Introduction
Endometrial cancer (EC) is the most commonly
occurring gynecological malignancy; the standard treatment includes
surgical interventions, followed by the administration of
anti-cancer therapeutics for high-risk patients (1,2).
Patient prognosis is good in the early stages; however, advanced or
recurrent disease is refractory (3). The use of a molecular targeting
drugs for the treatment of EC was approved for the first time last
year. Thus, the number of available molecular target therapeutics
for EC therapy is limited, in comparison with ovarian cancer
(3).
Regardless of the advances in cancer genome research
and the establishment of thorough databases such as The Cancer
Genome Atlas (TCGA), cancer treatment and therapy has yet to be
fully elucidated and overcome. Recently, it has been reported that
both genomic and epigenomic aberrations are critical in
carcinogenesis (4). The molecular
mechanisms of the cancer epigenome are mostly divided into DNA
methylation and histone modification. DNA methylation only
regulates a single gene by methylating a single base, whereas
histone modification regulates downstream genes in a multilayered
manner, through acetylation, methylation and the phosphorylation of
the histone proteins with histone modifying enzymes (5). DNA forms nucleosomes when wrapped
around histone proteins; histone modifications regulate
transcription by regulating the tightly packed (heterochromatin)
and relaxed lightly packed (euchromatin) forms of DNA (6).
Among the histone modifications, histone methylation
is carried out by histone methyltransferases and demethylases
(7). The histone methylation
sites are the lysine and arginine groups of amino acids and the
enzymes mediating histone methylation are lysine methyltransferases
and arginine methyltransferases. It has been demonstrated that
increased expression levels of histone methyltransferases are
involved in carcinogenesis and cancer progression (8,9).
For example, the lysine histone methyltransferase SUV39H2 has been
shown to be highly expressed in lung cancer and SUV39H2 knockdown
(KD) increases sensitivity to radiation and chemotherapy (10). In gynecological cancers, a histone
lysine methyltransferase, enhancer of zeste homolog 2 (EZH2) was
defined as a therapeutic target in EC (11), and Wolf-Hirschhorn syndrome
candidate gene-1, Su(var)3-9, enhancer of zeste, trithorax (SET)
domain-containing protein 8, and SET and myeloid, nervy, and DEAF-1
(MYND) domain containing 2 were highly expressed in ovarian cancer,
with their inhibition attenuating cell proliferation (12-15). Arginine methyltransferase is named
as a protein methyltransferase (PRMT) due to its ability to
methylate other proteins and not solely histones (16,17). As expected, PRMT and lysine
methyltransferase are therapeutic targets for the treatment of
various types of cancer, including breast, prostate, lung and blood
cancer (18). In a previous study
by the authors, it was reported that the expression of PRMT4 [also
known as coactivator-associated arginine methyltransferase 1
(CARM1)] was increased in EC, and treatment with a selective
inhibitor of CARM1 resulted in the apoptosis-induced suppression of
cell proliferation in EC cell lines (19).
PRMT6 is a member of the PRMT group and is highly
expressed in various types of cancer, including breast, prostate
and lung cancer (20-22) and is involved in cancer-related
mechanisms. The histone modification of PRMT6 has been reported to
asymmetrically dimethylate the histone H3R2 (H3R2me2a) and it has
been reported that H3K27 acetylation (H3K27ac) and H3K4 histone
trimethylation (H3K4me3) are affected through the methylation of
H3R2 (23). Jiang et al
(24) reported that the
expression of PRMT6 increased in EC, negatively correlated with
prognosis, and was associated with cell proliferation in EC cells.
However, the mechanism of PRMT6-mediated histone modification in EC
remains largely unknown.
Therefore, in the present study, the expression of
PRMT6 in EC was investigated, by using clinical specimens from the
University of Tokyo Hospital. A KD experiment was performed, in
order to examine the cell proliferation mechanism of PRMT6. In
addition, chromatin immunoprecipitation sequencing (ChIP-seq) was
performed, in order to investigate the genes regulated by PRMT6 via
histone modification.
Materials and methods
Clinical samples
EC tissue (n=55) and normal endometrial tissue
(n=20) were collected from patients who underwent surgery at the
University of Tokyo Hospital between 2010 and 2021. The list of
patients is presented in Table
SI and written informed consent was acquired from all patients.
The median age was 51 years (range, 31-80 years). The samples from
patients No. 1 to No. 52, N1 to N4, and N17 to N20 were used for
mRNA extraction, and the thin section samples were prepared from
patients No. 1 to No. 55 and N5 to N16. Clinical samples used in
additional experiments are highlighted in bold in Table SI. The present study was approved
by The Human Genome, Gene Analysis Research Ethics Committee of the
University of Tokyo (Approval no. G0683-22).
Cell lines and cell culture
A total of six EC cell lines (HEC1B, HEC50B, HEC265,
Ishikawa, HEC151A and HEC116) were used in the present study. All
cell lines were cultured in Eagle's minimal essential medium
(FUJIFILM Wako Pure Chemical Corporation) supplemented with 10%
heat-inactivated fetal bovine serum (Thermo Fisher Scientific,
Inc.) in a humidified incubator containing 5% CO2 at
37°C. All cell lines were not passaged >15 times.
Small interfering RNA (siRNA)
transfection
siRNA transfection was performed as previously
described (19). Briefly, siRNAs
were transfected at 37°C for 3.5 h using Lipofectamine RNA
interference (RNAi)MAX Transfection Reagent (Thermo Fisher
Scientific, Inc.), and the final concentration of siRNAs was 100
nM. PRMT6 siRNAs were as follows: siPRMT6#1 sense, 5′-CGG AAC AGG
UGG AUG CCA U-3′; siPRMT6#1 antisense, 5′-AUG GCA UCC ACC UGU UCC
G-3′; siPRMT6#2 sense, 5′-ACA GCA UAC CUA AGA AAC UCA GAA G-3′;
siPRMT6#2 antisense, 5′-CUU CUG AGU UUC UUA GGU AUG CUG U-3′
(MilliporeSigma). MISSION siRNA Universal Negative Control #1
(siNC) was used as a negative control (Merck KGaA). RNA extraction
for reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) was performed 72 h following siRNA transfection, and cell
viability assay, cell cycle analysis, apoptosis assay and protein
extraction for western blotting were performed 96 h following siRNA
transfection.
RNA extraction and RT-qPCR
RNA extraction and RT-qPCR were performed according
to a previously described procedure (19). Briefly, total RNA was extracted
from the cells using the RNeasy Mini kit (Qiagen, Inc.). Reverse
transcription was performed using ReverTra Ace qPCR Master Mix with
gDNA Remover (Toyobo Co., Ltd.), and single-strand complementary
DNA was synthesized. RT-qPCR was carried out using a One-Step SYBR
Prime Script RT-PCR kit (Takara Bio, Inc.) on a Light Cycler
instrument (Roche Diagnostics) and a QuantStudio instrument (Thermo
Fisher Scientific, Inc.). The Thermocycling conditions were as
follows: Initial denaturation step at 98°C for 2 min, followed by
45 cycles at 98°C for 10 sec, 60°C for 10 sec and 68°C for 30 sec.
β-actin was used as a reference gene, and relative gene expression
was analyzed using the 2-ΔΔCq method (25). The primer sequences are included
in Table SII. The primers for
the genes of endogenous retrovirus (ERV) were also designed for the
same sequence as previously reported, which was confirmed in the
gEVE database (http://geve.med.u-tokai.ac.jp) for the recognition of
ERVs (26).
Immunohistochemistry (IHC)
Clinical tissues were fixed with 20% neutral
buffered formalin at room temperature. The formalin fixation time
was usually within 24 h. Formalin-fixed paraffin-embedded sections
(thickness, 4-μm) were deparaffined and antigen retrieval
was performed with a citric acid buffer (Target Retrieval Solution
Citrate, pH6, 10X; Agilent Technologies, Inc.) using an autoclave.
Subsequently, the blocking of endogenous peroxidase was performed
using 200 μl/section of Dako REAL Peroxidase-Blocking
Solution (Agilent Technologies, Inc.) at 25°C for 10 min.
Anti-PRMT6 antibody (1:2,000; cat. no. 15395-1-AP; Proteintech
Group, Inc.) was applied as the primary antibody and incubated
overnight at 4°C. The following day, secondary antibody reaction
and detection were performed using Dako REAL EnVision Detection
System, Peroxidase/DAB, rabbit/mouse, and horseradish peroxidase
(HRP; Agilent Technologies, Inc.). Counterstaining was performed
with Meyer's hematoxylin solution (0.1% hematoxylin, FUJIFILM WAKO,
Tokyo, Japan) at 25°C for 2 min and dehydrated with ethanol and
xylene. The immunostained sections were observed using a biological
microscope (BX50F4; Olympus Corporation). The score of PRMT6
expression was calculated by the proportion of stained positive
cells [proportion score (PS)] and the intensity of staining
[intensity score (IS)], which are calculated as follows: Total
score (TS)=PS+IS. PS score 0, 0%; score 1, 1-20%; score 2, 21-40%;
score 3, 41-60%; score 4, 61-80%; score 5, >81%. IS score 0,
background; score 1, weak staining; score 2, moderate staining;
score 3, strong staining. Samples with a score of 0-5 were
designated as the PRMT6-low group and those with scores of 6-8 as
the PRMT6-high group. The results were recorded by two independent
observers, and the average score was calculated.
Cell viability assay
The cell viability assay was performed previously
demonstrated (19) and was
assessed using the Cell Counting Kit-8 (CCK-8; Dojindo
Laboratories, Inc.). The EC cells (HEC1B, HEC50B, HEC265, Ishikawa,
HEC151A and HEC116) were transfected with siRNAs were incubated at
37°C for 96 h. CCK-8 solution (10% amount of medium) was added to
each well and incubated at 37°C for 2 h. The absorbance of the
solution was measured at 450 nm using a Synergy LX multimode
microplate reader (BioTek Instruments, Inc.).
Cell cycle analysis
The cell cycle analysis was performed as previously
described (19). In brief, EC
cells were transfected with siRNAs as described earlier and
incubated at 37°C for 96 h. The cells were harvested with trypsin
and fixed with 70% ethanol at 4°C overnight and stained using
propidium iodide (MilliporeSigma) at 4°C for 15 min. The cell cycle
analysis was evaluated using fluorescence-activated cell sorting
(FACS) on a BD FACSCalibur HG Flow Cytometer Instrument (BD
Biosciences) and Cell Quest Pro software version 3.1 (BD
Biosciences). The data were calculated using FlowJo software
version 16 (FlowJo LLC).
Apoptosis assay
The apoptosis assay was carried out according to a
previously described protocol (19). Briefly, the EC cells were
transfected with siRNAs as previously described and incubated at
37°C for 96 h. The cells were harvested using trypsin and stained
with the Fluorescein isothiocyanate (FITC) Annexin V Apoptosis
Detection Kit II (BD Biosciences) at 25°C for 15 min. The cells
were evaluated using FACS on a BD FACSCalibur HG Flow Cytometer
Instrument (BD Biosciences) and Cell Quest Pro software version 3.1
(BD Biosciences), and the data were analyzed using FlowJo software
version 16 (FlowJo LLC).
Protein extraction and western
blotting
Protein extraction and western blotting were
performed as previously described (19). In brief, proteins from EC cells
(HEC1B, HEC50B, HEC265, Ishikawa, HEC151A and HEC116) were
extracted using RIPA Buffer (FUJIFILM Wako Pure Chemical
Corporation). Protein quantification was performed using Protein
Assay bicinchoninic acid (BCA) kit (Nalacai Tesque Inc.) The
samples were incubated with BCA working reagent at 37°C for 30 min.
The absorbance of them were measured at 562 nm using a Synergy LX
multimode microplate reader (BioTek Instruments, Inc.). The samples
(10 μg) were separated using sodium dodecyl
sulfate-polyacrylamide gel electrophoresis [SDS-PAGE; Mini-PROTEAN
TGX Precast Protein Gels (Any kD™); Bio-Rad Laboratories, Inc.] and
transferred by Trans-Blot Turbo Mini polyvinylidene difluoride
(PVDF) Transfer Packs (Bio-Rad Laboratories, Inc.). After blocking
process, the membrane was incubated with 5% skim milk as blocking
solution at 25°C for 60 min and incubated with the following
primary antibodies at 4°C overnight and incubated with the
following secondary antibodies at 25°C for 60 min. Proteins were
detected by Amersham ECL Select (Cytiva) and ImageQuant LAS 4000
(Cytiva). Rabbit anti-PRMT6 (1:1,000; cat. no. 15395-1-AP;
Proteintech Group, Inc.); rabbit anti-H3R2me2a (1:1,000; cat. no.
ab175007; Abcam); rabbit anti-Histone H3 (1:1,000, cat. no. ab1791;
Abcam), and mouse anti-β-actin (1:6,000; cat. no. A2228; Merck
KgaA) were used as the primary antibodies. Anti-mouse
immunoglobulin G (IgG) HRP-linked antibody (1:3,000; cat. no. 7076;
Cell Signaling Technology, Inc.) and anti-rabbit IgG HRP-linked
antibody (1:3,000; cat. no. 7074, Cell Signaling Technology, Inc.)
were used as secondary antibodies.
ChIP-seq
The EC cells were harvested with trypsin 24, 36, and
48 h following siRNA transfection. ChIP experiments were performed
based on the protocol of Maruyama et al (27) with certain modifications regarding
cell pellets freezing, and antibody concentration and chromatin
amount during immunoprecipitation. In brief, the cell pellets after
cross-linking were stored at -80°C. Chromatin was sonicated and
fragmented at 4°C using the Covaris S220 instrument (Covaris, LLC).
Dynabeads Protein G (Thermo Fisher Scientific, Inc.) bound to 2.5
μl of the anti-H3K27ac antibodies (cat. no. 8173; Cell
Signaling Technology, Inc.) was added to 50 μg chromatin and
incubated at 4°C overnight. DNA purification was performed using
Agencourt AMPure XP (Beckman Coulter, Inc.) and libraries for
ChIP-seq were prepared using ThruPLEX® DNA sequencing
(DNA-Seq) kit (Takara Bio, Inc.). All samples were sequenced on
NextSeq 550 (Illumina, Inc.) as paired-end reads. Five types of
anti-H3R2me2a antibodies used for immunoprecipitation did not work
well, and their details were as follows: Anti-histone H3
(asymmetric di methyl R2) antibody (2/25 μg chromatin; cat.
no. ab175007; Abcam), anti-dimethyl-histone H3 (Arg2) antibody
(10/100 μg chromatin; cat. no. 07-585; MilliporeSigma),
histone H3 [Asym-dimethyl Arg2] antibody (10/100 μg
chromatin; cat. no. NB21-1002; Novus Biologicals, LLC), histone
H3R2 dimethyl asymmetric (H3R2me2a) polyclonal antibody (0.1/50
μg chromatin, 0.5/50 μg chromatin, 2/50 μg
chromatin; cat. no. A-3714-050; Epigentek Group Inc.), anti-histone
H3 (asymmetric di methyl R2) antibody (0.1/50 μg chromatin,
0.5/50 μg chromatin, 1/50 μg chromatin; cat. no.
ab194706; Abcam).
RNA sequencing (RNA-seq)
Total RNA was extracted from the EC cells 48 h after
siRNA transfection using the RNeasy Mini Kit (Qiagen, Valencia,
Inc.). Libraries for RNA-seq were prepared using
SMARTer® Stranded Total RNA-Seq Kit v3 Pico Input
Mammalian (Takara Bio, Inc.). All samples were sequenced on NextSeq
550 (Illumina, Inc.) as paired-end reads.
ChIP-seq analysis
Low-quality reads with a quality value of <25 for
>90% of each base pair were first filtered by a
fastq_quality_filter. Filtered row reads were mapped to the human
genome (hg)38 by bowtie2 (v.2.4.2; https://bowtie-bio.sourceforge.net/bowtie2/index.shtml).
Multi-mapped reads and PCR duplicates were then removed using
Picard (v.2.25.3; https://broadinstitute.github.io/picard/). The reads
overlapped with the Encyclopedia of DNA Elements (ENCODE) black
list (https://www.encodeproject.org/files/ENCFF356LFX/)
were filtered out by bedtools (2.30.0; https://github.com/arq5x/bedtools2). Model-based
Analysis for ChIP-Seq (MACS)2 (v.2.2.7.1; https://github.com/macs3-project/MACS) was utilized
for calling peaks with the '-keep-dup auto' parameter.
Hypergeometric Optimization of Motif EnRichment (HOMER) (v.4.10;
http://homer.ucsd.edu/homer/motif/)
was used for identifying differential peaks between siRNA
experiments, and motif enrichment analysis. To obtain differential
peaks, the 'makeTagDirectory' was first used for creating tag
directories from each Binary Alignment Map (bam) file.
Subsequently, peak sets of each experiment were merged using
'mergePeaks'. The parameter 'getDifferentialPeaks' was then used
with the '-size given -F 2.0 -P 0.05' option. The H3K27ac peaks
were classified into seven patterns: i) The pattern one peak was
upregulated in siNC and downregulated in siPRMT6#1 and #2; ii) the
pattern two peaks were upregulated in siNC and siPRMT6#1 and
downregulated in siPRMT6#2; iii) the pattern three peaks were
upregulated in siNC and siPRMT6#2; iv) the pattern four peaks were
upregulated in siPRMT6#1 and downregulated in siNC and siPRMT6#2;
v) the pattern five peaks were upregulated commonly in siPRMT6 and
downregulated in siNC; vi) the pattern six peaks were upregulated
only in siPRMT6#2; and vii) the pattern seven peaks were commonly
upregulated in all samples. To focus on the effect on H3K27ac by
PRMT-KD, the pattern five peaks were analyzed further (Fig. S1). To obtain enriched motifs for
differential peaks, the 'findMotifsGenome.pl' parameter was used
with the '-size given' option. Gene Ontology (GO) analysis was
performed using Genomic Regions Enrichment of Annotations Tool
(GREAT) version 4.0.4.
RNA-seq analysis
To generate the expression count matrix, row reads
were trimmed to remove adaptor sequences by Skewer (v0.2.2;
https://github.com/relipmoc/skewer)
and mapped to the hg38 genome by Spliced Transcripts Alignment to a
Reference (STAR) (v.2.7.8a; https://github.com/alexdobin/STAR); the mapped reads
were then counted by featureCounts (v.2.0.10; https://subread.sourceforge.net/featureCounts.html).
Upregulated genes were defined as those satisfying the following
requirements: i) log2 fold change >1 and =1; and ii)
log2 average expression >1 and =1. Downregulated
genes were defined as those satisfying (1) log2 fold change <-1 and
=1, and (2) log2
average expression <1 and =1. Gene expression differences
between control and siPRMT6 experiments were visualized based on MA
plot; genes with (1)
log2 fold change >1 and = 1 (upregulated) or <-1
and =1 (downregulated), and (2)
log2 average expression >1 were identified as
differential genes. GO analysis was performed on these genes using
the Database for Annotation, Visualization and Integrated Discovery
(DAVID) 6.8. The target gene sets of nuclear factor of activated
T-cells 1 (NFATC1), SMAD2 and SMAD3 were obtained from ChIP
Enrichment Analysis (ChEA) Transcription Factor Targets (SMAD2,
SMAD3) and ENCODE Transcript Factor Targets (NFATC1) via
Harmonizome 3.0 (https://maayanlab.cloud/Harmonizome/).
Statistical analyses
Statistical analyses were conducted as previously
described (19). Comparisons of
two groups were performed using unpaired Student's t-test, and
comparisons of three groups were performed using one-way analysis
of variance (ANOVA) followed by Tukey's post hoc test. The data
were analyzed using Excel version 16 (Microsoft Corporation) and
John's Macintosh Project (JMP) Pro software version 16 (SAS
Institute, Inc.). Kaplan-Meier survival analysis (with the log-rank
test) of PRMT6 expression in IHC was analyzed using JMP Pro
software version 16 (SAS Institute, Inc.). P<0.05 was considered
to indicate a statistically significant difference. Each experiment
was performed at least in triplicate, and the data are presented as
the mean ± standard deviation (SD).
Results
PRMT6 overexpression results in a poor
prognosis in EC
In order to examine whether PRMT6 may be an
appropriate therapeutic target in EC, the expression of
PRMT6 in EC tissue (n=52, Table SI; patients No. 1 to No. 52) and
normal endometrial tissue (n=4; Table SI; N1 to N4 and N17 to N20) was
first investigated using RT-qPCR (Figs. 1A and B, and S2). Since there was
a statistically significant difference, PRMT6 was significantly
overexpressed in EC compared to normal endometrial tissues.
Secondly, IHC staining for PRMT6 was performed using clinical
specimens of EC (n=55; Table SI;
No. 1 to No. 55) and normal endometrial tissue (n=12; Table SI; N5 to N16). PRMT6 expression
using IHC was scored by the PS + IS sum [Fig. 1C and D(a)-D(d)] and was detected
to be increased in EC [Fig. 1C and
D(b)-D(d)] in comparison with the normal endometrial tissue
[Fig. 1C and D(a)]. To assess the
clinical significance of PRMT6 in EC, the overall survival (OS) and
disease-free survival (DFS) of patients with a high and low
expression of PRMT6 was analyzed. OS was significantly shorter in
the PRMT6 high expression group (Fig.
1E). There was no significant difference in DFS; however, a
trend towards a shorter DFS was observed in the PRMT6 high
expression group (Fig. 1F).
Finally, the histopathological classification using PRMT6
expression of the IHC score was evaluated. PRMT6 expression was
significantly higher in G3 tissues for the histological grade,
stage III and IV according to The International Federation of
Gynecology and Obstetrics (FIGO), than in G1 and G2, positive for
muscle invasion, FIGO stage I and II and negative for deep muscle
invasion stage III/IV (Fig.
1G-I). These data demonstrated that PRMT6 is overexpressed in
EC and that high PRMT6 expression levels affect EC progression,
resulting in a poor prognosis.
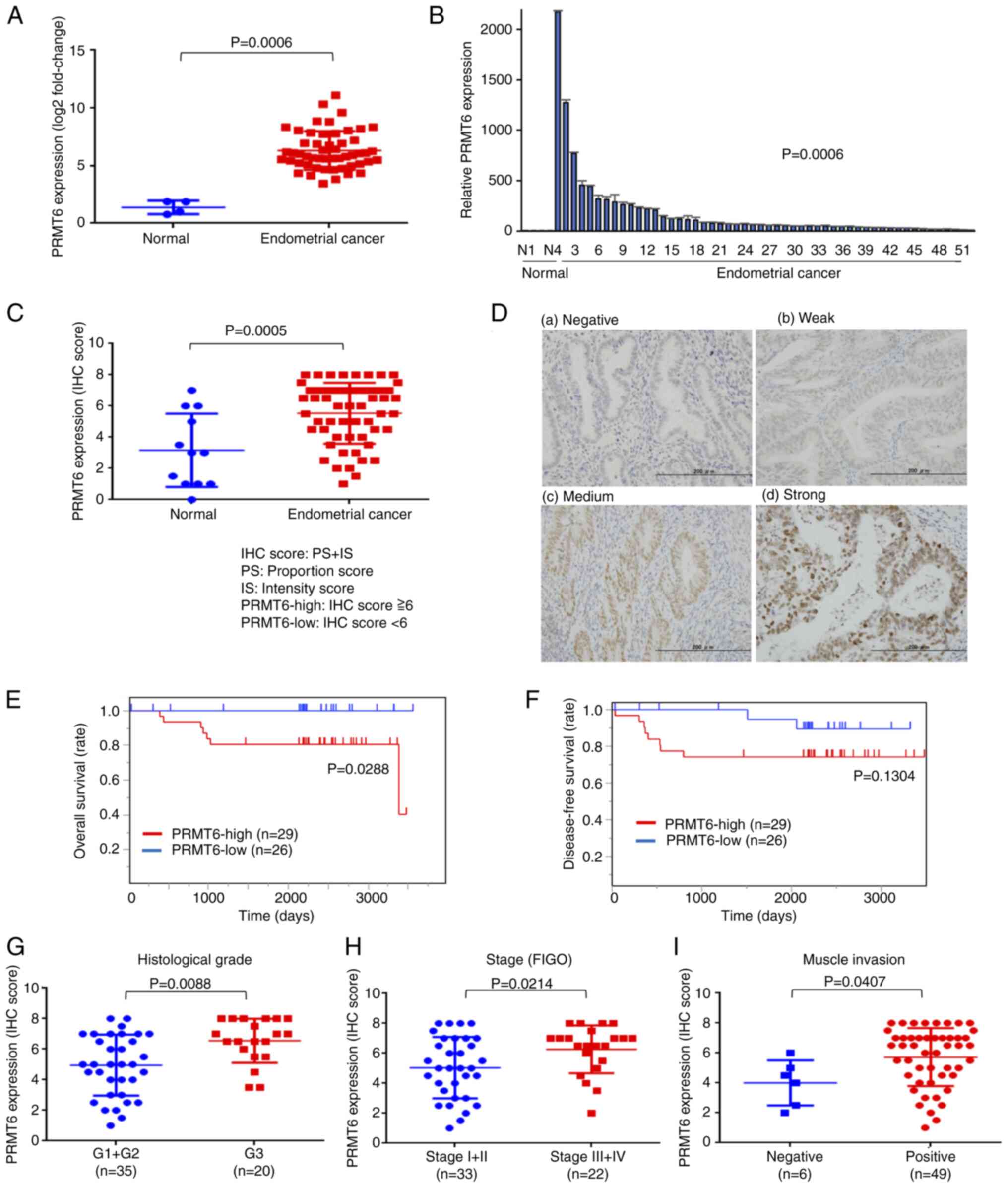 | Figure 1PRMT6 is overexpressed in endometrial
cancer and a high expression level is related to a poor prognosis.
(A and B) mRNA expression of PRMT6. The results of RT-qPCR
using clinical specimens of EC (n=52) and normal endometrial tissue
(n=4) are presented. (C) The IHC score of PRMT6. The results of IHC
using clinical specimens of EC (n=55) and normal endometrial tissue
(n=12) are presented. Total score (TS)=proportion score
(PS)+intensity score (IS). (D) The staining intensity of PRMT6 by
IHC is shown. (a) Normal endometrial tissue, (b-d) EC tissue. IS
score 0, background (negative); score 1, weak staining; score 2,
moderate staining; score 3, strong staining. Two groups are defined
using median as follows: Specimens with scores 0-5 are considered
PRMT6-low group and with 6-8 are considered as the PRMT6-high
group. The analysis of the OS (E) and DFS (F) in two groups using
the Kaplan-Meier method and the log-rank test are presented. The
expression of PRMT6 was evaluated using the IHC score (TS) for (G)
histological grade, (H) clinical FIGO stage, and (I) muscle
invasion. PRMT6, protein arginine methyltransferase 6; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction; IHC,
immunohistochemistry; EC, endometrial cancer; TS, total score; PS,
proportion score; IS, intensity score; OS, overall survival; DFS,
disease-free survival; FIGO, The International Federation of
Gynecology and Obstetrics. |
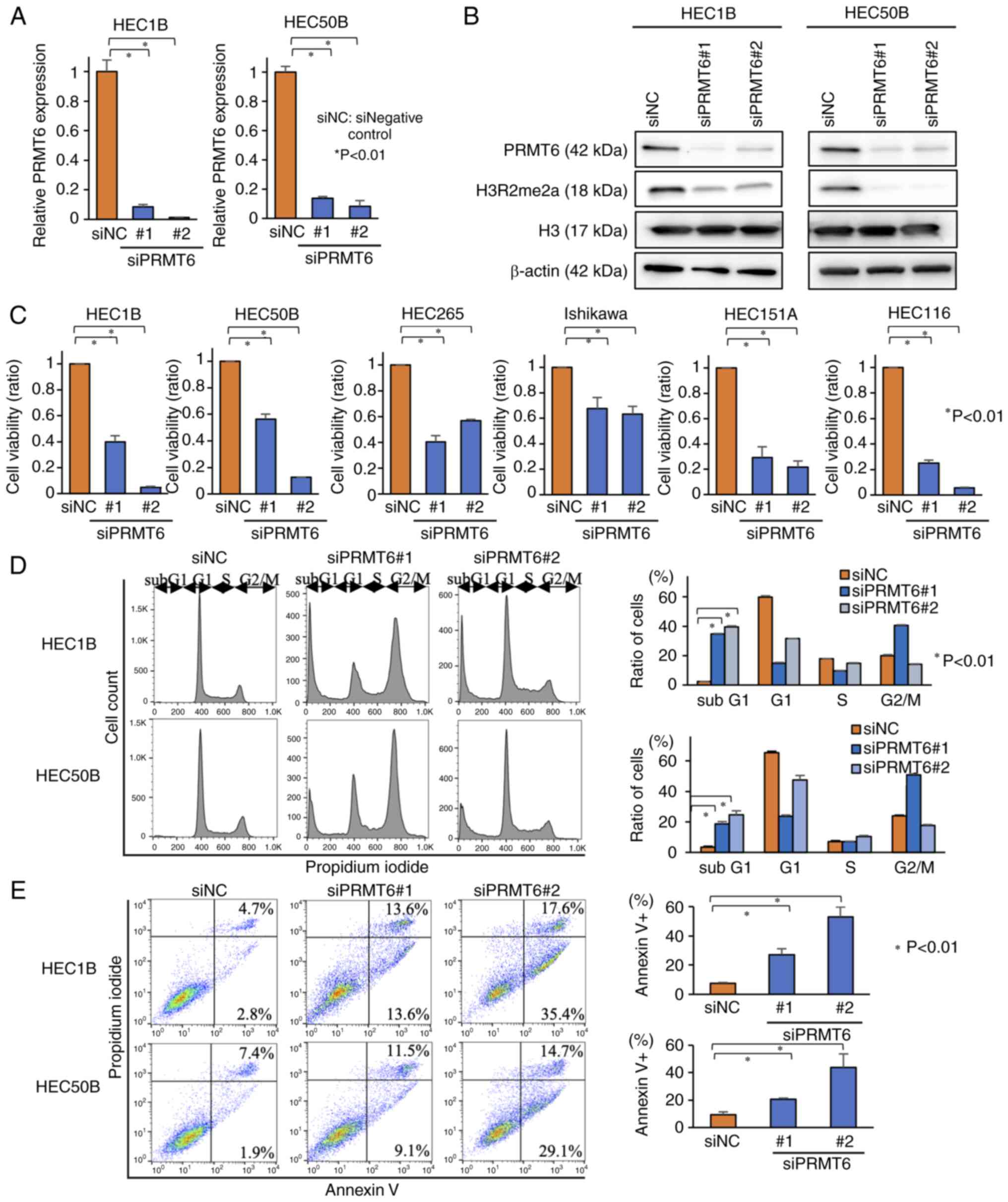
PRMT6 inhibition suppresses EC cell
proliferation accompanied by apoptosis with the hypomethylation of
H3R2me2a in EC cell lines
In the present study, mainly HEC1B and HEC50B were
selected, since these cells are fast-growing and difficult to
de-attach from the cell culture dish, rendering cell passaging less
demanding. To elucidate the antitumor effects of PRMT6 inhibition
in EC cells, PRMT6-KD EC cell lines (HEC1B and HEC50B) were first
established using siNC and two types of siPRMT6 (siPRMT6#1 and
siPRMT6#2). Subsequently, PRMT6 expression was evaluated using
RT-qPCR and western blotting and it was confirmed that PRMT6
expression was suppressed at the mRNA and protein level (Figs. 2A and B, and S3). The H3R2me2a
expression levels, catalyzed by PRMT6, were also attenuated by
PRMT6-KD (Fig. 2B). Subsequently,
cell viability assays were performed, by using six types of
PRMT6-KD EC cell lines. Cell viability was suppressed in all six
cell lines (Fig. 2C). Finally,
the apoptosis levels were evaluated in PRMT6-KD cells using cell
cycle analyses and flow cytometry assays. The proportion of cells
in the subG1 phase was increased in cell cycle analysis and the
proportion of Annexin-positive cells was also increased in the
Annexin assay due to PRMT6-KD (Fig.
2D and E). The results of flow cytometry revealed that PRMT6-KD
induced the apoptosis of EC cell lines. Hence, these data indicated
that PRMT6-KD attenuated the methylation of H3R2me2a and EC cell
growth was suppressed with apoptosis.
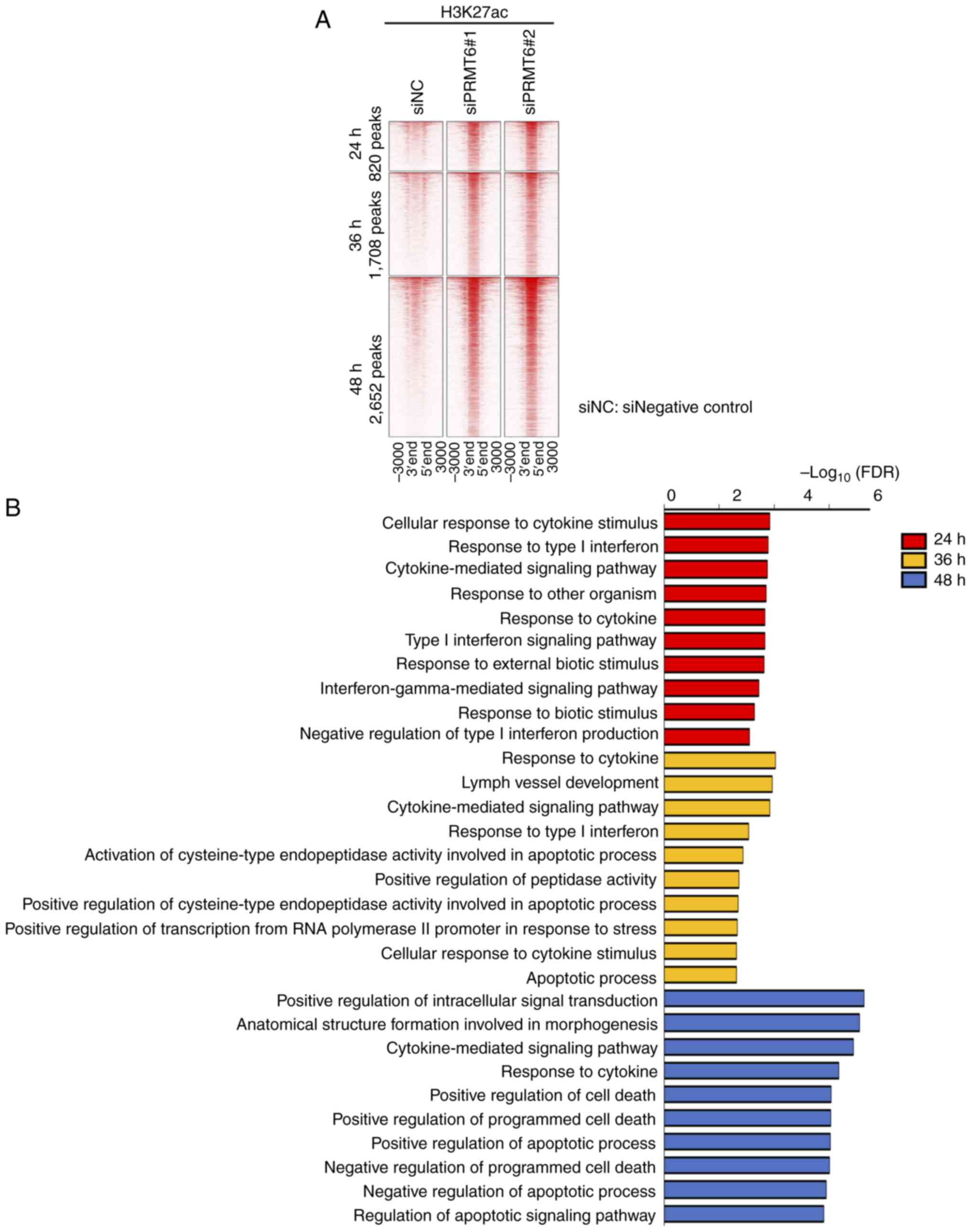
PRMT6 inhibition induces apoptosis via
interferon signaling through H3K27ac in EC cell lines
ChIP assays with H3R2me2a were first performed, in
order to clarify the mechanisms through which H3R2me2a, catalyzed
by PRMT6, is involved in the suppression of cell proliferation and
induction of apoptosis in EC cell lines. A total of five types of
commercially available anti-H3R2me2a antibodies were used and the
appropriate ChIP protocols were thoroughly considered; however, all
the antibodies did not perform adequately. Thus, ChIP experiments
with H3K27ac were conducted, which is an active histone mark, to
investigate the role of PRMT6 in histone modification using HEC1B.
As a preliminary experiment, it was confirmed that PRMT6 was
downregulated at the protein level 24 h following transfection with
siPRMT6 using western blotting (Fig. S4). Thus, H3K27ac ChIP-seq
was performed at three time points, 24, 36 and 48 h, following
PRMT6-KD.
In the analysis of H3K27ac ChIP-seq, the analysis
was focused on the peak pattern of the signals of H3K27ac enhanced
by PRMT6-KD, pattern 5, since H3K27ac is an active histone marker
(Fig. S1). The numbers of
H3K27ac peaks enhanced in common with two siPRMT6 types were 820,
1,708 and 2,562 peaks, at the time points 24, 36 and 48 h following
PRMT6-KD, respectively (Fig. 3A;
log2 FC >1 and P-value <0.01). PRMT6 inhibition increased the
number of genomic regions in which H3K27ac signals were enhanced
over time.
GO analysis of these peak sets with GREAT revealed
that interferon-related GO terms were enriched 24 h after PRMT6-KD,
apoptosis-related GO terms at 36 h, and cell death-related GO terms
at 48 h (Fig. 3B; top 10 enriched
terms are shown, FDR <0.01). These results suggested that
PRMT6-KD may activate interferon signaling, leading to apoptosis
and cell death. It was also observed that interferon-related
transcription factor motifs were enriched at all three time points
following PRMT6-KD by motif analysis using these peak sets
(q-value<0.05) (Fig. 4A). The
findings of the present study indicated that PRMT6 inhibition may
regulate the genomic regions related to interferon, apoptosis, and
cell death via H3K27ac signals.
PRMT6 regulates several interferon and
cancer-related transcription factors via H3K27ac in EC cell
lines
In order to evaluate the transcription factors that
PRMT6 regulates via H3K27ac in EC cell lines, motif analysis using
the peak sets presented in Fig.
3A were performed and the motifs were categorized into five
groups (Fig. 4A). Group 1
included transcription factor motifs significantly enriched in all
three time points peak sets after PRMT6-KD (q-value <0.05).
Groups 2, 3 and 4 were transcription factor motifs significantly
enriched at each time point 24, 36 and 48 h peak sets after
PRMT6-KD, respectively (q-value <0.05). Group 5 included
transcription factor motifs significantly enriched in the two time
point peak sets, 36 and 48 h following PRMT6-KD (q-value
<0.05).
It was considered that groups 1 and 2 included key
transcription factor motifs, since they were the first to change
triggered by PRMT6-KD. Thus, interferon-related transcription
factor motifs and PR/set domain 1 (PRDM1) were identified in group
1 and three transcription factor motifs in group 2, namely NFAT,
EBF transcription factor 2 (EBF2) and Tbox:Smad (Fig. 4B). NFAT and EBF2 each consist of
the transcription factors NFATC1 and EBF2. Tbox:Smad consists of
nodal growth differentiation factor (NODAL), SMAD2, SMAD3 and
SMAD4, which are members of the transforming growth factor β
(TGF-β) protein group, reported to be related to cancer (28). The changes in the mRNA expression
of these transcription factors corresponding to the aforementioned
motifs, PRDM1, NFATC1, EBF2, NODAL,
SMAD2, SMAD3 and SMAD4, were evaluated. The
transcription factors that presented with altered mRNA expression
levels were NFATC1, SMAD2 and SMAD3. No
significant changes were observed in the expression levels of the
other transcription factors (Fig.
4B). Motif analysis indicated that PRMT6 regulated several
interferon and cancer-related transcription factors via H3K27ac in
EC cell lines.
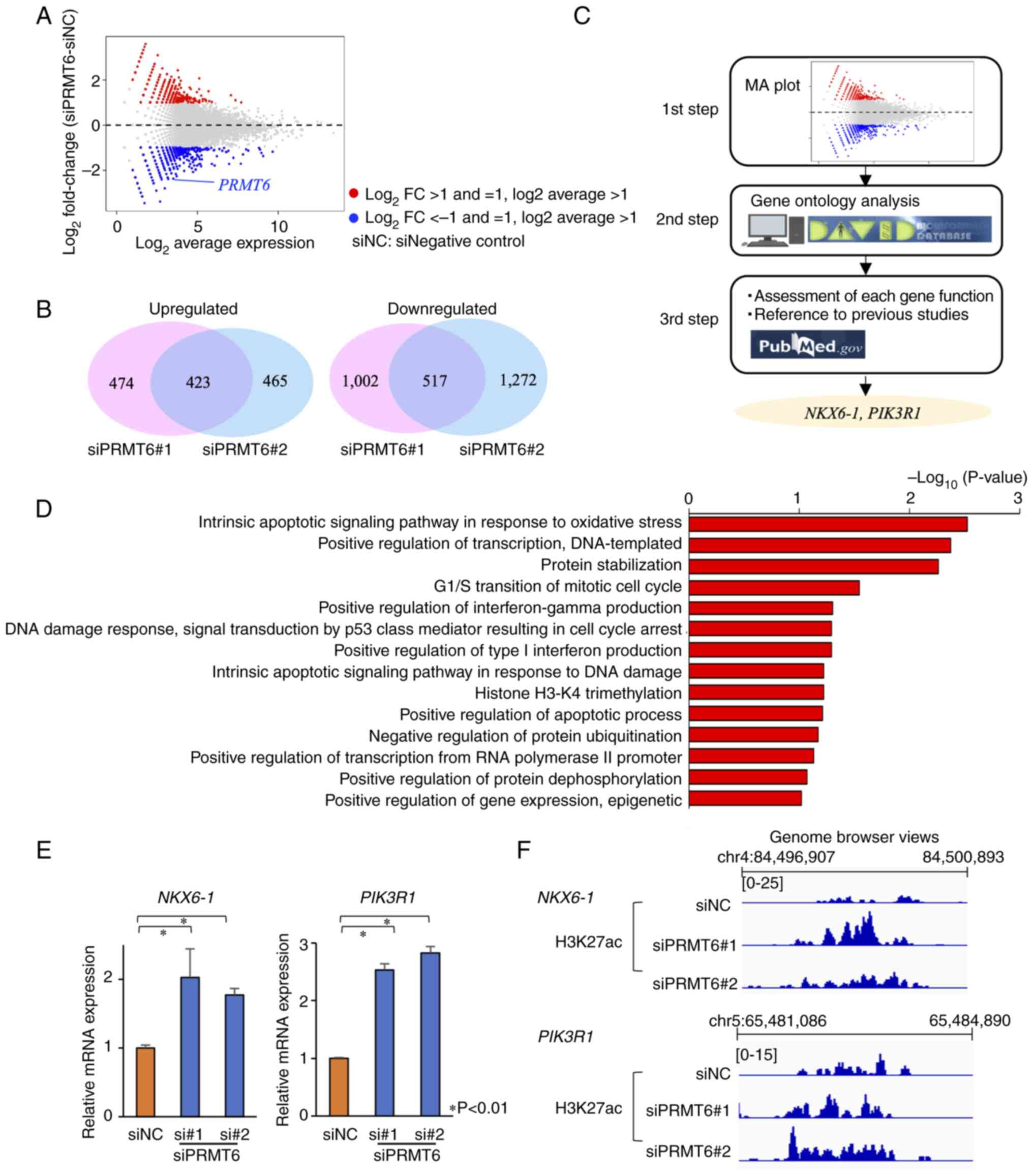
PRMT6 inhibition alters the expression of
940 genes and increases the expression of interferon-related gene
clusters in EC cell lines
In order to identify key downstream genes regulated
by PRMT6 in EC cells, RNA-seq was performed. The expression levels
of 940 genes were altered 48 h following PRMT6-KD (Fig. 5A and B). Furthermore, 423 genes
were upregulated and 517 genes were downregulated in common, among
the two siPRMT6 datasets (Fig.
5B). Firstly, GO analysis among the 423 upregulated genes was
performed using DAVID, to investigate the function of genes whose
expression was altered by PRMT6-KD (Fig. 5C and D). Interferon-related and
apoptosis-related GO terms which were enriched were identified
(P<0.05; Fig. 5D). These data
were consistent with the results of the GO analysis of ChIP-seq.
The findings of the present study suggested that PRMT6 inhibition
upregulated interferon-related and apoptosis-related genes.
Two tumor suppressor genes, NK6 homeobox
1 (NKX6-1) and phosphoinositide-3-kinase regulatory subunit 1
(PIK3R1), are upregulated following PRMT6 inhibition in EC cell
lines
In order to identify key downstream genes regulated
by PRMT6, the number of genes was curtailed according to the
methodology presented in Fig. 5C.
The GO terms enriched in the genes whose expression level was
increased in common in the two types of siPRMT6 datasets
(P<0.05), and GO terms related to cancer and epigenetic
regulation were first selected. Each gene function was then
considered using PubMed (Fig. 5C)
and two key genes were ultimately identified: NKX6-1, a tumor
suppressor gene (29-31), and PIK3R1, also reported to
be related to EC (32). The
expression levels of NKX6-1 and PIK3R1 were
significantly increased according to the results RT-qPCR (Fig. 5E) and the H3K27ac signals in the
two genes were also enhanced according to H3K27 ChIP-seq (Fig. 5F), using PRMT6-KD EC cell lines.
The data suggested that PRMT6 inhibition upregulated two tumor
suppressor genes, NKX6-1 and PIK3R1.
The expression of NKX6-1 and PIK3R1
was also investigated in EC tissues (n=10; Table SI) and normal endometrial tissues
(n=4; Table SI), using RT-qPCR
(Fig. S5). No significant changes were observed between the EC and
normal endometrial tissues.
Interferon-related and apoptosis-related
genes are identified downstream of the three transcription factor
motifs, NFATC1, SMAD2 and SMAD3
In order to identify interferon and
apoptosis-related genes regulated by the three transcription
motifs, NFATC1, SMAD2 and SMAD3, as identified using H3K27ac
ChIP-seq, an integrated ChIP-seq and RNA-seq analysis was
performed. The overlaps of 423 genes upregulated by PRMT6-KD and
genes expressing downstream targets of NFATC1, SMAD2 and SMAD3 were
evaluated. Among the 423 genes upregulated by PRMT6-KD, the numbers
of genes which expressed downstream targets of NFATC1, SMAD2 and
SMAD3 were 178, 39 and 63 genes, respectively (Fig. S6). According
to GO analysis with the inclusion of those 178, 39, and 63 genes,
three interferon-related genes were identified among NFATC1
downstream genes: Interferon regulatory factor 3 (IRF3),
interferon-stimulated gene 15 (ISG15) ubiquitin like
modifier (ISG15) and interferon regulatory factor 5
(IRF5). It was also revealed that multiple apoptosis-related
genes exist among the genes expressing targets downstream of
NFATC1, SMAD2 and SMAD3 (Figs. S6-S9).
This integrated analysis confirmed that PRMT6-KD may
regulate NFATC1, SMAD2 and SMAD3 transcription factors through
H3K27ac, and that they regulate in turn multiple interferon and
apoptosis-related genes.
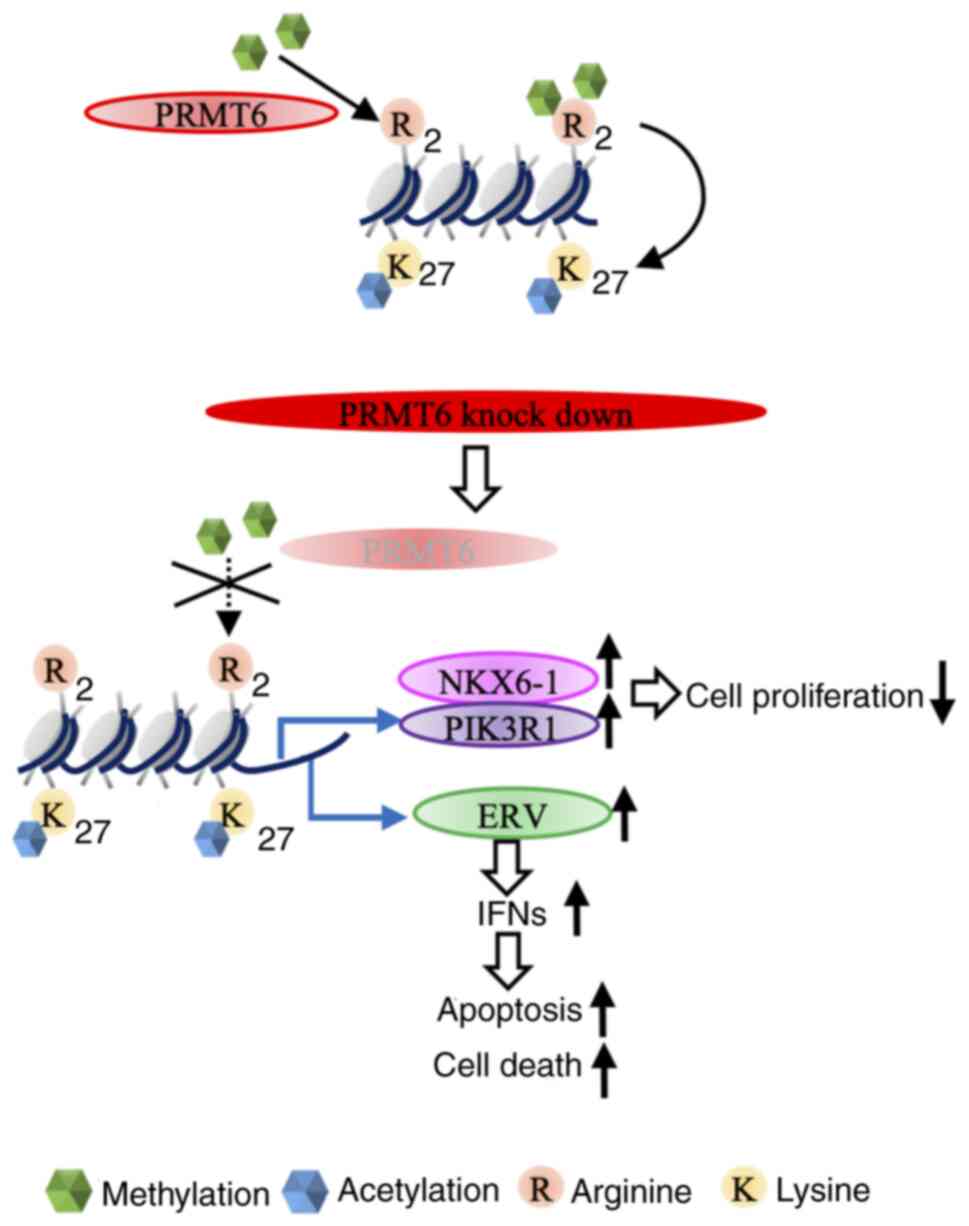
PRMT6 inhibition induces the activation
of ERV genes in EC cell lines
Recent studies have reported that ERV, whose
activation is normally suppressed in humans, is activated by the
epigenome and induces interferon secretion in various types of
cancer, including multiple myeloma and colorectal cancer (33,34). Therefore, herein, it was evaluated
whether PRMT6 inhibition also induces ERV gene activation in EC
cell lines. Similar to the ChIP-seq experiments, ERV expression was
investigated at the three time points: 24, 36 and 48 h following
PRMT6-KD. The expression levels of several ERV genes were
significantly increased in the PRMT6-KD EC cells (Fig. 6A) and the expression levels in the
majority of the ERV genes were increased 24 h following PRMT6-KD
(Fig. 6A and B). Interferon
signaling was also induced 24 h after PRMT6-KD by H3K27ac ChIP-seq
(Fig. 3B). On the whole, these
data indicated that PRMT6-KD may activate ERV genes and induce
interferon signaling (Fig.
7).
Finally, the expression of ERV genes in EC tissues
(n=10; Table SI) and normal
endometrial tissues (n=4, Table
SI) were also evaluated using RT-qPCR (Fig. S10). There were no
significant differences in ERV gene expression levels between the
EC and normal endometrial tissues.
Discussion
The present study investigated whether PRMT6 could
be designated as a therapeutic target for the treatment of EC. The
increased expression of PRMT6 was observed in clinical samples of
EC. KD of PRMT6 inhibited cell proliferation by inducing the
apoptosis of EC cell lines. Data from ChIP-seq and RNA-seq also
suggested that interferon and apoptotic pathways were activated in
PRMT6-KD EC cell lines. In addition, the activation of ERV was
identified as a cause of the interferon pathway activation.
PRMT6 has been reported to be highly expressed in a
variety of cancer types, including prostate, lung cancer and EC
(21,22,24). For example, in a previous study,
IHC using colorectal cancer clinical specimens demonstrated a
negative association between PRMT6 expression and DFS (35). In another study on gastric cancer,
it was demonstrated that both were DFS and OS negatively associated
with the expression of PRMT (36). Jiang et al (24) performed RT-qPCR and IHC analysis,
revealing increased PRMT6 expression in EC compared with normal
endometrium. That study also demonstrated, by using The Cancer
Genome Atlas database, that the expression of was PRMT6 positively
associated with the EC grade or the existence of a special tissue
type, including serous carcinoma. Additionally, the expression of
PRMT6 was negatively associated with OS (24). In the present study, RT-qPCR, IHC
and PRMT6 expression analyses were performed, using clinical
specimens of EC at the University of Tokyo Hospital. In comparison
with the normal endometrium, an increased PRMT6 expression was
observed in EC as previously reported (24), demonstrating a positive
association between PRMT6 expression and the pathological grade of
EC. The association between PRMT6 expression and prognosis was
identical to that of a previous report concerning OS. However,
there was no significant difference in DFS. A significant
difference in DFS could have been observed if the number of
patients was larger. In addition, the present study demonstrated an
association between PRMT6 expression, and stage and deep muscle
invasion.
KD experiments were also performed, in order to
investigate whether PRMT6 is a potential therapeutic target in EC
and to examine the antitumor mechanism of PRMT6 suppression.
Apoptosis-induced suppression of cell proliferation was observed in
EC cells under PRMT6-KD. The suppression of cell proliferation by
the inhibition of PRMT6 has been previously reported (37,38). There are some reports on the
mechanism of inhibition of cell proliferation by PRMT6. For
example, Kleinschmidt et al (38) revealed that the KD of PRMT6
inhibits the cell cycle by activating p21. PRMT6 has been reported
to enhance the phosphoinositide 3-kinase (PI3K)/AKT/mammalian
target of rapamycin (mTOR) pathway as an anti-apoptotic mechanism
in prostate cancer (21). It has
also been reported that PRMT6 promotes cell proliferation through
PI3K/AKT/mTOR pathway in EC (24). However, the effect of PRMT6 on
histone modification in EC had not been reported previously, at
least to the best of our knowledge. The histone modification of
PRMT6 has been reported to asymmetrically dimethylate H3R2 and that
H3K27ac and H3K4me3 are affected through methylation of H3R2me2a
(23). ChIP-seq analysis using
H3R2me2a antibodies is rarely mentioned in the literature and has
been solely reported previously by Bouchard et al (23), to the best of our knowledge.
Initially, ChIP-seq was also performed using H3R2me2a antibodies,
without any success (data not shown). This is due to the fact that
the ChIP-seq method is a delicate technique influenced by
antibodies and the type of cell line, among other factors.
Therefore, ChIP-seq for H3K27ac was also performed, which is
affected by H3R2me2a.
The KD of PRMT6 demonstrated an increase or decrease
in a number of peak patterns of H3K27ac and the present study
focused on the increased peak patterns. The time course of H3K27ac
was analyzed, using the ChIP-seq method on an hourly basis. The
acquired data suggested that histone acetylation, which controls
interferon-related, apoptosis-related, and cell death-related
genomic regions, was observed to change dynamically with time. When
significant transcription factors regulated by H3K27ac were
examined, NFATC1 and SMAD2/3 were identified.
NFAT is a transcription factor that regulates immune
system pathways. Among the NFAT family, NFATC1 and NFATC2 are
predominantly expressed in T-cells (39). Reppert et al (40) reported that NFATC1 promoted
interferon-γ. The data of the present study suggested that PRMT6-KD
activated interferon signaling and regulated NFATC1. Therefore,
these results suggest that NFATC1 is involved in the pathway
through which PRMT6-KD promotes the interferon pathway. As was
expected, three interferon-related genes and multiple
apoptosis-related genes were identified as genes expressing factors
downstream of NFATC1, by using integrated analysis of H3K27ac
ChIP-seq and upregulated genes by PRMT6-KD in RNA-seq (Figs. S6 and
S7).
TGF-β has been identified as a growth factor that
promotes cell transformation; however, it also contributes to the
inhibition of cell proliferation and the induction of apoptosis in
various types of cells, with its physiological effects being
diverse (41). Thus, while cell
growth suppression has been reported in cancer, the opposite has
been reported to induce the epithelial mesenchymal transition (EMT)
and promote metastasis (42). In
addition, SMAD2/3 is a downstream factor of TGF-β and is important
for transmitting TGF-β signaling. Kriseman et al (43) suggested that SMAD2/3 is
responsible for the normalization of endometrial function and that
the absence of SMAD may lead to abnormal cell proliferation and
progression in EC. According to the data of the present study,
multiple apoptosis-related genes were identified as genes
expressing factors downstream of SMAD2 and SMAD3, by using
integrated analysis as mentioned above (Figs. S6-S8). Therefore, in
view of the results of the present study, an increase in SMAD2/3
levels by PRMT6-KD is suggested as a possible therapeutic mechanism
for EC.
Changes in gene expression induced by PRMT6-KD were
also analyzed using the RNA-seq method and it was demonstrated that
PRMT6-KD altered the expression of various genes. The application
of GO analysis and the ChIP-seq method suggested that PRMT6
affected the interferon and apoptotic pathways. In addition, NKX6-1
and PIK3R1 were identified using the RNA-seq method. NKX6-1 is a
key transcription factor for pancreatic and neural development and
has been reported as a tumor suppressor gene in various types of
cancer. For example, it has been reported that NKX6 -1 suppresses
the EMT system in cervical and colorectal cancer (29-31). In the present study, the possible
involvement of NKX6-1 in EC was reported.
The PI3K/AKT/mTOR pathway is a major signaling
pathway required for the maintenance of normal cell functions such
as cell proliferation and differentiation. PIK3R1 also acts as a
suppressor in the PI3K/AKT/mTOR pathway and a functionally inactive
mutation in cancer causes activation of the PI3K/AKT/mTOR pathway
(44). Mutations in PIK3R1 are
detected in various types of cancer, including EC and colorectal
cancer (32). Jiang et al
(24) also reported that PRMT6
promoted phosphorylation of AKT and activated the PI3K/AKT/mTOR
pathway in EC cell lines. A different mechanism was revealed in the
present study, demonstrating that PRMT6 reduced PIK3R1 expression
level and activated the PI3K/AKT/mTOR pathway by affecting histone
modification.
In total, ~10% of mammalian genomes contain
sequences derived from ERVs (45). Although regulation of ERVs by
epigenome has not been previously reported, some studies have
revealed that ERVs, which are normally repressed in humans, are
activated by the epigenome, including histone modifications and DNA
methylations, to induce interferon (33,34,46,47). For example, Ishiguro et al
(33) revealed that the
inhibition of histone methyltransferase EZH2/G9a stimulated the
interferon response by increasing the expression of ERV genes.
Similar to previous research (33), the results of the present study
demonstrated significant increases in several ERVs by the KD of
PRMT6. In the time course of ERV expression, the expression was
highest at 24 h after PRMT6-KD, suggesting that ERVs are
synchronized with interferon activity. Therefore, the mechanism in
which PRMT6-KD stimulates apoptosis by activating interferon via
the activation of ERVs is considered.
The expression of ERV genes, NKX6-1, and
PIK3R1, was also examined using RT-qPCR using clinical
specimens from endometrial cancer and normal endometrium (Fig. S5).
No difference in expression of these genes between EC and the
normal endometrium was detected. Since the changes in expression of
these genes in the present study may be attributed to PRMT6
suppression, the absence of differences in expression between
endometrial carcinoma and normal endometrium is coherent.
The present study has several limitations. Firstly,
the ChIP-seq method using the antibody of H3R2me2a methylated by
PRMT6 could not be established. Therefore, it is difficult to prove
that PRMT6 directly regulates the ERV genes in the present study.
Secondly, in vivo experiments were not conducted. Several
PRMT6 inhibitors are in development at present; however, none of
the available inhibitors exhibited adequate specificity to be used
in the present study. Mouse experiments will be considered when a
prospective PRMT6 selective inhibitor is developed in the
future.
In the present study, it was revealed that PRMT6
may be a therapeutic target in EC by affecting multiple genes
through changes in histone modifications through the inhibition of
PRMT6. Novel tumor suppressor genes, to the best of our knowledge,
were also identified, including PIK3R1, transcription factors
related to interferon, and ERVs activation by PRMT6-KD through
histone modification.
Supplementary Data
Availability of data and materials
Sequencing data generated in the present study are
available at the Gene Expression Omnibus (GEO) repository (grant
no. GSE239296). The other datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
FI, KS, KKu and RM conceived and designed the
study. FI, KS, KKu and KKa designed the experiments. All
experiments were performed by FI, KKu, RH and ES. FI and KKu
acquired the data. The data were analyzed and interpreted by ST,
NT, KKa, YTo, YTa, AT, YM, MT, TI, MM, OWH, KO, HS, RM and YO. FI,
KS, KKu and RM prepared the manuscript and figures. ST, NT, KKa,
YTo, YTa, MK, AK, HH, AN, AT, YM, MT, TI, MM, OWH, KO, HS, RM, YO
reviewed and revised the manuscript for important intellectual
content. MK, AK, HH, AN interpreted the data, and provided
technical and material support. FI and KS confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
In the present study, all patients provided written
informed consent for the use of the tumor specimens for research
purposes. The present study was approved by the Human Genome, Gene
Analysis Research Ethics Committee of the University of Tokyo
(approval no. 683-19).
Patient consent for publication
Not applicable.
Competing interests
KO obtained research grants from Daiichi-Sankyo
Co., Ltd. and AstraZeneca plc, as well as lecture fees from Chugai
Pharmaceutical Co., Ltd. and AstraZeneca plc. All of the remaining
authors declare that they have no competing interests.
Abbreviations:
|
DAVID
|
Database for Annotation Visualization
and Integrated Discovery
|
|
EC
|
endometrial cancer
|
|
PRMT
|
protein arginine
methyltransferase
|
|
RNA-seq
|
RNA sequencing
|
|
IS
|
intensity score
|
|
PS
|
proportion score
|
|
PI
|
propidium iodide
|
|
TS
|
total score
|
|
FACS
|
fluorescence-activated cell
sorting
|
|
GO
|
Gene Ontology
|
|
GREAT
|
Genomic Regions Enrichment of
Annotations Tool
|
|
OS
|
overall survival
|
|
DFS
|
disease-free survival
|
|
NFATC1
|
nuclear factor of activated T-cells
1
|
|
EMT
|
epithelial-mesenchymal transition
|
|
SD
|
standard deviation
|
Acknowledgments
Not applicable.
Funding
The present study was supported by the Grant-in-Aid for
Scientific Research (B) (Grant no. 20H03820) from the Ministry of
Education, Culture, Sports, Science and Technology of Japan.
References
|
1
|
Anderson AS, Key TJ, Norat T, Scoccianti
C, Cecchini M, Berrino F, Boutron-Ruault MC, Espina C, Leitzmann M,
Powers H, et al: European code against cancer 4th edition: Obesity,
body fatness and cancer. Cancer Epidemiol. 39(Suppl 1): S34–S45.
2015.
|
|
2
|
Lachance JA, Darus CJ and Rice LW:
Surgical management and postoperative treatment of endometrial
carcinoma. Rev Obstet Gynecol. 1:97–105. 2008.
|
|
3
|
Makker V, Colombo N, Casado Herráez A,
Santin AD, Colomba E, Miller DS, Fujiwara K, Pignata S, Baron-Hay
S, Ray-Coquard I, et al: Lenvatinib plus pembrolizumab for advanced
endometrial cancer. N Engl J Med. 386:437–448. 2022.
|
|
4
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022.
|
|
5
|
Zhang L, Lu Q and Chang C: Epigenetics in
health and disease. Adv Exp Med Biol. 1253:3–55. 2020.
|
|
6
|
Margueron R, Trojer P and Reinberg D: The
key to development: Interpreting the histone code? Curr Opin Genet
Dev. 15:163–176. 2005.
|
|
7
|
Strahl BD and Allis CD: The language of
covalent histone modifications. Nature. 403:41–45. 2000.
|
|
8
|
Varier RA and Timmers HT: Histone lysine
methylation and demethylation pathways in cancer. Biochim Biophys
Acta. 1815:75–89. 2011.
|
|
9
|
Hamamoto R, Saloura V and Nakamura Y:
Critical roles of non-histone protein lysine methylation in human
tumorigenesis. Nat Rev Cancer. 15:110–124. 2015.
|
|
10
|
Sone K, Piao L, Nakakido M, Ueda K,
Jenuwein T, Nakamura Y and Hamamoto R: Critical role of lysine 134
methylation on histone H2AX for γ-H2AX production and DNA repair.
Nat Commun. 5:56912014.
|
|
11
|
Oki S, Sone K, Oda K, Hamamoto R, Ikemura
M, Maeda D, Takeuchi M, Tanikawa M, Mori-Uchino M, Nagasaka K, et
al: Oncogenic histone methyltransferase EZH2: A novel prognostic
marker with therapeutic potential in endometrial cancer.
Oncotarget. 8:40402–40411. 2017.
|
|
12
|
Kojima M, Sone K, Oda K, Hamamoto R,
Kaneko S, Oki S, Kukita A, Machino H, Honjoh H, Kawata Y, et al:
The histone methyltransferase WHSC1 is regulated by EZH2 and is
important for ovarian clear cell carcinoma cell proliferation. BMC
Cancer. 19:4552019.
|
|
13
|
Kukita A, Sone K, Oda K, Hamamoto R,
Kaneko S, Komatsu M, Wada M, Honjoh H, Kawata Y, Kojima M, et al:
Histone methyltransferase SMYD2 selective inhibitor LLY-507 in
combination with poly ADP ribose polymerase inhibitor has
therapeutic potential against high-grade serous ovarian carcinomas.
Biochem Biophys Res Commun. 513:340–346. 2019.
|
|
14
|
Kojima M, Sone K, Oda K, Hamamoto R,
Kaneko S, Oki S, Kukita A, Kawata A, Honjoh H, Kawata Y, et al: The
histone methyltransferase SMYD2 is a novel therapeutic target for
the induction of apoptosis in ovarian clear cell carcinoma cells.
Oncol Lett. 20:1532020.
|
|
15
|
Wada M, Kukita A, Sone K, Hamamoto R,
Kaneko S, Komatsu M, Takahashi Y, Inoue F, Kojima M, Honjoh H, et
al: Epigenetic modifier SETD8 as a therapeutic target for
high-grade serous ovarian cancer. Biomolecules. 10:16862020.
|
|
16
|
Blanc RS and Richard S: Arginine
methylation: The coming of age. Mol Cell. 65:8–24. 2017.
|
|
17
|
Bedford MT and Clarke SG: Protein arginine
methylation in mammals: Who, what, and why. Mol Cell. 33:1–13.
2009.
|
|
18
|
Yang Y and Bedford MT: Protein arginine
methyltransferases and cancer. Nat Rev Cancer. 13:37–50. 2013.
|
|
19
|
Inoue F, Sone K, Toyohara Y, Tanimoto S,
Takahashi Y, Kusakabe M, Kukita A, Honjoh H, Nishijima A, Taguchi
A, et al: Histone arginine methyltransferase CARM1 selective
inhibitor TP-064 induces apoptosis in endometrial cancer. Biochem
Biophys Res Commun. 601:123–128. 2022.
|
|
20
|
Dowhan DH, Harrison MJ, Eriksson NA,
Bailey P, Pearen MA, Fuller PJ, Funder JW, Simpson ER, Leedman PJ,
Tilley WD, et al: Protein arginine methyltransferase 6-dependent
gene expression and splicing: Association with breast cancer
outcomes. Endocr Relat Cancer. 19:509–526. 2012.
|
|
21
|
Almeida-Rios D, Graça I, Vieira FQ,
Ramalho-Carvalho J, Pereira-Silva E, Martins AT, Oliveira J,
Gonçalves CS, Costa BM, Henrique R and Jerónimo C: Histone
methyltransferase PRMT6 plays an oncogenic role of in prostate
cancer. Oncotarget. 7:53018–53028. 2016.
|
|
22
|
Avasarala S, Wu PY, Khan SQ, Yanlin S, Van
Scoyk M, Bao J, Di Lorenzo A, David O, Bedford MT, Gupta V, et al:
PRMT6 promotes lung tumor progression via the alternate activation
of tumor-associated macrophages. Mol Cancer Res. 18:166–178.
2020.
|
|
23
|
Bouchard C, Sahu P, Meixner M, Nötzold RR,
Rust MB, Kremmer E, Feederle R, Hart-Smith G, Finkernagel F,
Bartkuhn M, et al: Genomic location of PRMT6-dependent H3R2
methylation is linked to the transcriptional outcome of associated
genes. Cell Rep. 24:3339–3352. 2018.
|
|
24
|
Jiang N, Li QL, Pan W, Li J, Zhang MF, Cao
T, Su SG and Shen H: PRMT6 promotes endometrial cancer via AKT/mTOR
signaling and indicates poor prognosis. Int J Biochem Cell Biol.
120:1056812020.
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
26
|
Ohtani H, Liu M, Zhou W, Liang G and Jones
PA: Switching roles for DNA and histone methylation depend on
evolutionary ages of human endogenous retroviruses. Genome Res.
28:1147–1157. 2018.
|
|
27
|
Maruyama R, Choudhury S, Kowalczyk A,
Bessarabova M, Beresford-Smith B, Conway T, Kaspi A, Wu Z,
Nikolskaya T, Merino VF, et al: Epigenetic regulation of cell
type-specific expression patterns in the human mammary epithelium.
PLoS Genet. 7:e10013692011.
|
|
28
|
Kim SW, Yoon SJ, Chuong E, Oyolu C, Wills
AE, Gupta R and Baker J: Chromatin and transcriptional signatures
for nodal signaling during endoderm formation in hESCs. Dev Biol.
357:492–504. 2011.
|
|
29
|
Chang CC, Huang RL, Wang HC, Liao YP, Yu
MH and Lai HC: High methylation rate of LMX1A, NKX6-1, PAX1, PTPRR,
SOX1, and ZNF582 genes in cervical adenocarcinoma. Int J Gynecol
Cancer. 24:201–209. 2014.
|
|
30
|
Li HJ, Yu PN, Huang KY, Su HY, Hsiao TH,
Chang CP, Yu MH and Lin YW: NKX6.1 functions as a metastatic
suppressor through epigenetic regulation of the
epithelial-mesenchymal transition. Oncogene. 35:2266–2278.
2016.
|
|
31
|
Chung HH, Lee CT, Hu JM, Chou YC, Lin YW
and Shih YL: NKX6.1 represses tumorigenesis, metastasis and
chemoresistance in colorectal cancer. J Mol Sci. 21:51062020.
|
|
32
|
Cheung LW and Mills GB: Targeting
therapeutic liabilities engendered by PIK3R1 mutations for cancer
treatment. Pharmacogenomics. 17:297–307. 2016.
|
|
33
|
Ishiguro K, Kitajima H, Niinuma T,
Maruyama R, Nishiyama N, Ohtani H, Sudo G, Toyota M, Sasaki H,
Yamamoto E, et al: Dual EZH2 and G9a inhibition suppresses multiple
myeloma cell proliferation by regulating the interferon signal and
IRF4-MYC axis. Cell Death Discov. 7:72021.
|
|
34
|
Roulois D, Loo Yau H, Singhania R, Wang Y,
Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al:
DNA-demethylating agents target colorectal cancer cells by inducing
viral mimicry by endogenous transcripts. Cell. 162:961–973.
2015.
|
|
35
|
Lim Y, Yu S, Yun JA, Do IG, Cho L, Kim YH
and Kim HC: The prognostic significance of protein arginine
methyltransferase 6 expression in colon cancer. Oncotarget.
9:9010–9020. 2018.
|
|
36
|
Okuno K, Akiyama Y, Shimada S, Nakagawa M,
Tanioka T, Inokuchi M, Yamaoka S, Kojima K and Tanaka S: Asymmetric
dimethylation at histone H3 arginine 2 by PRMT6 in gastric cancer
progression. Carcinogenesis. 40:15–26. 2019.
|
|
37
|
Stein C, Riedl S, Rüthnick D, Nötzold RR
and Bauer UM: The arginine methyltransferase PRMT6 regulates cell
proliferation and senescence through transcriptional repression of
tumor suppressor genes. Nucleic Acids Res. 40:9522–9533. 2012.
|
|
38
|
Kleinschmidt MA, de Graaf P, van Teeffelen
HA and Timmers HT: Cell cycle regulation by the PRMT6 arginine
methyltransferase through repression of cyclin-dependent kinase
inhibitors. PLoS One. 7:e414462012.
|
|
39
|
Rao A, Luo C and Hogan PG: Transcription
factors of the NFAT family: Regulation and function. Annu Rev
Immunol. 15:707–747. 1997.
|
|
40
|
Reppert S, Zinser E, Holzinger C, Sandrock
L, Koch S and Finotto S: NFATc1 deficiency in T cells protects mice
from experimental autoimmune encephalomyelitis. Eur J Immunol.
45:1426–1440. 2015.
|
|
41
|
Zhang Y, Alexander PB and Wang XF: TGF-β
family signaling in the control of cell proliferation and survival.
Cold Spring Harb Perspect Biol. 9:a0221452017.
|
|
42
|
Ribatti D, Tamma R and Annese T:
Epithelial-mesenchymal transition in cancer: A historical overview.
Transl Oncol. 13:1007732020.
|
|
43
|
Kriseman M, Monsivais D, Agno J, Masand
RP, Creighton CJ and Matzuk MM: Uterine double-conditional
inactivation of Smad2 and Smad3 in mice causes endometrial
dysregulation, infertility and uterine cancer. Proc Natl Acad Sci
USA. 116:3873–3882. 2019.
|
|
44
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005.
|
|
45
|
European Bioinformatics Institute; Ewan B,
Nick G, Arkadiusz K, Emmanuel M, Alistair RG, Guy S, Arne S, Abel
UV, Simon W, et al: Initial sequencing and comparative analysis of
the mouse genome. Nature. 420:520–562. 2002.
|
|
46
|
Goodier JL: Restricting retrotransposons:
A review. Mob DNA. 7:162016.
|
|
47
|
Chiappinelli KB, Strissel PL, Desrichard
A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et
al: Inhibiting DNA methylation causes an interferon response in
cancer via dsRNA including endogenous retroviruses. Cell.
169:3612017.
|