Colorectal cancer (CRC) is the third most commonly
diagnosed type of cancer globally and ranks second in the cause of
cancer-related mortality (1). It
has a momentous effect on the lives and health of individuals. The
5-year survival rates are related to the stage at which the disease
is detected; patients with stage I disease have a 5-year survival
range >90%, whereas for patients with stage IV disease, the
survival rate slightly exceeds 10% in clinical observations.
Screening has been shown to reduce CRC morbidity and mortality
(2). However, for patients in
which the disease is not detected in time, follow-up treatment
remains the sole method which allows them to continue to
survive.
The majority of surgical treatments for patients
with stage I and II CRC are based on surgical resection and the
dissection of the surrounding lymph nodes. Apart from a small
number of patients who develop post-operative intestinal knotting
and wound dehiscence, the majority have a good prognosis and 5-year
survival rates. At present, the majority of patients who are
eligible for surgical treatment, based on health status and the
stage of the disease (3,4), receive open surgery (5). Simultaneously, minimally invasive
surgeries, such as laparoscopic surgeries (6,7)
and robotic surgeries (8) have
gradually gained a place in surgical treatment options due to their
characteristics of diminished hemorrhagic tendencies, expedited
post-operative recuperation, and lighter immune and inflammatory
reactions than open surgeries.
Patients with stage III CRC frequently undergo
chemotherapy to mitigate the risk of recurrence. In tandem,
individuals with stage II or III CRC for whom surgical
interventions prove intolerable may resort to a combined approach,
where chemotherapy is complemented by radiation therapy. For
patients with stage IV disease, the main treatment option is
chemotherapy. Furthermore, adjuvant chemotherapy, peri-operative
chemotherapy (9), and neoadjuvant
chemoradiotherapy (10,11) not only reduce the disease stage,
but also curtail the likelihood of recurrence and elevate the
overall survival prospects for these patients. However,
hepatotoxicity caused by chemotherapeutic drugs (12), particularly in frail or
susceptible elderly patients (13), limits their application.
An increasing number of targeted therapies are also
being applied for the treatment of CRC, which significantly enhance
the prognosis of patients with malignant tumors compared to other
treatments, particularly in individuals diagnosed with metastatic
CRC (3,13). Currently, the most frequently used
targeted therapies include angiogenesis inhibitors, such as
bevacizumab and epidermal growth factor (EGF) inhibitors, such as
panitumumab and cetuximab, alongside BRAF inhibitory therapy and
HER2 inhibitory therapy (14).
Neural precursor cell-expressed developmentally downregulated 8
(NEDD8) has attracted widespread attention for its function in CRC.
CRC tissues exhibit an elevated expression of NEDD8, and the
participation of NEDD8 in neddylation plays a crucial role in the
migration, proliferation and survival of cancer cells. The present
review summarizes and discusses the influence and potential of
targeted NEDD8 therapy in CRC.
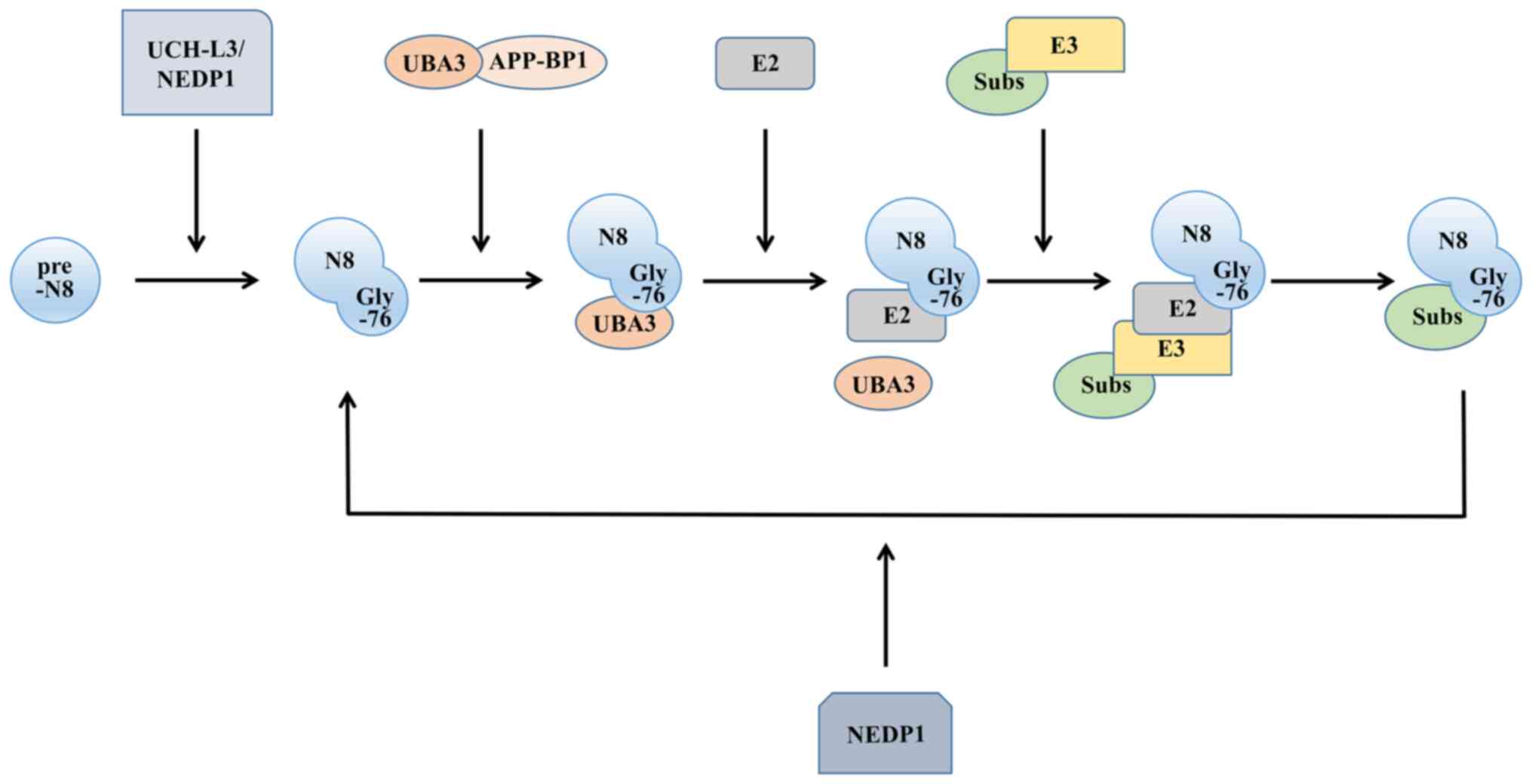
As a post-translational modification mechanism for
proteins, neddylation shares a number of similarities with
ubiquitination. There are four main enzymes involved in the
ubiquitin-like process: A precursor processing enzyme, NEDD
activating enzyme (E1), NEDD conjugating enzyme (E2) and NEDD
ligase (E3). The NEDD8 molecule is synthesized in a precursor form.
To expose the Gly-76 residue, the C-terminus of the protein
undergoes cleavage (17), where
the precursor processing enzymes activate, including ubiquitin
carboxyl-terminal hydrolase isozyme L3 (18), a common precursor processing
enzyme for NEDD8 and ubiquitin, and the NEDD8-specific precursor
processing enzyme NEDD8 protease 1 (19).
After processing the precursor, NEDD8 still needs to
be activated to function, similar to ubiquitin, and this process is
completed by E1. Functioning as a heterodimer, it comprises both
ubiquitin-like modifier activating enzyme 3 and amyloid beta
precursor protein binding protein 1. E1 then transfers NEDD8 to the
thiol lipid coupling intermediate through a transesterification
reaction (20). This forms the
E2-NEDD8 conjugates. The E2s discovered thus far mainly include
ubiquitin-conjugating enzyme (UBE)2M [also known as
ubiquitin-conjugating enzyme C12 (UBC12)] (21) and UBE2F (22). Finally, E3, with the function of
determining the specificity of the substrate, is required to
localize the NEDD8 molecule to the target molecule. Compared with
ubiquitination, neddylation has fewer types of E3s (Fig. 1).
NEDD8 exhibits specificity in binding to substrate
proteins. E3 ligases, in conjunction with their respective E2
binding enzymes, typically collaborate in recruiting specific
protein substrates to the substrate receptor domain of E3. NEDD8
then undergoes transfer from the independent active site of E3,
ultimately facilitating the interaction between NEDD8 and the
specific substrate proteins. For instance, ring box protein 1
demonstrates the capacity to use either UBE2M or UBE2F, affecting
the binding of NEDD8 to the specific lysine residue 720 within
cullin 1 (23). E2 also affects
the binding site of E3, which prevents inappropriate modifications
from occurring (21).
There are numerous substrates for neddylation, among
which the most characterized and well-documented are the cullin
family proteins (24). In
addition, other typical substrates are p53 (25), the Von Hippel-Lindau tumor
suppressor protein (pVHL) (26),
proliferating cell nuclear antigen (PCNA) (27), hypoxia-inducible factor (HIF)
(28), and murine double minute 2
(MDM2) (25).
Molecular scaffolds of cullin-ring ligase (CRL) E3
ubiquitin ligases involve cullin family proteins (29), which serve as adaptable
frameworks, connecting the active site of E2 enzymes with the
substrate binding site and facilitating the progression of the
ubiquitination process by bridging the spatial gap (30). There are seven types of cullins in
mammals: Cullin1, cullin2, cullin3, cullin4A, cullin4B, cullin5 and
cullin7 (31), of which cullin4A
was the first to be discovered (32). A crucial characteristic of CRL E3
is its dependence on the covalent modification of the cullin
protein by the NEDD8 molecule for its activation, resulting in a
relatively active conformation of the ring-type E3 ligase (33). Additionally, CRL E3 ubiquitin
ligases play crucial roles in coordinating the conjugation of
ubiquitin with substrate proteins. They are able to bind to the
target proteins and recruit E2 enzymes that are responsible for the
transfer of ubiquitin molecules (31). The neddylation of cullin proteins
can then instigate a profound transformation in their conformation,
promoting neddylation of the CRL substrate.
p53 is a tumor suppressor gene with transcription
factor activity related to DNA damage repair. In the case that its
function is inhibited, the capacity to efficiently mend DNA damage
is compromised, leading to its unabated accumulation. This, in
turn, paves the way for genome instability, ultimately fostering
the emergence of tumorigenesis. MDM2, a ring-type E3 ligase, is
mainly amplified in cancer. Notably, it holds the power to catalyze
the neddylation of p53, effectively curtailing the transcriptional
prowess of this vital transcription factor. At the same time, MDM2
can also undergo neddylation itself, thereby increasing its
stability and catalyzing p53 to undergo neddylation (25). Consequently, the functionality of
p53 is suppressed, leading to heightened genomic instability within
the malignant cells, thereby facilitating the progression of tumor
growth.
PCNA is involved in DNA synthesis and cell
proliferation. The molecular interplay between PCNA and NEDD8
possesses the capacity to counteract the process of ubiquitination.
However, PCNA that undergoes neddylation blocks the formation of
polη lesions, thereby inhibiting the recruitment of polη and
affecting DNA damage repair, which may cause genome instability and
lead to tumor occurrence (27).
Irregularities within the normal cellular
replication process cause the body to lose normal control of its
cell cycle and to prompt anomalous cell proliferation, -a decisive
sign of tumorigenesis (36). The
deficiency of enzymes involved in the neddylation process can
result in the inhibition of tumor proliferation. For example,
MLN4924, which selectively inhibits the E1 regulatory subunit [NEDD
activating enzyme (NAE)], can interfere with neddylation in a
number of types of cancer (37-39), particularly CRC (40), leading to cell cycle arrest or the
demise of the cell. In addition, SMAD ubiquitination regulatory
factor (Smurf)1, functioning as an E3 ligase, promotes its own
neddylation, a process which can subsequently enhance CRC
proliferation (41).
Metastasis encompasses the dissemination of cancer
cells from the initial location to neighboring tissues or other
organs in the body and their proliferation in the post-metastasis
site, which is the crucial cause of tumor occurrence and mortality
(36). MLN4924, an inhibitor of
neddylation, impedes the metastatic journey of neoplastic cells by
thwarting their intravascular persistence and the intricate
extravasation process (42).
NEDD8, via the s-phase kinase associated protein 2 (Skp2)/Slug
pathway, exerts a regulatory effect on the downregulation of
E-cadherin (43), consequently
triggering the activation of the epithelial-to-mesenchymal
transition (EMT) (44).
NEDD8 is inseparable from animal development and
plays a crucial regulatory role in cell metastasis and
proliferation. In addition, it is related to cell survival and
genetic changes. Previous research has confirmed that UBC12, highly
expressed in individuals with CRC, is involved in the activation
pathway of NEDD8. This upregulation in the expression of NEDD8 is
related to tumor cell proliferation and cloning ability (41).
CRC remains a significant contributor to
cancer-related mortality. Although patients can achieve certain
control of the disease through surgical resection and chemotherapy,
the majority of patients eventually succumb to the disease due to
tumor invasion and metastasis (68). The role of neddylation in CRC
primarily lies in fostering the proliferation and migration of
tumor cells, while concurrently protecting these cells from
apoptotic processes. It is evident that NEDD8 intricately
intertwines with the course of numerous cancers, including CRC, and
delving into the association between NEDD8 and CRC illustrates
paramount significance.
NEDD8 functions by covalently binding to substrates
in an ubiquitination-like manner (69). Its E3 not only has a ring scaffold
type, but also involves a HECT ligase. Smurf1 emerges as a
C2-WW-HECT ligase. Moreover, Smurf1 serves as a ubiquitin ligase
E3, whose expression is substantially heightened within CRC tissues
(41). Furthermore, in CRC
tissues, increased levels of Smurf1, NEDD8, NAE1 and UBC12
expression have been linked to the advancement of cancer and to
unfavorable clinical outcomes (41).
Smurf1 can be activated through neddylation,
promoting its association with E2. Additionally, Smurf1 possesses
the capacity to serve as an E3, thereby catalyzing its own
neddylation process (41). The
indispensable roles of NEDD8 and Smurf1 are evident in facilitating
the ubiquitination of RhoA, a protein mainly related to the
migration of tumor cells (70).
In breast cancer (71), elevated
Smurf1 expression has been substantiated as a consequence of
ERK-mediated phosphorylation, which results from the activation of
transforming growth factor β1 (TGF-β1) (72). It is worth noting that the
overexpression and activation of ERK have also been found in CRC
(73). Hence, it is conceivable
that a similar mechanism is enacted in CRC. However, some
researchers have illustrated that the expression of RhoA is
increased in CRC due to EGF (74).
Substantiated investigations have elucidated the
interconnection of RRP9 and NEDD8 through Smurf1 in CRC (79). RRP9, the integral element within
the U3 snoRNP complex, augments pre-rRNA processing via its
ubiquitination (79,81), an indispensable step in ribosome
biogenesis. This augmentation in ribosome biogenesis is frequently
associated with the increased proliferation observed in cancer as a
high production of ribosomes is necessary to maintain high levels
of cell growth and division (82,83). Therefore, Smurf1 exerts its E3
function and not only catalyzes RRP9 neddylation, but also
stimulates the proliferation and migration of CRC cells by
increasing pre-rRNA processing and upregulating ribosome biogenesis
(79). Therefore, Smurf1 exhibits
an elevated expression level and is activated through the process
of neddylation, promoting the development of colorectal tumors. In
the realm of CRC treatment, studies have illuminated that the
inhibition of Smurf1 in tumors manifests an amplified anti-tumor
efficacy in response to gemcitabine, cisplatin, and the
gemcitabine-cisplatin combination (84). This observation suggests that
addressing chemotherapy-resistant CRC by therapeutically targeting
NEDD8 to suppress Smurf1 activation holds substantial promise.
It has been found that the effect of NEDD8 on PTEN
is not derived from stabilizing its molecular structure, but from
facilitating the translocation of PTEN into the nucleus in breast
cancer cells. NEDD8 interacts with importin α, importin β and
importin 5 (IPO5), leading to the nuclear translocation of any PTEN
that has interacted with NEDD8 (87). Research on CRC cells has
demonstrated that a high expression of IPO5 was associated with
tumor development (90),
suggesting that NEDD8 promotes PTEN nuclear translocation in CRC
cells. NEDD8 stabilizes FASN through the NEDD8/XIAP/PTEN/FASN
pathway, thereby reducing FASN ubiquitination and promoting fat
synthesis (88). Elevated fat
synthesis affected by FASN stabilization is often related to tumor
cell proliferation (91). It has
also been confirmed that Smurf1 can ubiquitinate and degrade PTEN
in glioblastoma (92), which has
not yet been proven in CRC. In the domain of breast cancer therapy,
the targeted neddylation of PTEN has emerged as a highly promising
therapeutic strategy (93).
Building upon the analogous PTEN neddylation pathway, targeted
interventions along this route hold significant developmental
prospects in the context of CRC.
Abnormities in the Wnt/β-catenin signaling pathway
are prevalent within the majority of CRC cells. The constitutive
activation of this pathway promotes the propagation and survival of
CRC cells and is a main cause of adenoma formation (94). Principal among these anomalies is
the irregular expression of β-catenin, which serves as the primary
instigator of the irregular signaling of the Wnt/β-catenin pathway
in CRC cells. This perturbation exhibits an intricate association
with CSN5. CSN5 mainly affects an alternative degradation pathway
of β-catenin (95). In CRC cells,
the intricate web of interactions exists among β-catenin, CSN5, and
seven in absentia homolog 1 (SIAH-1), in which CSN5 plays a crucial
role in cellular processes, such as apoptosis and cell cycle
regulation. CRL activity can be regulated by deleting NEDD8
(96) to promote SIAH-1
degradation, causing β-catenin accumulation in CRC and promoting
the growth and multiplication of malignant cells (97). Adenoma genesis frequently emerges
as a pivotal risk factor in the onset of CRC (98). Consequently, the targeted
development of pharmaceutical agents addressing this pathway holds
the potential to contribute significantly to the prevention of CRC,
thereby mitigating the incidence of this malignancy.
Research has illustrated that inhibiting the NEDD8
pathway has a therapeutic effect on clinically invasive CRC and
that CRC cells exhibiting heightened sensitivity to NEDD8
inhibition often exhibit EMT transcriptional characteristics
(40). EMT is an important
prerequisite for CRC cells to metastasize (99), but the mechanisms of NEDD8 in
inducing CRC cell metastasis have not been fully elucidated. In
prostate cancer, which is also aggressive, it has been found that
NEDD8 can down-regulate E-cadherin and activate EMT through the
Skp2/Slug pathway to promote tumor cell metastasis (43). As a member of the substrate
recognition F-box protein family, Skp2 is a component of CRL1 whose
activity is regulated by NEDD8 (100). In other words, Skp2 depends on
NEDD8 for its function. NEDD8 combines with Skp2 to perform
neddylation, thereby activating the Skp2 downstream molecule Slug.
Slug activation may suppress EMT onset by downregulating E-cadherin
expression (101). Studies have
found that both Skp2 (102) and
Slug (103) exhibit a
significant upregulation within tissues afflicted by CRC. Moreover,
the downregulation of E-cadherin in these tissues also leads to the
occurrence of EMT (104). It can
be inferred that there is a mechanism in CRC tissues, whereby NEDD8
initiates the Skp2/Slug pathway, culminating in the downregulation
of E-cadherin. This, in turn, prompts neoplastic cells to undergo
the process of EMT, ultimately facilitating cellular
metastasis.
The process of DNA mismatch repair (MMR) is
evolutionarily conserved and serves to rectify mismatches arising
from DNA replication, evading proofreading mechanisms (105), in a key pathway in DNA repair.
The loss of MMR function induces a hypermutational phenotype, and
such cells are clinically identified through the genomic scarring
of MSI. It has been confirmed that the prognosis of individuals
with CRC is associated with the presence of MSI within the tumor
(106). Patients with MSI
develop intrinsic resistance to chemotherapy, making it difficult
to induce cancerous cell apoptosis by chemotherapy drugs, limiting
the number of effective treatments for patients (107). There are often a large number of
genome instabilities and mutations in MSI, resulting in cellular
proteome imbalances and aberrant protein accumulation in the cell.
To compensate for these proteins, NEDD8-mediated pathways are
indispensable for MSI tumors to clean misfolded aggregates, thereby
maintaining tumor cell survival (108).
Defects in apoptosis are the basis of tumorigenesis
and the main cause of chemotherapy failure (109). The role of the B-cell lymphoma 2
(Bcl-2) protein family in cellular apoptosis is of utmost
importance, particularly in mitochondrial apoptosis (110). Within the cellular proteome, the
Bcl-2 family can be segregated into two distinct categories:
Pro-apoptotic and anti-apoptotic. The stoichiometry of these two
categories determines whether the cell is apoptotic or not. When
the subtle balance is altered, a signal is transmitted through the
upstream molecule Bcl-2 homology domain only protein (BH3-only),
thereby activating Bcl-2-associated X protein (Bax) and
Bcl-2-associated K protein (Bak) on the mitochondrial surface,
leading to mitochondrial impairment and cell death (111). NOXA is not only a pro-apoptotic
protein, but is also a member of the BH3-only family. It plays a
key role in Bcl-2-mediated mitochondrial apoptosis, mainly by
selectively neutralizing anti-apoptotic Bcl-2 family proteins and
altering the balance of pro- and anti-apoptotic signals to cause
mitochondrial apoptosis (112).
Research has indicated an elevated protein expression of NOXA in
CRC; however, it has a short lifespan. The rapid degradation of
NOXA could potentially serve as a tactic employed by CRC cells to
evade the peril posed by elevated NOXA expression. Peroxiredoxin 1
exhibits a high expression level within CRC tissues, which enhances
cullin5 neddylation through the UBE2F/cullin5 pathway. This
neddylation can enhance the ubiquitination and degradation of NOXA
mediated by cullin5, resulting in increased tumor cell survival,
which is also reflected in the context of tumor cell resistance to
the chemotherapy drug etoposide (113). Addressing this pathway through
targeted interventions holds great promise in circumventing
resistance to etoposide in CRC, thereby presenting a novel
perspective for the chemotherapy of colorectal malignancies.
A central RNA-binding protein known as HuR plays a
crucial role in stabilizing cell proliferation-related mRNA to
regulate cell dedifferentiation, proliferation and survival
(114,115). A previous study validated the
significant association between the elevated expression of HuR and
the survival of cancerous cells in tumors (116), while the decreased expression of
HuR resulted in cell cycle arrest and promoted cellular apoptosis.
A previous study detected the upregulation of HuR and MDM2
expression in colorectal tumor tissues (117). Notably, an intricate correlation
has been established between HuR and MDM2. MDM2 has been previously
confirmed to be an E3 that inhibits the transcriptional activity of
p53 by catalyzing its neddylation (25). Moreover, MDM2 serves as an E3 in
CRC tissues. The catalytic substrate of MDM2 is converted into HuR
simultaneously, controlling the nuclear localization of HuR through
the NEDD8/MDM2/HuR pathway and protecting it from degradation. To
help tumor cells to survive, NEDD8 leads to the malignant
transformation of tumor cells (118).
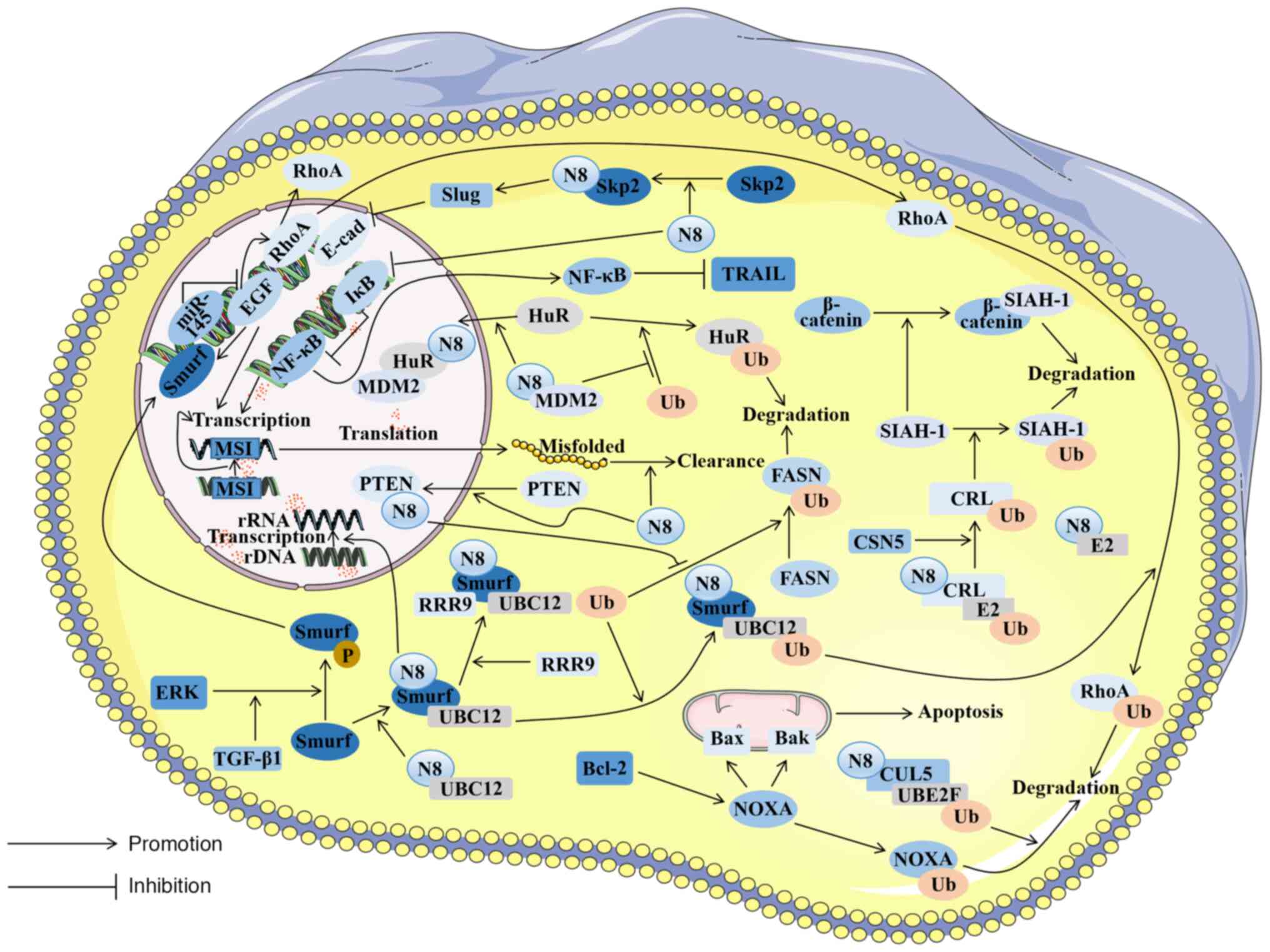
Chemoresistance is often related to patient
mortality and tumor metastasis in CRC; one form of chemoresistance
is resistance to oxaliplatin through the death receptor ligand [Fas
and tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL)] pathway (119). NF-κB
plays a pivotal role in the regulation of death receptor signaling
pathways (120). In
chemoresistant diffuse large B-cell lymphoma, it has been found
that neddylation resists tumor cell death by downregulating
inhibitory factor IκB and upregulating NF-κB. MLN4924 can reverse
such effects (121). The
abnormal expression of NF-κB has also been detected in CRC cells
(122). In an experiment
involving the intraperitoneal injection of mutant IκB colon cells
into mice suffering from metastatic CRC, it was discovered that
this approach improved the survival rate of the mice, suggesting
that NF-κB inhibition mediates the cell-killing effect caused by
TRAIL (123). Similarly, NEDD8
is abnormally activated in CRC tissues. It can be inferred that
NEDD8's presence in CRC inhibits IκB from activating the NF-κB
pathway, thereby inhibiting TRAIL-mediated cell killing and
allowing cancer cells to survive in the face of chemical drugs. In
light of the deleterious ramifications stemming from cancer cells'
resistance to platinum-based drugs, notably oxaliplatin, in the
majority of CRCs (123), the
targeted development of pharmaceutical agents along this pathway
holds immense promise. This endeavor has the potential to furnish
novel therapeutic strategies for treatment-resistant CRC (Fig. 2).
Neddylation is closely connected to the metastasis,
proliferation and survival of CRC cells. Previous research has
confirmed that inhibiting neddylation mediated by NEDD8 causes the
apoptosis of CRC cells (124).
Thus, targeting the neddylation pathway is expected to become a
novel treatment method for CRC. Concurrently, such targeted therapy
may also find application in combination with CRC treatment drugs
to improve the responsiveness of CRC cells to therapeutic
agents.
Neddylation plays a crucial role in the progression
of CRC. Therefore, the process of developing pharmaceutical
interventions targeting NEDD8 has prompted notable advancements in
the treatment of CRC. MLN4924 is a selective inhibitor of NAE1. It
was first confirmed that MLN4924 could affect the activity of E1 to
disrupt the cullin-ring ligase-mediated protein turnover as a NEDD8
substrate (124). Subsequently,
further research demonstrated that MLN4924 functions as a selective
inhibitor by competitively inhibiting the assembly of the
NEDD8-MLN4924 compound. When it is deprived of its capacity to
partake in subsequent enzymatic reactions, MLN4924 blocks
neddylation in CRC cells and ultimately ushers in the onset of
cellular death (125). As
research on MLN4924 progressed further, its role as a
radiosensitizer was demonstrated, illustrating that it could make
CRC cells more sensitive to radiation, primarily through p27
accumulation in cells significantly improving the arrest of
radiation-induced cell cycle G2/M phases, DNA damage responses and
cellular death (53). The method
of CRC cell death using MLN4924 was subsequently further studied.
MLN4924 stabilizes p53 to induce ribosome stress, leading to the
death of tumor cells. Mitoxantrone 1 also has similar action
characteristics (126). As a
typical p53 target gene death receptor, TRAIL receptor 2 expression
is also significantly upregulated during the process (127). On the other hand, MLN4924
induces cell death by activating Bax and Bak on the outer
mitochondrial membrane to initiate mitochondrial outer membrane
permeabilization (127). During
this biological process, there is also the upregulation in the
expression of BH3 interacting domain death agonist (BID). As a
member of the pro-apoptotic Bcl2 family, BID activates the
downstream targets of caspase-8 in the exogenous pathway (128), causing CRC tumor cell death.
Currently, the application of MLN4924 in the treatment of CRC is
still being studied in the phase I/II clinical stages. These
experimental results demonstrate that this drug can control the
progression of cancer through the apoptosis of cancer cells
(129,130). MLN4924 is additionally being
used in synergy with conventional chemotherapy agents to enhance
their efficacy significantly. For example, MLN4924 can induce DNA
damage response and upregulate cell cycle checkpoint kinase 2
protein expression levels to render CRC cells more sensitive to
oxaliplatin (131). At the same
time, MLN4924 can also be combined with irinotecan as a
chemotherapeutic method to combat chemotherapy-refractory p53
mutant CRC (127).
However, with the use of MLN4924, CRC tissues will
inevitably become resistant. This may be related to FLICE
inhibitory protein in MLN4924-treated cells, which leads to
resistance to standard care therapies (127). It may also be associated with
the variation of NAE, which reduces the affinity of the enzyme to
MLN4924 and ATP and increases NEDD8 activation (132).
Beyond its anticipated therapeutic effects in
targeted therapy, MLN4924 has demonstrated certain unintended
consequences that may foster cancer cell proliferation. These
outcomes are primarily associated with the activation of
intracellular ERK and JNK signaling, leading to activator protein-1
activation (133).
Simultaneously, the inhibitory effects of MLN4924-induced
neddylation suppression can upregulate T-cell negative regulator
programmed death-ligand 1 (PD-L1) expression (134). Cancer cells exhibiting a high
expression of PD-L1 can attenuate T-cell cytotoxicity through the
activation of the PD-1/PD-L1 axis, inducing immune suppression
(135). Combining anti-PD-L1
antibodies or mitogen-activated protein kinase inhibitors with
MLN4924 administration offers a potential avenue to address these
challenges effectively (133).
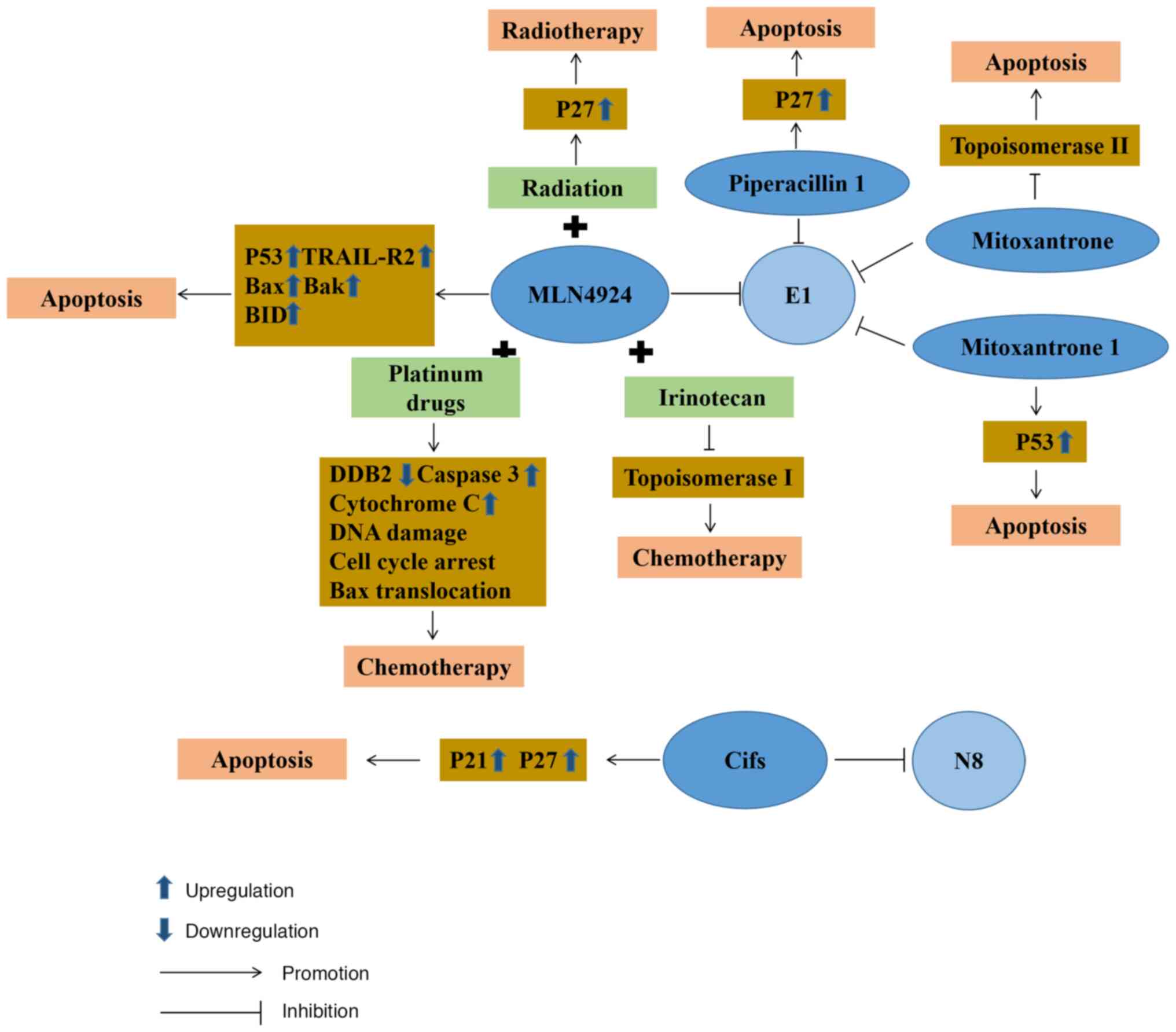
Since developing new drugs remains costly, there is
a trend to seek to repurpose existing approved and investigational
drugs given their known safety profile and to reduce costs
(136). Piperacillin, known as a
β-lactam antibiotic, is often used for the management of suspected
bacterial infections relying on empirical treatment combined with
tazobactam (137). It is
frequently used in cancer patients to treat fever attacks following
chemotherapy and neutropenia (138,139). Based on the integrated virtual
screening method, it has been found that piperacillin 1 (140) can inhibit the degradation of
p27, known as a downstream protein substrate of NAE1, the
regulatory subunit of E1. It possesses an ATP-competitive inhibitor
without the ability to form the covalent bonds of E1 in the future.
Mitoxantrone, as a DNA topoisomerase II poison, causes tumor cell
apoptosis by inhibiting DNA synthesis and delaying cell cycle
progression, which can improve the therapeutic properties of
anthracyclines (141,142). Through an FDA-approved drug
database analysis by virtual screening, mitoxantrone 1 (126) was found to have highly selective
NAE activity and to compete with ATP, with great potential as an
inhibitor of E1.
Cifs are capable of impeding the activity of
cullin-ring E3 ubiquitin ligases, resulting in cell cycle arrest.
At the same time, Cifs also participate in cancer progression by
deamidating NEDD8 (143). In CRC
cells, cells that regulate Cif expression by doxycycline lead to
tumor apoptosis through p21 and p27 accumulation (144). In terms of specific use,
attenuated Salmonella typhimurium VNP20009 can be used to
deliver Cif genes to tumor cells, inducing the expression and
intracellular accumulation of proteins p27 and p21, thereby
inhibiting the growth of tumor cells (145).
Platinum-based pharmaceuticals can be employed in
the management of CRC and are amenable to concomitant
administration with various classes of antineoplastic agents.
Commonly used platinum drugs include oxaliplatin, carboplatin and
cisplatin, all of which have received global approval (146). Cisplatin is a first-generation
platinum drug and interferes with DNA repair by crosslinking with
the purine bases of DNA, leading to DNA damage and stimulating
tumor cell death (147).
Nevertheless, cisplatin lacks the specificity to selectively target
CRC cells, consequently leading to diminished accumulative
concentrations and a discernible impact on the therapeutic efficacy
(148). During the analysis of
tissues from patients with squamous cell carcinoma in the head and
neck, it was discovered that MLN4924 possesses the capacity to
impede the transcription of DDB2 facilitated by E2F1 (149). Since DDB2 demonstrates a crucial
function in modulating sensitivity to cisplatin, the combined
application of MLN4924 and cisplatin can increase the activity of
cisplatin. Moreover, tissues with a suppressed expression of
cullin4A exhibit higher susceptibility to cisplatin, which
potentially relates to its connection with DNA repair. In addition,
the enhancing effect of MLN4924 on cisplatin has been found in
breast cancer (150), pancreatic
cancer (151) and cervical
tumors (152). It can be
concluded that the combined application of MLN4924 and cisplatin in
CRC tissues may also enhance the sensitivity of cisplatin to
compensate for the impact of its low accumulation concentration.
Carboplatin is a cisplatin derivative and a second-generation
platinum drug whose mode of action is similar to that of cisplatin.
It induces tumor cell apoptosis by damaging DNA. Compared with
cisplatin, carboplatin produces inter-chain and intra-chain
doublets or single adducts following application (153). In individuals with advanced
solid tumors, a clinical phase I trial revealed that E1 inhibitor,
MLN4924, combined with carboplatin was well-tolerated and stood as
a promising benchmark for forthcoming drug development against CRC
(130).
Oxaliplatin, a third-generation platinum therapeutic
agent, is a first-line chemotherapy drug. As one of the more
commonly used chemotherapeutic drugs following surgical resection,
it plays a pivotal role in the management of CRC. It initiates the
death of malignant cells by orchestrating a halt in cell cycle
progression at the critical juncture of G2/M, thus initiating an
apoptotic cascade that includes Bax mitochondrial translocation,
the release of cytochrome c and caspase-3 catalytic
activation (154). In a previous
a study on CRC tissues, it was found that MLN4924 combined with
oxaliplatin increased oxaliplatin-induced apoptosis (131). Although it has not yet entered
clinical trials for CRC, phase I trials have been carried out in
the therapeutic management of gastric carcinoma (155). CRC cells have also developed
some drug resistance to platinum agents, such as the inhibition of
drug accumulation in tumor cells, as well as the acquisition of
EMT. A large number of studies on drug resistance have found that
exosomes loaded with miR-128-3p (156) and the application of
nanoparticles (157) can
re-sensitize CRC cells to platinum drugs in vivo.
Irinotecan is used as a second-line
chemotherapeutic agent employed in the treatment of patients with
advanced stages of CRC in the event that first-line chemotherapy
drug treatment fails. It not only selectively inhibits
topoisomerase I, causing tumor cell death by inhibiting DNA
function, but also has potent anticancer functions following
intracellular modification in cells (158). SN38, the metabolite of
irinotecan in vivo, has a synergistic effect with MLN4924.
Apoptosis induced by this pathway proceeds in a manner that does
not depend on the presence of p53 and circumvents the effects of
TP53 mutations in advanced-stag CRC cells (127). This reveals a novel pathway for
managing patients grappling with advanced CRC, particularly those
battling chemotherapy-resistant p53 mutant CRC. However, it has not
yet entered the clinical trial stage. In terms of drug resistance,
epigenetic alterations are likely to cause resistance to
irinotecan, such as the acetylation of histones in CRC. Therefore,
partially drug-resistant CRC can be combated by combining histone
deacetylase inhibitors with irinotecan (159) (Fig. 3).
In recent years, post-translational modifications
of proteins have gradually attracted wide attention. Owing to this,
a more in-depth understanding of the ubiquitin-like modification of
neddylation has been obtained. Existing research evidence indicates
that NEDD8 is primarily related to the proliferation, migration,
survival and genetic alterations of tumor cells, and to the
microenvironment of tumorigenesis. In CRC, NEDD8 is of paramount
significance in the onset and progression of the disease,
particularly within the realms of tumor cell migration,
proliferation and viability. It is through these mechanisms that
anticancer pharmaceuticals directed toward NEDD8 offer novel
insight into therapeutic approaches for CRC. They mainly target
NAEs for anticancer treatment. There are also drugs, such as cycle
inhibitors that target the NEDD8 substrate cullin-ring E3 ubiquitin
ligase for anticancer effects. Simultaneously, amalgamated
pharmacological interventions, exemplified by the synergistic
deployment of MLN4924 alongside irinotecan or by the combination of
MLN4924 and oxaliplatin, represent promising strategies in the
battle against partially resistant or recalcitrant CRC.
CRC poses one of the greatest threats to human life
and health worldwide. Consequently, relentless exploration into the
etiology and therapeutic modalities for CRC is imperative. As NEDD8
emerges as a nascent frontier in the arena of anticancer targets,
it necessitates a more profound investigation. At present,
treatments mainly target NAEs. Regrettably, limited attention has
been devoted to agents targeting E2, E3 and NEDD8 substrates of the
NEDD8 pathway. The potential emergence of NEDD8 substrates,
including Smurf, IκB, cullin 5 and PTEN, as novel targets for
therapeutic intervention, holds promise. Simultaneously, the
deneddylation of CSN5 exhibits pharmaceutical design potential.
Henceforth, other more effective targeted drugs need to be designed
for the pathways that cause CRC. At the same time, for some NEDD8
target drugs that have been used, drug resistance issues should
also be paid investigated during the period of treatment, such as
the emergence of MLN4924 resistance. In future research, an
exploration of novel therapeutic analogs or strategies to combat
drug resistance is paramount. Consequently, studying the
pathogenesis of CRC based on NEDD8 and unearthing drug targets for
CRC are of utmost importance for the prognosis and survival of
patients with CRC.
Not applicable.
TW and XL contributed equally to the present study;
they were involved in the conception and design of the study, and
in the drafting of the manuscript. RM and JS were involved in the
analysis of the literature. SH, ZS and MW conceived and supervised
the study, and directed the writing. All the authors have read and
approved the final version of this review. Data authentication is
not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
The present study was supported by the Shandong Natural Science
Foundation (grant no. ZR2021QH046).
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
3
|
Miller KD, Siegel RL, Lin CC, Mariotto AB,
Kramer JL, Rowland JH, Stein KD, Alteri R and Jemal A: Cancer
treatment and survivorship statistics, 2016. CA Cancer J Clin.
66:271–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roque-Castellano C, Fariña-Castro R,
Nogués-Ramia EM, Artiles-Armas M and Marchena-Gómez J: Colorectal
cancer surgery in selected nonagenarians is relatively safe and it
is associated with a good long-term survival: An observational
study. World J Surg Oncol. 18:1202020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Salibasic M, Pusina S, Bicakcic E, Pasic
A, Gavric I, Kulovic E, Rovcanin A and Beslija S: Colorectal cancer
surgical treatment, our experience. Med Arch. 73:412–414. 2019.
View Article : Google Scholar
|
|
6
|
Luo W, Wu M and Chen Y: Laparoscopic
versus open surgery for elderly patients with colorectal cancer: A
systematic review and meta-analysis of matched studies. ANZ J Surg.
92:2003–2017. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ustuner MA, Deniz A and Simsek A:
Laparoscopic <em>versus</em> open surgery in colorectal
cancer: Is laparoscopy safe enough? J Coll Physicians Surg Pak.
32:1170–1174. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu G, Zhang S, Zhang Y, Fu X and Liu X:
Robotic surgery in rectal cancer: Potential, challenges, and
opportunities. Curr Treat Options Oncol. 23:961–979. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Riesco-Martinez MC, Modrego A,
Espinosa-Olarte P, La Salvia A and Garcia-Carbonero R:
Perioperative chemotherapy for liver metastasis of colorectal
cancer: Lessons learned and future perspectives. Curr Treat Options
Oncol. 23:1320–1337. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Habr-Gama A, Perez RO, São Julião GP,
Proscurshim I and Gama-Rodrigues J: Nonoperative approaches to
rectal cancer: A critical evaluation. Semin Radiat Oncol.
21:234–239. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsu YJ, Chern YJ, Lai IL, Chiang SF, Liao
CK, Tsai WS, Hung HY, Hsieh PS, Yeh CY, Chiang JM, et al:
Usefulness of close surveillance for rectal cancer patients after
neoadjuvant chemoradiotherapy. Open Med (Wars). 17:1438–1448. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McWhirter D, Kitteringham N, Jones RP,
Malik H, Park K and Palmer D: Chemotherapy induced hepatotoxicity
in metastatic colorectal cancer: A review of mechanisms and
outcomes. Crit Rev Oncol Hematol. 88:404–415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim JH: Chemotherapy for colorectal cancer
in the elderly. World J Gastroenterol. 21:5158–5166. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Piawah S and Venook AP: Targeted therapy
for colorectal cancer metastases: A review of current methods of
molecularly targeted therapy and the use of tumor biomarkers in the
treatment of metastatic colorectal cancer. Cancer. 125:4139–4147.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumar S, Tomooka Y and Noda M:
Identification of a set of genes with developmentally
down-regulated expression in the mouse brain. Biochem Biophys Res
Commun. 185:1155–1161. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kumar S, Yoshida Y and Noda M: Cloning of
a cDNA which encodes a novel ubiquitin-like protein. Biochem
Biophys Res Commun. 195:393–399. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kamitani T, Kito K, Nguyen HP and Yeh ET:
Characterization of NEDD8, a developmentally down-regulated
ubiquitin-like protein. J Biol Chem. 272:28557–28562. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wada H, Kito K, Caskey LS, Yeh ET and
Kamitani T: Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem
Biophys Res Commun. 251:688–692. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mendoza HM, Shen LN, Botting C, Lewis A,
Chen J, Ink B and Hay RT: NEDP1, a highly conserved cysteine
protease that deNEDDylates Cullins. J Biol Chem. 278:25637–25643.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gong L and Yeh ET: Identification of the
activating and conjugating enzymes of the NEDD8 conjugation
pathway. J Biol Chem. 274:12036–12042. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang DT, Zhuang M, Ayrault O and Schulman
BA: Identification of conjugation specificity determinants unmasks
vestigial preference for ubiquitin within the NEDD8 E2. Nat Struct
Mol Biol. 15:280–287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang DT, Ayrault O, Hunt HW, Taherbhoy
AM, Duda DM, Scott DC, Borg LA, Neale G, Murray PJ, Roussel MF and
Schulman BA: E2-RING expansion of the NEDD8 cascade confers
specificity to cullin modification. Mol Cell. 33:483–495. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baek K, Scott DC and Schulman BA: NEDD8
and ubiquitin ligation by cullin-RING E3 ligases. Curr Opin Struct
Biol. 67:101–109. 2021. View Article : Google Scholar :
|
|
24
|
Lydeard JR, Schulman BA and Harper JW:
Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO
Rep. 14:1050–1061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xirodimas DP, Saville MK, Bourdon JC, Hay
RT and Lane DP: Mdm2-mediated NEDD8 conjugation of p53 inhibits its
transcriptional activity. Cell. 118:83–97. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stickle NH, Chung J, Klco JM, Hill RP,
Kaelin WG Jr and Ohh M: pVHL modification by NEDD8 is required for
fibronectin matrix assembly and suppression of tumor development.
Mol Cell Biol. 24:3251–3261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guan J, Yu S and Zheng X: NEDDylation
antagonizes ubiquitination of proliferating cell nuclear antigen
and regulates the recruitment of polymerase η in response to
oxidative DNA damage. Protein Cell. 9:365–379. 2018.
|
|
28
|
Ryu JH, Li SH, Park HS, Park JW, Lee B and
Chun YS: Hypoxia-inducible factor α subunit stabilization by NEDD8
conjugation is reactive oxygen species-dependent. J Biol Chem.
286:6963–6970. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brown JS and Jackson SP: Ubiquitylation,
neddylation and the DNA damage response. Open Biol. 5:1500182015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu J and Nussinov R: Flexible cullins in
cullin-RING E3 ligases allosterically regulate ubiquitination. J
Biol Chem. 286:40934–40942. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sarikas A, Hartmann T and Pan ZQ: The
cullin protein family. Genome Biol. 12:2202011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Osaka F, Kawasaki H, Aida N, Saeki M,
Chiba T, Kawashima S, Tanaka K and Kato S: A new NEDD8-ligating
system for cullin-4A. Genes Dev. 12:2263–2268. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Petroski MD and Deshaies RJ: Function and
regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol.
6:9–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nam SY, Ko YS, Jung J, Yoon J, Kim YH,
Choi YJ, Park JW, Chang MS, Kim WH and Lee BL: A hypoxia-dependent
upregulation of hypoxia-inducible factor-1 by nuclear factor-κB
promotes gastric tumour growth and angiogenesis. Br J Cancer.
104:166–174. 2011. View Article : Google Scholar
|
|
35
|
Semenza GL: HIF-1 and mechanisms of
hypoxia sensing. Curr Opin Cell Biol. 13:167–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kittai AS, Danilova OV, Lam V, Liu T,
Bruss N, Best S, Fan G and Danilov AV: NEDD8-activating enzyme
inhibition induces cell cycle arrest and anaphase catastrophe in
malignant T-cells. Oncotarget. 12:2068–2074. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lan H, Tang Z, Jin H and Sun Y:
Neddylation inhibitor MLN4924 suppresses growth and migration of
human gastric cancer cells. Sci Rep. 6:242182016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu H, Bei Q and Luo X: MLN4924 inhibits
cell proliferation by targeting the activated neddylation pathway
in endometrial carcinoma. J Int Med Res.
49:30006052110185922021.PubMed/NCBI
|
|
40
|
Picco G, Petti C, Sassi F, Grillone K,
Migliardi G, Rossi T, Isella C, Di Nicolantonio F, Sarotto I,
Sapino A, et al: Efficacy of NEDD8 pathway inhibition in
preclinical models of poorly differentiated, clinically aggressive
colorectal cancer. J Natl Cancer Inst. 109:djw2092017. View Article : Google Scholar
|
|
41
|
Xie P, Zhang M, He S, Lu K, Chen Y, Xing
G, Lu Y, Liu P, Li Y, Wang S, et al: The covalent modifier Nedd8 is
critical for the activation of Smurf1 ubiquitin ligase in
tumorigenesis. Nat Commun. 5:37332014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jiang Y, Liang Y, Li L, Zhou L, Cheng W,
Yang X, Yang X, Qi H, Yu J, Jeong LS, et al: Targeting neddylation
inhibits intravascular survival and extravasation of cancer cells
to prevent lung-cancer metastasis. Cell Biol Toxicol. 35:233–245.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mickova A, Kharaishvili G, Kurfurstova D,
Gachechiladze M, Kral M, Vacek O, Pokryvkova B, Mistrik M, Soucek K
and Bouchal J: Skp2 and slug are coexpressed in aggressive prostate
cancer and inhibited by neddylation blockade. Int J Mol Sci.
22:28442021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tan KL and Pezzella F: Inhibition of NEDD8
and FAT10 ligase activities through the degrading enzyme NEDD8
ultimate buster 1: A potential anticancer approach. Oncol Lett.
12:4287–4296. 2016. View Article : Google Scholar
|
|
45
|
Watson IR, Blanch A, Lin DC, Ohh M and
Irwin MS: Mdm2-mediated NEDD8 modification of TAp73 regulates its
transactivation function. J Biol Chem. 281:34096–34103. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Aoki I, Higuchi M and Gotoh Y: NEDDylation
controls the target specificity of E2F1 and apoptosis induction.
Oncogene. 32:3954–3964. 2013. View Article : Google Scholar
|
|
47
|
Halazonetis TD, Gorgoulis VG and Bartek J:
An oncogene-induced DNA damage model for cancer development.
Science. 319:1352–1355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Garvin AJ: Beyond reversal: ubiquitin and
ubiquitin-like proteases and the orchestration of the DNA double
strand break repair response. Biochem Soc Trans. 47:1881–1893.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Meir M, Galanty Y, Kashani L, Blank M,
Khosravi R, Fernández-Ávila MJ, Cruz-García A, Star A, Shochot L,
Thomas Y, et al: The COP9 signalosome is vital for timely repair of
DNA double-strand breaks. Nucleic Acids Res. 43:4517–4530. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gâtel P, Piechaczyk M and Bossis G:
Ubiquitin, SUMO, and Nedd8 as therapeutic targets in cancer. Adv
Exp Med Biol. 1233:29–54. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36:522017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sun Y, Baechler SA, Zhang X, Kumar S,
Factor VM, Arakawa Y, Chau CH, Okamoto K, Parikh A, Walker B, et
al: Targeting neddylation sensitizes colorectal cancer to
topoisomerase I inhibitors by inactivating the DCAF13-CRL4
ubiquitin ligase complex. Nat Commun. 14:37622023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wan J, Zhu J, Li G and Zhang Z:
Radiosensitization of human colorectal cancer cells by MLN4924: An
Inhibitor of NEDD8-Activating Enzyme. Technol Cancer Res Treat.
15:527–534. 2016. View Article : Google Scholar
|
|
54
|
Shao Y, Liu Z, Song X, Sun R, Zhou Y,
Zhang D, Sun H, Huang J, Wu C, Gu W, et al: ALKBH5/YTHDF2-mediated
m6A modification of circAFF2 enhances radiosensitivity of
colorectal cancer by inhibiting Cullin neddylation. Clin Transl
Med. 13:e13182023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Whiteside TL: The tumor microenvironment
and its role in promoting tumor growth. Oncogene. 27:5904–5912.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhou L, Jiang Y, Luo Q, Li L and Jia L:
Neddylation: A novel modulator of the tumor microenvironment. Mol
Cancer. 18:772019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chang FM, Reyna SM, Granados JC, Wei SJ,
Innis-Whitehouse W, Maffi SK, Rodriguez E, Slaga TJ and Short JD:
Inhibition of neddylation represses lipopolysaccharide-induced
proinflammatory cytokine production in macrophage cells. J Biol
Chem. 287:35756–35767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li L, Liu B, Dong T, Lee HW, Yu J, Zheng
Y, Gao H, Zhang Y, Chu Y, Liu G, et al: Neddylation pathway
regulates the proliferation and survival of macrophages. Biochem
Biophys Res Commun. 432:494–498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhou L, Jiang Y, Liu X, Li L, Yang X, Dong
C, Liu X, Lin Y, Li Y, Yu J, et al: Promotion of tumor-associated
macrophages infiltration by elevated neddylation pathway via
NF-κB-CCL2 signaling in lung cancer. Oncogene. 38:5792–5804. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Joyce JA and Fearon DT: T cell exclusion,
immune privilege, and the tumor microenvironment. Science.
348:74–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jin HS, Liao L, Park Y and Liu YC:
Neddylation pathway regulates T-cell function by targeting an
adaptor protein Shc and a protein kinase Erk signaling. Proc Natl
Acad Sci USA. 110:624–629. 2013. View Article : Google Scholar :
|
|
62
|
Jiang Y, Li L, Li Y, Liu G, Hoffman RM and
Jia L: Neddylation regulates macrophages and implications for
cancer therapy. Front Cell Dev Biol. 9:6811862021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Best S, Lam V, Liu T, Bruss N, Kittai A,
Danilova OV, Murray S, Berger A, Pennock ND, Lind EF and Danilov
AV: Immunomodulatory effects of pevonedistat, a NEDD8-activating
enzyme inhibitor, in chronic lymphocytic leukemia-derived T cells.
Leukemia. 35:156–168. 2021. View Article : Google Scholar :
|
|
64
|
Maishi N and Hida K: Tumor endothelial
cells accelerate tumor metastasis. Cancer Sci. 108:1921–1926. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tang N, Wang L, Esko J, Giordano FJ, Huang
Y, Gerber HP, Ferrara N and Johnson RS: Loss of HIF-1alpha in
endothelial cells disrupts a hypoxia-driven VEGF autocrine loop
necessary for tumorigenesis. Cancer Cell. 6:485–495. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Shi CS, Kuo KL, Lin WC, Chen MS, Liu SH,
Liao SM, Hsu CH, Chang YW, Chang HC and Huang KH: Neddylation
inhibitor, MLN4924 suppresses angiogenesis in huvecs and solid
cancers: in vitro and in vivo study. Am J Cancer Res. 10:953–964.
2020.PubMed/NCBI
|
|
67
|
Yao WT, Wu JF, Yu GY, Wang R, Wang K, Li
LH, Chen P, Jiang YN, Cheng H, Lee HW, et al: Suppression of tumor
angiogenesis by targeting the protein neddylation pathway. Cell
Death Dis. 5:e10592014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Tauriello DVF, Palomo-Ponce S, Stork D,
Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M,
Ibiza S, Cañellas A, Hernando-Momblona X, et al: TGFβ drives immune
evasion in genetically reconstituted colon cancer metastasis.
Nature. 554:538–543. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kerscher O, Felberbaum R and Hochstrasser
M: Modification of proteins by ubiquitin and ubiquitin-like
proteins. Annu Rev Cell Dev Biol. 22:159–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
He S, Cao Y, Xie P, Dong G and Zhang L:
The Nedd8 Non-covalent Binding Region in the Smurf HECT Domain is
Critical to its Ubiquitn Ligase Function. Sci Rep. 7:413642017.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zheng J, Shi Z, Yang P, Zhao Y, Tang W, Ye
S, Xuan Z, Chen C, Shao C, Wu Q and Sun H: ERK-Smurf1-RhoA
signaling is critical for TGFβ-drived EMT and tumor metastasis.
Life Sci Alliance. 5:e2021013302022. View Article : Google Scholar
|
|
72
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-beta1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Fang JY and Richardson BC: The MAPK
signalling pathways and colorectal cancer. Lancet Oncol. 6:322–327.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cardoso AP, Pinto ML, Pinto AT, Oliveira
MI, Pinto MT, Gonçalves R, Relvas JB, Figueiredo C, Seruca R,
Mantovani A, et al: Macrophages stimulate gastric and colorectal
cancer invasion through EGFR Y(1086), c-Src, Erk1/2 and Akt
phosphorylation and smallGTPase activity. Oncogene. 33:2123–2133.
2014. View Article : Google Scholar
|
|
75
|
Price JT, Wilson HM and Haites NE:
Epidermal growth factor (EGF) increases the in vitro invasion,
motility and adhesion interactions of the primary renal carcinoma
cell line, A704. Eur J Cancer. 32A:1977–1982. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li S, Wu X, Xu Y, Wu S, Li Z, Chen R,
Huang N, Zhu Z and Xu X: miR-145 suppresses colorectal cancer cell
migration and invasion by targeting an ETS-related gene. Oncol Rep.
36:1917–1926. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kwon A, Lee HL, Woo KM, Ryoo HM and Baek
JH: SMURF1 plays a role in EGF-induced breast cancer cell migration
and invasion. Mol Cells. 36:548–555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ridley AJ, Schwartz MA, Burridge K, Firtel
RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR: Cell
migration: Integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Du MG, Liu F, Chang Y, Tong S, Liu W, Chen
YJ and Xie P: Neddylation modification of the U3 snoRNA-binding
protein RRP9 by Smurf1 promotes tumorigenesis. J Biol Chem.
297:1013072021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Barrios-Rodiles M, Brown KR, Ozdamar B,
Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW,
et al: High-throughput mapping of a dynamic signaling network in
mammalian cells. Science. 307:1621–1625. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Clerget G, Bourguignon-Igel V,
Marmier-Gourrier N, Rolland N, Wacheul L, Manival X, Charron C,
Kufel J, Méreau A, Senty-Ségault V, et al: Synergistic defects in
pre-rRNA processing from mutations in the U3-specific protein Rrp9
and U3 snoRNA. Nucleic Acids Res. 48:3848–3868. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Pecoraro A, Pagano M, Russo G and Russo A:
Ribosome Biogenesis and Cancer: Overview on Ribosomal Proteins. Int
J Mol Sci. 22:54962021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Pelletier J, Thomas G and Volarević S:
Ribosome biogenesis in cancer: New players and therapeutic avenues.
Nat Rev Cancer. 18:51–63. 2018. View Article : Google Scholar
|
|
84
|
Guo J, Xu G, Mao C and Wei R: Low
Expression of Smurf1 Enhances the Chemosensitivity of Human
Colorectal Cancer to Gemcitabine and Cisplatin in Patient-Derived
Xenograft Models. Transl Oncol. 13:1008042020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Salmena L, Carracedo A and Pandolfi PP:
Tenets of PTEN tumor suppression. Cell. 133:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Xie P, Peng Z, Chen Y, Li H, Du M, Tan Y,
Zhang X, Lu Z, Cui CP, Liu CH, et al: Neddylation of PTEN regulates
its nuclear import and promotes tumor development. Cell Res.
31:291–311. 2021. View Article : Google Scholar :
|
|
88
|
Finicle BT, Jayashankar V and Edinger AL:
Nutrient scavenging in cancer. Nat Rev Cancer. 18:619–633. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Murphy N, Song M, Papadimitriou N,
Carreras-Torres R, Langenberg C, Martin RM, Tsilidis KK, Barroso I,
Chen J, Frayling TM, et al: Associations Between Glycemic Traits
and Colorectal Cancer: A Mendelian Randomization Analysis. J Natl
Cancer Inst. 114:740–752. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhang W, Lu Y and Li X, Zhang J, Lin W,
Zhang W, Zheng L and Li X: IPO5 promotes the proliferation and
tumourigenicity of colorectal cancer cells by mediating RASAL2
nuclear transportation. J Exp Clin Cancer Res. 38:2962019.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Xia Q, Zhang H, Zhang P, Li Y, Xu M, Li X,
Li X and Dong L: Oncogenic Smurf1 promotes PTEN wild-type
glioblastoma growth by mediating PTEN ubiquitylation. Oncogene.
39:5902–5915. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Du MG, Peng ZQ, Gai WB, Liu F, Liu W, Chen
YJ, Li HC, Zhang X, Liu CH, Zhang LQ, et al: The Absence of PTEN in
Breast Cancer Is a Driver of MLN4924 Resistance. Front Cell Dev
Biol. 9:6674352021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Phelps RA, Chidester S, Dehghanizadeh S,
Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW and
Jones DA: A two-step model for colon adenoma initiation and
progression caused by APC loss. Cell. 137:623–634. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Schütz AK, Hennes T, Jumpertz S, Fuchs S
and Bernhagen J: Role of CSN5/JAB1 in Wnt/β-catenin activation in
colorectal cancer cells. FEBS Lett. 586:1645–1651. 2012. View Article : Google Scholar
|
|
96
|
Cope GA and Deshaies RJ: COP9 signalosome:
A multifunctional regulator of SCF and other cullin-based ubiquitin
ligases. Cell. 114:663–671. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Jumpertz S, Hennes T, Asare Y, Vervoorts
J, Bernhagen J and Schütz AK: The β-catenin E3 ubiquitin ligase
SIAH-1 is regulated by CSN5/JAB1 in CRC cells. Cell Signal.
26:2051–2059. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Sninsky JA, Shore BM, Lupu GV and Crockett
SD: Risk Factors for Colorectal Polyps and Cancer. Gastrointest
Endosc Clin N Am. 32:195–213. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Aiello NM, Maddipati R, Norgard RJ, Balli
D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EE, et al:
EMT Subtype Influences Epithelial Plasticity and Mode of Cell
Migration. Dev Cell. 45:681–695.e84. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Asmamaw MD, Liu Y, Zheng YC, Shi XJ and
Liu HM: Skp2 in the ubiquitin-proteasome system: A comprehensive
review. Med Res Rev. 40:1920–1949. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Serrano-Gomez SJ, Maziveyi M and Alahari
SK: Regulation of epithelial-mesenchymal transition through
epigenetic and post-translational modifications. Mol Cancer.
15:182016. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yu X, Zhou L, Liu W, Liu L, Gao F, Li W
and Liu H: Skp2 stabilizes Mcl-1 and confers radioresistance in
colorectal cancer. Cell Death Dis. 13:2492022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Chen P, Li X, Zhang R, Liu S, Xiang Y,
Zhang M, Chen X, Pan T, Yan L, Feng J, et al: Combinative treatment
of β-elemene and cetuximab is sensitive to KRAS mutant colorectal
cancer cells by inducing ferroptosis and inhibiting
epithelial-mesenchymal transformation. Theranostics. 10:5107–5119.
2020. View Article : Google Scholar :
|
|
104
|
Wang L, Li S, Luo H, Lu Q and Yu S: PCSK9
promotes the progression and metastasis of colon cancer cells
through regulation of EMT and PI3K/AKT signaling in tumor cells and
phenotypic polarization of macrophages. J Exp Clin Cancer Res.
41:3032022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Kunkel TA and Erie DA: DNA mismatch
repair. Annu Rev Biochem. 74:681–710. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Guastadisegni C, Colafranceschi M, Ottini
L and Dogliotti E: Microsatellite instability as a marker of
prognosis and response to therapy: A meta-analysis of colorectal
cancer survival data. Eur J Cancer. 46:2788–2798. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
de la Chapelle A and Hampel H: Clinical
relevance of microsatellite instability in colorectal cancer. J
Clin Oncol. 28:3380–3387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
McGrail DJ, Garnett J, Yin J, Dai H, Shih
DJH, Lam TNA, Li Y, Sun C, Li Y, Schmandt R, et al: Proteome
Instability Is a Therapeutic Vulnerability in Mismatch
Repair-Deficient Cancer. Cancer Cell. 37:371–386.e12. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: A link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kim H, Rafiuddin-Shah M, Tu HC, Jeffers
JR, Zambetti GP, Hsieh JJ and Cheng EH: Hierarchical regulation of
mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell
Biol. 8:1348–1358. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wong WW and Puthalakath H: Bcl-2 family
proteins: The sentinels of the mitochondrial apoptosis pathway.
IUBMB Life. 60:390–397. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ploner C, Kofler R and Villunger A: Noxa:
At the tip of the balance between life and death. Oncogene.
27(Suppl 1): S84–S92. 2008. View Article : Google Scholar
|
|
113
|
Xu S, Ma Y, Tong Q, Yang J, Liu J, Wang Y,
Li G, Zeng J, Fang S, Li F, et al: Cullin-5 neddylation-mediated
NOXA degradation is enhanced by PRDX1 oligomers in colorectal
cancer. Cell Death Dis. 12:2652021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Brennan CM and Steitz JA: HuR and mRNA
stability. Cell Mol Life Sci. 58:266–277. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang W, Caldwell MC, Lin S, Furneaux H and
Gorospe M: HuR regulates cyclin A and cyclin B1 mRNA stability
during cell proliferation. EMBO J. 19:2340–2350. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Abdelmohsen K and Gorospe M:
Posttranscriptional regulation of cancer traits by HuR. Wiley
Interdiscip Rev RNA. 1:214–229. 2010. View
Article : Google Scholar
|
|
117
|
McLarnon A: Cancer: Mdm2-regulated
stabilization of HuR by neddylation in HCC and colon cancer-a
possible target for therapy. Nat Rev Gastroenterol Hepatol.
9:42011.
|
|
118
|
Embade N, Fernández-Ramos D, Varela-Rey M,
Beraza N, Sini M, Gutiérrez de Juan V, Woodhoo A, Martínez-López N,
Rodríguez-Iruretagoyena B, Bustamante FJ, et al: Murine double
minute 2 regulates Hu antigen R stability in human liver and colon
cancer through NEDDylation. Hepatology. 55:1237–1248. 2012.
View Article : Google Scholar
|
|
119
|
Greenlee JD, Lopez-Cavestany M,
Ortiz-Otero N, Liu K, Subramanian T, Cagir B and King MR:
Oxaliplatin resistance in colorectal cancer enhances TRAIL
sensitivity via death receptor 4 upregulation and lipid raft
localization. Elife. 10:e677502021. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Lee SJ, Lee DE, Choi SY and Kwon OS:
OSMI-1 Enhances TRAIL-Induced Apoptosis through ER Stress and NF-κB
Signaling in Colon Cancer Cells. Int J Mol Sci. 22:110732021.
View Article : Google Scholar
|
|
121
|
Paiva C, Godbersen JC, Rowland T, Danilova
OV, Danes C, Berger A and Danilov AV: Pevonedistat, a
Nedd8-activating enzyme inhibitor, sensitizes neoplastic B-cells to
death receptor-mediated apoptosis. Oncotarget. 8:21128–21139. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Sakamoto K, Maeda S, Hikiba Y, Nakagawa H,
Hayakawa Y, Shibata W, Yanai A, Ogura K and Omata M: Constitutive
NF-kappaB activation in colorectal carcinoma plays a key role in
angiogenesis, promoting tumor growth. Clin Cancer Res.
15:2248–2258. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Linares J, Sallent-Aragay A,
Badia-Ramentol J, Recort-Bascuas A, Méndez A, Manero-Rupérez N, Re
DL, Rivas EI, Guiu M, Zwick M, et al: Long-term platinum-based drug
accumulation in cancer-associated fibroblasts promotes colorectal
cancer progression and resistance to therapy. Nat Commun.
14:7462023. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Soucy TA, Smith PG, Milhollen MA, Berger
AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP,
Critchley S, et al: An inhibitor of NEDD8-activating enzyme as a
new approach to treat cancer. Nature. 458:732–736. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Brownell JE, Sintchak MD, Gavin JM, Liao
H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt
AL, et al: Substrate-assisted inhibition of ubiquitin-like
protein-activating enzymes: The NEDD8 E1 inhibitor MLN4924 forms a
NEDD8-AMP mimetic in situ. Mol Cell. 37:102–111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Wu KJ, Zhong HJ, Li G, Liu C, Wang HD, Ma
DL and Leung CH: Structure-based identification of a
NEDD8-activating enzyme inhibitor via drug repurposing. Eur J Med
Chem. 143:1021–1027. 2018. View Article : Google Scholar
|
|
127
|
Ferris J, Espona-Fiedler M, Hamilton C,
Holohan C, Crawford N, McIntyre AJ, Roberts JZ, Wappett M, McDade
SS, Longley DB and Coyle V: Pevonedistat (MLN4924): Mechanism of
cell death induction and therapeutic potential in colorectal
cancer. Cell Death Discov. 6:612020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Sekeres MA, Watts J, Radinoff A, Sangerman
MA, Cerrano M, Lopez PF, Zeidner JF, Campelo MD, Graux C, Liesveld
J, et al: Randomized phase 2 trial of pevonedistat plus azacitidine
versus azacitidine for higher-risk MDS/CMML or low-blast AML.
Leukemia. 35:2119–2124. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zhou X, Sedarati F, Faller DV, Zhao D,
Faessel HM, Chowdhury S, Bolleddula J, Li Y, Venkatakrishnan K and
Papai Z: Phase I study assessing the mass balance,
pharmacokinetics, and excretion of [14C]-pevonedistat, a
NEDD8-activating enzyme inhibitor in patients with advanced solid
tumors. Invest New Drugs. 39:488–498. 2021. View Article : Google Scholar
|
|
131
|
Zheng W, Luo Z, Zhang J, Min P, Li W, Xu
D, Zhang Z, Xiong P, Liang H and Liu J: Neural precursor cell
expressed, developmentally downregulated 8-activating enzyme
inhibitor MLN4924 sensitizes colorectal cancer cells to oxaliplatin
by inducing DNA damage, G2 cell cycle arrest and apoptosis. Mol Med
Rep. 15:2795–2801. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Toth JI, Yang L, Dahl R and Petroski MD: A
gatekeeper residue for NEDD8-activating enzyme inhibition by
MLN4924. Cell Rep. 1:309–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zhang S, You X, Xu T, Chen Q, Li H, Dou L
and Sun Y, Xiong X, Meredith MA and Sun Y: PD-L1 induction via the
MEK-JNK-AP1 axis by a neddylation inhibitor promotes
cancer-associated immunosuppression. Cell Death Dis. 13:8442022.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Zhou S, Zhao X, Yang Z, Yang R, Chen C,
Zhao K, Wang W, Ma Y, Zhang Q and Wang X: Neddylation inhibition
upregulates PD-L1 expression and enhances the efficacy of immune
checkpoint blockade in glioblastoma. Int J Cancer. 145:763–774.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Gong J, Chehrazi-Raffle A, Reddi S and
Salgia R: Development of PD-1 and PD-L1 inhibitors as a form of
cancer immunotherapy: A comprehensive review of registration trials
and future considerations. J Immunother Cancer. 6:82018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Issa NT, Stathias V, Schürer S and
Dakshanamurthy S: Machine and deep learning approaches for cancer
drug repurposing. Semin Cancer Biol. 68:132–142. 2021. View Article : Google Scholar
|
|
137
|
Gin A, Dilay L, Karlowsky JA, Walkty A,
Rubinstein E and Zhanel GG: Piperacillin-tazobactam: A
beta-lactam/beta-lactamase inhibitor combination. Expert Rev Anti
Infect Ther. 5:365–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Maarbjerg SF, Thorsted A, Friberg LE,
Nielsen EI, Wang M, Schrøder H and Albertsen BK: Continuous
infusion of piperacillin-tazobactam significantly improves target
attainment in children with cancer and fever. Cancer Rep (Hoboken).
5:e15852022. View Article : Google Scholar
|
|
139
|
Rosanova MT, Cuellar-Pompa L and Lede R:
Efficacy and safety of empirical treatment with
piperacillin/tazobactan as monotherapy in episodes of neutropenia
and fever in children with cancer: Systematic review and
meta-analysis. Rev Chilena Infectol. 38:488–494. 2021.In Spanish.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zhong HJ, Liu LJ, Chan DS, Wang HM, Chan
PW, Ma DL and Leung CH: Structure-based repurposing of FDA-approved
drugs as inhibitors of NEDD8-activating enzyme. Biochimie.
102:211–215. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Evison BJ, Sleebs BE, Watson KG, Phillips
DR and Cutts SM: Mitoxantrone, more than just another topoisomerase
II poison. Med Res Rev. 36:248–299. 2016. View Article : Google Scholar
|
|
142
|
Faulds D, Balfour JA, Chrisp P and Langtry
HD: Mitoxantrone. A review of its pharmacodynamic and
pharmacokinetic properties, and therapeutic potential in the
chemotherapy of cancer. Drugs. 41:400–449. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Taieb F, Nougayrède JP and Oswald E: Cycle
inhibiting factors (cifs): Cyclomodulins that usurp the
ubiquitin-dependent degradation pathway of host cells. Toxins
(Basel). 3:356–368. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Liu L, Ni J, Zhang J and He X:
Construction and characterization of regulated cycle inhibiting
factors induced upon Tet-On system in human colon cancer cell
lines. Anticancer Drugs. 29:854–860. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Liu L, Zhang J, Gu M, Li G, Ni J and Fan
M: Antitumor effect of cycle inhibiting factor expression in colon
cancer via salmonella VNP20009. Anticancer Agents Med Chem.
20:1722–1727. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Wheate NJ, Walker S, Craig GE and Oun R:
The status of platinum anticancer drugs in the clinic and in
clinical trials. Dalton Trans. 39:8113–8127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Tchounwou PB, Dasari S, Noubissi FK, Ray P
and Kumar S: Advances in our understanding of the molecular
mechanisms of action of cisplatin in cancer therapy. J Exp
Pharmacol. 13:303–328. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Li W, Sun Y, Chen J, Jiang Z and Yang J:
PEGylated cisplatin nanoparticles for treating colorectal cancer in
a pH-Responsive manner. J Immunol Res. 2022:80239152022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Jones TM, Espitia CM, Ooi A, Bauman JE,
Carew JS and Nawrocki ST: Targeted CUL4A inhibition synergizes with
cisplatin to yield long-term survival in models of head and neck
squamous cell carcinoma through a DDB2-mediated mechanism. Cell
Death Dis. 13:3502022. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Misra S, Zhang X, Wani NA, Sizemore S and
Ray A: Both BRCA1-wild type and -mutant triple-negative breast
cancers show sensitivity to the NAE inhibitor MLN4924 which is
enhanced upon MLN4924 and cisplatin combination treatment.
Oncotarget. 11:784–800. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Zeng Y, Iv YS, Pan QH, Zhou YG and Li H:
An overactive neddylation pathway serves as a therapeutic target
and MLN4924 enhances the anticancer activity of cisplatin in
pancreatic cancer. Oncol Lett. 18:2724–2732. 2019.PubMed/NCBI
|
|
152
|
Lin WC, Kuo KL, Shi CS, Wu JT, Hsieh JT,
Chang HC, Liao SM, Chou CT, Chiang CK, Chiu WS, et al: MLN4924, a
Novel NEDD8-activating enzyme inhibitor, exhibits antitumor
activity and enhances cisplatin-induced cytotoxicity in human
cervical carcinoma: In vitro and in vivo study. Am J Cancer Res.
5:3350–3362. 2015.
|
|
153
|
Ho GY, Woodward N and Coward JI: Cisplatin
versus carboplatin: Comparative review of therapeutic management in
solid malignancies. Crit Rev Oncol Hematol. 102:37–46. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Arango D, Wilson AJ, Shi Q, Corner GA,
Arañes MJ, Nicholas C, Lesser M, Mariadason JM and Augenlicht LH:
Molecular mechanisms of action and prediction of response to
oxaliplatin in colorectal cancer cells. Br J Cancer. 91:1931–1946.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Shoji H, Takahari D, Hara H, Nagashima K,
Adachi J and Boku N: A phase I study of pevonedistat plus
capecitabine plus oxaliplatin in patients with advanced gastric
cancer refractory to platinum (NCCH-1811). Future Sci OA.
7:FSO7212021. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Liu T, Zhang X, Du L, Wang Y, Liu X, Tian
H, Wang L, Li P, Zhao Y, Duan W, et al: Exosome-transmitted
miR-128-3p increase chemosensitivity of oxaliplatin-resistant
colorectal cancer. Mol Cancer. 18:432019. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Buyana B, Naki T, Alven S and Aderibigbe
BA: Nanoparticles loaded with platinum drugs for colorectal cancer
therapy. Int J Mol Sci. 23:112612022. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Hicks LD, Hyatt JL, Stoddard S, Tsurkan L,
Edwards CC, Wadkins RM and Potter PM: Improved, selective, human
intestinal carboxylesterase inhibitors designed to modulate
7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin
(Irinotecan; CPT-11) toxicity. J Med Chem. 52:3742–3752. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Meisenberg C, Ashour ME, El-Shafie L, Liao
C, Hodgson A, Pilborough A, Khurram SA, Downs JA, Ward SE and
El-Khamisy SF: Epigenetic changes in histone acetylation underpin
resistance to the topoisomerase I inhibitor irinotecan. Nucleic
Acids Res. 45:1159–1176. 2017.PubMed/NCBI
|