Introduction
Ovarian cancer, one of the most lethal gynecological
malignancies, affects 240,000 women worldwide annually, with a
five-year survival rate <45% (1). Ovarian cancers can be classified
into different types based on clinical behavior, histopathology,
and molecular and genetic analyses. These include type I (low-grade
serous carcinomas, low-grade endometrioid carcinomas, clear cell
carcinomas and sero-mucinous carcinomas) and type II (high-grade
serous carcinomas, high-grade endometrioid carcinomas and
undifferentiated carcinomas) tumors, with epithelial ovarian
malignancies accounting for the majority (2). Previous epidemiological studies and
meta-analyses have identified several risk factors for ovarian
cancer, including family genetic history (for example, mutations in
BRCA1 and BRCA2 genes), contraceptive use, short lactation
duration, a body mass index ≥30 kg/m2, and other
gynecological diseases such as vaginitis and polycystic ovary
syndrome (1,3).
Curative and survival trends in ovarian cancer have
not significantly improved owing to the challenges of early
diagnosis, including the lack of clear screening tools, and
indistinct signs and symptoms. Moreover, high metastasis and
recurrence rates and drug resistance to chemotherapy are also
important reasons for the poor prognosis of patients with ovarian
cancer (4). Therefore, it is
important to identify potential ovarian cancer targets and clarify
their roles and molecular mechanisms in the malignant biological
behavior of ovarian cancer.
Cancer stem cells (CSCs), a subpopulation within
tumors, possess self-renewal and differentiation capacities akin to
those of stem cells, thus sustaining tumor growth and the
regeneration of an heterogeneous tumor mass (5,6).
Accumulating evidence indicates that CSCs exist in numerous types
of tumors, including leukemia, breast, rectal and ovarian cancer. A
previous study revealed that CSCs have become the significant
drivers of chemoresistance in ovarian cancer (7). Hu et al (8) in 2010 also found that the CSCs
residing in ovarian epithelial tumors are not targeted by
chemotherapy, which is the primary cause of chemotherapy failure.
Therefore, a therapeutic approach is required to eliminate rapidly
proliferating differentiated cancer cells and slow-proliferating
drug-resistant CSCs. The generation of CSCs is highly regulated by
the molecular process of epithelial-mesenchymal transition (EMT),
which plays a key role in the growth and metastasis of tumors
(9,10), Therefore, determining the key
mediators participating in the regulation of CSC function, such as
EMT, could provide a potential therapeutic target for the treatment
of ovarian cancer metastasis.
The Hippo signaling pathway controls tissue growth
and cell fate, and the dysregulation of Hippo activity leads to the
development of tumors, including ovarian cancer. In mammals, the
core components of the Hippo pathway are a pair of related
serine/threonine kinases, including mammalian STE20-like protein
kinase 1 (MST1) and 2 (MST2) and the large tumor suppressor kinases
(LATS1/2) (11). Activation of
Hippo signaling restricts tissue growth by promoting
LATS1/2-dependent phosphorylation of the homologous oncoproteins,
including Yes-associated protein (YAP) and transcriptional
co-activator with PDZ-binding motif (TAZ). The phosphorylated (p-)
YAP and TAZ accumulate in the cytoplasm, where they are degraded by
ubiquitination-dependent proteasome (12). By contrast, the inhibition of
Hippo signaling results in tissue growth and cell viability via YAP
and TAZ, which translocate to the nucleus to transactivate targets
in cooperation with the TEAD transcription factor.
As a member of the SWItch/Sucrose Non-Fermenting
(SWI/SNF) chromatin remodeling complex, AT-rich binding domain 1A
(ARID1A) uses the energy of ATP hydroxylation to reshape the
chromatin structure and can slide nucleosomes along the DNA
template (13). Abnormal ARID1A
expression occurs at a high frequency in congenital conditions and
various cancers, including ovarian cancer (14,15). ARID1A participates in regulating
the expression of various target genes in nucleus, whose alteration
gives rise to tumor progression. For example, in bladder cancer,
co-mutations in the ARID1A, GPRC5A and MLL2 genes enhance the
self-renewal and tumorigenesis of bladder cancer non-stem cells,
which contribute to the characteristics of cancer cell stemness
(16), suggesting the role of
ARID1A as a stemness mediator in cancer cells.
Different from the previous findings that ARID1A
commonly functions by affecting gene expression in nucleus, in the
present study, it was reported that ARID1A inhibits EMT and
stemness in ovarian cancer cells by participating in the regulation
of Hippo signaling activity, with the downregulated cell viability,
migration and colony formation. This suggests that ARID1A functions
as a tumor suppressor in ovarian cancer. In addition, the current
data exhibited that Hippo activity reciprocally exerts effects on
ARID1A expression, indicating a feedback regulation between Hippo
and ARID1A in ovarian cancer cells. Thus, ARID1A could be a novel
prognostic molecule for ovarian cancer and a potential target for
drug development for the treatment of ovarian cancer.
Materials and methods
Cell lines and cell culture
Human ovarian cancer cell lines SK-OV-3 and A2780
and 293T cells were obtained from the American Type Culture
Collection (ATCC), and were maintained in high glucose DMEM (Thermo
Fisher Scientific, Inc.) supplemented with 10% (v/v) fetal bovine
serum (FBS; Thermo Fisher Scientific, Inc.) and 1% (v/v)
penicillin/streptomycin (Beyotime Institute of Biotechnology) as
previously described (17-19).
All the cells were cultured at 37°C in a humidified chamber with 5%
CO2.
Generation of SK-OV-3-derived ovarian
cancer stem-like cells (OCSCs)
The SK-OV-3 derived OCSCs were generated as
previously described (20).
Briefly, SK-OV-3 cells were harvested and washed with FBS-free DMEM
medium twice, then re-suspended and maintained under stem cell
conditions by serum-free in DMEM medium supplemented with 5 mg/ml
insulin (Sigma-Aldrich; Merck KGaA), 10 ng/ml human recombinant
epidermal growth factor (Invitrogen; Thermo Fisher Scientific,
Inc.), 10 ng/ml basic fibroblast growth factor (Invitrogen; Thermo
Fisher Scientific, Inc.) and 0.3% bovine serum albumin
(Sigma-Aldrich; Merck KGaA). Culture media were changed every 2
days by centrifuging the cells at 60 × g for 5 min at room
temperature to remove the dead cell debris and SK-OV-3 cells
proliferated as non-adherent spheres consequently in this
condition. The cells of non-adherent spheres were subjected to
identification of stem cell-like properties before following
experiments.
Antibodies and reagents
Antibodies for ARID1A (mouse; cat. no. sc-32761),
YAP (cat. no. sc-101199), CTGF (cat. no. sc-373936), CYR61 (cat.
no. sc-374129), Nanog (cat. no. sc-293121), Sox2 (cat. no.
sc-365823), Oct3/4 (cat. no. sc-5279), GAPDH (cat. no. sc-32233)
and normal mouse IgG (cat. no. sc-2025) were purchased from Santa
Cruz Biotechnology, Inc. Antibodies for p-YAP antibody (cat. no.
AF5965), α-Tubulin (cat. no. AF0001), N-cadherin (cat. no. AF5237)
and E-cadherin (cat. no. AF0138) were obtained from Beyotime
Institute of Biotechnology. p-MST1 (cat. no. bs-3294R), MST1 (cat.
no. bs-3504R), p-LATS1 (cat. no. bs-3245R), LATS1 (cat. no.
bs-2904R), TAZ (cat. no. bs-12367R) and Vimentin (cat. no.
bs-23063R) antibodies were purchased from BIOSS. Normal rabbit IgG
(cat. no. 2729) and p-TAZ antibody (cat. no. 59971) were obtained
from Cell Signaling Technology, Inc.
Short hairpin RNA (shRNA) sequences,
viruses and infection
Plasmids expressing ARID1A-shRNA were generated by
insertion with the hairpin shRNA templates of complementary
oligonucleotides at the sites of XbaI and NotI into the shRNA
expression vector, Pll3.7. ShRNA sequences used are as follows:
shRNA sequence targeting ARID1A: 5'-AAC CAA AGT TAC TGT TGT TTA-3';
and a scrambled shRNA sequence (5'-TTT GTA CTA CAC AAA AGT ACTG-3')
was used as control. Sequences were obtained from Shanghai Sangon
Biotechnology Co. Ltd. Lentiviruses expressing ARID1A-shRNA or
scrambled-shRNA were generated by co-transfecting the HEK293T
packaging cells with lentiviral shRNA expression vector, and
lentiviruses-containing supernatants with the titers greater than
1×106 cfu/ml was used for infection of cells in the
presence of 8 μg/ml polybrene (Sigma-Aldrich; Merck KGaA) as
previously described (21).
Transfection
Transient transfection was performed using
Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) with Opti-MEM (cat. no, 31985-062; Invitrogen; Thermo Fisher
Scientific, Inc.) medium according to the manufacturer's
instructions. Briefly, SK-OV-3 or A2780 cells were transfected with
10 μg indicated plasmids. At 12 h after transfection, the
cells were cultured in fresh medium, and the cells were collected
48 h after transfection. Then the cellular lysates were subjected
to reverse transcription-quantitative PCR (RT-qPCR), western
blotting (WB) or other assays.
RNA isolation and RT-qPCR
Total RNA was extracted from SK-OV-3 cells and A2780
cells by using a TRIzol reagent (Takara Biotechnology Co., Ltd.)
according to the manufacturer's instructions. 5 μg of total
RNA was reversely transcribed by using SuperScript III reagent
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol, and the oligo-(deoxythymidine) primer with incubation at
42°C for 1 h. After the termination of cDNA synthesis, mRNA levels
of target genes were determined by RT-qPCR as previously described
(21). Briefly, the initial
denaturation was 95°C for 5 min; followed by 40 cycles of
denaturation (95°C, 10 sec), annealing and extension (60°C, 30
sec). All reactions were conducted in a 20 μl reaction
volume in triplicate. The relative expression of the mRNA levels
was normalized to the GAPDH levels, and the relative difference in
mRNA levels was calculated by the 2-ΔΔCq method
(22). The primers used were as
follows: ARID1A forward, 5'-TCA TGC CCA ACC TTC GTA TC-3' and
reverse, 5'-GAT GGC TGC TGG GAG TATG-3'; CTGF forward, 5'-CCT GTG
CAG CAT GGA CGTT-3' and reverse, 5'-GGA CCA GGC AGT TGG CTC TAA-3';
CYR61 forward, 5'-CTC CCT GTT TTT GGA ATG GA-3' and reverse, 5'-TGG
TCT TGC TGC ATT TCT TG-3'; Nanog forward, 5'-CCA TCC TTG CAA ATG
TCT TCTG-3' and reverse, 5'-CTT TGG GAC TGG TGG AAG AATC-3'; Sox2
forward, 5'-GTG GTT ACC TCT TCC TCC CACT-3' and reverse, 5'-AGT GCT
GGG ACA TGT GAA GTCT-3'; Oct3/4 forward, 5'-GTG GAG GAA GCT GAC AAC
AATG-3' and reverse, 5'-AAT TCT CCA GGT TGC CTC TCA CT-3';
E-Cadherin forward, 5'-ACC AAC GAT AAT CCT CCG AT-3' and reverse,
5'-TCA GTG TGG TGA TTA CGA CG-3'; N-Cadherin forward, 5'-AAT CCT
CCA GAG TTT ACT GC-3' and reverse, 5'-TCC TTA TCG GTC ACA GTT
AG-3'; Vimentin forward, 5'-GAG AGG AAG CCG AAA ACAC-3' and
reverse, 5'-TGC GTT CAA GGT CAA GACG-3'; and GAPDH forward, 5'-CCT
GTT CGA CAG TCA GCCG-3' and reverse, 5'-CGA CCA AAT CCG TTG ACT
CC-3'.
WB
WB was performed using standard protocols as
previously described (23).
Briefly, cells were harvested and rinsed with pre-chilled PBS on
ice, and then cell lysates were prepared using RIPA buffer
(Beyotime Institute of Biotechnology). BCA protein assay kit was
conducted to detect the concentration of protein. Generally, 50
μg of total protein was subjected to 8-15% sodium dodecyl
sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto PVDF membrane (MilliporeSigma). Membranes were
then blocked with 5% non-fat milk at room temperature for 1 h
followed by incubation with different primary antibodies (1:1,000)
at 4°C overnight. The membranes were then incubated with
corresponding HRP-labeled Goat Anti-Rabbit IgG (H + L) (1:1,000
cat. no. A0208΄ Beyotime Institute of Biotechnology) for 1 h at
room temperature the following day. Signals were subsequently
detected using an ECL Kit (cat. no. P0018AS; Beyotime Institute of
Biotechnology) according to the manufacturer's protocol.
Immunoprecipitation
SK-OV-3 or A2780 cells were washed with PBS and
lysed with ice-cold extraction buffer (1% Triton-X 100, 10 mM EDTA,
1 mM PMSF, 1% Cocktail, pH=7.4). The soluble fraction was obtained
by centrifuging cell lysates at 13,523 x g for 10 min at 4°C. The
prepared supernatants were incubated at 4°C overnight with an
ARID1A antibody, a TAZ antibody or control IgG. Then the mixture
was incubated with 50 μl protein-A-agarose beads (Beyotime
Institute of Biotechnology) for 2 h at 4°C, followed by three times
of washing using lysis buffer. Immunoprecipitants were eluted with
SDS loading buffer and resolved in SDS-PAGE gels. The proteins were
transferred onto PVDF membranes and were further probed with
appropriate antibodies correspondingly, referring to the a
forementioned method in WB.
Cell Counting Kit-8 (CCK-8) assay
CCK-8 assay was performed as per the manufacturer's
instructions (Shanghai Yeasen Biotechnology Co., Ltd.) as
previously described (24).
Briefly, 24 h after transfection, cells were seeded into 96-well
plates at a cell density of ~4×103 cells/well. At the
selected time course after attachment, cells were incubated with 10
μl CCK-8 reagent for 2 h in the dark at 37°C, and optical
density was consequently measured at a wavelength of 450 nm using a
microplate reader (Tecan Group, Ltd.).
Wound healing assay
Wound healing assay was performed as previously
described (15). Briefly, SK-OV-3
cells were seeded in six-well plate at 2×105 cells/well
and cultured for 24 h to confluence. Subsequently, cells were
incubated with serum-free medium for 12 h. A sterile tip was used
to scratch a straight line in each well. Then the wound gaps were
monitored by light microscopy after 24 h, and were calculated using
ImageJ software (National Institutes of Health). Experiments were
performed for three independent experiments.
Colony formation assay
The colony formation assay was performed as
previously described (23). A
cell colony formed by the offspring of a single cell for >6
consecutive generations in vitro is called a colony,
containing >50 cells. Briefly, SK-OV-3 cells were placed into a
six-well plate at a density of ~500 cells/well, and were cultured
at 37°C for two weeks. Colonies were then fixed with 4%
paraformaldehyde (PFA) for 15 min and stained with 0.5% crystal
violet (Sigma-Aldrich; Merck KGaA) in 2% ethanol for 10 min. After
rinsing and drying, colonies were counted manually and images were
captured. Each experiment was performed in triplicate.
Xenograft assay
Animal experiments were approved (approval no.
21045) by the Animal Ethics Committee of Zhejiang University
(Zhejiang, China) and performed according to the Guide for the Care
and Use of Laboratory Animals (NIH Publication no. 85-23, revised
1996). A total of 10 BALB/c nude mice (age, 4-5 weeks; weight,
18-20 g) were purchased from Vital River Laboratory Animal
Technology Co., Ltd. and were bred under pathogen-free conditions
(temperature, 18-22°C; humidity, 50-60%; 12/12-h light/dark cycle).
The mice bedding, feed and water were replaced every 2 days. Mice
were allocated into two groups: Control group and ARID1A group (n=5
in each group). Stably ARID1A-expressing viruses-infected (ARID1A)
or control viruses-infected (Control) SK-OV-3 cells
(1×106 cells in 100 μl PBS) were injected into
the right flank of mice (5×106 cells per mouse) via
subcutaneous injection. The volumes (V) of the xenograft tumors
were examined every 3 days as follows: V = 0.5 x a x b2,
where 'a' indicates the long axis and 'b' indicates the short axis.
After 21 days, the mice began to succumb and were immobile and
rigid, and were not in a favorable mental state, the body weight
was very low, and certain tumors reached the 1-1.5 cm in diameter.
To reduce suffering, the mice were euthanized via intraperitoneal
injection of 120 mg/kg pentobarbital sodium. Verification of death
included cardiac and respiratory arrest, lack of reflexes and
changes in mucosal color. The subcutaneous tumor tissues were
consequently dissected and collected, and were weighed and used for
subsequent examinations.
Immunohistochemistry for Ki67
The collected xenograft tumor tissues were fixed in
4% PFA, embedded in paraffin, cut into sections with the thickness
of 4 μm, dewaxed with xylene (Sinopharm Chemical Reagent
Co., Ltd.) and rehydrated (100% ethanol for 3 min, 95% ethanol for
2 min, 80% ethanol for 2 min, 75% ethanol for 2 min, H2O
for 1 min). Then, the sections were treated with 0.01 M citrate
buffer for antigen retrieval, incubated with 3%
H2O2 solution at room temperature for 10 min,
and incubated with 5% goat serum (Beyotime Institute of
Biotechnology) at 37°C for 30 min. Next, the sections were
incubated with the primary antibody against Ki67 (1:100; cat. no.
ER1706-46; HUABIO) at 4°C overnight, and then with goat anti-rabbit
IgG H&L (HRP) secondary antibody diluted at 1:1,000 at room
temperature for 30 min next day. After DAB and hematoxylin
staining, the sections were sealed with neutral resin and observed
under a light microscope.
Statistical analysis
The results are presented as the mean ± SD. The SPSS
19.0 software program (IBM Corp.) and Excel (Excel 2016; Microsoft
Corporation) were used for statistical analysis. Statistical
significance of the data was analyzed by unpaired Student's t-test
between two groups or with one-way ANOVA among multiple groups,
followed by Dunnett's post hoc test. All P-values were two-sided,
and P<0.05 was considered to indicate a statistically
significant difference. All the experiments were repeated for a
minimum of three times independently.
Results
ARID1A regulates EMT and stemness in
ovarian cancer cells
To explore the role of ARID1A in ovarian cancer
cells, the ovarian cancer cell line SK-OV-3 was transfected with an
ARID1A-expressing vector to ectopically express ARID1A and a vector
expressing ARID1A-shRNA to suppress endogenous ARID1A expression.
The results revealed that ARID1A overexpression significantly
inhibited SK-OV-3 cell viability (Fig. 1A), whereas silencing ARID1A
increased SK-OV-3 cell viability (Fig. 1B). Moreover, ARID1A overexpression
suppressed SK-OV-3 cell migration and decreased colony numbers
(Fig. 1C, D, G and H), whereas
ARID1A knockdown significantly enhanced the migratory and colony
formation abilities of SK-OV-3 cells (Fig. 1E, F, I and J). Consistent with
these results, ARID1A-overexpression increased the expression of
epithelial markers such as E-cadherin and decreased the expression
of mesenchymal markers (including N-Cadherin and Vimentin), and
stemness markers (such as Nanog, Oct3/4 and Sox2) (Fig. 1K and L). ARID1A knockdown resulted
in the opposite effects (Fig. 1M and
N), illustrating that ARID1A regulates EMT and stemness in
ovarian cancer cells.
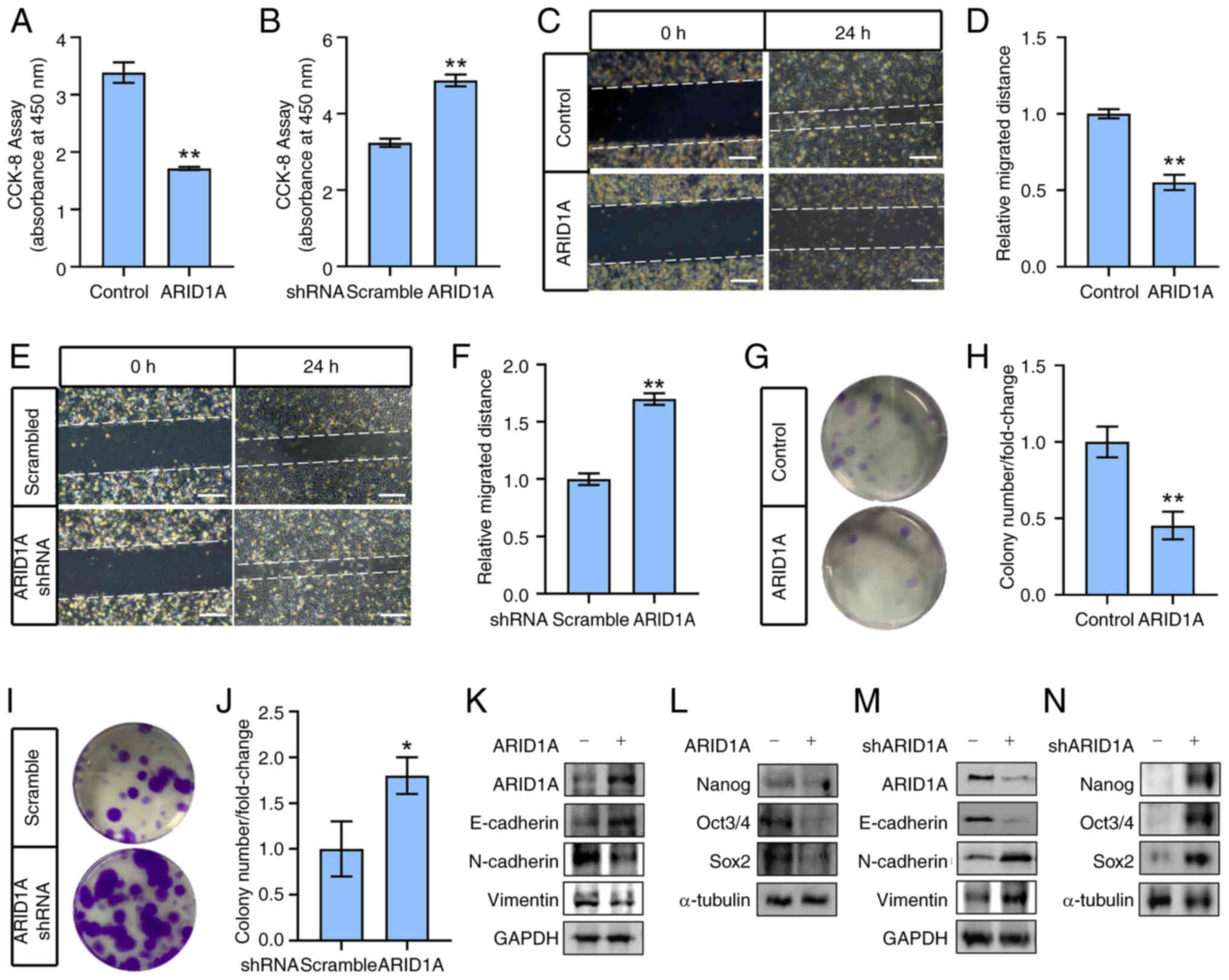 | Figure 1ARID1A regulates
epithelial-mesenchymal transition and stemness in ovarian cancer
cells. (A) SK-OV-3 cells were transfected with
ARID1A-overexpressing or control vector (Control) and cell
viability was measured after 48 h by CCK-8 assay. (B) SK-OV-3 cells
were transfected with ARID1A-shRNA or control shRNA (Scramble) and
cell viability was measured after 72 h by CCK-8 assay. (C) SK-OV-3
cells were transfected with ARID1A-overexpressing or control vector
(Control) and cell migration was measured at 24 h after scratch by
wound healing assay. (D) Quantitative statistics of panel C. (E)
SK-OV-3 cells were transfected with ARID1A-shRNA or control shRNA
(Scramble) and cell migration was measured at 24 h after scratch by
wound healing assay. (F) Quantitative statistics of panel E. (G)
SK-OV-3 cells were transfected with ARID1A-overexpressing or
control vector (Control) and cell colony formation assay was
performed. (H) Quantitative statistics of panel G. (I) SK-OV-3
cells were transfected with ARID1A-shRNA or control shRNA
(Scramble) and cell colony formation assay was performed. (J)
Quantitative statistics of panel I. (K) SK-OV-3 cells were
transfected with ARID1A-overexpressing (ARID1A+) or
control vector (ARID1A-) and protein levels of ARID1A, E-Cadherin,
N-Cadherin and Vimentin were measured by WB after 48 h. GAPDH was
used as loading control. (L) SK-OV-3 cells were transfected with
ARID1A-overexpressing (ARID1A+) or control vector
(ARID1A-) and protein levels of Nanog, Oct3/4 and Sox2
were measured by WB after 48 h. α-tubulin was used as loading
control. (M) SK-OV-3 cells were transfected with ARID1A-shRNA
(shARID1A+) or control shRNA (shARID1A-) and protein
levels of ARID1A, E-Cadherin, N-Cadherin and Vimentin were measured
by WB after 72 h. GAPDH was used as loading control. (N) SK-OV-3
cells were transfected with ARID1A-shRNA (shARID1A+) or
control shRNA (shARID1A-) and protein levels of Nanog, Oct3/4 and
Sox2 were measured by WB after 72 h. α-tubulin was used as loading
control. Scale bars in C and E, 50 μm. *P<0.05
and **P<0.01 (n=3). ARID1A, AT-rich binding domain
1A; CCK-8 Cell Counting Kit-8; shRNA, short hairpin RNA; WB,
western blotting. |
ARID1A controls Hippo signaling activity
in ovarian cancer cells
The Hippo pathway is closely associated with EMT and
stemness in cancer cells (7,25).
The potential effect of ARID1A on Hippo signaling was investigated
to gain insight into the molecular mechanism connecting ARID1A
expression with EMT and stemness in ovarian cancer cells. The
protein levels and phosphorylation status of the main components of
the Hippo pathway (MST1/2, LATS1/2, TAZ and YAP) (26) were analyzed in ovarian cancer cell
lines expressing ARID1A or ARID1A-shRNA. The present results
revealed that ARID1A overexpression resulted in a more active Hippo
pathway in SK-OV-3 ovarian cancer cells, with an increased ratio of
p-MST1/total MST1 and p-LATS1/total LATS1, compared with that in
the control (Fig. 2A).
Conversely, ARID1A-shRNA-expressing SK-OV-3 cells displayed a less
active Hippo pathway with a decreased p-MST1/total MST1 to
p-LATS1/total LATS1 ratio compared with the control cells (Fig. 2B). Consistent with this, p-TAZ and
p-YAP levels were upregulated, but total TAZ and YAP expression was
downregulated upon ARID1A-overexpression (Fig. 2C), whereas suppression of ARID1A
expression by ARID1A-shRNA exerted the opposite effects (Fig. 2D). As expected, the mRNA and
protein expression of TAZ/YAP-mediated target genes, including CTGF
and CYR61, was significantly reduced by ARID1A overexpression and
induced by ARID1A knockdown (Fig.
2E-H). This result was verified using the human ovarian cancer
A2780 cell line, demonstrating that ARID1A affected CTGF and CYR61
mRNA expression (Fig. 2I and J).
Thus, Hippo signaling activity was revealed to be regulated by
ARID1A in ovarian cancer cells.
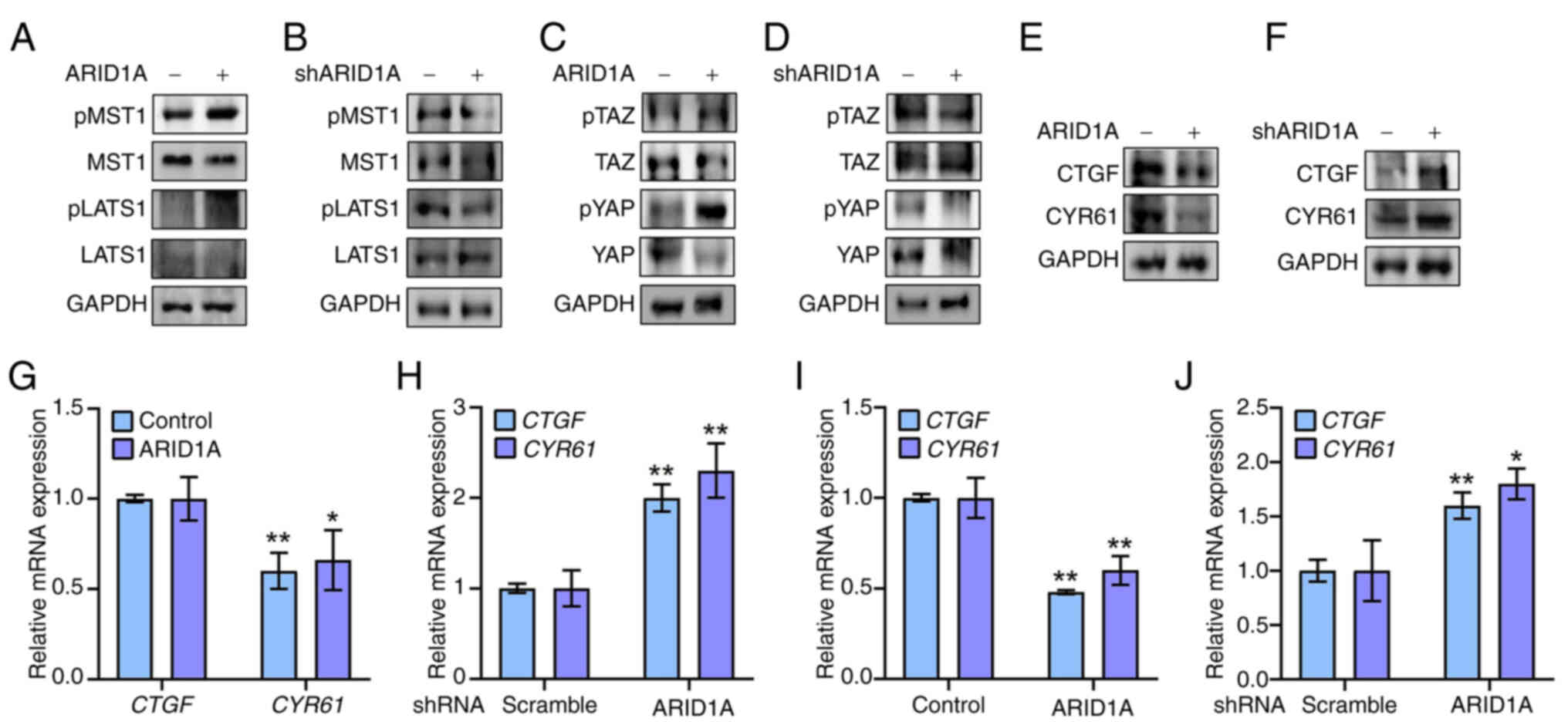 | Figure 2ARID1A controls Hippo signaling
activity in ovarian cancer cells. (A) SK-OV-3 cells were
transfected with ARID1A-overexpressing (ARID1A+) or
control vector (ARID1A-) and protein levels of pMST1,
MST1, pLATS1 and LATS1 were measured by WB after 12 h. GAPDH was
used as loading control. (B) SK-OV-3 cells were transfected with
ARID1A-shRNA (shARID1A+) or control shRNA
(shARID1A-) and protein levels of pMST1, MST1, pLATS1
and LATS1 were measured by WB after 24 h. GAPDH was used as loading
control. (C) SK-OV-3 cells were transfected with
ARID1A-overexpressing (ARID1A+) or control vector
(ARID1A-) and protein levels of p-TAZ, TAZ, p-YAP and YAP were
measured by WB after 12 h. GAPDH was used as loading control. (D)
SK-OV-3 cells were transfected with ARID1A-shRNA
(shARID1A+) or control shRNA (shARID1A-) and
protein levels of pTAZ, TAZ, pYAP and YAP were measured by WB after
24 h. GAPDH was used as loading control. (E) SK-OV-3 cells were
transfected with ARID1A-overexpressing (ARID1A+) or
control vector (ARID1A-) and protein levels of CTGF and
CYR61 were measured by WB after 12 h. GAPDH was used as loading
control. (F) SK-OV-3 cells were transfected with ARID1A-shRNA
(shARID1A+) or control shRNA (shARID1A-) and protein
levels of CTGF and CYR61 were measured by WB after 24 h. GAPDH was
used as loading control. (G) SK-OV-3 cells were transfected with
ARID1A-overexpressing (ARID1A) or control vector (Control) and mRNA
levels of CTGF and CYR61 were measured by RT-qPCR. (H) SK-OV-3
cells were transfected with ARID1A-shRNA or control shRNA
(Scramble) and mRNA levels of CTGF and CYR61 were measured by
RT-qPCR. (I) A2780 cells were transfected with
ARID1A-overexpressing (ARID1A) or control vector (Control) and mRNA
levels of CTGF and CYR61 were measured by RT-qPCR. (J) A2780 cells
were transfected with ARID1A-shRNA or control shRNA (Scramble) and
mRNA levels of CTGF and CYR61 were measured by RT-qPCR.
*P<0.05 and **P<0.01 (n=3). ARID1A,
AT-rich binding domain 1A; p-, phosphorylated; shRNA, short hairpin
RNA; TAZ, transcriptional co-activator with PDZ-binding motif;
RT-qPCR, reverse transcription-quantitative PCR; WB, western
blotting. |
Hippo activation induces ARID1A
expression and inhibits EMT and stemness in ovarian cancer
cells
Cell density mediates Hippo pathway activity in
cultured cells (27). To test the
possible reciprocal effect of Hippo activity on ARID1A expression,
SK-OV-3 cells were seeded at either low or high densities to
deactivate or activate the Hippo pathway, respectively, which was
estimated by examining the target gene CTGF and CYR61 expression
(Fig. 3A). Activation of Hippo
signaling by high cell density upregulated ARID1A protein
expression (Fig. 3B), whereas
ARID1A mRNA levels remained unchanged (Fig. 3B), suggesting that Hippo regulates
ARID1A at a translational or post-translational level. As expected,
activation of the Hippo pathway at high density weakened EMT and
stemness in SK-OV-3 cells. This was associated with upregulated
E-cadherin expression and downregulated expression of N-cadherin,
Vimentin (Fig. 3C) and stemness
markers (Nanog, Oct3/4 and Sox2) at both the mRNA and protein
levels (Fig. 3D and E). By
contrast, overexpression of TAZ, a key effector of the Hippo
pathway, apparently reduced ARID1A protein expression (Fig. 3F) despite a slight alteration at
the mRNA level (data not shown). At a culture condition with medium
cell density, the TAZ-overexpressing ovarian cancer cells displayed
upregulated EMT and stemness, including decreased E-cadherin
expression and increased expression of N-Cadherin and Vimentin
(Fig. 3F) and of stemness
markers, including Nanog, Oct3/4 and Sox2 (Fig. 3G). Co-immunoprecipitation assays
were performed to validate the possible regulation of ARID1A
protein expression by Hippo. The present results showed an
interaction between endogenous ARID1A and TAZ in SK-OV-3 cells,
which could be verified using an ARID1A- or TAZ-antibody in A2780
cells (Fig. 3H-J), suggesting the
potential mediation of ARID1A by TAZ. These results indicated that
activation of Hippo signaling induces ARID1A expression and
suppresses EMT and stemness via TAZ inhibition in ovarian cancer
cells.
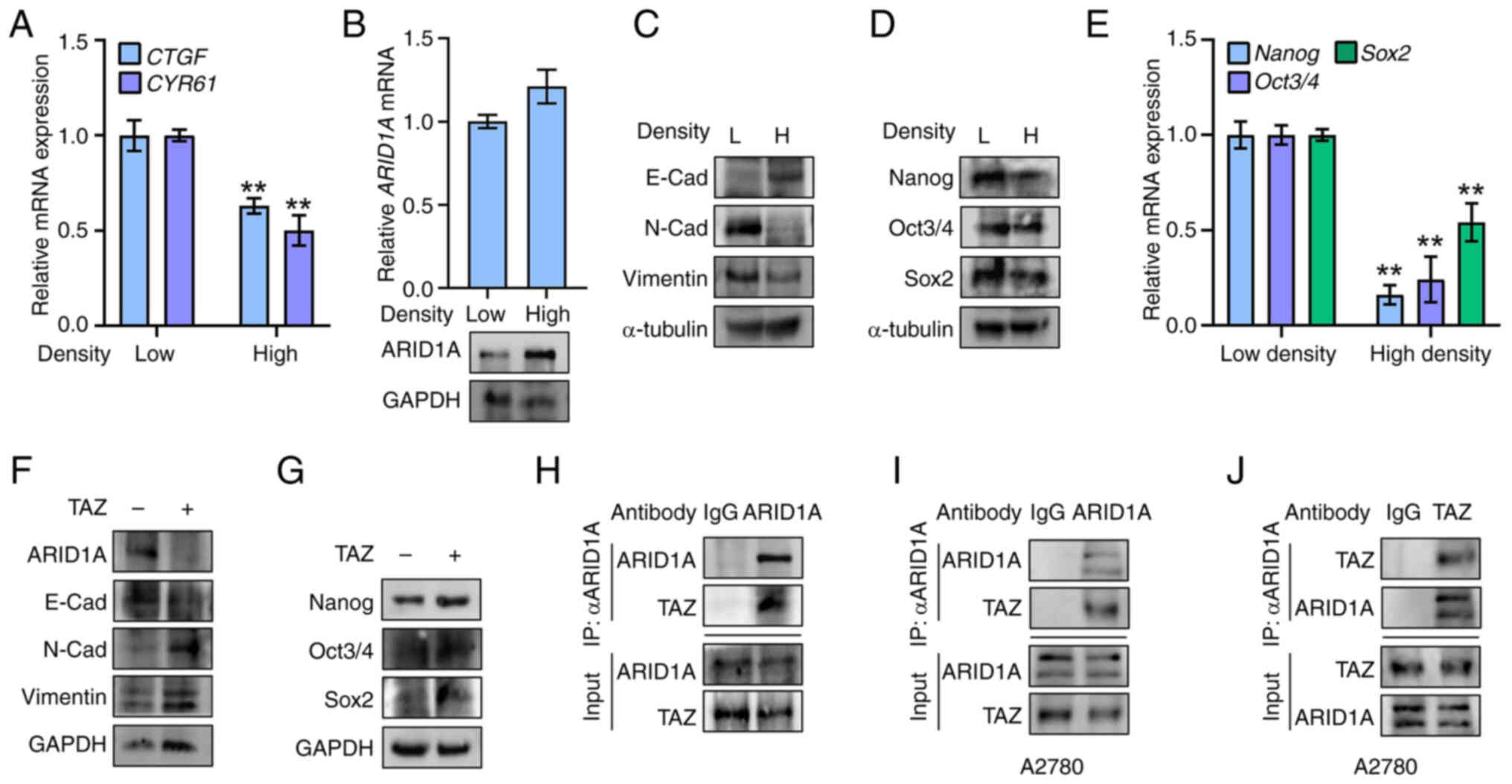 | Figure 3Hippo activation induces ARID1A
expression and inhibits epithelial-mesenchymal transition and
stemness in ovarian cancer cells. (A) SK-OV-3 cells were cultured
in low density (Low) or high density (High) and mRNA levels of CTGF
and CYR61 were measured by RT-qPCR. (B) SK-OV-3 cells were cultured
in low density (Low) or high density (High) and mRNA levels and
protein of ARID1A were measured by RT-qPCR and WB, respectively.
(C) SK-OV-3 cells were cultured in low density (L) or high density
(H) and protein levels of E-Cadherin, N-Cadherin and Vimentin were
measured by WB. α-tubulin was used as loading control. (D) SK-OV-3
cells were cultured in low density (L) or high density (H) and
protein levels of Nanog, Oct3/4 and Sox2 were measured by WB.
α-tubulin was used as loading control. (E) SK-OV-3 cells were
cultured in low density or high density and mRNA levels of Nanog,
Oct3/4 and Sox2 were measured by RT-qPCR. (F) SK-OV-3 cells were
seeded at a density of 5×106 cells in a 100-mm cell
culture dish and were transfected with TAZ-expressing plasmid
(TAZ+) or control vector (TAZ-). Protein
levels of ARID1A, E-Cadherin, N-Cadherin and Vimentin were measured
by WB. GAPDH was used as loading control. (G) SK-OV-3 cells were
seeded at a density of 5×106 cells in a 100-mm cell
culture dish and were transfected with TAZ-expressing plasmid
(TAZ+) or control vector (TAZ-). Protein
levels of Nanog, Oct3/4 and Sox2 were measured by WB. GAPDH was
used as loading control. (H) Co-immunoprecipitation of endogenous
TAZ and ARID1A in SK-OV-3 cells. IP: ARID1A, WB: TAZ. (I) Co-IP of
endogenous ARID1A and TAZ in A2780 cells. IP: ARID1A, WB: TAZ. (J)
Co-immunoprecipitation of endogenous ARID1A and TAZ in A2780 cells.
IP: TAZ, WB: ARID1A. **P<0.01 (n=3). ARID1A, AT-rich
binding domain 1A; RT-qPCR, reverse transcription-quantitative PCR;
TAZ, transcriptional co-activator with PDZ-binding motif; IP,
immunoprecipitation; WB, western blotting. |
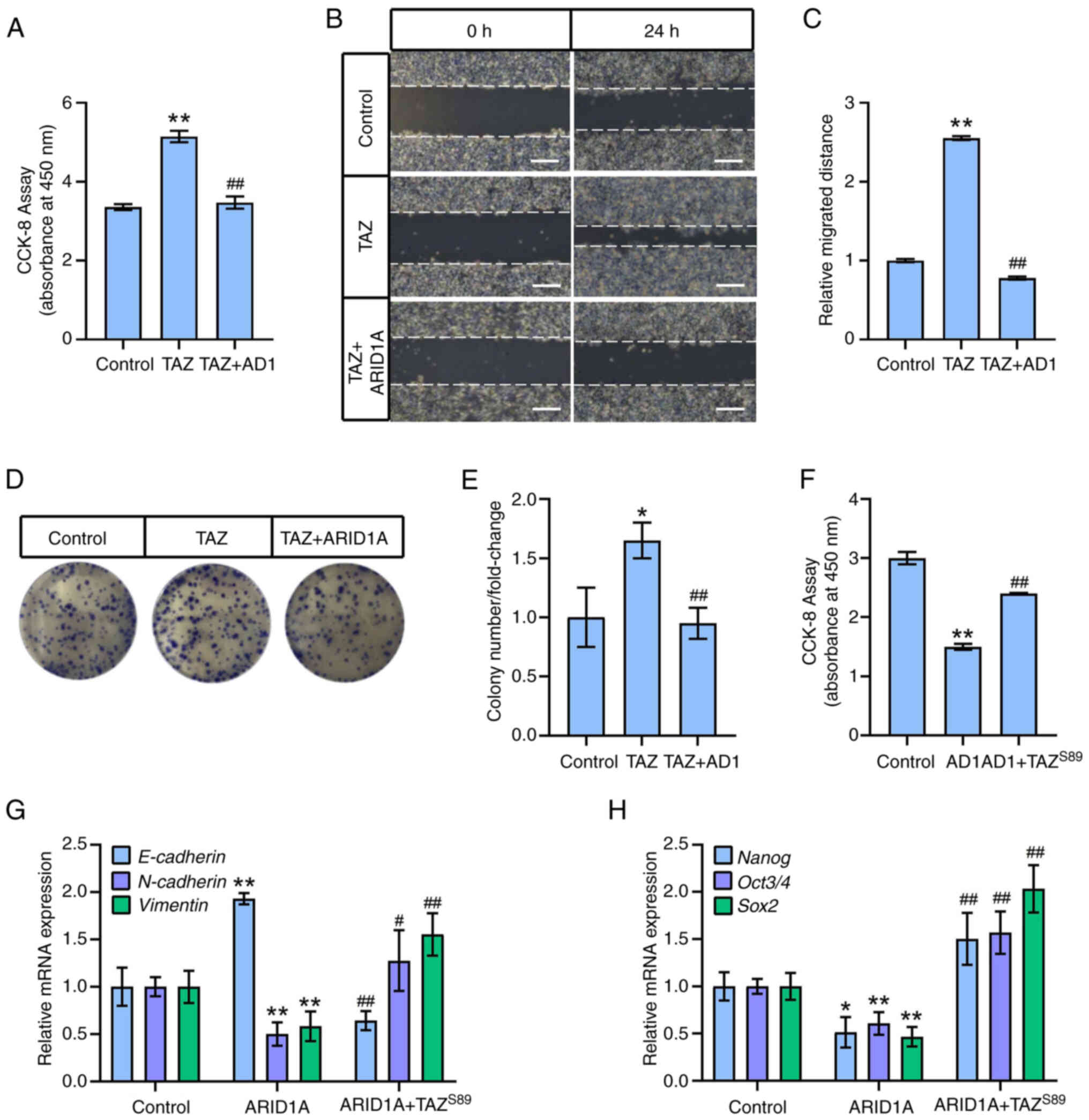
ARID1A inhibits EMT and stemness by
activating the Hippo pathway in ovarian cancer cells
Given that ARID1A negatively regulates EMT and
stemness in ovarian cancer cells but positively mediates the Hippo
pathway, it was hypothesized that ARID1A affects EMT and stemness
in ovarian cancer cells through the Hippo pathway. ARID1A
overexpression significantly reduced TAZ-mediated viability,
migration and colony formation in SK-OV-3 cells (Fig. 4A-E). Furthermore, overexpression
of a gain-of-function mutant of TAZ (TAZ-S89) effectively countered
the effects derived from ARID1A in ovarian cancer cells. This
mutant has a mutation at Ser 89 (to Ala), rendering it resistant to
phosphorylation by the upstream kinase LATS, and consequently,
TAZ-S89 stably enters the nucleus and transactivates the downstream
targets (28). The current data
revealed that TAZ-S89 not only effectively reversed the
ARID1A-suppressd cell viability (Fig.
4F) but mitigated the expression alterations of EMT and
stemness markers induced by ARID1A overexpression, including the
downregulation of E-cadherin and the upregulation of N-cadherin,
Vimentin, Nanog, Oct3/4 and Sox2, compared with cells
overexpressing ARID1A alone (Fig. 4G
and H). Thus, activation of TAZ could attenuate the
ARID1A-trigged suppression of EMT and stemness, reinforcing the
notion that ARID1A negatively regulates EMT and stemness by
activating the Hippo pathway.
ARID1A-overexpressing OCSCs exhibit less
stemness in vitro and in vivo
SK-OV-3-derived OCSCs were isolated to further test
the role of ARID1A in ovarian cancer stemness, as previously
described (20). The isolated and
cultured OCSCs displayed sphere-forming and stem cell-like
phenotypes (Fig. 5A). The OCSCs
possessed higher viability (Fig.
5B) and exhibited increased mRNA and protein expression of
stemness markers Nanog, Oct3/4 and Sox2 (Fig. 5C and D), compared with the parent
SK-OV-3 cells. By contrast, ARID1A expression was reduced in the
OCSCs (Fig. 5E). However,
overexpression of ARID1A in the OCSCs significantly downregulated
the mRNA and protein expression of CTGF and CYR61 (Fig. 5F and G). It also lowered the EMT
ability and stemness as determined by measuring the expression of
E-cadherin, N-cadherin, Vimentin, and stemness markers Nanog,
Oct3/4 and Sox2 (Fig. 5H and I).
By contrast and as expected, ARID1A overexpression reduced OCSC
migration and colony numbers (Fig.
5J-M).
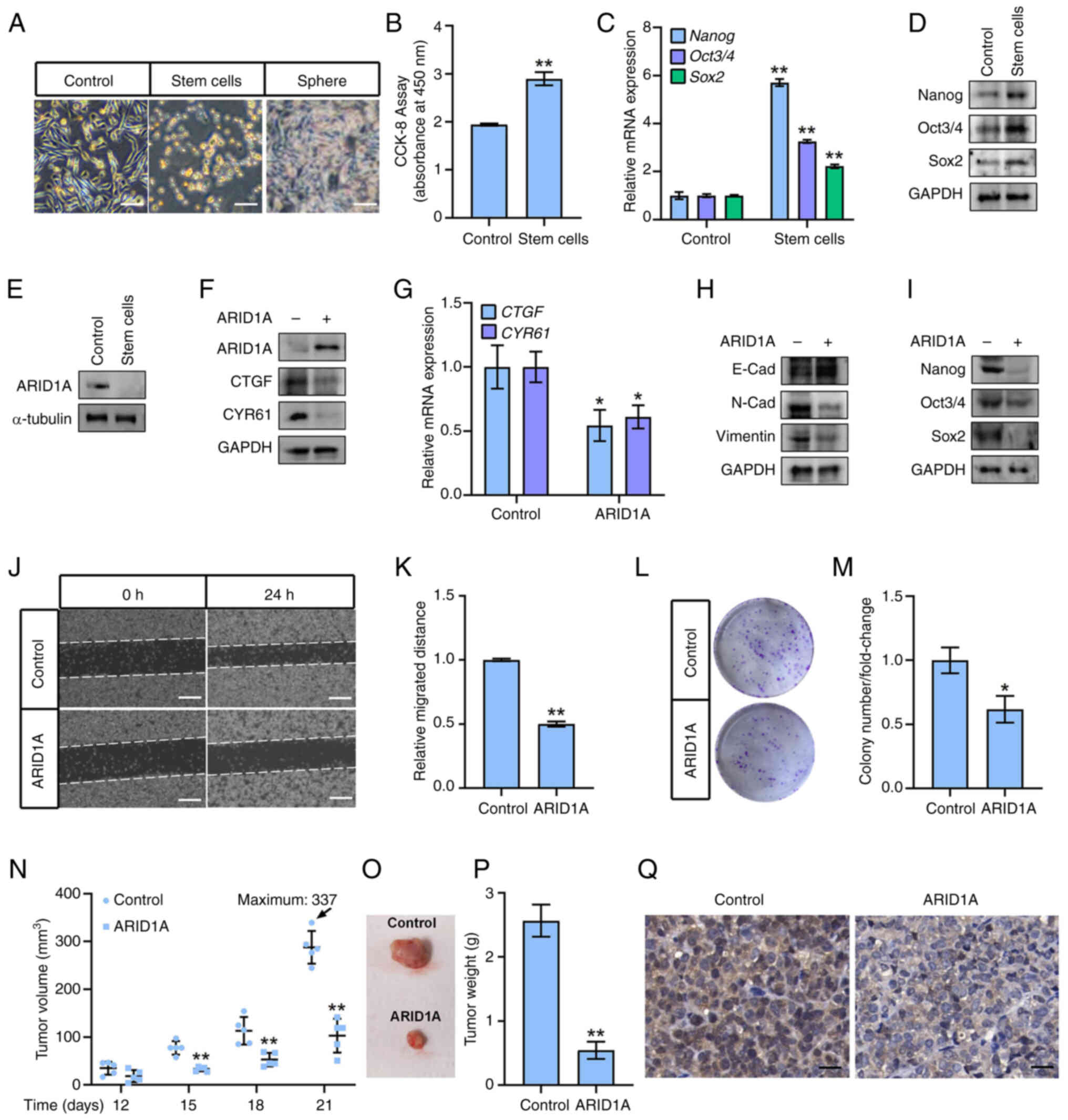 | Figure 5ARID1A-overexpressing OCSCs show less
stemness in vitro and in vivo. (A) Observation with
microscope of isolated OCSCs. (B) Viability of OCSCs was measured
by CCK-8 assay. (C) mRNA levels of Nanog, Oct3/4 and Sox2 were
measured by WB in OCSCs. (D) Protein levels of Nanog, Oct3/4 and
Sox2 were measured by WB in OCSCs. GAPDH was used as loading
control. (E) Protein levels of ARID1A were measured by WB in OCSCs.
α-tubulin was used as loading control. (F) OCSCs were infected with
ARID1A-expressing lentiviruses and protein levels of ARID1A, CTGF
and CYR61 were measured by WB. GAPDH was used as loading control.
(G) OCSCs were infected with ARID1A-expressing lentiviruses and
mRNA levels of CTGF and CYR61 were measured by reverse
transcription-quantitative PCR. (H) OCSCs were infected with
ARID1A-expressing lentiviruses and protein levels of E-Cadherin,
N-Cadherin and Vimentin were measured by WB. GAPDH was used as
loading control. (I) OCSCs were infected with ARID1A-expressing
lentiviruses and protein levels of Nanog, Oct3/4 and Sox2 were
measured by WB. GAPDH was used as loading control. (J) OCSCs were
infected with ARID1A-expressing lentiviruses and cell migration was
measured at 24 h after scratch by wound healing assay. (K)
Quantitative statistics of panel J. (L) OCSCs were infected with
ARID1A-expressing lentiviruses and cell colony formation assay was
performed. (M) Quantitative statistics of panel L. (N) Volumes of
xenografts derived from control OCSCs and ARID1A-expressing OCSCs
at 12, 15, 18 and 21 days post-inoculation. The maximum tumor
volume is indicated. (O) Image showing the size of the
representative tumor xenografts from two groups. (P) Weights of
xenografts derived from control OCSCs and ARID1A-expressing OCSCs
at 21 days post-inoculation. (Q) Ki67 staining for tumor tissues
derived from control OCSCs and ARID1A-expressing OCSCs. Scale bars,
100 μm in A, 50 μm in J, 100 μm in P.
*P<0.05 and **P<0.01 (n=3). OCSCs,
ovarian cancer stem cells; CCK-8, Cell Counting Kit-8; WB, western
blotting; ARID1A, AT-rich binding domain 1A. |
Finally, ARID1A expression was determined by
ARID1A-expressing lentiviruses in OCSCs (ARID1AOCSCs), which were
subsequently injected into nude mice to form OCSCs-derived
xenografts, to evaluate the roles of ARID1A in OCSCs' stemness
in vivo. The volumes of control OCSCs and ARID1A-OCSC
xenografts increased in a time-dependent manner within 21 days
post-inoculation (Fig. 5N).
However, no significant difference was observed between the body
weights of mice bearing different xenografts (data not shown).
Notably, the volumes of xenografts derived from control OCSCs were
significantly higher than those of ARID1A-OCSCs from 15 to 21 days
post-inoculation (Fig. 5N and O),
and the xenograft-derived control OCSCs were heavier than the
ARID1A-OCSCs group (Fig. 5P).
Ki67 staining was then performed to investigate the proliferation
of ARID1A-OCSCs xenografts. The present results demonstrated that
ARID1A overexpression in OCSC-derived xenografts led to a
significant decrease in the proliferation of tumor cells compared
with that in the control OCSC-derived xenografts (Fig. 5Q). Thus, ARID1A was shown to
inhibit stemness in OCSC xenografts.
Discussion
In the present study, using an in vitro cell
culture model and an in vivo xenograft approach, it was
found that ARID1A targets the Hippo/TAZ pathway to decrease the
stemness of ovarian CSCs.
ARID1A encodes a large subunit (250 kDa) of
mammalian SWI/SNF chromatin remodeling complexes. The ARID1A gene
is evolutionarily conserved, and mutations in ARID1A have been
found in a broad array of tumor types, including ovarian cancer
(13). Previous studies have
shown that the loss of ARID1A is associated with the oncogenic
transformation of ovarian clear cell adenocarcinoma, and decreased
ARID1A expression is correlated with the chemoresistance of ovarian
cancer cells (29), indicating
the key role of ARID1A in ovarian cancer (30). In the present study, it was
revealed that ARID1A negatively regulates ovarian cancer cell
viability, migration and colony formation, and contributes to the
suppression of EMT and stemness in ovarian cancer cells, further
demonstrating the important role of ARID1A in the progression of
ovarian cancer. Further studies are needed to identify and define
the key amino acid sites in ARID1A that regulate ovarian
cancer.
Emerging evidence indicates that several cancers are
correlated with aberrant levels of YAP, TAZ, and dysregulated Hippo
pathway activity (31,32). Hippo signaling is inhibited in
numerous tumors; YAP and TAZ translocate into the nucleus to
regulate target genes involved in cell proliferation and the
control of cell metastasis and stemness (23). Therefore, the Hippo-YAP/TAZ
pathway is considered a key mediator in multiple types of cancers.
It was previously reported that overexpression of TAZ promotes the
proliferation and migration of R182 human epithelial ovarian cancer
cells (33) and that
overexpression of YAP elevates the malignant behavior of SK-OV-3
and ES-2 ovarian cancer cells. Consistent with this, it was
demonstrated that ectopic expression of TAZ enhances EMT and
stemness of SK-OV-3 ovarian cancer cells, reinforcing the notion
that TAZ participates in ovarian cancer progression (34). While a previous study demonstrated
that ARID1A-containing SWI/SNF complex (ARID1A-SWI/SNF) operates as
an inhibitor of the pro-oncogenic transcriptional coactivators
YAP/TAZ (35), in the present
study it was additionally revealed that ARID1A also exerts negative
effects on YAP/TAZ-mediated downstream targets by upregulating
phosphorylation levels of MST1, an upstream regulator of YAP/TAZ,
providing novel linking regarding to ARID1A and Hippo signaling. In
addition, the present study it was reported that the altered Hippo
activity reciprocally makes a difference in ARID1A expression,
further identifying the potential 'cross-talk' between ARID1A and
Hippo pathway. Intriguingly, overexpression of a gain-of-function
TAZ mutant with a mutation at Ser89 effectively negated the
negative regulation caused by ARID1A, indicating that
ARID1A-mediated downregulation of cell function in SK-OV-3 cells is
dependent on TAZ suppression, which is in contrast to a recently
published paper exhibiting that the downregulation of ARID1A
promoted migration and invasion in human triple-negative breast
cancer cells through the Hippo/YAP signaling axis (36), indicating different mechanisms may
exist in multiple cancers with particular contexts. Nevertheless,
the current data illustrated at least that TAZ may be a potential
novel therapeutic target for the treatment of ovarian cancer,
particularly in cases where ARID1A is dysregulated.
Hippo signaling is sensitive to extracellular
stimuli such as cell density and mechanical stresses that trigger
Hippo activation or inactivation (37). In the past few years, multiple
factors have been shown to affect Hippo activity in regulating
carcinogenesis, expanding knowledge of the association between the
Hippo pathway and tumors. Huang et al (38) in 2020 showed that loss of PDLIM1
impedes Hippo signaling to activate YAP in hepatocellular
carcinoma. Zhang et al (39) in 2019 found that metformin
attenuates PD-L1 expression by activating the Hippo signaling
pathway in colorectal cancer. In addition, the present study
reported that ARID1A modulates the Hippo pathway by regulating the
activity of upstream kinases to control ovarian cancer cell
function. Given that ARID1A is commonly known as a chromatin
remodeling factor and is mainly distributed in the nucleus
(15), it would be interesting to
understand the molecular mechanisms by which ARID1A regulates the
core kinases of the Hippo pathway.
The precise regulation of ARID1A expression is
pivotal for maintaining body homeostasis. Dysregulation of ARID1A
and mutations in ARID1A are closely associated with various
diseases, including cancer (13).
It has been found that ARID1A mutation occurs at the early stages
of cancer from endometriosis to endometriosis-associated carcinoma
in ovarian cancer and also from a typical endometrial hyperplasia
to endometrioid adenocarcinoma in endometrial cancer (40). In addition, it was previously
identified that ARID1A expression is dramatically decreased and, in
some cases, is even lost in ovarian cancer tissues (41-43), indicating ARID1A deficiency has
potential as a biomarker for precision medicine of ovarian cancer
in clinical settings. In the present study, it was found that
activation of the Hippo pathway resulted in a noticeable increase
in ARID1A protein expression, whereas overexpression of TAZ-S89 had
the opposite effect, which also suggests frequent aberrant ARID1A
expression in ovarian cancer cells. Considering that TAZ affects
ARID1A protein expression rather than mRNA levels and that TAZ
binds with ARID1A, it is worth investigating how TAZ regulates
ARID1A protein, and determining whether TAZ-ARID1A protein-protein
binding is essential for ARID1A expression alteration.
Nevertheless, in addition to the present data, considering the
complexity of cancer biology, ARID1A might also regulate EMT and
stemness in ovarian cancer through other cell pathways, functioning
synergistically with other molecules/genes in ovarian cancer
progression, which is worthy of further investigation. Moreover, it
is worth verifying the results presented herein in clinical ovarian
cancer tissue samples in future, which would provide new target for
drug development of ovarian cancer therapy.
In conclusion, developing novel ARID1A-specific
agonists may be very effective against ovarian cancer initiation,
metastasis and relapse by activating the Hippo/TAZ pathway and
attenuating the stemness of ovarian cancer cells.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
ShoX performed all the experiments and prepared the
figures. CZ, QX, ZA and ShuX analyzed the data. GX and CL
contributed to the acquisition of data. CT designed the study and
wrote the manuscript. All authors read and approved the final
manuscript. ShoX and CT confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Animal experiments were approved (approval no.
21045) by the Animal Ethics Committee of Zhejiang University
(Zhejiang, China) and performed according to the Guide for the Care
and Use of Laboratory Animals (NIH Publication no. 85-23, revised
1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Starting Research
Foundation of The Children's Hospital, Zhejiang University School
of Medicine (grant no. 481), the Foundation for The Top-Notch Youth
Talent Cultivation Project of Independent Design Project of
National Clinical Research Center for Child Health (grant no.
Q21A0006), the National Natural Science Foundation of China (grant
no. 31801207) to Chao Tang and the National Natural Science
Foundation of China (grant no. 32100560).
References
|
1
|
Webb PM and Jordan SJ: Epidemiology of
epithelial ovarian cancer. Best Pract Res Clin Obstet Gynaecol.
41:3–14. 2017. View Article : Google Scholar
|
|
2
|
Kurman RJ and Shih IeM: The dualistic
model of ovarian carcinogenesis: Revisited, revised, and expanded.
Am J Pathol. 186:733–747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alsop K, Fereday S, Meldrum C, deFazio A,
Emmanuel C, George J, Dobrovic A, Birrer MJ, Webb PM, Stewart C, et
al: BRCA mutation frequency and patterns of treatment response in
BRCA mutation-positive women with ovarian cancer: A report from the
Australian Ovarian Cancer Study Group. J Clin Oncol. 30:2654–2663.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gadducci A, Guarneri V, Peccatori FA,
Ronzino G, Scandurra G, Zamagni C, Zola P and Salutari V: Current
strategies for the targeted treatment of high-grade serous
epithelial ovarian cancer and relevance of BRCA mutational status.
J Ovarian Res. 12:92019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beck B and Blanpain C: Unravelling cancer
stem cell potential. Nat Rev Cancer. 13:727–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Colak S and Medema JP: Cancer stem
cells-important players in tumor therapy resistance. FEBS J.
281:4779–4791. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Munoz-Galvan S, Felipe-Abrio B,
Verdugo-Sivianes EM, Perez M, Jiménez-García MP, Suarez-Martinez E,
Estevez-Garcia P and Carnero A: Downregulation of MYPT1 increases
tumor resistance in ovarian cancer by targeting the Hippo pathway
and increasing the stemness. Mol Cancer. 19:72020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu L, McArthur C and Jaffe RB: Ovarian
cancer stem-like side-population cells are tumourigenic and
chemoresistant. Br J Cancer. 102:1276–1283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY,
Kim CH, Park HG, Han SI and Kang HS: Induction of metastasis,
cancer stem cell phenotype, and oncogenic metabolism in cancer
cells by ionizing radiation. Mol Cancer. 16:102017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou P, Li B, Liu F, Zhang M, Wang Q, Liu
Y, Yao Y and Li D: The epithelial to mesenchymal transition (EMT)
and cancer stem cells: Implication for treatment resistance in
pancreatic cancer. Mol Cancer. 16:522017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng Y and Pan D: The hippo signaling
pathway in development and disease. Dev Cell. 50:264–282. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang C, Wang J, Yao M, Ji X, Shi W, Xu C,
Zeng LH and Wu X: Hippo signaling activates hedgehog signaling by
Taz-driven Gli3 processing. Cell Regen. 12:32023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu S and Tang C: The Role of ARID1A in
Tumors: Tumor initiation or tumor suppression? Front Oncol.
11:7451872021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mathur R: ARID1A loss in cancer: Towards a
mechanistic understanding. Pharmacol Ther. 190:15–23. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin M, Xu S, Li J, Li L and Tang C: Role
of ARID1A in the regulation of human trophoblast migration and
invasion. Reprod Sci. 29:2363–2373. 2022. View Article : Google Scholar
|
|
16
|
Yang Z, Li C, Fan Z, Liu H, Zhang X, Cai
Z, Xu L, Luo J, Huang Y, He L, et al: Single-cell Sequencing
Reveals Variants in ARID1A, GPRC5A and MLL2 Driving Self-renewal of
human bladder cancer stem cells. Eur Urol. 71:8–12. 2017.
View Article : Google Scholar
|
|
17
|
Ding DC, Liu HW and Chu TY: Interleukin-6
from ovarian mesenchymal stem cells promotes proliferation, sphere
and colony formation and tumorigenesis of an ovarian cancer cell
line SKOV3. J Cancer. 7:1815–1823. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu H, Ren Q, Yan Z, Xu S, Luo J, Wu X and
Tang C: Human HAND1 inhibits the conversion of cholesterol to
steroids in trophoblasts. J Genet Genomics. 49:350–363. 2022.
View Article : Google Scholar
|
|
19
|
Zhang LL, Xu YL, Tang ZH, Xu XH, Chen X,
Li T, Ding CY, Huang MQ, Chen XP, Wang YT, et al: Effects of alisol
B 23-acetate on ovarian cancer cells: G1 phase cell cycle arrest,
apoptosis, migration and invasion inhibition. Phytomedicine.
23:800–809. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lai D, Wang F, Chen Y, Wang C, Liu S, Lu
B, Ge X and Guo L: Human ovarian cancer stem-like cells can be
efficiently killed by үδ T lymphocytes. Cancer Immunol Immunother.
61:979–989. 2012. View Article : Google Scholar
|
|
21
|
Tang C, Jin M, Ma B, Cao B, Lin C, Xu S,
Li J and Xu Q: RGS2 promotes estradiol biosynthesis by trophoblasts
during human pregnancy. Exp Mol Med. 55:240–252. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang C, Takahashi-Kanemitsu A, Kikuchi I,
Ben C and Hatakeyama M: Transcriptional Co-activator Functions of
YAP and TAZ Are inversely regulated by tyrosine phosphorylation
status of parafibromin. iScience. 1:1–15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Jin M, Cao B, Lin C, Li J, Xu Q, Ren Q, Xu
S and Tang C: Tianma gouteng decoction exerts pregnancy-protective
effects against preeclampsia via regulation of oxidative stress and
NO Signaling. Front Pharmacol. 13:8490742022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Akrida I, Bravou V and Papadaki H: The
deadly cross-talk between Hippo pathway and epithelial-mesenchymal
transition (EMT) in cancer. Mol Biol Rep. 49:10065–10076. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fu M, Hu Y, Lan T, Guan KL, Luo T and Luo
M: The Hippo signalling pathway and its implications in human
health and diseases. Signal Transduct Target Ther. 7:3762022.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang J, Liu S, Heallen T and Martin JF:
The Hippo pathway in the heart: Pivotal roles in development,
disease, and regeneration. Nat Rev Cardiol. 15:672–684. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Q, Stel WV, Noord VEV, Leegwater H,
Coban B, Elbertse K, Pruijs JTM, Béquignon OJM, Westen GV, Dévédec
SEL and Danen EHJ: Hypoxia Triggers TAZ Phosphorylation in Basal A
Triple Negative Breast Cancer Cells. Int J Mol Sci. 23:101192022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yokoyama Y, Matsushita Y, Shigeto T,
Futagami M and Mizunuma H: Decreased ARID1A expression is
correlated with chemoresistance in epithelial ovarian cancer. J
Gynecol Oncol. 25:58–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
De P and Dey N: Mutation-Driven Signals of
ARID1A and PI3K pathways in ovarian carcinomas: Alteration is an
opportunity. Int J Mol Sci. 20:57322019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dey A, Varelas X and Guan KL: Targeting
the Hippo pathway in cancer, fibrosis, wound healing and
regenerative medicine. Nat Rev Drug Discov. 19:480–494. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cunningham R and Hansen CG: The Hippo
pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in
cancer. Clin Sci (Lond). 136:197–222. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jeong GO, Shin SH, Seo EJ, Kwon YW, Heo
SC, Kim KH, Yoon MS, Suh DS and Kim JH: TAZ mediates
lysophosphatidic acid-induced migration and proliferation of
epithelial ovarian cancer cells. Cell Physiol Biochem. 32:253–263.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei X, Jia Y, Lou H, Ma J, Huang Q, Meng
Y, Sun C, Yang Z, Li X, Xu S, et al: Targeting YAP suppresses
ovarian cancer progression through regulation of the PI3K/Akt/mTOR
pathway. Oncol Rep. 42:2768–2776. 2019.PubMed/NCBI
|
|
35
|
Chang L, Azzolin L, Di Biagio D, Zanconato
F, Battilana G, Lucon Xiccato R, Aragona M, Giulitti S, Panciera T,
Gandin A, et al: The SWI/SNF complex is a mechanoregulated
inhibitor of YAP and TAZ. Nature. 563:265–269. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang Y, Chen X, Qiao X, Xie Y, Guo D, Li
B, Cao J, Tao Z and Hu X: Chromatin Remodelling Molecule ARID1A
determines metastatic heterogeneity in triple-negative breast
cancer by competitively binding to YAP. Cancers (Basel).
15:24472023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma S, Meng Z, Chen R and Guan KL: The
hippo pathway: Biology and pathophysiology. Annu Rev Biochem.
88:577–604. 2019. View Article : Google Scholar
|
|
38
|
Huang Z, Zhou JK, Wang K, Chen H, Qin S,
Liu J, Luo M, Chen Y, Jiang J, Zhou L, et al: PDLIM1 inhibits tumor
metastasis through activating hippo signaling in hepatocellular
carcinoma. Hepatology. 71:1643–1659. 2020. View Article : Google Scholar
|
|
39
|
Zhang JJ, Zhang QS, Li ZQ, Zhou JW and Du
J: Metformin attenuates PD-L1 expression through activating Hippo
signaling pathway in colorectal cancer cells. Am J Transl Res.
11:6965–6976. 2019.PubMed/NCBI
|
|
40
|
Takeda T, Banno K, Okawa R, Yanokura M,
Iijima M, Irie-Kunitomi H, Nakamura K, Iida M, Adachi M, Umene K,
et al: ARID1A gene mutation in ovarian and endometrial cancers
(Review). Oncol Rep. 35:607–613. 2016. View Article : Google Scholar :
|
|
41
|
Takahashi K, Takenaka M, Okamoto A,
Bowtell DDL and Kohno T: Treatment Strategies for ARID1A-Deficient
ovarian clear cell carcinoma. Cancers (Basel). 13:17692021.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Winarto H, Tan MI, Sadikin M and Wanandi
SI: ARID1A expression is down-regulated by oxidative stress in
endometriosis and endometriosis-associated ovarian cancer. Transl
Oncogenomics. 9:11772727166898182017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kawahara N, Yamada Y and Kobayashi H:
CCNE1 is a putative therapeutic target for ARID1A-Mutated ovarian
clear cell carcinoma. Int J Mol Sci. 22:58692021. View Article : Google Scholar : PubMed/NCBI
|