and Enz inhibits the proliferation and migration of TNBC cells in vitro
AR-positive TNBC cells were treated with Enz monotherapy, Chid monotherapy or combination therapy at appropriate concentrations according to the IC50 values for the corresponding cells. Morphological changes resulted from treatment with Enz (10 μM for MDA-MB-453, 20 μM for CAL-51 and 20 μM for MDA-MB-231) and Chid (1 μM for MDA-MB-453, 2 μM for CAL-51 and 10 μM for MDA-MB-231), including cell shrinkage, cell size reduction, rounder cell morphology, nuclear chromatin condensation and fragmentation, cytoplasmic vacuolar changes and reduced synapses (Fig. 2A). The most marked changes in morphology were observed in response to the combination therapy, followed by Enz or Chid monotherapy in MDA-MB-231, CAL-51 and MDA-MB-453 cells. The colony formation assay showed that the combination therapy with Enz and Chid for 48 h significantly inhibited colony formation compared with that in the control and monotherapy groups (P<0.05; Fig. 2B). In addition, Enz + Chid inhibited cell migration more efficiently than either drug alone in AR-positive TNBC cell lines (P<0.05; Fig. 2C).
 |
Figure 2.
Chid combined with Enz inhibits proliferation and migration of triple-negative breast cancer cells in vitro. (A) Morphological changes of cells treated with Enz and/or Chid (magnification, ×100; scale bar, 200 μm). Combination treatment of cells with Enz and Chid for 48 h significantly inhibited (B) cell colony formation (magnification, ×40; scale bar, 1,000 μm) and (C) cell migration (magnification, ×200; scale bar, 100 μm). *P<0.05, **P<0.01, ****P<0.001. Chid, chidamide; Con, control; Enz, enzalutamide; ns, not significant.
|
Combination of Chid and Enz induces cell cycle arrest at G2/M phase
Tumour cell proliferation is partially caused by cell cycle dysregulation (29). Based on the aforementioned inhibitory effects of Enz and Chid combination therapy on proliferation, flow cytometry was used to evaluate the effect of Enz and Chid on the cell cycle. Cells were treated with Enz or Chid, alone or in combination at a certain concentration, for 48 h. The results showed that Enz or Chid monotherapy slightly increased the proportion of cells in G2/M phase, whereas the elevation was more pronounced in the combination group compared with that in the control and monotherapy groups (Fig. 3A). This resulted in a significant decrease in the proportion of cells in the G1/S phase.
 |
Figure 3.
Combination of Chid and Enz induces cell cycle arrest at G2/M phase. (A) Cell cycle evaluation after 48 h of single-drug or combined treatment in MDA-MB-231, CAL-51 and MDA-MB-453 cells detected an increase in the number of cells in G2/M phase with combination treatment. (B) Western blotting detected the downregulation of cyclin D1 and cyclin E1, and the upregulation of the inhibitor of cyclin-dependent kinase 4/6, p16INK4a. Chid, chidamide; Con, control; Enz, enzalutamide.
|
Multiple molecules, including cyclin-cyclin-dependent kinase (CDK) complexes, have been identified as regulators of cell proliferation (30). The cell cycle is precisely regulated by multiple CDKs at the corresponding cell cycle checkpoint (31). Western blotting was used to detect Enz- and Chid-induced changes in cell cycle progression. Compared with in the control and monotherapy groups, decreased cell proliferation was associated with downregulation of cyclin D1 and cyclin E1 upon the combination treatment of Enz and Chid, whereas the individual drugs had a limited effect on AR-positive TNBC cell lines (Fig. 3B). In addition, an inhibitor of CDK4/6, p16INK4a, was markedly upregulated in the combination treatment group (Fig. 3B) (32).
Combination of Chid and Enz promotes the expression of IRS4 and inhibits the PI3K/AKT/mTOR pathway by blocking KRAS signalling
The present study then explored the potential molecular mechanisms underlying the effects of the combination therapy. Transcriptome sequencing-based analysis was performed to examine the transcriptomes of CAL-51 cells treated with vehicle, Enz, Chid, or a combination of the two for 48 h. KEGG functional enrichment analysis showed that the 'PI3K-Akt' signalling pathway was significantly enriched (P=0.00053, count=66) (Fig. 4A). Western blot analysis confirmed the inhibition of the expression of key proteins in the PI3K/Akt signalling pathway in response to the combination treatment (Fig. 4B). GSEA revealed that several important tumour suppressor pathways were upregulated only in response to combined drug treatment (NES >1.7, NOM P<0.05, FDR q<0.05), whereas the change was not significant with Enz alone (NES <1.5, NOM P>0.05, FDR q>0.05) (Figs. 4C, D and S2).
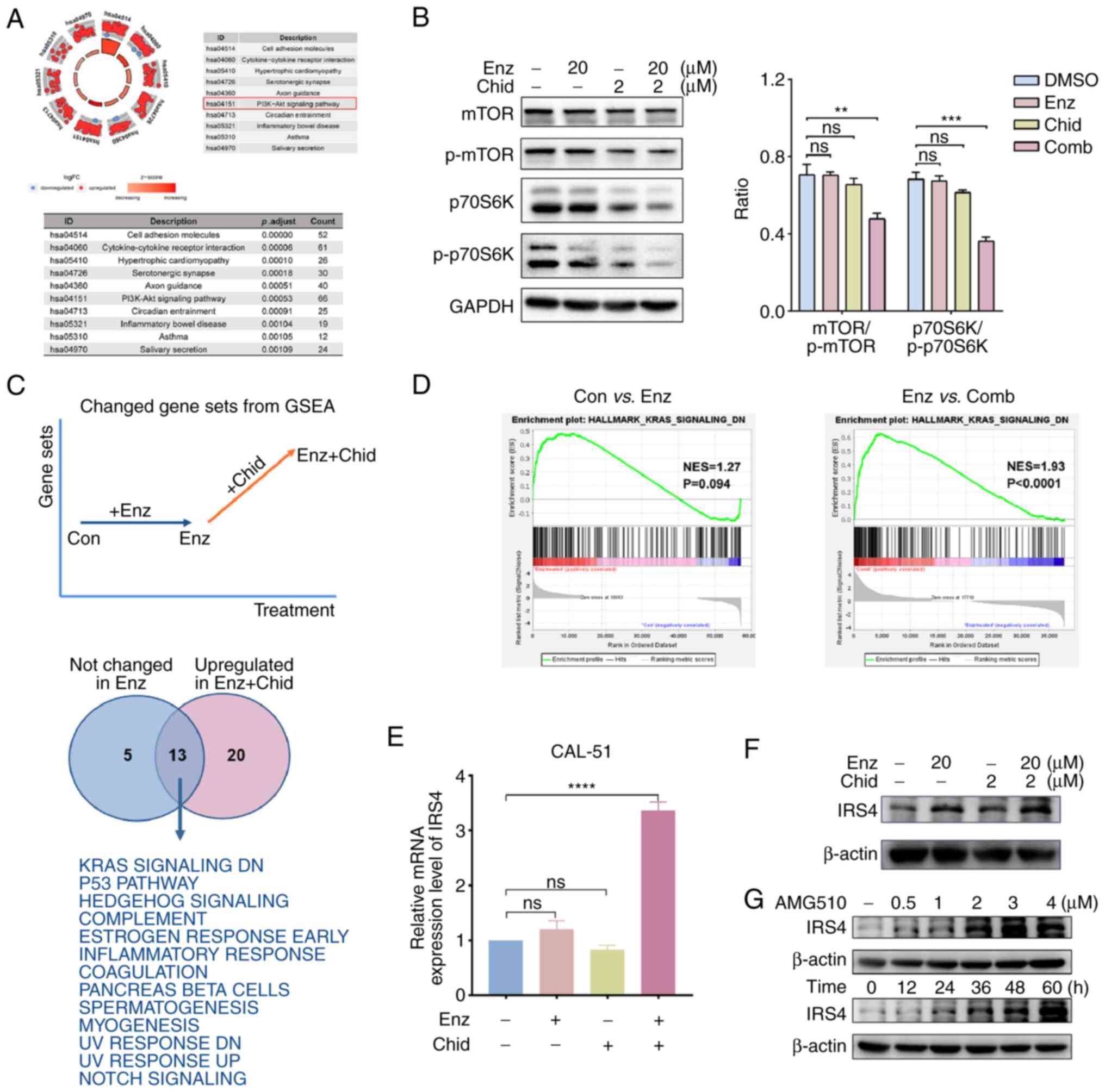 |
Figure 4.
Combination of Chid and Enz promotes expression of IRS4 to block KRAS signalling activation and to inhibit PI3K/AKT/mTOR. (A) Functional enrichment analysis of RNA-sequencing data revealed that the PI3K/AKT/mTOR signalling pathway was significantly enriched in CAL-51 cells treated with Enz and Chid. (B) Western blotting determined the inhibition of mTOR and p70S6k when cells were treated with Enz and Chid combination treatment. (C) GSEA revealed several tumour suppressor pathways were enriched only upon combination treatment of Enz and Chid, and (D) regulation of genes downstream of KRAS may have an important role. The upregulation of IRS4 was determined by (E) reverse transcription-quantitative PCR and (F) western blotting. (G) IRS4 was upregulated under the inhibition of KRAS with time and increasing concentration of KRAS inhibitor. **P<0.01, ***P<0.005 and ****P<0.001. Chid, chidamide; Con, control; Comb, combination; Enz, enzalutamide; GSEA, Gene Set Enrichment Analysis; IRS4, insulin receptor substrate 4; p-, phosphorylated.
|
RAS has been shown to efficiently activate the 'PI3K-Akt' signalling pathway (33,34). In CAL-51 cells, genes that were downregulated by KRAS activation were upregulated only upon combined treatment with Enz and Chid (NES=1.93, P<0.0001), whereas the change was not significant with Enz alone (NES=1.27, P=0.094; Fig. 4D). These genes may be involved in the regulation of the downstream PI3K/Akt/mTOR signalling pathway in response to combination treatment. A list of genes was screened, including CHST2, TEX15, CALCB, IRS4, ADRA2C, CAMK1D, KLHDC8A, RYR2, WNT16, GDNF and BRDT, to further elucidate the association between KRAS activation and the PI3K/Akt/mTOR signalling pathway. It was revealed that the expression levels of these genes were also upregulated in the combination group compared with the other groups (Table SI). Subsequent RT-qPCR verification showed that only IRS4 was significantly upregulated by the combination treatment of Enz and Chid compared with in the control and monotherapy groups (Fig. 4E). The protein expression levels of IRS4 were then detected in CAL-51 cells treated with Enz and/or Chid, and it was revealed that IRS4 was markedly upregulated following combined treatment with Enz and Chid compared with in the control and monotherapy groups (Fig. 4F). Marked overexpression of IRS1 has been shown to reduce the growth of lung cancer, and IRS1 deficiency can drive a proinflammatory phenotype in lung adenocarcinoma with KRAS mutations (35,36). In the present study, the KRAS inhibitor AMG510 was used to treat CAL-51 for 1, 6, 12, 24 and 48 h at 37°C. The results showed that IRS4 expression increased over time. In addition, IRS4 was upregulated as a result of increased AMG510 concentration for 48 h (Fig. 4G).
Combination of Chid and Enz increases the in vivo therapeutic effect in nude mice through inhibiting the PI3K/AKT/mTOR pathway
The present study investigated the antitumour efficacy of combination treatment with Enz and Chid in a xenograft mouse model of CAL-51. Mice were treated with Enz or Chid, alone or in combination, after 30 days of CAL-51 xenograft model construction. The frequency of administration in each group is shown in Fig. 5A. The treatment lasted for 14 days due to the heavy tumour burden of the control group (the maximum tumour volume detected was 500 mm3). The tumour growth curves for each group are presented in Fig. 5B. It was observed that the combination of Enz and Chid significantly slowed tumour growth compared with that in the control and monotherapy groups, displaying excellent antitumour efficacy higher than that of either monotherapy (Fig. 5C). Notably, no obvious adverse drug reactions, such as vomiting and diarrhoea, were observed, indicating that the combination regimen of Enz and Chid was tolerable to these mice.
 |
Figure 5.
Chid enhances the anti-AR therapeutic effect in nude mice. (A) Construction of a subcutaneous xenograft model and drug administration regimen. (B) Tumour growth curve and (C) representative images of subcutaneous tumours with the indicated treatment. (D) Representative immunohistochemical staining of AR, Ki-67, mTOR and IRS4 protein expression in a subcutaneous xenograft model (magnification, ×200). ***P<0.05 and ****P<0.01. AR, androgen receptor; BW, body weight; Chid, chidamide; Con, control; Enz, enzalutamide; H&E, haematoxylin and eosin; IRS4, insulin receptor substrate 4.
|
As confirmed by histological analysis, the decreased proliferation and increased area of necrosis may account for the observed response to the combination therapy (Fig. 5D). IHC staining showed decreased expression of Ki-67 and mTOR in the combination group compared with that in the control and monotherapy groups (Fig. 5D), which is consistent with the results of the in vitro experiment. These findings indicated that Chid, in combination with Enz, may sufficiently block the activation of the PI3K/Akt/mTOR signalling pathway to inhibit tumour growth and upregulate the expression of IRS4 (Fig. 6).
 |
Figure 6.
HDAC inhibitors target IRS4 to enhance anti-AR therapy in AR-positive triple-negative breast cancer. Proposed working model. The activation of KRAS inhibits the expression of IRS4. The low expression of IRS4 can promote the activation of PI3K/AKT/mTOR signalling to promote tumour proliferation. However, the combination of chidamide and enzalutamide can increase the expression of IRS4, resulting in blockade of the transmission of KRAS signals, and thus inhibit the PI3K/AKT/mTOR signalling pathway. Finally, tumour proliferation can be inhibited through cell cycle arrest. IRS4, insulin receptor substrate 4.
|
Discussion
Both prostate and breast cancer rely on steroid hormones to drive growth, and anti-androgen therapy has been approved for prostate cancer. Approximately one-third of patients with TNBC also express AR and high AR levels predict poor patient outcomes (3,37). When binding with circulating androgens, the receptor-hormone complex translocates into the nucleus to promote transcription of target genes (8). AR can also be activated in an ERK-dependent or independent manner. Cytoplasmic AR interacts with PI3K, RAS GTPase and Src proteins, and serves a role in the phosphorylation of mTOR, FOXO1 and PKA, which results in increased cell proliferation (1). The AR-related signalling pathways are considered to be potential therapeutic targets for TNBC, and potential treatment strategies targeting AR are being actively investigated. For patients with LAR TNBC, AR-target therapy should be considered due to the oestrogen receptor-regulated gene transcription in these patients (3). Several clinical trials have demonstrated that a number of patients with TNBC can achieve clinical benefit from anti-AR therapy (38,39). To further improve the outcome of patients with AR-positive TNBC, clinical trials combining anti-AR drugs and other agents, including palbociclib, pembrolizumab and paclitaxel, are being conducted (40,41).
The combination of Enz and Chid, an HDACi, has the potential to improve the efficacy of anti-AR therapy in AR-positive patients. Chid can selectively inhibit the activity of class I (HDAC1, 2, 3) and class II (HDAC10) HDACs, and has shown promising results in multiple tumour types (17). Enz itself is an antagonist of AR, which can effectively inhibit the protein expression of AR. Notably, the present study also verified that Chid could effectively reduce the protein expression levels of AR. HDACs positively modulate AR transactivation and expression, highlighting the rationale of using HDACis for AR suppression. HDACis disrupt the AR axis at multiple levels, as follows: i) HDACis may induce AR protein degradation, leading to AR ubiquitination and subsequent proteasome degradation; ii) HDACis may lower AR protein levels by decreasing the production of AR mRNA; iii) HDACis may suppress the transcription of AR-regulated genes (42).
A combination of Chid and exemestane has been demonstrated to improve progression-free survival for postmenopausal patients with advanced, hormone receptor-positive, HER2-negative breast cancer (20). Furthermore, Chid has been shown to increase the expression of PD-L1 through the transcription factor STAT1 to increase therapeutic response in soft tissue sarcoma. In addition, Chid combined with anti-PD-1 can improve survival in patients with advanced and metastatic sarcoma (43). In patients with nasopharyngeal carcinoma (NPC), DNTTIP1 has been reported to decrease DUSP2 expression by recruiting HDAC1 to its promoter, resulting in activation of the ERK signalling pathway. Targeting DNTTIP1/HDAC1 with Chid has been shown to benefit patients with metastasized NPC (44).
In our preclinical model, it was revealed that the combination of Enz and Chid produced a synergistic effect on AR-positive TNBC cell lines and a xenograft mouse model, suppressing proliferation and tumour growth via cell cycle arrest. Crucially, high AR levels are associated with low IC50 values. Further exploration of the underlying mechanism showed that treatment blocked the KRAS signalling pathway, which resulted in inhibition of the PI3K/AKT/mTOR signalling pathway. Based on GSEA and western blotting validation, the combination of Enz and Chid may inhibit tumour growth by increasing the expression of IRS4, which blocks the KRAS signalling pathway. IRS4 is an IRS that participates in the signal transduction of insulin and other cytokines; at present, there are more studies on IRS1 and IRS2 (36,45,46), but few on IRS4. Different IRS proteins seem to have different functions at the cellular level (43). IRS1 expression is reduced in more poorly differentiated, higher grade breast cancer tumours (46). In a lung cancer study, neutrophil elastase was shown to mediate IRS1 degradation, thereby tilting the PI3K axis toward tumour cell proliferation (36). In ductal carcinoma in situ, high IRS1 with low IRS2 expression could predict the responsiveness and effectiveness of chemotherapy in breast cancer (47). In addition, in a KRAS mutant lung adenocarcinoma subgroup, reduced IRS1 expression has been reported to result in an obvious survival disadvantage. IRS1 activates JAK/STAT signalling through the IL-22 receptor, resulting in enhanced tumour-promoting inflammation (35). The present study suggested that KRAS signalling was blocked by the upregulation of IRS4 expression in response to combination treatment with Enz and Chid, which suppressed the growth of AR-positive TNBC. The expression of IRS4 was also significantly upregulated following treatment with the KRAS inhibitor AMG510 in a concentration- and time-dependent manner.
The mammalian cell cycle is regulated by several activators (cyclins) and inhibitors (Ink4 and Cip), and the dysregulation of the cell cycle is a common characteristic of cancer (48). The present study showed that the combination of Enz and Chid induced cell cycle arrest at the G2/M phase. This effect was accompanied by the downregulation of cyclin D1 and cyclin E1, as well as the upregulation of p16INK4a. Cyclin D1 and cyclin E1 mediate the transition from G2/M to G1/S phase, whereas p16INK4a inhibits the CDK (CDK4 for example) associated with cyclin D1 (49). Overall, the present findings indicated that combination treatment suppresses cell proliferation by arresting the cell cycle at the G2/M phase.
Elevated expression of HDACs, particularly class I HDACs (HDAC1, 2, and 3), is frequently associated with a poor prognosis in prostate cancer, the inhibition of HDAC1 and HDAC3 can lead to changes in the expression of genes related to AR regulation (50). HDAC3 has been reported to be associated with the development of various types of cancer (51), and KDELR2 has been shown to promote breast cancer proliferation through HDAC3-mediated cell cycle progression (52). Therefore, HDAC3 may be a promising candidate target for combination therapy.
In the present study, the expression of IRS4 protein was observed in the TNBC subtype; therefore, our future studies will focus on AR-positive TNBC breast cancer, investigating the biological function of IRS4. Notably, the combination of Enz and Chid exhibited positive effects in both in vitro and in vivo experiments. In a follow-up study, we plan to conduct an early clinical trial to evaluate patient responses to this treatment. In the stage of designing the clinical trial, we will reasonably consider patient screening. The biggest challenge may arise from potential adverse reactions when these drugs are used in combination. We will determine the optimal combination dosage based on established dosing regimens and insights gained from Phase I trials. This research may yield promising outcomes and provide patients with an effective option.
In conclusion, the present study demonstrated the synergistic effect of Enz and Chid on the treatment of AR-positive TNBC, as the combination was more effective than each drug alone. Preliminary exploration of the underlying mechanism revealed that combination treatment with Enz and Chid may effectively block the KRAS signalling pathway by targeting IRS4, subsequently inhibiting the PI3K/Akt/mTOR pathway. This approach holds promise for the treatment of AR-positive TNBC. Moreover, IRS4 expression may serve as a valuable biomarker for patient selection for the combination of Chid and Enz.
Supplementary Data
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the Figshare repository, figshare.com/ (accession no. 10.6084/m9.figshare.24525037, ttps://figshare.com/search?q=10.6084%2Fm9.figshare.24525037).
Authors' contributions
YH, WZ and ZT were involved in the conception of the study. YH, YM, YZ, JZ, SZ, DZ, DX and YL conducted the experiments or analysis. YH and YM were involved in drafting the manuscript. WZ and ZT revised the manuscript for important intellectual content. WZ and ZT supervised the study. YH and YM confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study design was approved by the Administrative Ethics Committee of Tianjin Medical University Cancer Institute and Hospital (approval no. HSTF-AE-2023023; Tianjin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Abbreviations:
|
AR
|
androgen receptor
|
|
Chid
|
chidamide
|
|
Enz
|
enzalutamide
|
|
GSEA
|
Gene Set Enrichment Analysis
|
|
HDAC
|
histone deacetylase
|
|
HDACi
|
HDAC inhibitor
|
|
HER2
|
human epidermal growth factor receptor 2
|
|
IRS4
|
insulin receptor substrate 4
|
|
TNBC
|
triple-negative breast cancer
|
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 81472183), the Natural Science Foundation of Tianjin (grant no. 18JCYBJC91600), the Tianjin Key Medical Discipline (Specialty) Construction Project, and the Tianjin Medical University Cancer Hospital '14th Five-Year' Peak Discipline Support Program Project.
References
|
1
|
Gerratana L, Basile D, Buono G, De Placido S, Giuliano M, Minichillo S, Coinu A, Martorana F, De Santo I, Del Mastro L, et al: Androgen receptor in triple negative breast cancer: A potential target for the targetless subtype. Cancer Treat Rev. 68:102–110. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vagia E, Mahalingam D and Cristofanilli M: The landscape of targeted therapies in TNBC. Cancers (Basel). 12:9162020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 121:2750–2767. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, Xiao Y, Yu KD, Liu YR, Yu Y, et al: Genomic and transcriptomic landscape of triple-negative breast cancers: Subtypes and treatment strategies. Cancer Cell. 35:428–440.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kono M, Fujii T, Lim B, Karuturi MS, Tripathy D and Ueno NT: Androgen receptor function and androgen receptor-targeted therapies in breast cancer: A review. JAMA Oncol. 3:1266–1273. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, et al: The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 7:114792016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Network: Comprehensive molecular portraits of human breast tumours. Nature. 490:61–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Narayanan R and Dalton JT: Androgen receptor: A complex therapeutic target for breast cancer. Cancers (Basel). 8:1082016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, et al: Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 371:424–433. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al: Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 367:1187–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caiazza F, Murray A, Madden SF, Synnott NC, Ryan EJ, O'Donovan N, Crown J and Duffy MJ: Preclinical evaluation of the AR inhibitor enzalutamide in triple-negative breast cancer cells. Endocr Relat Cancer. 23:323–334. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Traina TA, Miller K, Yardley DA, Eakle J, Schwartzberg LS, O'Shaughnessy J, Gradishar W, Schmid P, Winer E, Kelly C, et al: Enzalutamide for the treatment of androgen receptor-expressing triple-negative breast cancer. J Clin Oncol. 36:884–890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang YZ, Liu Y, Xiao Y, Hu X, Jiang L, Zuo WJ, Ma D, Ding J, Zhu X, Zou J, et al: Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic triple-negative breast cancer: The FUTURE trial. Cell Res. 31:178–186. 2021. View Article : Google Scholar :
|
|
14
|
Soldi R, Cohen AL, Cheng L, Sun Y, Moos PJ and Bild AH: A genomic approach to predict synergistic combinations for breast cancer treatment. Pharmacogenomics J. 13:94–104. 2013. View Article : Google Scholar
|
|
15
|
Bates SE: Epigenetic therapies for cancer. N Engl J Med. 383:650–663. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fedele P, Orlando L and Cinieri S: Targeting triple negative breast cancer with histone deacetylase inhibitors. Expert Opin Investig Drugs. 26:1199–1206. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, Zhang J, Dong M, Du X and Lu XP: Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol. 69:901–909. 2012. View Article : Google Scholar
|
|
18
|
Shi Y, Dong M, Hong X, Zhang W, Feng J, Zhu J, Yu L, Ke X, Huang H, Shen Z, et al: Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol. 26:1766–1771. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi Y, Jia B, Xu W, Li W, Liu T, Liu P, Zhao W, Zhang H, Sun X, Yang H, et al: Chidamide in relapsed or refractory peripheral T cell lymphoma: A multicenter real-world study in China. J Hematol Oncol. 10:692017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang Z, Li W, Hu X, Zhang Q, Sun T, Cui S, Wang S, Ouyang Q, Yin Y, Geng C, et al: Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20:806–815. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tu K, Yu Y, Wang Y, Yang T, Hu Q, Qin X, Tu J, Yang C, Kong L and Zhang Z: Combination of chidamide-mediated epigenetic modulation with immunotherapy: Boosting tumor immunogenicity and response to PD-1/PD-L1 blockade. ACS Appl Mater Interfaces. 13:39003–39017. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
National Institutes of Health: Guide for the care and use of laboratory animals. 7th edition. National Academy Press; Washington DC: 1996
|
|
23
|
Guidelines for Euthanasia of Rodents Using Carbon Dioxide. 2020 Edition. https://oacu.oir.nih.gov/system/files/media/file/2021-06/b5_euthanasia_of_rodents_using_carbon_dioxide.pdf.
|
|
24
|
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al: clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2:1001412021.PubMed/NCBI
|
|
25
|
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
27
|
Davis ID, Martin AJ, Stockler MR, Begbie S, Chi KN, Chowdhury S, Coskinas X, Frydenberg M, Hague WE, Horvath LG, et al: Enzalutamide with standard first-line therapy in metastatic prostate cancer. N Engl J Med. 381:121–131. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sternberg CN, Fizazi K, Saad F, Shore ND, De Giorgi U, Penson DF, Ferreira U, Efstathiou E, Madziarska K, Kolinsky MP, et al: Enzalutamide and survival in nonmetastatic, castration-resistant prostate cancer. N Engl J Med. 382:2197–2206. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu YW, Xia R, Lu K, Xie M, Yang F, Sun M, De W, Wang C and Ji G: LincRNAFEZF1-AS1 represses p21 expression to promote gastric cancer proliferation through LSD1-Mediated H3K4me2 demethylation. Mol Cancer. 16:392017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma X, Wang L, Huang D, Li Y, Yang D, Li T, Li F, Sun L, Wei H, He K, et al: Polo-like kinase 1 coordinates biosynthesis during cell cycle progression by directly activating pentose phosphate pathway. Nat Commun. 8:15062017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pack LR, Daigh LH and Meyer T: Putting the brakes on the cell cycle: Mechanisms of cellular growth arrest. Curr Opin Cell Biol. 60:106–113. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sherr CJ, Beach D and Shapiro GI: Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 6:353–367. 2016. View Article : Google Scholar :
|
|
33
|
Webb CP, Van Aelst L, Wigler MH and Vande Woude GF: Signaling pathways in Ras-mediated tumorigenicity and metastasis. Proc Natl Acad Sci USA. 95:8773–8778. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marte BM, Rodriguez-Viciana P, Wennström S, Warne PH and Downward J: R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr Biol. 7:63–70. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Metz HE, Kargl J, Busch SE, Kim KH, Kurland BF, Abberbock SR, Randolph-Habecker J, Knoblaugh SE, Kolls JK, White MF and Houghton AM: Insulin receptor substrate-1 deficiency drives a proinflammatory phenotype in KRAS mutant lung adenocarcinoma. Proc Natl Acad Sci USA. 113:8795–8800. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR, et al: Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 16:219–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu R, Dawood S, Holmes MD, Collins LC, Schnitt SJ, Cole K, Marotti JD, Hankinson SE, Colditz GA and Tamimi RM: Androgen receptor expression and breast cancer survival in postmenopausal women. Clin Cancer Res. 17:1867–1874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, Blackwell K, Rugo H, Nabell L, Forero A, et al: Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin Cancer Res. 19:5505–5512. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Traina TA, Miller K, Yardley DA, O'Shaughnessy J, Cortes J, Awada A, Kelly CM, Trudeau ME, Schmid P, Gianni L, et al: Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). J Clin Oncol. 33(Suppl 15): S10032015. View Article : Google Scholar
|
|
40
|
Gucalp A, Proverbs-Singh TA, Corben A, Moynahan ME, Patil S, Boyle LA, Hudis CA and Traina TA: Phase I/II trial of palbociclib in combination with bicalutamide for the treatment of androgen receptor (AR)+ metastatic breast cancer (MBC). J Clin Oncol. 34(15 Suppl): TPS11032016. View Article : Google Scholar
|
|
41
|
Robles AJ, Cai S, Cichewicz RH and Mooberry SL: Selective activity of deguelin identifies therapeutic targets for androgen receptor-positive breast cancer. Breast Cancer Res Treat. 157:475–488. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hu WY, Xu L, Chen B, Ou S, Muzzarelli KM, Hu DP, Li Y, Yang Z, Vander Griend DJ, Prins GS and Qin Z: Targeting prostate cancer cells with enzalutamide-HDAC inhibitor hybrid drug 2-75. Prostate. 79:1166–1179. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Que Y, Zhang XL, Liu ZX, Zhao JJ, Pan QZ, Wen XZ, Xiao W, Xu BS, Hong DC, Guo TH, et al: Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J Immunother Cancer. 9:e0016962021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ding S, Gao Y, Lv D, Tao Y, Liu S, Chen C, Huang Z, Zheng S, Hu Y, Chow LKY, et al: DNTTIP1 promotes nasopharyngeal carcinoma metastasis via recruiting HDAC1 to DUSP2 promoter and activating ERK signaling pathway. EBioMedicine. 81:1041002022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saltiel AR and Kahn CR: Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 414:799–806. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mardilovich K, Pankratz SL and Shaw LM: Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun Signal. 7:142009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Porter HA, Perry A, Kingsley C, Tran NL and Keegan AD: IRS1 is highly expressed in localized breast tumors and regulates the sensitivity of breast cancer cells to chemotherapy, while IRS2 is highly expressed in invasive breast tumors. Cancer Lett. 338:239–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Malumbres M and Barbacid M: Cell cycle, CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, Sage J, Kõivomägi M and Skotheim JM: Cyclin D-Cdk4,6 drives cell-cycle progression via the retinoblastoma protein's C-terminal helix. Mol Cell. 74:758–770.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M and Kristiansen DG: Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 98:604–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ramaiah MJ, Tangutur AD and Manyam RR: Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 277:1195042021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wei H, Ma W, Lu X, Liu H, Lin K, Wang Y, Ye Z, Sun L, Huang Z, Pan T, et al: KDELR2 promotes breast cancer proliferation via HDAC3-mediated cell cycle progression. Cancer Commun (Lond). 41:904–920. 2021. View Article : Google Scholar : PubMed/NCBI
|














