Glioblastoma (GBM) is a fatal primary cancer of the
brain. Unlike other cancers, such as breast and lung, where
therapies have improved overall survival (OS) and progression-free
survival (PFS), the same trend is observed in GBM (1). GBM is indeed a difficult disease due
to its complex biology, anatomical barriers and poor functional
status of the affected patient population (2).
The median age of diagnosis for GBM is ~64 years,
with the average incidence rate being 3.19/100,000 of the
population. These patients have a median OS time of ~15 months
(3). GBM occurs more frequently
in men than in women (1,3). The only confirmed risk factor for
the development of GBM is exposure to high doses of ionizing
radiation (4). Clinical
presentation of GBM varies greatly depending on the brain location
and can be manifested as weakness, numbness, loss of vision,
slurring of words, mood disorders, fatigue, memory disorders and
seizures (5). Upon presentation,
magnetic resonance imaging (MRI) of the brain is preferred for
radiographic assessment (1),
followed by either biopsy or surgical resection. Upon confirmation
of the diagnosis, treatment includes radiotherapy (RT) and
temozolomide (TMZ) (6). This
regimen provides suboptimal outcomes with the median survival of
~15 months (6).
Pathologically, GBM is derived from the unregulated
growth of cells of astrocytic origin and is the most common primary
brain neoplasm accounting for >60% of all brain tumors in adults
(7,4). WHO 2021 reclassified GBMs as grade 4
primary brain tumors that are IDH-wild type (wt) with or without
histological features of GBM (micro-vascularization and/or
necrosis) encompassing one or more of the following molecular
alterations, such as hTERT promoter mutation, EGFR amplification or
alteration, the combined gain of chromosome 7 and the complete loss
of chromosome 10 (7+/10-) (8). GBM can also be classified further
into two distinct subtypes: Primary and secondary. Between the two
subtypes of GBM, primary GBM occurs mainly in older patients at an
average age of 62 years, accounting for 80% of GBM cases, while
secondary GBM mainly occurs in younger patients at an average age
of 45 years (3). Primary tumors,
at the time of diagnosis, show little to no evidence of the tumor's
origin coming from a lower grade glioma, while secondary GBM
develops from a lower grade glioma (WHO grade 2 or 3) into a grade
4 glioma (9). Primary GBM can be
further classified as either neural, classical, mesenchymal or
proneural transcriptional profiles whereas secondary GBM tends to
have a more proneural transcriptional profile and a
hypermethylation phenotype (10).
Signs and symptoms arising from GBM vary depending
on its location within the brain. Focal neurological/cognitive
deficits, headaches, seizures and increased intracranial pressure
are common and often progress within days to weeks (11). Less commonly, ~25% of patients
present with seizures and benefit from anticonvulsant medication
administration (12).
Corticosteroids may also be prescribed for symptom alleviation
related to peritumoral edema (PTE) (13). Brain MRI typically reveals
enhanced tumor regions with central mass necrosis and increased
T2/FLAIR signal intensity in PTE (14-16). Pathological confirmation after
biopsy or maximal safe surgical resection leads to diagnosis of GBM
per WHO 2021 classification.
Following surgical resection, the current standard
of care for high-grade gliomas includes adjuvant RT with
concomitant and maintenance systemic TMZ. TMZ is an FDA-approved
DNA alkylating agent, and when combined with RT, it has shown to
increase OS in patients with GBM; with a median OS time of 14.6
months compared with 12.1 months with radiation alone (17-19). Treating patients with
tumor-treating fields (TTF), or alternating electric fields, in
combination with maintenance TMZ, has been shown to increase both
PFS and OS time. The former increases to 6.7 months compared with 4
months with TMZ alone, while the latter increases to 20.9 months
compared with 16 months with TMZ alone (20).
Upon recurrence of GBM, there are two main pathways
that begin to differentiate the treatment: i) local tumor
recurrence and ii) diffuse/multiple tumors outside of the initial
resection cavity. With local tumor recurrence, the patient must be
assessed with brain MRI for the feasibility of additional surgical
resection. After resection, non-invasive treatment should be
considered. If the tumor is unable to be resected, not recommended,
or not elected by the patient, then the patient is directly
diverted to non-invasive treatments. Such treatments include
clinical trials, systemic chemotherapy, reirradiation, alternating
electric field therapy and palliative care. For diffuse/multiple
tumors outside of the initial resection cavity, the treatments are
the same as the non-invasive therapy with the addition of surgical
resection of symptomatic or large lesions. Resection of the
recurrent tumor also allows for genetic and histologic analysis to
understand the progression of the tumor from the genetics
identified at initial diagnosis.
Preferred regimens of systemic chemotherapy in
recurrent high-grade gliomas include re-challenging with TMZ,
lomustine or carmustine, PCV (procarbazine, carmustine and
vincristine), regorafenib, or bevacizumab. If there has been a long
interval between initial treatment with TMZ and recurrence of the
glioma, then it is reasonable to consider rechallenging with TMZ
therapy at variable proposed dosages, which was shown to have a
response rate of 64% in the study reported by Perry et al
(21). Another therapeutic option
is the use of a nitrosourea such as lomustine or carmustine
(22-25). This therapy should be particularly
considered in those with methylguanine-DNA methyltransferase (MGMT)
unmethylated promoter status, whose response to TMZ is suboptimal
(26,27). In PCV therapy, carmustine may be
substituted for lomustine. In a randomized, placebo-controlled
clinical trial, traditional carmustine administration was compared
with a carmustine-biodegradable polymer placed surgically at the
tumor site in order to understand its impact on OS and effects of
systemic toxicities in recurrent gliomas. The median survival time
of the treatment group was 31 weeks compared with 23 weeks in the
placebo group and 6-month survivability was 50% higher in the
carmustine-polymer treatment group (28,29). Regorafenib was compared with
lomustine in a randomized, open-label phase II clinical trial
studying its effectiveness in treating recurrent gliomas and
increased average survival time by 1.8 months compared with
lomustine (30).
Bevacizumab, an FDA-approved, anti-VEGF monoclonal
antibody, has failed to show a survival advantage against recurrent
GBMs while it has led to significantly improved PFS (31-33). The increase in PFS may be due to
its mechanism of action, which alters the blood-brain barrier
(BBB), and it may also play a role in the prevention or improvement
of rapid neurologic deterioration (34,35).
RT should be considered if there is a sufficient
time period since the last RT in order to prevent additional
RT-related complications, or if there was a favorable response to
RT in the past. RT may be used in combination with bevacizumab even
when bevacizumab monotherapy fails, if retaining the
steroid-sparing effects of bevacizumab are desired. An FDA-approved
biosimilar agent may be used in place of bevacizumab. A
meta-analysis reviewed 50 eligible non-comparative studies
including 2,095 patients to determine the efficacy and toxicity of
reirradiation of recurrent GBM (36). The meta-analysis demonstrated an
OS at 6 and 12 months of 73 and 36%, respectively. A PFS 6-month
rate of 43% and PFS 12-month rate of 17% was also observed.
Alternating electric field therapy has been
FDA-approved for safety in recurrent glioma therapy based on the
results of the EF-11 clinical trial (37). This treatment is a non-invasive,
low-intensity and intermediate frequency electric field therapy
which interferes with cell division by interrupting microtubule
formation during mitosis (38).
This trial compared chemotherapy-free treatment using alternating
electric field therapy vs. traditional chemotherapy in recurrent
GBM. There was no increase of survival between the two groups.
Median survival time was 6.6 vs. 6.0 months, respectively [hazard
ratio, 0.86; 95% confidence interval (CI), 0.66-1.12; P=0.27]. The
analysis of the Patient Registry Dataset on all patients with
recurrent GBM receiving TTF from 2011-2013 showed 1/3 of patients
received treatment at first recurrence as opposed to 9% in the
EF-11 clinical trial. The overall median survival in the study was
9.6 months, displaying a significant improvement as compared with
the results of the EF-11 trial.
One of the most profound characteristics of GBM is
its high intra- and inter-tumor heterogeneity. Classical, neural,
proneural and mesenchymal subtypes further differ in their
subclonal evolution which is time, location and treatment dependent
(39). Snuderl et al
(40) showed evidence of these
distinct clonal populations within the same tumor in relationship
to receptor tyrosine kinases (RTKs) and that while these
relationships were mutually exclusive, they shared common mutations
suggesting that these clones arise from the same precursor cells.
Sottoriva et al (41) also
reported their genetic analyses which showed that a single
precursor cell gives rise to different subclonal populations. Their
work highlights the significance of understanding intratumoral
heterogeneity and its implications in a clinical setting. It also
sheds light on the idea that intratumoral heterogeneity may allow
these subclones to survive initial standard of care treatment, with
increase in genetic/epigenetic aberrations in response to therapy
aiding in therapeutic resistance and in turn leading to recurrence
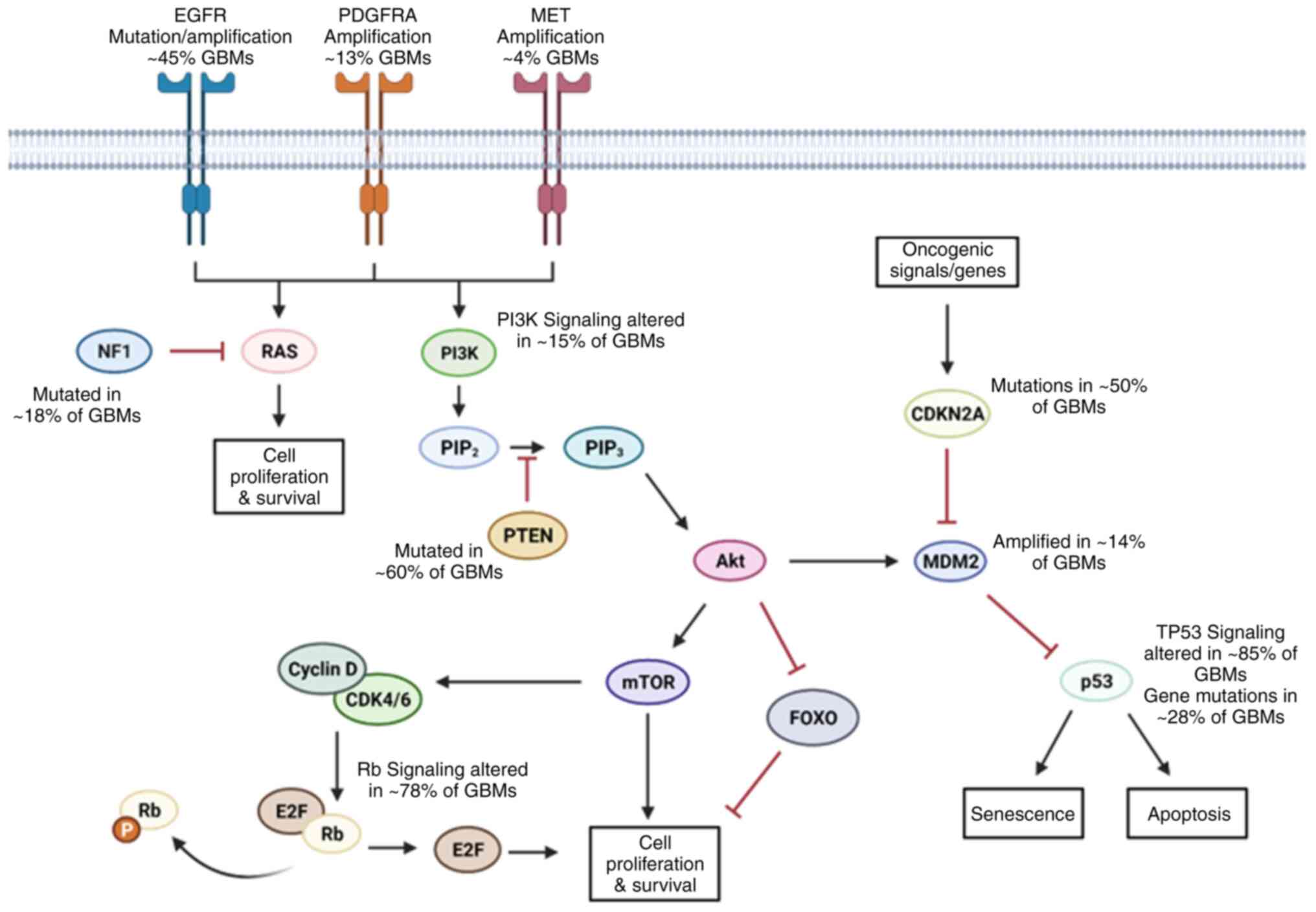
(41). In GBM, the most
clinically relevant alterations are: Epidermal growth factor
receptor (EGFR), retinoblastoma (Rb), TP53 and RTK/Ras/PI3K
signaling pathways (42) as shown
in Fig. 1.
EGFR is the most common amplification/mutation in
GBMs, observed in ~58% of cases (43). Tumors with EGFR amplification have
also been identified to harbor the EGFRvIII mutation. This mutation
is caused by the deletion of exons 2-7, producing a constitutively
active form of EGFR (44). This
form of EGFR has direct consequences on cell proliferation, tumor
initiation and resistance to apoptosis due to continuous
autophosphorylation of downstream signaling proteins (45-47).
The p53 protein isoforms are referred to as the
'guardians of the genome' and are encoded by the TP53 gene.
P53 proteins protect DNA by promoting cell cycle arrest and DNA
repair mechanisms when damaged. They also initiate apoptosis if DNA
damage is beyond repair. It is one of the most frequently mutated
genes in all cancers (53). In
GBM, the pathway is altered in 85% of patients. The p53 protein
alone is modified in 28% of GBM samples, but its frequency varies
depending on the molecular subtype: Proneural, 54%; mesenchymal,
32%; neural, 21%; and classical, 0% (54). In GBMs with an amplification of
PDGFRA, which is a marker of the proneural classification, a loss
of wt-p53 has been revealed to constitute a more invasive tumor
(55). Most p53 mutations are
gain-of-function (GOF) missense mutations in the DNA binding domain
(55). This gene signature has
also been associated with the increased inflammation and decreased
OS in patients (56). Some
studies have reported that reactivating p53 in GBM cell lines can
increase drug sensitivity and inhibit growth in vitro
(57,58). These studies have shown that the
loss of wt-p53 and/or GOF of p53 aids in the progression of GBM and
may be an effective clinical target.
PI3K is a family of kinases upstream of the
PI3K/Akt/mTOR pathway. This pathway coordinates pro-survival
signaling throughout cells. PI3K is regulated by both PTEN and
PIK3R1 (59). The function of
PTEN is to prevent uncontrolled proliferation in healthy cells
(60). In GBM, PI3K frequently
has GOF mutations in its catalytic domain, promoting Akt
overactivation and cell growth (61). The loss of heterozygosity mutation
in PTEN is found in ~60% of GBMs. Mutations in PTEN tend to occur
more frequently at later stages of cancer development and have a
similar effect as the PI3K GOF mutations (62). The repressive subunit of PI3K,
PIK3R1, has also been demonstrated to be mutated/altered in GBM
samples at a higher frequency than PI3K alterations (59,63,64). Numerous studies have demonstrated
that inhibition of the PI3K/Akt pathway in GBM cell lines inhibits
growth in vitro and in vivo (65,66). However, clinical trials have shown
that inhibitors of this pathway are ineffective in improving the
long-term survival of patients (67).
There are several other genetic and epigenetic
alterations that lead to propagation of growth, viability and
invasion of GBM and carry prognostic and therapeutic implications.
For the sake of discussion in this paper, we will be focusing on
these aforementioned pathways as they are most significantly
upregulated or downregulated in GBMs. Altogether, the complex
heterogeneity of GBM leads to the survival and proliferation of
malignant cells. These alterations/mutations not only affect the
tumorigenicity of cells, but allow them to evade chemotherapeutics
and RT. The intricate web of interactions these pathways have on
one another and have independent of one another indicates the dire
need for novel therapeutic targets. It also strengthens the need
for multimodal treatment strategies to combat the heterogenous
nature of GBM. The literature shows that not all cells harbor the
same mutations within the same tumor. Consequently, by targeting a
specific protein, this treatment strategy may only be targeting a
subpopulation of tumor cells present and lead to inevitable relapse
and treatment-resistant tumors.
TMZ, which was granted FDA approval in 2005, is the
standard chemotherapeutic drug used concurrently with RT for
patients with GBM. At physiological pH, TMZ is converted into
methyl-diazonium ions. Methyl-diazonium ions cause formation of DNA
adducts by transferring a methyl group to DNA, which ultimately
causes cytotoxicity. Methylation of the O3 position of
adenine and the N7 and O6 position of guanine
causes DNA breaks, leading to G2/M cell cycle arrest and apoptosis
(68).
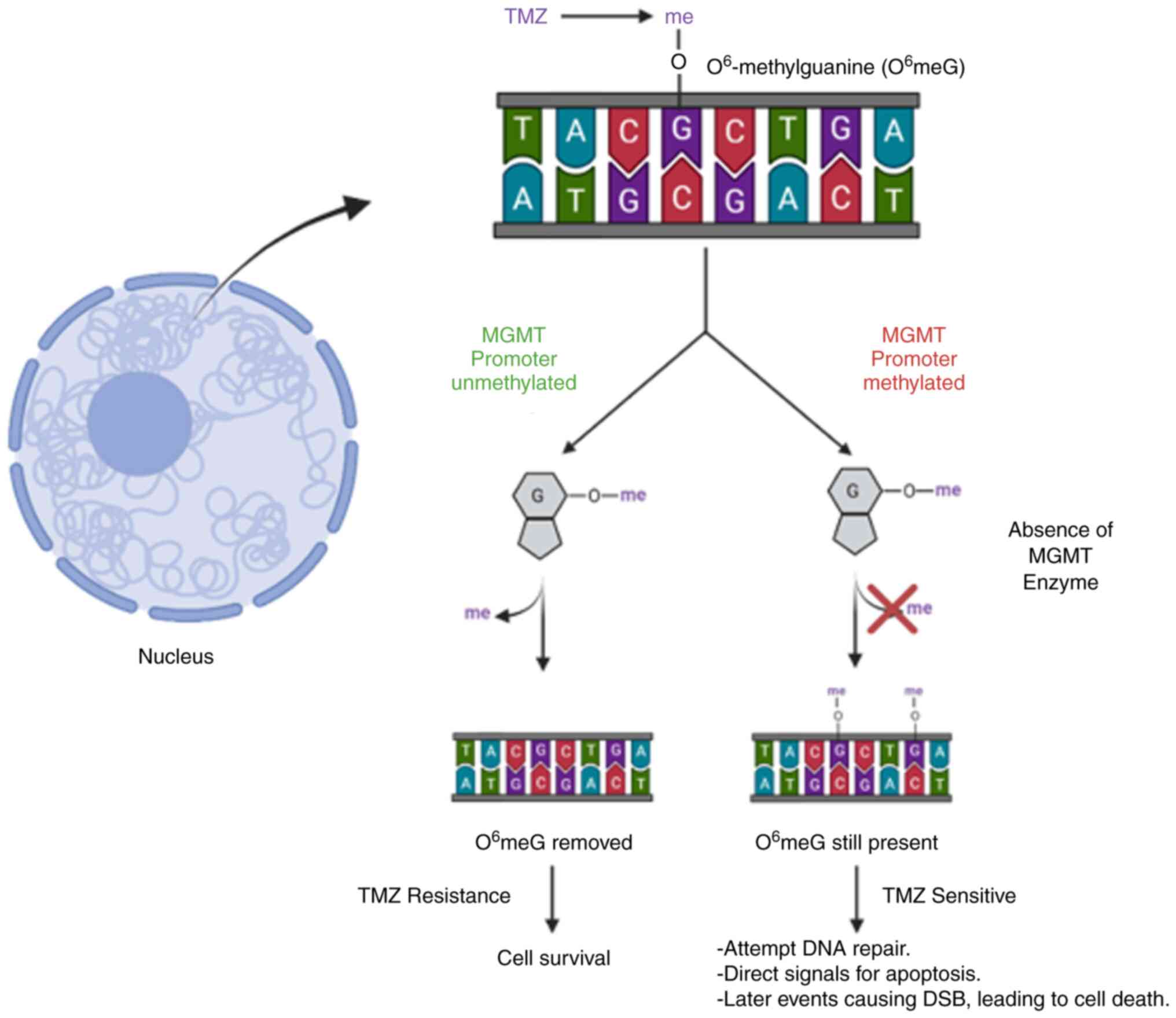
Although TMZ is an important part of GBM therapy,
resistance to this drug is common. After exposure, almost 50%
patients exposed to TMZ stop responding positively. MGMT is the
primary contributor to resistance to TMZ (Fig. 2). MGMT is involved in direct
repair of DNA in the presence of methylated residues. If MGMT is
present, it directly removes O6-methylguanine residues,
essentially rendering the drug ineffective. Hypermethylation of
MGMT's promoter region leads to decreased expression of MGMT,
preventing the removal of O6-methylguanine residues and
resulting in cells being sensitive to TMZ treatment. Stupp et
al (19) showed the clinical
implications of MGMT promoter methylation status while diagnosing
and treating patients. They demonstrated that when comparing
methylation status alone, patients who had the MGMT promoter region
methylated had an increase of OS by 6 months compared with patients
with an unmethylated MGMT status. The effects of methylation status
in response to treatment were also reported and an improved
response was observed to both TMZ and radiation therapy in patients
who harbored a methylated MGMT promoter status (69). Several published clinical trials
have stratified treatment plans according to MGMT methylation
status, signifying its importance in drug efficacy and patient
response (69-71).
Besides DNA repair proteins, cancer stem cells also
contribute to TMZ resistance. Ligands of the Wnt/β-catenin pathway
such as Wnt3a, Wnt7a and Wnt1 are known to induce stemness and have
been reported to be highly expressed in GBM cells. As compared with
healthy cells, Wnt signaling is upregulated in glioma stem cells
(GSCs). This upregulation results in higher self-renewal
capabilities, motility and altered epithelial-to-mesenchymal
transition (EMT) activator expression (72). A previous study showed that EMT in
GBM promotes resistance to chemotherapy, including TMZ, which can
be reversed by knocking out β-catenin. Moreover, β-catenin has been
linked to genes involved in EMT such as ZEB1, Snail,
Slug and Twist (73).
Bevacizumab, a humanized monoclonal antibody against
VEGF-A, has received FDA approval for the treatment of a variety of
cancers (74). The FDA approved
bevacizumab in 2009 to treat recurrent GBMs (75,76). While the use of anti-angiogenetic
therapies provides some benefit to PFS, patients ultimately become
resistant to these anti-angiogenic therapies (77).
The use of bevacizumab leads to the creation of
intratumoral hypoxia due to a decrease in blood vessel formation
(78). This creation of a hypoxic
microenvironment results in an increased expression of
hypoxia-inducible factor 1-alpha (HIF1α), which increases the
expression of VEGF (79).
Alongside an increase in HIF1α and VEGF, hypoxia also causes
upregulation of c-Met and phospho-c-Met (p-Met). Increases in
expression of these proteins have been reported to have direct
consequences on downstream signaling pathways that are involved in
resistance to anti-angiogenic treatments, leading to more invasive
tumors. The hepatocyte growth factor (HGF) and c-Met pathway has
been extensively examined in the context of
anti-angiogenic-resistant tumors (78). Multiple studies have shown the
effects of the HGF/c-Met pathway on invasiveness and tumor growth.
HGF interacts with c-Met, causing activation of several pathways
such as the pathways mentioned previously as well as the MAPK/ERK
and NF-κB pathways (78,80,81).
Approved by the FDA in the 1980s, carboplatin has
been used for different cancers over the years. The drug creates
DNA lesions, thus disrupting replication and transcription which
ultimately leads to cell death. Although carboplatin is an
established drug, resistance after persistent use is common. In
total, three primary mechanisms are involved: Decreased drug
availability, DNA repair mechanism alteration and changes in
microenvironmental responses. CTR1 downregulation, ATP7A/7B and
MRP2 upregulation help cancer cells in keeping the intracellular
concentration of the drug low by reduced uptake and increased
efflux. Any drug that reaches the tumor site and gets inside of the
cell is neutralized by high levels of glutathione (GSH).
Carboplatin-resistant cells have high levels of GSH and
GSH-supporting proteins such as γ-glutamyl-cysteine synthetase and
glutathione s-transferases (90,91).
Similar to other DNA-damaging chemotherapeutics, a
key resistance mechanism involves DNA repair. Most
carboplatin-induced lesions are excised by nucleotide excision
repair (NER). Proteins involved in NER such as ERCC1, ERCC4 and XPF
are observed to be upregulated in resistant tumors. High levels of
ERCC1 are a marker of poor prognosis in numerous cancers, rendering
drugs such as carboplatin and cisplatin ineffective. Other enzymes
including MMR proteins MSH2 and MLH 1, and specific DNA polymerases
such as REV1 and REV3 also play a part, albeit an indirect one, in
chemoresistance (92). High
expression levels of these proteins cause an increase in DNA
replication, which can prevent tumor recurrence. However, in the
event of relapse, they make DNA-damaging chemotherapies
obsolete.
The initial therapy for GBM consists of maximal
surgical resection followed by adjuvant RT to a dose of 60 Gy with
concurrent TMZ, per the so-called Stupp protocol, followed by
maintenance TMZ and the use of TTF. This regimen became the
standard of care following a series of landmark trials which
established the role for the respective adjuvant therapies. The
benefit of adjuvant RT was initially demonstrated in trials
conducted by the Brain Tumor Study Group, which found that the
addition of RT roughly doubled survival time compared with
post-operative observation (93).
This benefit was also found to be dose-dependent, with patients
receiving higher RT doses living longer (94). After preclinical data demonstrated
reduced tumor cell survival with the combination of TMZ and RT,
this combination was investigated clinically (95).
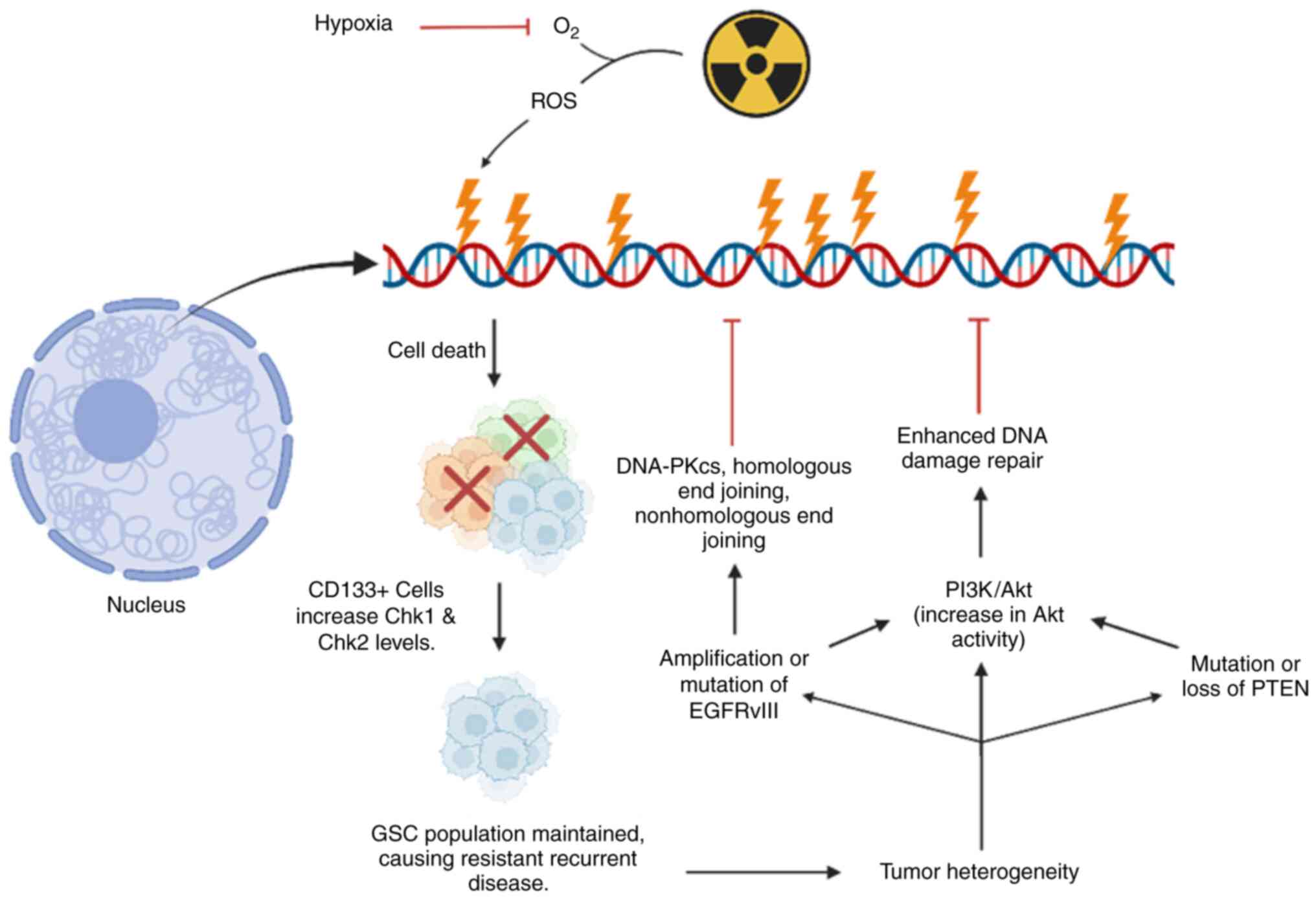
Solid tumors have abnormal vasculature, and
consequently varying degrees of oxygenation. GBM is a rapidly
growing, hypoxic tumor, and the degree of hypoxia is further
associated with increased neoangiogenesis and accelerated
endothelial proliferation. In turn, this neoangiogenesis causes
remodeling of the extracellular matrix, and increased overall
invasiveness of tumor cells (98). Furthermore, hypoxia also reduces
the lethal effect of irradiation by reducing the generation of the
reactive oxygen species that mediate RT-induced DNA damage
(99,100). Thus, tumor hypoxia is a crucial
driver of aggressiveness of GBM and reduces RT effectiveness
(Fig. 3). Hypoxia also results in
downstream signaling via HIFs, with multiple targets including
vascular endothelial growth factor A (VEGF) (101).
Given these findings, agents targeting this pathway
have been developed, most prominently bevacizumab. This humanized
monoclonal antibody binds the circulating VEGF-A ligand, reducing
its ability to bind to receptors, altering the kinetics of ligand
binding to endothelial cells and downregulating angiogenesis.
Despite compelling preclinical data of the benefits of RT with
bevacizumab, real-world outcomes have been less than optimal. A
total of two phase III clinical trials both failed to demonstrate
significant improvements in OS, though improvements in PFS were
noted (102,75). Interestingly, the use of
bevacizumab actually reduced survival time in the most favorable
subgroup (MGMT-methylated with favorable gene signatures from the
9-gene molecular profile) from 25 months to 16.7 months. The 9-gene
molecular profile used for stratification in the trial came from
the work of Colman et al (103), who reported the development of a
9-gene array that can be used as a predictor of survival in
patients with GBM. The 9 genes that were selected as prognostic
indicators were: Aquaporin 1 (AQP1), YKL-40 (CHI3L1),
epithelial membrane protein 3 (EMP3), glycoprotein
(GPNMB), insulin-like growth factor binding protein 2
(IGFBP2), galectin 3 (LGALS3), oligodendrocyte
lineage transcription factor 2 (OLIG2), podoplanin
(PDPN) and reticulon 1 (RTN1). These genes were shown
to give survival time predictions independently of MGMT status in
patients and were observed to provide similar predictions of
survival as MGMT methylation status (103). Further investigation is
necessary to successfully exploit hypoxia therapeutically.
CD133 (prominin-1) is identified as a putative
hallmark of stem cells, both in tumors and neural progenitor cells
(106,110,111). CD133-positive
(CD133+) GSCs have more robust DNA repair mechanisms and
greater growth checkpoint activation following DNA damage vs.
CD133-negative (CD133-) GBM tumor cells (112). Consequently, irradiation exerts
a potent evolutionary pressure that inadvertently selects for the
survival of CD133+ cells. This ultimately contributes to
eventual repopulation of the surviving niche of tumor cells, which
likely underlies the observation of the distinct genetic profile of
recurrent GBM (113). More
specifically, exome sequencing has demonstrated that some recurrent
tumors appear to originate from clonal expansion of specific
subpopulations of the original tumor (113).
Other molecular signaling pathways have also been
shown to contribute to radioresistance, both in the de novo
and recurrent setting. Alterations of the EGFR are among the most
common mutations in GBM, present in >50% of tumors (115). Specifically, mutations of
EGFR-wt to a specific, constitutively active variant, EGFRvIII are
highly oncogenic (116).
EGFRvIII has been demonstrated to confer radioresistance compared
with EGFR-wt by multifold activation of pro-proliferative signaling
via mitogen-activated protein kinase (MAPK). It has also been shown
to cause robust stimulation of anti-apoptotic pathways via the
Akt/phosphatidylinositol-3-kinase pathways (PI3K-Akt) in response
to irradiation (117). This
hyperactivation of the PI3K/Akt signaling pathway by EGFRvIII
reduces radiosensitivity via enhanced repair of DNA DSBs (118). Unfortunately, response to EGFR
inhibition has generally been modest (119). This has been attributed to poor
tumor penetrance as well as due to redundant mutations of these
downstream signaling cascades. However, inhibition of downstream
PI3K-Akt has been demonstrated to radio-sensitize glioma cells
in vitro (120). Ongoing
investigations of the Akt pathways and other molecular cascades
downstream of EGFR may prove productive.
While targeting the mechanisms underlying the
radioresistance of GBM remains a largely preclinical endeavor, the
inevitability of recurrence has prompted clinical efforts to
improve the efficacy of RT. In the upfront setting, this was
primarily investigated from the perspective of RT target
delineation. It has been demonstrated that GBM exists as distinct
subpopulations of cells with unique roles in the growth, signaling
and invasiveness of the tumor (121). Thus, one area of inquiry is that
of incorporating novel imaging modalities to localize and
characterize GBM more granularly. For example, multiparametric MRI
sequences to assess hyper-cellularity and hyper-perfusion has been
shown to be predictive of subsequent sites of failure (122). Similarly, hypoxia imaging is an
ongoing area of investigation, given its association with increased
invasiveness and radioresistance (123). Imaging that more robustly
correlates the known intratumoral heterogeneity with location and
may allow for more optimal, biologically-driven, RT dose
distribution, often referred to as 'dose-painting'. Novel PET
agents such as [11C] methionine-PET (MET-PET);
[18F] fluoro-ethyl-L-tyrosine (FET-PET), and
[18F]-FDOPA-PET are being investigated in
dose-escalation trials (124).
In the recurrent setting, several reirradiation
approaches have been attempted as salvage options. These are
complicated by considerations of the location of the recurrent
lesion relative to the initial course (in-field, marginal,
out-of-field), prior RT dose to adjacent organs-at-risk, volume of
recurrent/progressive disease and changes in tumor biology. GBMs
fail overwhelmingly in-field, and thus additional radiation doses
often overlap significantly with the initial course (125). Consequently, this may increase
the risk of radio-necrosis or other toxicity if full-dose,
conventionally fractionated reirradiation, namely 60 Gy/30
fractions, was attempted (125).
The use of more conformal, stereotactic approaches, whether
single-fraction radiosurgery or fractionated stereotactic RT
(SRS/SRT), may limit the toxicity of reirradiation (126). For larger volume recurrences,
more conservative hypo-fractionated approaches should be utilized
to reduce toxicity (127).
Additionally, the changes in biology at the time of recurrence may
dictate response to RT. Specifically, recurrent GBM has been
observed to shift to a more aggressive, mesenchymal phenotype
(126). Intriguingly, it has
been observed that RT itself (as well as other therapies) may play
a crucial role in the mesenchymal transition of recurrent GBM,
which in turn is more treatment-resistant (128).
Regardless of a patient's response to prior
treatment, ~90% of patients will show disease recurrence within the
first 2 years of treatment (18).
This, alongside the poor survival rate of GBM itself, is what fuels
the search for novel targets and therapeutic strategies. Histone
deacetylases (HDAC) have become a target of extreme interest in
drug development for cancer. Wang et al (129) showed that overexpression of
HDAC6 promotes proliferation and treatment resistance in GBM.
Further studies by Yang et al (130) revealed an increase in activity
of the HDAC1/2/6 and Sp1 axis that leads to tumor growth and drug
resistance in GBM. It was revealed that inhibiting HDAC1/2/6
significantly reduced the proliferative abilities of both GBM and
TMZ-resistant GBM cells. The greatest efficacy in their
TMZ-resistant orthotopic GBM model was observed when comparing OS
between TMZ and TMZ plus their HDAC inhibitor (MPT0B291) (130). These results indicated that HDAC
pathways may be a valuable target in the fight against GBM and
recurrent disease.
Another area that has gained interest in the fight
against cancer is immunotherapy. Programmed cell death protein 1,
programmed death-ligand 1 (PD-L1) and T-cell immunoglobulin mucin
receptor 3 have been found to be overexpressed on GBM tissues
(131-133). While this suggests that
immunotherapy may be a great asset in the fight against GBM, it has
suboptimal results due to the extreme immunosuppressive nature of
GBM and the immune-privileged environment of the CNS (134). Tong et al (135) reported that the use of ACT001,
which is currently in a phase I/II clinical trial (NCT05053880),
significantly reduces the expression of PD-L1 in GBM. It inhibits
the phosphorylation of STAT3, preventing transcription of PD-L1.
This was shown to cause a decrease in a protumor immune responses
and an increase in antitumor immune responses (135).
There have been multiple studies on how GBM and
other malignancies evade the response of anti-angiogenic therapies
such as bevacizumab. These studies highlight the need for the
development of therapeutic strategies to target these evasive
mechanisms either alone or in combination with other therapies.
Scholz et al (136)
investigated the potential use of targeting angiopoietin-2 (Ang-2)
in both treatment-naïve and bevacizumab-resistant GBM. The
aforementioned study showed an increase in survival when targeting
both VEGF and Ang-2 (136).
Other studies have also looked at targeting pathways in response to
the hypoxic environment caused by bevacizumab treatment. Piao et
al (137) showed that using
altiratinib, an inhibitor of MET, VEGFR2, TIE2 and tropomyosin
receptor kinases, was significantly effective in decreasing cell
viability in vitro. It was also identified that altiratinib
in combination with bevacizumab provided the best overall results
in reducing tumor volume, invasiveness and mesenchymal markers
compared with bevacizumab treatment alone. It was also demonstrated
in their xenograft models that the combination treatment provided
the greatest benefit to OS (137). Carbonell et al (138) exploited β1 integrins in
bevacizumab-resistant GBM cells. The group reported that β1
integrin expression was increased after becoming resistant to
bevacizumab, in their bevacizumab-resistant clinical and xenograft
samples. It was reported that targeting β1 integrins had a
significant effect on the proliferation and mesenchymal-like
properties of bevacizumab-resistant cells (138). While these findings are
optimistic, markedly further investigation is required regarding
the use of immunotherapies in treating CNS malignancies such as
GBM. Emphasis must focus on understanding the tumor
microenvironment (TME) in these tumors in order to develop more
effective single-agent or combination therapies.
With EGFR and EGFRvIII being the most common
alterations in GBM, they would appear to be valuable targets in
treating GBM. However, EGFR inhibitors have been shown to be less
effective than anticipated. Zanca et al (139) reported that EGFRvIII-positive
cells secreted interleukin-6 (IL-6) that activated NFκB, which in
turn activated survivin and decreased the sensitivity to EGFR
inhibitors (139). Liu et
al (140) tested a
third-generation EGFR inhibitor, AZD9291 (Osimertinib), and
compared the response to erlotinib and gefitinib. Osimertinib
easily crosses the BBB, making it an attractive compound for
treating GBM. Compared with earlier versions of EGFR inhibitors,
AZD9291 continued to inhibit the EGFR/ERK pathway, leading to an
improved response in their murine models and an increase in OS
(140). While EGFR again would
be a sound target for GBM, treatment is suboptimal until multiple
aspects are inhibited by the therapy. These results indicate the
need for innovative next-generation compounds that can inhibit
multiple aspects of the pathways.
Another fast-growing field in the treatment of
cancers is the examination of different classes of RNA molecules
such as long non-coding RNAs (lncRNAs) and microRNAs (miRs).
LncRNAs do not code for protein and are >200 base pairs (bp) in
length (141). Lu et al
(142) revealed that small
nucleolar RNA host gene 12 (SNHG12) was upregulated in
TMZ-resistant cells compared with non-treated cells. It was found
that the promoter region of this lncRNA had a decrease in
methylation, allowing easier access by transcription factors such
as Sp1. It was later showed that this lncRNA does indeed play a
role in TMZ resistance when expression was knocked down using short
hairpin RNA (142). By knocking
down expression of Sp1, an increased sensitivity to TMZ compared
with the control was revealed. It was also found that lncRNA SNHG12
interacts with miR-129-5p, and this interaction stops miR-129-5p
from inhibiting MAPK1 or E2F7 (142). By preventing this inhibition,
the MAPK signaling pathway has an increased level of activity,
allowing for the inhibition of apoptotic proteins. The combined
activity of E2F7 and MAPK allows for cell proliferation and
survival through G1/S phase transitions. Mazor et al
(143) showed that the presence
of lncRNA TP73-AS1 correlates to TMZ resistance in GSCs. It was
demonstrated that when lncRNA TP73-AS1 was knocked down, there was
a significant decrease in cell viability when treating with TMZ
compared with the control cells. It was also reported that
following knockdown of this lncRNA, metabolic processes were
affected via RNA sequencing data. It was shown that one of the
major proteins regulated by lncRNA, TP73-AS1, was aldehyde
dehydrogenase 1 family member A1, which has been previously
reported as a stem cell marker in cancers and corresponds to
treatment resistance (143).
MicroRNAs have also gained traction in
understanding the mechanisms behind therapeutic resistance. MiRs
are small, single stranded RNA sequences which bind to
3'-untranslated regions (UTR) of mRNA that effect gene expression
post-transcriptionally. Li et al (144) demonstrated that miR-1268a
regulates the expression of ABCC1 in GBM cells. ABCC1, also known
as MRP1, is a drug efflux pump that removes drugs from the cells.
It was identified that upon treatment with TMZ, miR-1268a was
downregulated while protein expression of ABCC1 was upregulated.
This was also confirmed in patient samples which compared primary
tumors to recurrent tumors. When miR-1268a mimics were
overexpressed, a decrease in ABCC1 protein levels was observed. The
mimics also allowed the cells to become sensitive to TMZ treatment
both in vitro and in vivo (144). Luo et al (72) reported another miR, miR-126-3p,
that is involved in TMZ resistance in GBM. It was shown that in
patient samples, miR-126-3p was decreased in TMZ-resistant samples
compared with TMZ-sensitive samples. TMZ-resistant cell lines were
also created and it was revealed that compared with their
TMZ-sensitive parental cell lines, miR-126-3p was decreased. When
miR-126-3p mimics were transfected into the TMZ-resistant cell
lines, it was observed that the expression of miR-126-3p made the
cells sensitive to TMZ compared with controls by affecting cell
viability and proliferative abilities. It was later demonstrated
that miR-126-3p binds to the 3'-UTR of SOX2 and
downregulates its expression while a decrease in miR-126-3p showed
an increase in SOX2 protein levels. Following this discovery, it
was found that when miR-126-3p decreases SOX2 levels, the
Wnt/β-catenin signaling was inhibited. These results suggested that
miR-126-3p promotes TMZ sensitivity by inhibiting SOX2 expression,
which prevents Wnt/β-catenin signaling (72).
As shown in the literature, GBM is notorious for
having multiple mechanisms at its disposal to evade current
treatment strategies (145-147). In brief, multiple studies have
reported that to overcome this treatment-resistant characteristic
of GBM, it is needed to find ways to target pathways and/or
proteins that are involved in these resistance mechanisms. Using
multimodal treatment strategies has shown to re-sensitize cells to
therapies and increase OS preclinically.
While preclinical investigations appear promising,
clinical trial results have been dismal when it comes to GBM.
Currently there are 320 actively enrolling clinical trials for GBM
according to clinicaltrials.gov. Of these 320, 117 of them include
recurrent GBM. In the fight against GBM and recurrent GBM, novel
treatment strategies are a must. It has been shown preclinically
that some of the best responses come from a multimodal treatment
approach. Investigators must start incorporating these therapeutic
resistance mechanisms into consideration for their trials or the
outcomes will continue to be suboptimal.
Another area that has hindered the progress of
successful clinical trials is the presence of the blood-brain
barrier (BBB). There is a current phase I/II clinical trial
(NCT04440358) where the aim is to establish the safety and efficacy
of using microbubbles in order to disrupt the BBB in patients with
recurrent GBM undergoing intravenous carboplatin therapy. The
primary goal of this trial is to open up the BBB prior to
chemotherapy administration, allowing for improved drug
delivery.
With advancements made in the ability to deliver
therapies more effectively to GBM tumors and by targeting these
resistance mechanisms, it is hopeful that current and future
clinical trials will lead to improved outcomes with regard to PFS
and OS. Current clinical trials that are actively recruiting
patients with a focus on targeting aspects of therapeutic
resistance are presented in Table
I (clinicaltrials.gov).
GBM is a highly aggressive tumor characterized by
poor patient survival. One of the leading causes of the dismal
outcome is the heterogeneous biology of the TME and mutations in
regulatory signaling pathways. Collectively, these promote
resistance to radiation and standard drug treatments. Dysregulation
is observed in tumor signaling pathways, including PI3K/Akt, Tp53,
Rb, STAT/Notch, CDKN2A and reelin (146-148). Altered signaling promotes
tumorigenesis by enhancing migration, proliferation and invasion
and prevents apoptosis in tumor cells (145,146). Profiling the transcriptional,
genetic and epigenetic changes within the TME has led to new
insights in the diagnosis of GBM. Cellular variation rising from
intratumoral and intertumoral mutations have led to investigations
into novel subtype specific therapies. Ongoing clinical trials for
drugs which target specific molecular markers and genes involve
patients having neurofibromin 1, EGFRvIII and BRAFv600 mutations,
as well as EGFR gene amplification (39). In most cases, patients within
these trials are classified based on the mutation of hTERT
promoter gene, MGMT promoter methylation status, IDH1/2
status, and aberration of EGFR/PDGFR signaling. However, the
clinical translation of targeted treatment remains unknown.
Furthermore, the effect of such therapies on the host immune system
and secondary neuroinflammatory responses needs to be
elucidated.
GBM stem cells are hypothesized to influence
intratumoral cellular variation due to their high tumorigenic
potential. Preliminary studies demonstrated that GBM stem cells
impact cell growth dynamics and evade cell death mediated by
radiation and chemotherapy. This indicates that it may be essential
to target GBM stem cells with genetic and molecular tumor subtypes.
Investigation into multimodal therapy has also given rise to novel
therapeutic regimens to treat GBM. One such treatment involves
using electric TTFs that interfere with cell division through
misalignment of mitotic spindles. Another modality uses focused
ultrasound to disrupt the BBB (low-intensity) or ablate the tumor
mass (high-intensity). These techniques seek to aid drug delivery,
overcome resistance and increase drug efficacy for tumors. New
treatment modalities in conjunction with targeted
immunotherapy/chemotherapy may be essential for improving the
outcomes of patients with GBM.
No matter the response to initial treatment,
patients will ultimately succumb to recurrent disease. Recurrent
disease tends to be more aggressive and resistant to treatment
compared with the initial tumor. This leaves first-line therapies
ineffective and give patients only a limited number of second-line
treatment options. The continued poor OS indicates the dire need
for novel targeted therapeutic strategies to overcome these
resistance mechanisms. This review has highlighted key mechanisms
behind treatment resistance in GBM, indicating the dire need for
novel treatment strategies against these key resistance
mechanisms.
Not applicable.
SA conceived the study. JS, TA, AB and SA wrote the
original draft of the manuscript. JS and SA wrote, reviewed and
edited the manuscript. PL and SA supervised the study. PL and SA
conducted project administration. SA acquired funding. All authors
read and approved the final manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
No funding was received.
|
1
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hale JS, Sinyuk M, Rich JN and Lathia JD:
Decoding the cancer stem cell hypothesis in glioblastoma. CNS
Oncol. 2:319–330. 2013. View Article : Google Scholar :
|
|
3
|
Thakkar JP, Dolecek TA, Horbinski C,
Ostrom QT, Lightner DD, Barnholtz-Sloan JS and Villano JL:
Epidemiologic and molecular prognostic review of glioblastoma.
Cancer Epidemiol Biomark Amp Prev. 23:1985–1996. 2014. View Article : Google Scholar
|
|
4
|
Hanif F, Muzaffar K, Perveen K, Malhi SM
and Simjee ShU: Glioblastoma Multiforme: A review of its
epidemiology and pathogenesis through clinical presentation and
treatment. Asian Pac J Cancer Prev. 18:3–9. 2017.PubMed/NCBI
|
|
5
|
Alexander BM and Cloughesy TF: Adult
Glioblastoma. J Clin Oncol. 35:2402–2409. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Vleeschouwer S: Glioblastoma. Codon
Publications; Brisbane, QLD: 2017, View Article : Google Scholar
|
|
7
|
Young RM, Jamshidi A, Davis G and Sherman
JH: Current trends in the surgical management and treatment of
adult glioblastoma. Ann Transl Med. 3:1212015.PubMed/NCBI
|
|
8
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohgaki H and Kleihues P: Genetic pathways
to primary and secondary glioblastoma. Am J Pathol. 170:1445–1453.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ohgaki H and Kleihues P: The Definition of
primary and secondary glioblastoma. Clin Cancer Res. 19:764–772.
2013. View Article : Google Scholar
|
|
11
|
Valentinis L, Tuniz F, Valent F, Mucchiut
M, Little D, Skrap M, Bergonzi P and Zanchin G: Headache attributed
to intracranial tumours: A prospective cohort study. Cephalalgia.
30:389–398. 2010. View Article : Google Scholar
|
|
12
|
Chaichana KL, Parker SL, Olivi A and
Quiñones-Hinojosa A: Long-term seizure outcomes in adult patients
undergoing primary resection of malignant brain astrocytomas:
Clinical article. J Neurosurg. 111:282–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Davis ME: Glioblastoma: Overview of
disease and treatment. Clin J Oncol Nurs. 20(5 Suppl): S2–S8. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wen PY, Weller M, Lee EQ, Alexander BM,
Barnholtz-Sloan JS, Barthel FP, Batchelor TT, Bindra RS, Chang SM,
Chiocca EA, et al: Glioblastoma in adults: A Society for
Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO)
consensus review on current management and future directions.
Neuro-Oncol. 22:1073–1113. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wen PY, Macdonald DR, Reardon DA,
Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert
MR, Lassman AB, et al: Updated response assessment criteria for
high-grade gliomas: Response assessment in neuro-oncology working
group. J Clin Oncol. 28:1963–1972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu LS, Eschbacher JM, Dueck AC, Heiserman
JE, Liu S, Karis JP, Smith KA, Shapiro WR, Pinnaduwage DS, Coons
SW, et al: Correlations between perfusion MR imaging cerebral blood
volume, microvessel quantification, and clinical outcome using
stereotactic analysis in recurrent high-grade glioma. Am J
Neuroradiol. 33:692012. View Article : Google Scholar
|
|
17
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar
|
|
18
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stupp R, Taillibert S, Kanner A, Read W,
Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K,
et al: Effect of tumor-treating fields plus maintenance
temozolomide vs maintenance temozolomide alone on survival in
patients with glioblastoma: A randomized clinical trial. JAMA.
318:2306–2316. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perry JR, Rizek P, Cashman R, Morrison M
and Morrison T: Temozolomide rechallenge in recurrent malignant
glioma by using a continuous temozolomide schedule. Cancer.
113:2152–2157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brandes AA, Tosoni A, Amistà P, Nicolardi
L, Grosso D, Berti F and Ermani M: How effective is BCNU in
recurrent glioblastoma in the modern era? Neurology. 63:12812004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Reithmeier T, Graf E, Piroth T, Trippel M,
Pinsker MO and Nikkhah G: BCNU for recurrent glioblastoma
multiforme: Efficacy, toxicity and prognostic factors. BMC Cancer.
10:302010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wick W, Puduvalli VK, Chamberlain MC, van
den Bent MJ, Carpentier AF, Cher LM, Mason W, Weller M, Hong S,
Musib L, et al: Phase III study of enzastaurin compared with
lomustine in the treatment of recurrent intracranial glioblastoma.
J Clin Oncol. 28:1168–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taal W, Oosterkamp HM, Walenkamp AM,
Dubbink HJ, Beerepoot LV, Hanse MC, Buter J, Honkoop AH, Boerman D,
de Vos FY, et al: Single-agent bevacizumab or lomustine versus a
combination of bevacizumab plus lomustine in patients with
recurrent glioblastoma (BELOB trial): A randomised controlled phase
2 trial. Lancet Oncol. 15:943–953. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Glas M, Happold C, Rieger J, Wiewrodt D,
Bähr O, Steinbach JP, Wick W, Kortmann RD, Reifenberger G, Weller M
and Herrlinger U: Long-term survival of patients with glioblastoma
treated with radiotherapy and lomustine plus temozolomide. J Clin
Oncol. 27:1257–1261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Herrlinger U, Rieger J, Koch D, Loeser S,
Blaschke B, Kortmann RD, Steinbach JP, Hundsberger T, Wick W,
Meyermann R, et al: Phase II trial of lomustine plus temozolomide
chemotherapy in addition to radiotherapy in newly diagnosed
glioblastoma: UKT-03. J Clin Oncol. 24:4412–4417. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brem H, Piantadosi S, Burger PC, Walker M,
Selker R, Vick NA, Black K, Sisti M, Brem S, Mohr G, et al:
Placebo-controlled trial of safety and efficacy of intraoperative
controlled delivery by biodegradable polymers of chemotherapy for
recurrent gliomas. Lancet. 345:1008–1012. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McGirt MJ and Brem H: Carmustine wafers
(Gliadel) plus concomitant temozolomide therapy after resection of
malignant astrocytoma: Growing evidence for safety and efficacy.
Ann Surg Oncol. 17:1729–1731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lombardi G, De Salvo GL, Brandes AA, Eoli
M, Rudà R, Faedi M, Lolli I, Pace A, Daniele B, Pasqualetti F, et
al: Regorafenib compared with lomustine in patients with relapsed
glioblastoma (REGOMA): A multicentre, open-label, randomised,
controlled, phase 2 trial. Lancet Oncol. 20:110–119. 2019.
View Article : Google Scholar
|
|
31
|
Friedman HS, Prados MD, Wen PY, Mikkelsen
T, Schiff D, Abrey LE, Yung WKA, Paleologos N, Nicholas MK, Jensen
R, et al: Bevacizumab alone and in combination with irinotecan in
recurrent glioblastoma. J Clin Oncol. 27:4733–4740. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wick W, Gorlia T, Bendszus M, Taphoorn M,
Sahm F, Harting I, Brandes AA, Taal W, Domont J, Idbaih A, et al:
Lomustine and bevacizumab in progressive glioblastoma. N Engl J
Med. 377:1954–1963. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ameratunga M, Pavlakis N, Wheeler H, Grant
R, Simes J and Khasraw M: Anti-angiogenic therapy for high-grade
glioma. Cochrane Database Syst Rev. 11:CD0082182018.PubMed/NCBI
|
|
34
|
Kaley T, Nolan C, Carver A and Omuro A:
Bevacizumab for acute neurologic deterioration in patients with
glioblastoma. CNS Oncol. 2:413–418. 2013. View Article : Google Scholar
|
|
35
|
Wick W, Weller M, van den Bent M and Stupp
R: Bevacizumab and recurrent malignant gliomas: A european
perspective. J Clin Oncol. 28:e188–e189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kazmi F, Soon YY, Leong YH, Koh WY and
Vellayappan B: Re-irradiation for recurrent glioblastoma (GBM): A
systematic review and meta-analysis. J Neurooncol. 142:79–90. 2019.
View Article : Google Scholar
|
|
37
|
Stupp R, Wong ET, Kanner AA, Steinberg D,
Engelhard H, Heidecke V, Kirson ED, Taillibert S, Liebermann F,
Dbalý V, et al: NovoTTF-100A versus physician's choice chemotherapy
in recurrent glioblastoma: A randomised phase III trial of a novel
treatment modality. Eur J Cancer. 48:2192–2202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee SX, Tunkyi A, Wong E and Swanson KD:
Mitosis interference of cancer cells during anaphase by electric
field from NovoTTF-100A: An update. J Clin Oncol. 30(15_Suppl):
e21078. 2012. View Article : Google Scholar
|
|
39
|
Lee E, Yong RL, Paddison P and Zhu J:
Comparison of glioblastoma (GBM) molecular classification methods.
Semin Cancer Biol. 53:201–211. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Snuderl M, Fazlollahi L, Le LP, Nitta M,
Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD,
Betensky RA, et al: Mosaic amplification of multiple receptor
tyrosine kinase genes in glioblastoma. Cancer Cell. 20:810–817.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sottoriva A, Spiteri I, Piccirillo SG,
Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C and Tavaré
S: Intratumor heterogeneity in human glioblastoma reflects cancer
evolutionary dynamics. Proc Natl Acad Sci. 110:4009–4014. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Aldape K, Zadeh G, Mansouri S,
Reifenberger G and von Deimling A: Glioblastoma: Pathology,
molecular mechanisms and markers. Acta Neuropathol (Berl).
129:829–848. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
An Z, Aksoy O, Zheng T, Fan QW and Weiss
WA: Epidermal growth factor receptor and EGFRvIII in glioblastoma:
Signaling pathways and targeted therapies. Oncogene. 37:1561–1575.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Arteaga CL and Engelman JA: ERBB
receptors: From oncogene discovery to basic science to
mechanism-based cancer therapeutics. Cancer Cell. 25:282–303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Narita Y, Nagane M, Mishima K, Huang HJS,
Furnari FB and Cavenee WK: Mutant Epidermal growth factor receptor
signaling Down-Regulates p27 through activation of the
phosphatidylinositol 3-Kinase/Akt pathway in glioblastomas. Cancer
Res. 62:6764–6769. 2002.PubMed/NCBI
|
|
46
|
Huang HJS, Nagane M, Klingbeil CK, Lin H,
Nishikawa R, Ji XD, Huang CM, Gill GN, Wiley HS and Cavenee WK: The
enhanced tumorigenic activity of a mutant epidermal growth factor
receptor common in human cancers is mediated by threshold levels of
constitutive tyrosine phosphorylation and unattenuated signaling. J
Biol Chem. 272:2927–2935. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nagane M, Levitzki A, Gazit A, Cavenee WK
and Huang HJS: Drug resistance of human glioblastoma cells
conferred by a tumor-specific mutant epidermal growth factor
receptor through modulation of Bcl-XL and caspase-3-like proteases.
Proc Natl Acad Sci. 95:5724–5729. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Inda M del M, Bonavia R, Mukasa A, Narita
Y, Sah DW, Vandenberg S, Brennan C, Johns TG, Bachoo R, Hadwiger P,
et al: Tumor heterogeneity is an active process maintained by a
mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev.
24:1731–1745. 2010. View Article : Google Scholar
|
|
49
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nakamura M, Yonekawa Y, Kleihues P and
Ohgaki H: Promoter Hypermethylation of the RB1 gene in
Glioblastomas. Lab Invest. 81:77–82. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cerami E, Demir E, Schultz N, Taylor BS
and Sander C: Automated network analysis identifies core pathways
in glioblastoma. PLoS One. 5:e89182010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Muñoz-Hidalgo L, San-Miguel T, Megías J,
Monleón D, Navarro L, Roldán P, Cerdá-Nicolás M and López-Ginés C:
Somatic copy number alterations are associated with EGFR
amplification and shortened survival in patients with primary
glioblastoma. Neoplasia. 22:10–21. 2020. View Article : Google Scholar
|
|
53
|
Aubrey BJ, Strasser A and Kelly GL:
Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb
Perspect Med. 6:a0260622016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Verhaak RGW, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Integrated genomic analysis identifies clinically relevant subtypes
of glioblastoma characterized by abnormalities in PDGFRA, IDH1,
EGFR, and NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang Y, Dube C, Gibert M Jr, Cruickshanks
N, Wang B, Coughlan M, Yang Y, Setiady I, Deveau C, Saoud K, et al:
The p53 pathway in glioblastoma. Cancers (Basel). 10:2972018.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ham SW, Jeon HY, Jin X, Kim EJ, Kim JK,
Shin YJ, Lee Y, Kim SH, Lee SY, Seo S, et al: TP53 gain-of-function
mutation promotes inflammation in glioblastoma. Cell Death Differ.
26:409–425. 2019. View Article : Google Scholar :
|
|
57
|
Forte IM, Indovina P, Iannuzzi CA, Cirillo
D, Di Marzo D, Barone D, Capone F, Pentimalli F and Giordano A:
Targeted therapy based on p53 reactivation reduces both
glioblastoma cell growth and resistance to temozolomide. Int J
Oncol. 54:2189–2199. 2019.PubMed/NCBI
|
|
58
|
Verreault M, Schmitt C, Goldwirt L, Pelton
K, Haidar S, Levasseur C, Guehennec J, Knoff D, Labussière M, Marie
Y, et al: Preclinical efficacy of the MDM2 inhibitor RG7112 in
MDM2-smplified and TP53 Wild-type glioblastomas. Clin Cancer Res.
22:1185–1196. 2016. View Article : Google Scholar
|
|
59
|
Mizoguchi M, Nutt CL, Mohapatra G and
Louis DN: Genetic alterations of phosphoinositide 3-kinase subunit
genes in human glioblastomas. Brain Pathol. 14:372–377. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Papa A and Pandolfi PP: The PTEN-PI3K axis
in cancer. Biomolecules. 9:1532019. View Article : Google Scholar
|
|
61
|
Lino MM and Merlo A: PI3Kinase signaling
in glioblastoma. J Neurooncol. 103:417–427. 2011. View Article : Google Scholar :
|
|
62
|
Koul D: PTEN signaling pathways in
glioblastoma. Cancer Biol Ther. 7:1321–1325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 26:3212008.
|
|
64
|
Rao SK, Edwards J, Joshi AD, Siu IM and
Riggins GJ: A survey of glioblastoma genomic amplifications and
deletions. J Neurooncol. 96:169–179. 2010. View Article : Google Scholar
|
|
65
|
von Achenbach C, Weller M, Kaulich K,
Gramatzki D, Zacher A, Fabbro D, Reifenberger G and Szabó E:
Synergistic growth inhibition mediated by dual PI3K/mTOR pathway
targeting and genetic or direct pharmacological AKT inhibition in
human glioblastoma models. J Neurochem. 153:510–524. 2020.
View Article : Google Scholar
|
|
66
|
Lin F, de Gooijer MC, Hanekamp D,
Chandrasekaran G, Buil LC, Thota N, Sparidans RW, Beijnen JH,
Würdinger T and van Tellingen O: PI3K-mTOR Pathway Inhibition
exhibits efficacy against high-grade glioma in clinically relevant
mouse models. Clin Cancer Res. 23:12862017. View Article : Google Scholar
|
|
67
|
Wen PY, Lee EQ, Reardon DA, Ligon KL and
Alfred Yung WK: Current clinical development of PI3K pathway
inhibitors in glioblastoma. Neuro Oncol. 14:819–829. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Cohen MH, Johnson JR and Pazdur R: Food
and drug administration drug approval summary: Temozolomide plus
radiation therapy for the treatment of newly diagnosed glioblastoma
multiforme. Clin Cancer Res. 11:67672005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kitange GJ, Carlson BL, Schroeder MA,
Grogan PT, Lamont JD, Decker PA, Wu W, James CD and Sarkaria JN:
Induction of MGMT expression is associated with temozolomide
resistance in glioblastoma xenografts. Neuro Oncol. 11:281–291.
2009. View Article : Google Scholar :
|
|
71
|
Alnahhas I, Alsawas M, Rayi A, Palmer JD,
Raval R, Ong S, Giglio P, Murad MH and Puduvalli V: Characterizing
benefit from temozolomide in MGMT promoter unmethylated and
methylated glioblastoma: A systematic review and meta-analysis.
Neuro Oncol Adv. 2:vdaa0822020. View Article : Google Scholar
|
|
72
|
Luo W, Yan D, Song Z, Zhu X, Liu X, Li X
and Zhao S: miR-126-3p sensitizes glioblastoma cells to
temozolomide by inactivating Wnt/β-catenin signaling via targeting
SOX2. Life Sci. 226:98–106. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Garg M: Epithelial-mesenchymal
transition-activating transcription factors-multifunctional
regulators in cancer. World J Stem Cells. 5:188–195. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Diaz RJ, Ali S, Qadir MG, De La Fuente MI,
Ivan ME and Komotar RJ: The role of bevacizumab in the treatment of
glioblastoma. J Neurooncol. 133:455–467. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gilbert MR, Dignam JJ, Armstrong TS, Wefel
JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S,
Won M, et al: A randomized trial of bevacizumab for newly diagnosed
glioblastoma. N Engl J Med. 370:699–708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kreisl TN, Kim L, Moore K, Duic P, Royce
C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, et al:
Phase II Trial of Single-Agent Bevacizumab Followed by Bevacizumab
Plus Irinotecan at Tumor Progression in Recurrent Glioblastoma. J
Clin Oncol. 5:740–745. 2009. View Article : Google Scholar
|
|
77
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Haibe Y, Kreidieh M, El Hajj H, Khalifeh
I, Mukherji D, Temraz S and Shamseddine A: Resistance mechanisms to
Anti-angiogenic therapies in cancer. Front Oncol. 10:2212020.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Forsythe JA, Jiang BH, Iyer NV, Agani F,
Leung SW, Koos RD and Semenza GL: Activation of vascular
endothelial growth factor gene transcription by hypoxia-inducible
factor 1. Mol Cell Biol. 16:4604–4613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Trusolino L, Bertotti A and Comoglio PM:
MET signalling: Principles and functions in development, organ
regeneration and cancer. Nat Rev Mol Cell Biol. 11:834–848. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lu KV, Chang JP, Parachoniak CA, Pandika
MM, Aghi MK, Meyronet D, Isachenko N, Fouse SD, Phillips JJ,
Cheresh DA, et al: VEGF inhibits tumor cell invasion and
mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell.
22:21–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hande KR: Etoposide: Four decades of
development of a topoisomerase II inhibitor. Eur J Cancer.
34:1514–1521. 1998. View Article : Google Scholar
|
|
84
|
Montecucco A, Zanetta F and Biamonti G:
Molecular mechanisms of etoposide. EXCLI J. 14:95–108.
2015.PubMed/NCBI
|
|
85
|
Biasoli D, Kahn SA, Cornélio TA, Furtado
M, Campanati L, Chneiweiss H, Moura-Neto V and Borges HL:
Retinoblastoma protein regulates the crosstalk between autophagy
and apoptosis, and favors glioblastoma resistance to etoposide.
Cell Death Dis. 4:e767. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
McLendon R, Friedman A, Bigner D, Van Meir
EG, Brat DJ, Marie Mastrogianakis G, Olson JJ, Mikkelsen T, Lehman
N, Aldape A, et al: Comprehensive genomic characterization defines
human glioblastoma genes and core pathways. Nature. 455:1061–1068.
2008. View Article : Google Scholar
|
|
87
|
Senturk JC, Bohlman S and Manfredi JJ:
Mdm2 selectively suppresses DNA damage arising from inhibition of
topoisomerase II independent of p53. Oncogene. 36:6085–6096. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Conradt L, Henrich A, Wirth M, Reichert M,
Lesina M, Algül H, Schmid RM, Krämer OH, Saur D and Schneider G:
Mdm2 inhibitors synergize with topoisomerase II inhibitors to
induce p53-independent pancreatic cancer cell death. Int J Cancer.
132:2248–2257. 2013. View Article : Google Scholar
|
|
89
|
Kondo S, Kondo Y, Hara H, Kaakaji R,
Peterson JW, Morimura T, Takeuchi J and Barnett GH: mdm2 gene
mediates the expression of mdr1 gene and P-glycoprotein in a human
glioblastoma cell line. Br J Cancer. 74:1263–1268. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar
|
|
91
|
Pénzváltó Z, Lánczky A, Lénárt J,
Meggyesházi N, Krenács T, Szoboszlai N, Denkert C, Pete I and
Győrffy B: MEK1 is associated with carboplatin resistance and is a
prognostic biomarker in epithelial ovarian cancer. BMC Cancer.
14:8372014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ahmad A, Robinson AR, Duensing A, van
Drunen E, Beverloo HB, Weisberg DB, Hasty P, Hoeijmakers JH and
Niedernhofer LJ: ERCC1-XPF endonuclease facilitates DNA
double-strand break repair. Mol Cell Biol. 28:5082–5092. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Walker MD, Alexander E Jr, Hunt WE,
MacCarty CS, Mahaley MS Jr, Mealey J Jr, Norrell HA, Owens G,
Ransohoff J, Wilson CB, et al: Evaluation of BCNU and/or
radiotherapy in the treatment of anaplastic gliomas: A cooperative
clinical trial. J Neurosurg. 49:333–343. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Walker MD, Strike TA and Sheline GE: An
analysis of dose-effect relationship in the radiotherapy of
malignant gliomas. Int J Radiat Oncol. 5:1725–1731. 1979.
View Article : Google Scholar
|
|
95
|
van Rijn J, Heimans JJ, van den Berg J,
van der Valk P and Slotman BJ: Survival of human glioma cells
treated with various combination of temozolomide and X-rays. Int J
Radiat Oncol. 47:779–784. 2000. View Article : Google Scholar
|
|
96
|
Blumenthal DT, Gorlia T, Gilbert MR, Kim
MM, Burt Nabors L, Mason WP, Hegi ME, Zhang P, Golfinopoulos V,
Perry JR, et al: Is More better? The impact of extended adjuvant
temozolomide in newly diagnosed glioblastoma: A secondary analysis
of EORTC and NRG Oncology/RTOG. Neuro Oncol. 19:1119–1126. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
van Linde ME, Brahm CG, de Witt Hamer PC,
Reijneveld JC, Bruynzeel AME, Vandertop WP, van de Ven PM,
Wagemakers M, van der Weide HL, Enting RH, et al: Treatment outcome
of patients with recurrent glioblastoma multiforme: A retrospective
multicenter analysis. J Neurooncol. 135:183–192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Monteiro AR, Hill R, Pilkington GJ and
Madureira PA: The role of hypoxia in glioblastoma invasion. Cells.
6:452017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Gray LH, Conger AD, Ebert M, Hornsey S and
Scott OCA: The concentration of oxygen dissolved in tissues at the
time of irradiation as a factor in radiotherapy. Br J Radiol.
26:638–648. 1953. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Horsman MR, Mortensen LS, Petersen JB,
Busk M and Overgaard J: Imaging hypoxia to improve radiotherapy
outcome. Nat Rev Clin Oncol. 9:674–687. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ikeda E, Achen MG, Breier G and Risau W:
Hypoxia-induced transcriptional activation and increased mRNA
stability of vascular endothelial growth factor in C6 glioma cells.
J Biol Chem. 270:19761–19766. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chinot OL, Wick W, Mason W, Henriksson R,
Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea
D, et al: Bevacizumab plus Radiotherapy-Temozolomide for newly
diagnosed glioblastoma. N Engl J Med. 370:709–722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Colman H, Zhang L, Sulman EP, McDonald JM,
Shooshtari NL, Rivera A, Popoff S, Nutt CL, Louis DN, Cairncross
JG, et al: A multigene predictor of outcome in glioblastoma. Neuro
Oncol. 12:49–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Li Z, Bao S, Wu Q, Wang H, Eyler C,
Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al:
Hypoxia-Inducible factors regulate tumorigenic capacity of glioma
stem cells. Cancer Cell. 15:501–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: Accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Prieto-Vila M, Takahashi RU, Usuba W,
Kohama I and Ochiya T: Drug resistance driven by cancer stem cells
and their niche. Int J Mol Sci. 18:25742017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Batlle E and Clevers H: Cancer stem cells
revisited. Nat Med. 23:1124–1134. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Roninson IB, Broude EV and Chang BD: If
not apoptosis, then what? Treatment-induced senescence and mitotic
catastrophe in tumor cells. Drug Resist Updat. 4:303–313. 2001.
View Article : Google Scholar
|
|
109
|
Chen J, Li Y, Yu TS, McKay RM, Burns DK,
Kernie SG and Parada LF: A restricted cell population propagates
glioblastoma growth after chemotherapy. Nature. 488:522–526. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Peh GSL, Lang RJ, Pera MF and Hawes SM:
CD133 expression by neural progenitors derived from human embryonic
stem cells and its use for their prospective isolation. Stem Cells
Dev. 18:269–282. 2009. View Article : Google Scholar
|
|
111
|
Kim YS, Kaidina AM, Chiang JH, Yarygin KN
and Lupatov AY: Cancer stem cell molecular markers verified in
vivo. Biochem Mosc Suppl Ser B Biomed Chem. 11:43–54. 2017.
View Article : Google Scholar
|
|
112
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Kim H, Zheng S, Amini SS, Virk SM,
Mikkelsen T, Brat DJ, Grimsby J, Sougnez C, Muller F, Hu J, et al:
Whole-genome and multisector exome sequencing of primary and
post-treatment glioblastoma reveals patterns of tumor evolution.
Genome Res. 25:316–327. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Singh SK, Clarke ID, Terasaki M, Bonn VE,
Hawkins C, Squire J and Dirks PB: Identification of a cancer stem
cell in human brain tumors. Cancer Res. 63:58212003.PubMed/NCBI
|
|
115
|
Brennan CW, Verhaak RGW, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: The somatic genomic landscape of glioblastoma.
Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wong AJ, Ruppert JM, Bigner SH, Grzeschik
CH, Humphrey PA, Bigner DS and Vogelstein B: Structural alterations
of the epidermal growth factor receptor gene in human gliomas. Proc
Natl Acad Sci. 89:29651992. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lammering G, Hewit TH, Valerie K, Contessa
JN, Amorino GP, Dent P and Schmidt-Ullrich RK: EGFRvIII-mediated
radioresistance through a strong cytoprotective response. Oncogene.
22:5545–5553. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Mukherjee B, McEllin B, Camacho CV,
Tomimatsu N, Sirasanagandala S, Nannepaga S, Hatanpaa KJ, Mickey B,
Madden C, Maher E, et al: EGFRvIII and DNA double-strand break
repair: A molecular mechanism for radioresistance in glioblastoma.
Cancer Res. 69:42522009. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chakravarti A, Wang M, Robins HI,
Lautenschlaeger T, Curran WJ, Brachman DG, Schultz CJ, Choucair A,
Dolled-Filhart M, Christiansen J, et al: RTOG 0211: A Phase 1/2
study of radiation therapy with concurrent gefitinib for newly
diagnosed glioblastoma patients. Int J Radiat Oncol. 85:1206–1211.
2013. View Article : Google Scholar
|
|
120
|
Kao GD, Jiang Z, Fernandes AM, Gupta AK
and Maity A: Inhibition of phosphatidylinositol-3-OH Kinase/Akt
signaling impairs DNA repair in glioblastoma cells following
ionizing radiation. J Biol Chem. 282:21206–21212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Charles NA, Holland EC, Gilbertson R,
Glass R and Kettenmann H: The brain tumor microenvironment. Glia.
60:502–514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wahl DR, Kim MM, Aryal MP, Hartman H,
Lawrence TS, Schipper MJ, Parmar HA and Cao Y: Combining perfusion
and High B-value diffusion MRI to inform prognosis and predict
failure patterns in glioblastoma. Int J Radiat Oncol Biol Phys.
102:757–764. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Gérard M, Corroyer-Dulmont A, Lesueur P,
Collet S, Chérel M, Bourgeois M, Stefan D, Limkin EJ, Perrio C,
Guillamo JS, et al: Hypoxia imaging and adaptive radiotherapy: A
State-of-the-Art approach in the management of glioma. Front Med.
6:1172019. View Article : Google Scholar
|
|
124
|
Drake LR, Hillmer AT and Cai Z: Approaches
to PET imaging of glioblastoma. Molecules. 25:5682020. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wallner KE, Galicich JH, Krol G, Arbit E
and Malkin MG: Patterns of failure following treatment for
glioblastoma multiforme and anaplastic astrocytoma. Int J Radiat
Oncol. 16:1405–1409. 1989. View Article : Google Scholar
|
|
126
|
Niranjan A, Monaco EAI, Kano H, Flickinger
JC and Lunsford LD: Stereotactic radiosurgery in the multimodality
management of residual or recurrent glioblastoma multiforme. Prog
Neurol Surg. 31:48–61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Shaw E, Scott C, Souhami L, Dinapoli R,
Kline R, Loeffler J and Farnan N: Single dose radiosurgical
treatment of recurrent previously irradiated primary brain tumors
and brain metastases: Final report of RTOG protocol 90-05. Int J
Radiat Oncol. 47:291–298. 2000. View Article : Google Scholar
|
|
128
|
Kim Y, Varn FS, Park SH, Yoon BW, Park HR,
Lee C, Verhaak RGW and Paek SH: Perspective of mesenchymal
transformation in glioblastoma. Acta Neuropathol Commun. 9:502021.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wang Z, Hu P, Tang F, Lian H, Chen X,
Zhang Y, He X, Liu W and Xie C: HDAC6 promotes cell proliferation
and confers resistance to temozolomide in glioblastoma. Cancer
Lett. 379:134–142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Yang WB, Hsu CC, Hsu TI, Liou JP, Chang
KY, Chen PY, Liu JJ, Yang ST, Wang JY, Yeh SH, et al: Increased
activation of HDAC1/2/6 and Sp1 underlies therapeutic resistance
and tumor growth in glioblastoma. Neuro Oncol. 22:1439–1451. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Liu S, Wang Z, Wang Y, Fan X, Zhang C, Ma
W, Qiu X and Jiang T: PD-1 related transcriptome profile and
clinical outcome in diffuse gliomas. OncoImmunology.
7:e13827922018. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wang Z, Zhang C, Liu X, Wang Z, Sun L, Li
G, Liang J, Hu H, Liu Y, Zhang W and Jiang T: Molecular and
clinical characterization of PD-L1 expression at transcriptional
level via 976 samples of brain glioma. OncoImmunology.
5:e11963102016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Li G, Wang Z, Zhang C, Liu X, Cai J, Wang
Z, Hu H, Wu F, Bao Z, Liu Y, et al: Molecular and clinical
characterization of TIM-3 in glioma through 1,024 samples.
OncoImmunology. 6:e13283392017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
McGranahan T, Therkelsen KE, Ahmad S and
Nagpal S: Current state of immunotherapy for treatment of
glioblastoma. Curr Treat Options Oncol. 20:242019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Tong L, Li J, Li Q, Wang X, Medikonda R,
Zhao T, Li T, Ma H, Yi L, Liu P, et al: ACT001 reduces the
expression of PD-L1 by inhibiting the phosphorylation of STAT3 in
glioblastoma. Theranostics. 10:5943–5956. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Scholz A, Harter PN, Cremer S, Yalcin BH,
Gurnik S, Yamaji M, Di Tacchio M, Sommer K, Baumgarten P, Bähr O,
et al: Endothelial cell-derived angiopoietin-2 is a therapeutic
target in treatment-naive and bevacizumab-resistant glioblastoma.
EMBO Mol Med. 8:39–57. 2016. View Article : Google Scholar
|
|
137
|
Piao Y, Liang J, Holmes L, Henry V, Sulman
E and Groot JF: Acquired resistance to Anti-VEGF therapy in
glioblastoma is associated with a mesenchymal transition. Clin
Cancer Res. 19:4392–4403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Carbonell WS, DeLay M, Jahangiri A, Park
CC and Aghi MK: β1 Integrin targeting potentiates antiangiogenic
therapy and inhibits the growth of bevacizumab-resistant
glioblastoma. Cancer Res. 73:3145–3154. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Zanca C, Villa GR, Benitez JA, Thorne AH,
Koga T, D'Antonio M, Ikegami S, Ma J, Boyer AD, Banisadr A, et al:
Glioblastoma cellular cross-talk converges on NF-κB to attenuate
EGFR inhibitor sensitivity. Genes Dev. 31:1212–1227. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Liu X, Chen X, Shi L, Shan Q, Cao Q, Yue
C, Li H, Li S, Wang J, Gao S, et al: The third-generation EGFR
inhibitor AZD9291 overcomes primary resistance by continuously
blocking ERK signaling in glioblastoma. J Exp Clin Cancer Res.
38:2192019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Sigova AA, Mullen AC, Molinie B, Gupta S,
Orlando DA, Guenther MG, Almada AE, Lin C, Sharp PA, Giallourakis
CC and Young RA: Divergent transcription of long noncoding RNA/mRNA
gene pairs in embryonic stem cells. Proc Natl Acad Sci.
110:2876–2881. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Lu C, Wei Y, Wang X, Zhang Z, Yin J, Li W,
Chen L, Lyu X, Shi Z, Yan W and You Y: DNA-methylation-mediated
activating of lncRNA SNHG12 promotes temozolomide resistance in
glioblastoma. Mol Cancer. 19:282020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Mazor G, Levin L, Picard D, Ahmadov U,
Carén H, Borkhardt A, Reifenberger G, Leprivier G, Remke M and
Rotblat B: The lncRNA TP73-AS1 is linked to aggressiveness in
glioblastoma and promotes temozolomide resistance in glioblastoma
cancer stem cells. Cell Death Dis. 10:2462019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Li Y, Liu Y, Ren J, Deng S, Yi G, Guo M,
Shu S, Zhao L, Peng Y and Qi S: miR-1268a regulates ABCC1
expression to mediate temozolomide resistance in glioblastoma. J
Neurooncol. 138:499–508. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Biamonte F, Sica G, Filippini A and
D'Alessio A: Evidence of reelin signaling in GBM and its derived
cancer stem cells. Brain Sci. 11:7452021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Tulip IJ, Kim SO, Kim EJ, Kim J, Lee JY,
Kim H and Kim SC: Combined inhibition of STAT and Notch signalling
effectively suppresses tumourigenesis by inducing apoptosis and
inhibiting proliferation, migration and invasion in glioblastoma
cells. Anim Cells Syst (Seoul). 25:161–170. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Lavanya C, Venkataswamy MM, Sibin MK,
Srinivas Bharath MM and Chetan GK: Down regulation of human
telomerase reverse transcriptase (hTERT) expression by BIBR1532 in
human glioblastoma LN18 cells. Cytotechnology. 70:1143–1154. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Sibin MK, Bhat DI, Narasingarao KV,
Lavanya CH and Chetan GK: CDKN2A (p16) mRNA decreased expression is
a marker of poor prognosis in malignant high-grade glioma. Tumour
Biol. 36:7607–7614. 2015. View Article : Google Scholar : PubMed/NCBI
|