Introduction
Liver cancer is one of the most common types of
primary cancer and is among the leading causes of cancer-related
deaths worldwide. In addition, it is associated with ~110,000
deaths each year in China (1,2).
After staging assessments, only 20% of patients with early-stage
liver cancer qualify for potentially curative treatments, such as
liver resection, transplantation and local ablation (3). By contrast, the majority of patients
with liver cancer, particularly those in the advanced stages,
receive palliative or symptomatic care, resulting in a 3-year
survival rate of <30% with most patients not surviving past 3
months (4). Accumulated clinical
and epidemiological research has demonstrated that non-alcoholic
fatty liver disease (NAFLD) is an independent risk factor for liver
cancer that has emerged as a major cause due to its rapidly growing
incidence (5-7). These findings indicate that altered
lipid metabolism may serve a critical role in liver cancer, and
that the identification of novel agents to treat liver cancer is
urgently needed.
Dysregulation of lipid metabolism occurs in both
cancer tissues and cancer cells (8). A growing body of evidence has
indicated that activation of the fatty acid synthesis pathway may
serve a pivotal role in cancer initiation (9,10).
Previous studies have consistently linked abnormal lipogenesis to
cancer development, since uncontrolled lipogenesis is vital for
providing cancer cells with an abundant supply of lipid components
and facilitating their uncontrolled proliferation (11,12). Notably, increased lipid
biosynthesis promotes cancer progression in liver cancer cells.
Furthermore, inhibition of fatty acid synthase (FASN), a key enzyme
governing lipogenesis, is considered a promising strategy for
blocking the proliferation of human liver cancer cells (13). Previous investigations have
indicated that sterol regulatory element-binding protein 1
(SREBP1), a well-established transcriptional master regulator
involved in lipogenesis, contributes to the progression of liver
cancer by stimulating cancer cell proliferation and metastasis
(14-16). Notably, mammalian target of
rapamycin (mTOR) is crucial for SREBP1 regulation (17). Inhibition of the mTOR/SREBP1
pathway has been shown to negatively affect lipogenesis (18,19). Additionally, heightened
lipogenesis can affect the sensitivity of liver cancer cells to
anticancer drugs (20). These
findings indicate the crucial role of elevated lipogenesis as a
driving force of liver cancer progression; however, the precise
molecular mechanisms underlying increased lipogenesis in liver
cancer remain to be elucidated.
Ferroptosis is a novel form of cell death, and
depletion of glutathione (GSH), inactivation of GSH peroxidase 4
(GPX4), and accumulation of cellular iron and lipid reactive oxygen
species (ROS) are the typical molecular events of this type of cell
death (21). Ferroptosis serves
an important role in the development of liver cancer (22), and the precise targeting of tumor
cells to induce ferroptosis is a new approach for the treatment of
this type of cancer. In addition, sorafenib resistance in liver
cancer has been reported to be associated with ferroptosis
(23) through the upregulation of
antioxidant pathways [such as nuclear factor erythroid 2-related
factor 2 (Nrf2)], altered GPX4 expression, increased ferritin and
decreased free iron levels, and reduced polyunsaturated fatty acids
(24-27).
Stearoyl-CoA desaturase 1 (SCD1), which is regulated
by SREBP1, is a critical modulator of fatty acid metabolism
(28). SCD1 desaturates
palmitoyl-CoA or stearoyl-CoA to form palmitoleic acid and oleic
acid, respectively (28).
Upregulation of SCD1 can increase the synthesis of monounsaturated
fatty acids (MUFAs) and protect cancer cells against ferroptosis
(29,30). Moreover, SCD1 has been reported to
be significantly upregulated in liver cancer cells (28,31,32), and SCD1 expression has been linked
to a poor prognosis in several types of cancer, such as
hepatocellular, gastric and ovarian cancer (29,31,33,34). RSL3 is a small-molecule compound
that inhibits the activity of GPX4, promoting lipid peroxidation
and triggering ferroptosis in susceptible cells (35). Inhibition of SCD1 has been shown
to promote RSL3-induced ferroptosis in colorectal cancer cells
(36). Collectively, these
studies suggest that a combination of SCD1 inhibitors and
ferroptosis inducers may exert promising therapeutic effects in
cancer treatment.
Cassiae semen, which refers to the dried mature
seeds of Cassie obtusifolia L. or Cassia toral L.,
and is commonly known as sicklepod, belongs to the Leguminosae
family. This plant product is widely cultivated in Korea and China,
and is easily grown and often used as a popular roasted tea
(37). Cassiae semen has a
longstanding tradition of being used as a herbal remedy for liver
and eye-related disorders. Aurantio-obtusin (AO), the primary
bioactive compound derived from Cassiae semen, is characterized by
its unique anthraquinone structure (38). Notably, the backbone of
anthraquinones is a pivotal structural blocker in the development
of anticancer drugs (39). AO
exhibits a wide range of pharmacological effects, including
neuroprotective, hepatoprotective, anti-hyperlipidemic,
antioxidant, antimicrobial and anti-allergic activities (37,38,40,41). In addition, AO has been shown to
reduce SREBP1c, FASN and SCD1 levels in the white adipose tissue of
obese mice (40); however, in the
brown adipose tissue of obese mice, AO can significantly enhance
mitochondrial metabolism and uncoupling protein 1 expression by
activating PPARα (42).
Additionally, AO may attenuate NAFLD by inhibiting de novo
lipid synthesis, which is regulated by SREBP1, and by promoting
autophagy flux to alleviate liver steatosis in a mouse model of
NAFLD (42). Several studies have
demonstrated a close link between the inhibition of lipogenesis and
the induction of ferroptosis in overcoming chemotherapy resistance
(20,36). However, few investigations have
focused on the relationship between AO and ferroptosis, which could
represent a novel approach to sensitizing tumors to chemotherapy
(24,26,43). Given that AO exhibits strong
lipogenesis-inhibiting capabilities, the present study hypothesized
that the regulatory effects of AO on lipogenesis may serve a
crucial role in liver tumor growth, survival and sensitivity to
ferroptosis.
The present study explored the anticancer activities
of AO, when administered either alone or in combination with the
ferroptosis inducer RSL3, and the underlying mechanisms were
investigated. The present findings may pioneer the synergistic use
of AO with a ferroptosis inducer as an innovative strategy for
inhibiting liver cancer cell proliferation, with potential
implications for future clinical use.
Materials and methods
Reagents
AO, RSL3 and ferrostatin-1 (Fer-1, ferroptosis
inhibitor) were purchased from Shanghai Macklin Biochemical Co.,
Ltd. A939572 (SCD1 inhibitor) was purchased from Sigma-Aldrich;
Merck KGaA. Palmitic acid (PA) was obtained from Beijing Solarbio
Science & Technology Co., Ltd. (cat. no. SP8060). MK2206 (AKT
inhibitor; cat. no. S1078) was purchased from Selleck Chemicals.
Anti-GAPDH (cat. no. 2118; 1:1,000), anti-phosphorylated (p)-AKT
(cat. no. 4060; 1:1,000), anti-AKT (cat. no. 9272; 1:1,000),
anti-AMP-activated protein kinase (AMPK)α (cat. no. 2532; 1:1,000),
anti-p-AMPKα (cat. no. 50081; 1:1,000), anti-p-mTOR (cat. no. 5536;
1:1,000), anti-mTOR (cat. no. 2983; 1:1,000), anti-FASN (cat. no.
3180; 1:1,000), anti-Nrf2 (cat. no. 12721; 1:1,000), anti-heme
oxygenase 1 (HO-1; cat. no. 43966; 1:1,000), anti-GPX4 (cat. no.
52455; 1:1,000) and anti-SCD1 (cat. no. 2794; 1:1,000) antibodies
were purchased from Cell Signaling Technology, Inc., and
anti-SREBP1 (cat. no. sc-365513; 1:500) was purchased from Santa
Cruz Biotechnology, Inc. All other reagents used in the experiments
met or exceeded analytical grade standards. The clinical liver
cancer sample data used in the present study were obtained from The
Cancer Gene Atlas (TCGA) project available in the ULCAN database
(https://ualcan.path.uab.edu) (44). A total of 371 primary liver tumor
samples and 50 normal liver tissue samples from healthy controls
were used in the analysis. Pearson correlation analysis was used to
assess the correlations between samples.
Specimen collection and patient
information
Liver cancer specimens and adjacent normal tissues
were collected from patients who underwent surgical resection at
Hunan Provincial People's Hospital (Changsha, China). The present
study was approved by The Ethics Committee of Hunan Provincial
People's Hospital [approval no. (2023)-178]. A total of 15 patients
were included in the study. The average age of the patients was
61.7 years (age range, 53-71 years), and the cohort consisted of
nine men and six women. The normal specimens used in the present
study were adjacent healthy tissues from the same patients from
whom the liver cancer specimens were obtained.
Cell lines and culture conditions
Human liver cancer cells lines SK-Hep1 and HepG2
cells were provided by the Medical College of Hunan Normal
University (Changsha, China). SK-Hep1 cells were maintained in
basic RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc.),
whereas HepG2 cells were cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.). Both culture media were supplemented with 10%
fetal bovine serum (FBS; Suzhou ExCell Biology, Inc.) and 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
The cell cultures were incubated at 37°C in a humidified atmosphere
containing 5% CO2.
Cell viability and clonogenic assays
Cell viability was assessed using the Cell Counting
Kit (CCK)-8 Cell Proliferation and Cytotoxicity Assay Kit (cat. no.
CA1210; Beijing Solarbio Science & Technology Co., Ltd.).
Briefly, cells were seeded at a density of 8x103
cells/well in 96-well culture plates and cultured in medium
containing 10% FBS at 37°C. After 24 h, the cells were treated with
10-600 μM AO, 5-800 μM PA or 0.05-8 nM RSL3, for 48 h
at 37°C. For the combined treatment, the cells were treated with AO
(50 μM), PA (SK-Hep1: 10 μM, HepG2: 100 μM),
MK2206 (1 μM), A9 (8 μM), Fer-1 (5 nM), RSL3
(SK-Hep1: 0.01 nM, HepG2: 1 nM) or their combination at the
indicated concentrations for 48 h at 37°C. A 10% CCK-8 solution was
prepared in medium and added to each well, and the plates were
incubated for a further 1.5 h at 37°C. Finally, the absorbance was
measured at 450 nm using a microplate reader (Synergy HTX; BioTek;
Agilent Technologies, Inc.). Dose-response curves were generated
and the half-maximal inhibitory concentration was determined using
SPSS (version 16.0; SPSS, Inc.).
Clonogenic survival was assessed by determining the
colony-forming capacity of cells. Briefly, 8×103 cells
were seeded into 24-well dishes in 0.5 ml medium. After 24 h, cells
were treated with varying concentrations of AO (0, 100 and 200
μM) daily for 7 consecutive days. For the combination
therapy, the cells were treated with AO (0, 100 and 200 μM),
PA (100 μM for HepG2 cells) or treated with RSL3 (0, 0.01
and 0.02 nM for SK-Hep1 cells and 0, 1 and 2 nM for HepG2 cells),
A9 (8 μM for SK-Hep1), AO (50 μM for SK-Hep1) daily
for 7 consecutive days in 1 ml medium containing 10% FBS at 37°C.
Subsequently, cells were fixed with 10% formaldehyde (0.5 ml) for
30 min at room temperature and stained with 0.1% crystal violet for
2 h at room temperature. Colonies consisting of >50 cells were
detected. Using a microplate reader (BioTek; Agilent Technologies,
Inc.), absorbance was measured at 550 nm using the area scanning
function to quantify the number of cell colonies.
EdU staining
An EdU Cell Proliferation Kit with Alexa Fluor 488
(cat. no. KGA331; Nanjing KeyGen Biotech Co., Ltd.) was used to
detect proliferating cells according to the manufacturer's
instructions. Briefly, cells were seeded at a density of
8×103 cells/well in 96-well culture plates and cultured
in medium containing 10% FBS at 37°C. After 24 h, the cells were
treated with AO (0, 100 and 200 μM) for 24 h at 37°C. For
the combination therapy, the cells were treated with A9 (8
μM), RSL3 (SK-Hep1: 0.01 nM, HepG2: 1 nM), AO (50 μM)
or their combination at the indicated concentrations for 24 h at
37°C. Then pre-warmed EdU working solution was added to the treated
cells for EdU labeling and was incubated for 2 h at 37°C. After
incubation, the medium was removed and 50 μl 4% neutral
paraformaldehyde was added to each well, followed by incubation at
room temperature for 30 min. The fixing solution was then removed
and 50 μl 2 mg/ml glycine solution was added to each well,
and incubated at room temperature for 5 min. Subsequently, cells
were washed twice with 0.1 ml/well 3% bovine serum albumin (cat.
no. ST2254-5g; Beyotime Institute of Biotechnology) in PBS,
followed by incubation at room temperature for 20 min. Click-iT
reaction solution (100 μl) was then added to each well and
incubated at room temperature for 30 min in the dark. Finally, 0.1
ml 1X Hoechst 33342 solution was added to each well, followed by
incubation at room temperature for 15-30 min in the dark. After
staining, images were captured using an Olympus BX51 fluorescence
microscope (Olympus Corporation).
Lipid peroxidation
Lipid peroxidation was assessed using the Lipid
Peroxide (LPO) Content Assay Kit (cat. no. BC5245; Beijing Solarbio
Science & Technology Co., Ltd.), according to the
manufacturer's instructions. Briefly, cells were seeded at a
density of 5×106 cells/100-mm culture dish and were
incubated at 37°C in a 5% CO2 incubator. The next day,
cells were treated with varying concentrations of AO (50, 100 and
200 μM) for 24 h at 37°C. For the combined treatment, the
cells were treated with A9 (8 μM), RSL3 (SK-Hep1: 0.01 nM,
HepG2: 1 nM), AO (50 μM), or their combination at the
indicated concentrations for 24 h at 37°C. The cells
(~5x107) were then harvested and treated with the
extraction solution. Subsequently, cells were disrupted using
ultrasonic waves in an ice bath (frequency, 20 kHZ; power, 200 W;
ultrasonication, 3 sec; interval, 7 sec; total time, 3 min) and
centrifuged at 8,000 × g and 4°C for 10 min. The resulting
supernatant was collected, and reagent solutions were added
according to the manufacturer's instructions. Finally, the
absorbance of each sample was measured at 532 nm, then at 600
nm.
Small interfering RNA (siRNA)
transfection
Cells were transfected with commercially available
SCD1 siRNAs (siSCD1), with the following sequences: siSCD1-1, sense
5'-GAG ACG AUG CCC CUC UAC UUG G-3', antisense 5'-CCA AGU AGA GGG
GCA UCG UCU C-3'; siSCD1-2, sense 5'-GGA GAA ACA UCA UCC UUA
UUU-3', antisense 5'-AAA UAA GGA UGA UGU UUC UCC-3', or with the
following negative control siRNA: sense 5'-UUC UCC GAA CGU GUC ACG
UTT-3', anti-sense 5'-ACG UGA CAC GUU CGG AGA ATT-3' (all from
Guangzhou RiboBio Co., Ltd.) using the transfection reagent
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Briefly, cells were seeded in 6-well plates at a
density of 3×105 cells/dish. When cells reached 30-50%
confluence, they were transfected with 50 nM siSCD1 or 50 nM
negative control siRNA using Lipofectamine 2000 in the presence of
1% penicillin-streptomycin and 10% FBS, for 6 h at 37°C. After
washing with PBS, the medium was replaced with RPMI-1640 or DMEM.
After 36 h, cell proteins were harvested and the effectiveness of
silencing was confirmed through western blot analysis.
Western blotting
Cells were seeded at a density of 5x105
cells/well in 6-well culture plates and cultured in medium
containing 10% FBS at 37°C. After 24 h, the cells were treated with
AO (100 μM) for 6, 12 or 24 h, or with AO (50, 100 and 200
μM) for 24 h at 37°C. For the combined treatment, the cells
were treated with AO (50 μM), MK2206 (1 μM), A9 (8
μM), RSL3 (0.01 nM for SK-Hep1, 1 nM for HepG2), or their
combination at the indicated concentrations for 24 h at 37°C. Cells
were then suspended in ice-cold RIPA buffer (Beyotime Institute of
Biotechnology) containing 2% protease and phosphatase inhibitor
cocktail (Beyotime Institute of Biotechnology). The protein
concentration of lysates was then measured using the BCA assay.
Homogenates containing 20 μg total protein were separated by
SDS-PAGE on gradient gels (10-15%) and were transferred to
polyvinylidene fluoride membranes (MilliporeSigma). The blocking
reagent used was 5% non-fat milk at room temperature for 1 h,
followed by three washes with TBS-0.1% Tween. The membranes were
blotted with specific primary antibodies overnight at 4°C with
shaking, followed by incubation with HRP-conjugated secondary
anti-rabbit or anti-mouse antibodies (cat. nos. L3032 and L3012;
1:10,000; Signalway Antibody LLC) for 1 h at room temperature. The
membranes were then visualized using enhanced chemiluminescence
(cat. no. PMK0448; Wuhan Pumoke Biotechnology Co., Ltd.) and a Gel
Doc 2000 system (Bio-Rad Laboratories, Inc.). Band intensities were
semi-quantified using ImageJ version 1.8.0 software (National
Institutes of Health).
Transwell migration assay
Cell migration was assessed using Transwell plates
(24-well insert; pore size, 8 μm; Corning, Inc.).
Approximately 4x104 cells/well in 200 μl
serum-free medium were seeded into the upper chambers, while 700
μl medium supplemented with 20% FBS served as a
chemoattractant in the lower chamber. The next day, cells were
treated with AO (100 and 200 μM) for 24 h at 37°C in 5%
CO2, the medium in the upper chamber was aspirated and
the cells were fixed with 4% paraformaldehyde for 30 min at room
temperature and stained with 0.1% crystal violet for 2 h at room
temperature. Non-migratory cells located on the upper side of the
membrane were gently removed using cotton wool and cells on the
lower side of the membrane were semi-quantified. Representative
images were captured by inverted fluorescence microscopy. Five
random fields (×200 magnification) were selected and the average
was calculated. The data are presented as the mean ± standard error
from three independent experiments.
Xenograft tumor mouse model
Female BALB/c nude mice were purchased from Hunan
SJA Laboratory Animal Co., Ltd. All animal experiments were
conducted in strict accordance with guidelines approved by the
Institutional Animal Care and Use Committee at Hunan Normal
University (approval no. D2021059; Changsha, China). Female BALB/c
nude mice (age, 6 weeks; weight, 18.0±2.0 g; n=6 mice/group) were
subcutaneously injected with single-cell HepG2 lines into the right
flanks of nude mice (1×106 cells in 100 μl PBS)
on day 1. The mice were housed under controlled environmental
conditions; the temperature was maintained at 22±2°C and the
relative humidity was kept at 50±10%. In addition, the mice had
ad libitum access to food and water and were maintained
under a 12-h light/dark cycle. Tumor volumes and mouse weight were
determined every 2 days. Tumor size was measured using calipers,
and tumor volume was calculated using the formula: 1/2 × [length ×
(width)2]. After 15 days, all mice were deeply
anesthetized with 1% pentobarbital sodium (40 mg/kg) to minimize
any potential pain or distress during the procedure, followed by
cervical dislocation to sacrifice the mice. Then tumors and major
organs (liver and kidney) were removed for subsequent use in
experiments. The humane experimental endpoints included tumor
weight being >10% of animal body weight, tumor volume exceeding
2,000 mm3, and weight loss of >20% animal body
weight; no animals reached these criteria during the
experiment.
Oil red O staining
Cells were seeded at a density of 4×104
cells/well in 24-well culture plates. After 24 h, the cells were
treated with AO (100 and 200 μM) for another 24 h at 37°C.
For the combined treatment, the cells were treated with AO (50
μM), PA (SK-Hep1: 10 μM, HepG2: 100 μM) or
their combination at the indicated concentrations for 24 h at 37°C.
Then, cells in 24-cell dishes were washed with PBS and fixed with
4% paraformaldehyde for 20-30 min at room temperature, followed by
another wash with PBS. Subsequently, the cells were incubated with
Oil red O (0.5% in isopropanol; cat. no. 01391; Sigma-Aldrich;
Merck KGaA) for 15 min at room temperature, were rinsed with 60%
isopropanol for 30 sec and washed twice with distilled water. After
washing, the cells were stained with hematoxylin (cat. no. BA40211;
Baso Diagnostic Inc.) for 5 min at room temperature. After
staining, the cells were washed with water to remove any unbound
dye and were examined under a light microscope.
Histology
Tumor tissues from mice and human patients were
fixed in 4% paraformaldehyde (Wuhan Servicebio Technology Co.,
Ltd.) for 24 h at room temperature, followed by embedding in
paraffin and sectioning to a thickness of 7 μm. For
histological analysis, the sections were rinsed three times with
distilled water for 3 min. The sections were stained with
hematoxylin for 3 min and eosin for 15-30 sec at room temperature
(both from Wuhan Servicebio Technology Co., Ltd.) and evaluated for
disease grading by a certified pathologist. For
immunohistochemistry, the sections were deparaffinized and
rehydrated using a sequence of xylene, 100, 95 and 75% ethanol. The
sections were incubated with 3% H2O2 for 20
min at room temperature to quench endogenous peroxidase activity,
washed with PBS, and boiled in Tris-EDTA retrieval solution for 5
min in a pressure cooker for antigen retrieval. After naturally
cooling to room temperature, the sections were blocked with 10%
goat serum (Beijing Solarbio Science & Technology Co., Ltd.) at
room temperature for 20 min and incubated overnight at 4°C with the
anti-GPX4 (1:200; cat. no. DF6701; Affinity Biosciences) and
anti-SCD1 (1:100; cat. no. A16429; Abclonal Biotech Co., Ltd.)
primary antibodies. The sections were then washed with PBS and
incubated with Reagent 2 (from the Goat Hypersensitivity Two-Step
Detection Kit; cat. no. PV-9005; OriGene Technologies, Inc.) for 20
min at 37°C. Subsequently, the sections were washed with PBS and
incubated for 20 min at 37°C with Reagent 3 (from the Goat
Hypersensitivity Two-Step Detection Kit) following the
manufacturer's instructions. The sections were washed again with
PBS and stained using the DAB substrate kit (Cell Signaling
Technology, Inc.), followed by counterstaining with Gill's
hematoxylin (Beijing Solarbio Science & Technology Co., Ltd.).
Finally, the sections were dehydrated and mounted using neutral
resin (Beijing Solarbio Science & Technology Co., Ltd.).The
slides were captured using an Olympus BX51 fluorescence microscope
(Olympus Corporation).
Statistical analyses
All data are presented as the mean ± SD, or as the
mean ± standard error of the mean for the Transwell assay. Each
experiment was performed three times. Statistical analyses was
performed using SPSS (version 16.0). Significance between two
groups was evaluated using the unpaired Student's t-test, whereas
the significance among multiple groups was determined using one-way
analysis of variance followed by the Bonferroni significant
difference test. Graphs were created using GraphPad Prism 6.0
(Dotmatics). P<0.05 was considered to indicate a statistically
significant difference.
Results
AO inhibits liver cancer cell
proliferation, colony formation and migration
To assess the anticancer activities of AO in liver
cancer cells, its effect on the proliferation, colony formation and
migration of liver cancer cells were investigated. As shown in
Fig. 1A and B, AO treatment
resulted in a concentration-dependent reduction in the viability
and proliferation of liver cancer cells. The results of the colony
formation assay demonstrated a dose-dependent inhibitory effect of
AO (100-200 μM) (Fig. 1C).
Notably, there was a significant difference in the sensitivity to
AO between these two liver cancer cell lines, AO treatment exerted
a more pronounced inhibitory effect on SK-Hep1 cells than on HepG2
cells (Fig. 1A). Subsequently,
the present study assessed the effect of AO on cell migration. As
shown in Fig. 1D, 200 μM
AO exerted a significant inhibitory effect on the migration of
these two cell lines compared with in the control group. These data
suggested that AO effectively inhibited proliferation, colony
formation and migration in liver cancer cells.
AO downregulates lipogenesis via SREBP1
and FASN
To assess the effect of AO treatment on lipid
accumulation, Oil red O staining was employed to detect the
alterations of lipids following various treatments. A marked
reduction in the number of lipid bodies was observed in both
SK-Hep1 and HepG2 cells after AO treatment (Fig. 2A). SREBP1 consists of both a
precursor form (pSREBP1) and a mature form (mSREBP1). In the
present study, AO significantly reduced the expression levels of
both forms of SREBP1 and also inhibited the downstream expression
of FASN (Fig. 2B). PA is a
synthetic product of FASN, and the addition of PA can partially
compensate for the function of FASN (45). The present study examined whether
AO could reduce lipid levels in PA-treated liver cancer cells.
First, the effect of PA on the viability of the liver cancer cell
lines SK-Hep1 and HepG2 was examined (Fig. 2C), which indicated that there were
significant differences in viability between cells treated with
varying concentrations of PA and the control group. Furthermore,
there were notable differences in the sensitivity of these two
liver cancer cell lines to PA treatment; the IC50 value
of PA in SK-Hep1 cells was markedly lower at 42.68 μM,
compared with the substantially higher value of 348.71 μM in
HepG2 cells. To prevent excessive lipid accumulation toxicity, 10
μM PA was selected for inducing lipid accumulation in
SK-Hep1 cells and 100 μM PA was selected for HepG2 cells. PA
induced lipid accumulation in both HepG2 and SK-Hep1 cells, whereas
AO effectively reduced the number of lipid bodies induced by PA
(Fig. 2D). Furthermore, the
inhibitory effects of AO on the viability and colony formation of
SK-Hep1 and HepG2 cells were partially reversed by PA (Fig. 2E and F). These results indicated
that AO may inhibit the proliferation of liver cancer cells by
suppressing fatty acid synthesis.
 | Figure 2AO downregulates lipogenesis via the
SREBP1 and FASN axis. (A) SK-Hep1 and HepG2 cells were exposed to
AO for 24 h and then subjected to Oil red O assay for the detection
of lipid droplets. Lipid droplets are indicated with red arrows.
(B) SK-Hep1 and HepG2 cells were treated with 100 μM AO for
0, 6, 12 and 24 h, and western blot analysis was used to examine
the expression of pSREBP1, mSREBP1 and FASN; GAPDH was included as
a loading control. *P<0.05, **P<0.01,
***P<0.001 vs. 0 h. (C) CCK-8 assay evaluated the
effects of PA on SK-Hep1 and HepG2 cell viability.
*P<0.05, #P<0.0001 vs. 0 μM. (D)
SK-Hep1 and HepG2 cells were treated with 10 and 100 μM PA,
respectively, and 100 μM AO for 24 h. Lipid droplets were
visualized using Oil red O staining. Lipid droplets are indicated
with red arrows. (E) SK-Hep1 and HepG2 cells were treated with
different concentrations of AO combined with PA for 48 h, and cell
viability was measured using the CCK-8 assay. (F) HepG2 cells were
treated with different concentrations of AO combined with PA (100
μM) for 7 days, and colony formation was measured.
**P<0.01, ***P<0.001,
#P<0.0001. n=3. AO, aurantio-obtusin; CCK-8, Cell
Counting Kit-8; CTRL, control; FASN, fatty acid synthase; mSREBP1,
mature SREBP1; ns, not significant; PA, palmitic acid; pSREBP1,
precursor SREBP1; SREBP1, sterol regulatory element-binding protein
1. |
AO decreases SREBP1 expression by
inactivating the AKT/mTOR signaling pathway
It has previously been shown that AKT and AMPK serve
contrasting roles in fatty acid synthesis (46). The present study investigated the
effect of AO on the protein expression levels of p-AKT, p-mTOR and
p-AMPKα. AO treatment simultaneously reduced the protein expression
levels of p-AKT, p-mTOR and p-AMPK. (Fig. 3A and B). Activation of AMPK can
inhibit fatty acid synthesis and promote catabolism, thereby
restoring cellular energy homeostasis; by contrast, inhibition of
AMPK generally reduces its inhibitory effect on fatty acid
synthesis (46). These findings
suggested that AO may hinder SREBP1 expression by inactivating the
AKT/mTOR pathway rather than activating AMPK. Furthermore, the
viability of SK-Hep1 and HepG2 cells treated with a combination of
an AKT inhibitor (MK2206) and AO was markedly reduced compared with
that of cells treated with either MK2206 or AO alone (Fig. 3C and D). MK2206 also amplified the
AO-induced downregulation of p-AKT (Fig. 3E).
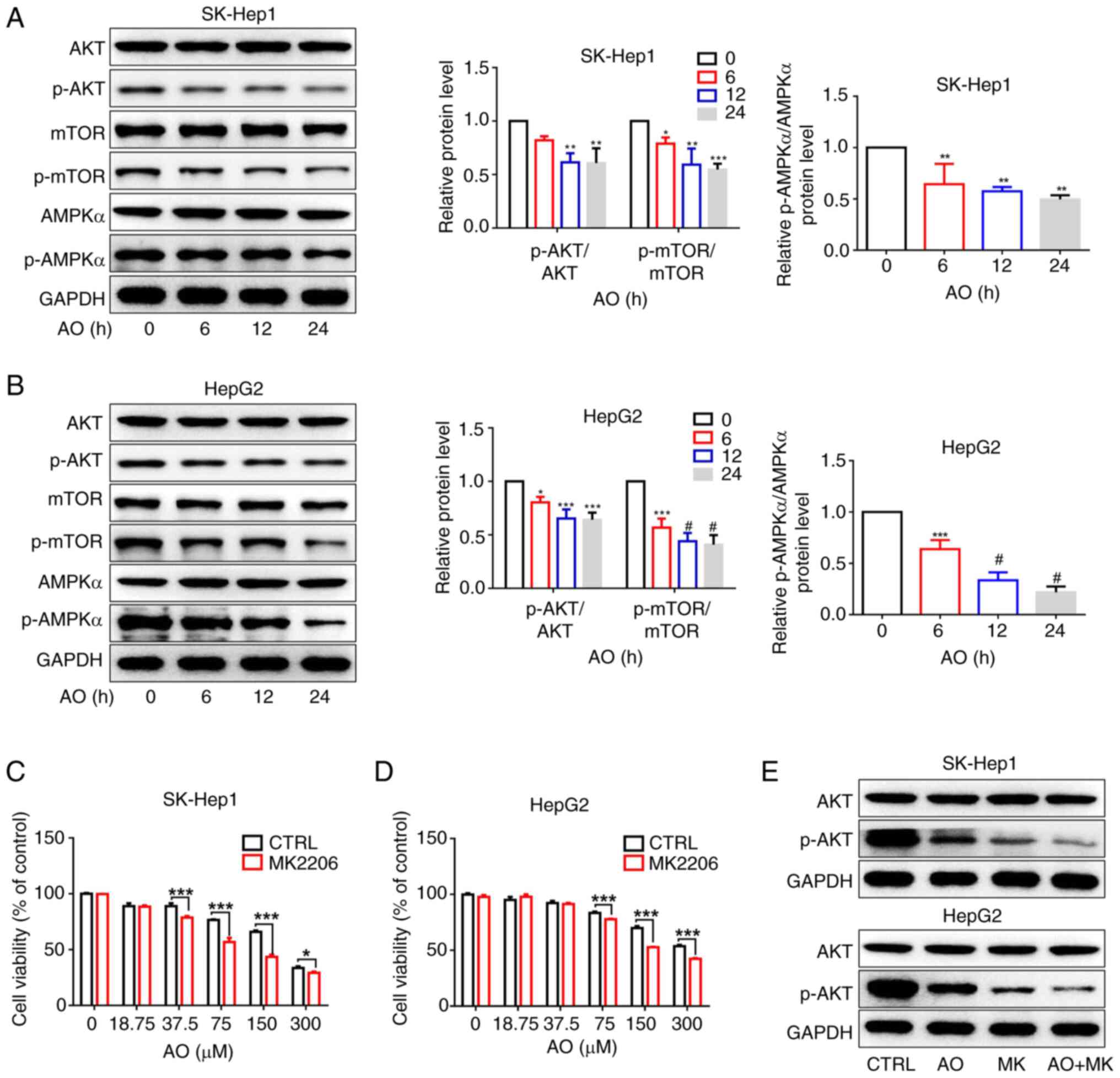 | Figure 3AO inhibits the expression of SREBP1
by inactivating the AKT/mTOR signaling pathway. (A) Protein
expression levels of p-AKT, p-mTOR and p-AMPKα in SK-Hep1 and HepG2
cells were detected by western blotting after treatment with AO
(100 μM) for different durations (0, 6, 12 and 24 h).
*P<0.05, **P<0.01,
***P<0.001 vs. 0 h. (B) Protein expression levels of
p-AKT, p-mTOR and p-AMPKα were semi-quantified.
*P<0.05, ***P<0.001,
#P<0.0001 vs. 0 h. (C) SK-Hep1 and (D) HepG2 cells
were treated with different concentrations of AO combined with
MK2206 (1 μM) for 48 h, and cell viability was measured
using the cell counting Kit-8 assay. (E) Protein expression levels
of p-AKT were detected by western blotting in SK-Hep1 and HepG2
cells treated with AO (50 μM), MK2206 (1 μM) or the
combination for 24 h. *P<0.05,
***P<0.001. n=3. AMPKα, AMP-activated protein kinase
α; AO, aurantio-obtusin; mTOR, mammalian target of rapamycin; p-,
phosphorylated. |
AO induces ferroptosis by inactivating
Nrf2/HO-1/GPX4 signaling
Ferroptosis is an iron-dependent form of cell death
triggered by lipid peroxidation, which is suppressed by GPX4, a key
enzyme in mammals capable of reducing esterified phospholipid
hydroperoxides (21,35). To elucidate the relationship
between AO and ferroptosis, the effect of AO on the expression of
GPX4 was investigated. The results demonstrated that AO effectively
suppressed the expression of GPX4 in both SK-Hep1 and HepG2 cells
in a dose-dependent manner (Fig. 4A
and B). Furthermore, the intracellular levels of LPO in these
cells were assessed. After treatment with AO, there was a
significant increase in LPO levels (Fig. 4C and D). These results suggested
that AO may have the potential to induce ferroptosis. Increasing
evidence has highlighted the pivotal role of the Nrf2/HO-1/GPX4
axis in mediating ferroptosis (47). To determine the effects of AO on
the Nrf2/HO-1 signaling pathway, western blot analysis was
conducted. The results showed that AO significantly suppressed the
expression of Nrf2 and HO-1 in SK-Hep1 and HepG2 cells (Fig. 4E-G). These findings suggested that
AO may downregulate GPX4 expression and induce ferroptosis in liver
cancer cells by inhibiting the Nrf2/HO-1 signaling pathway.
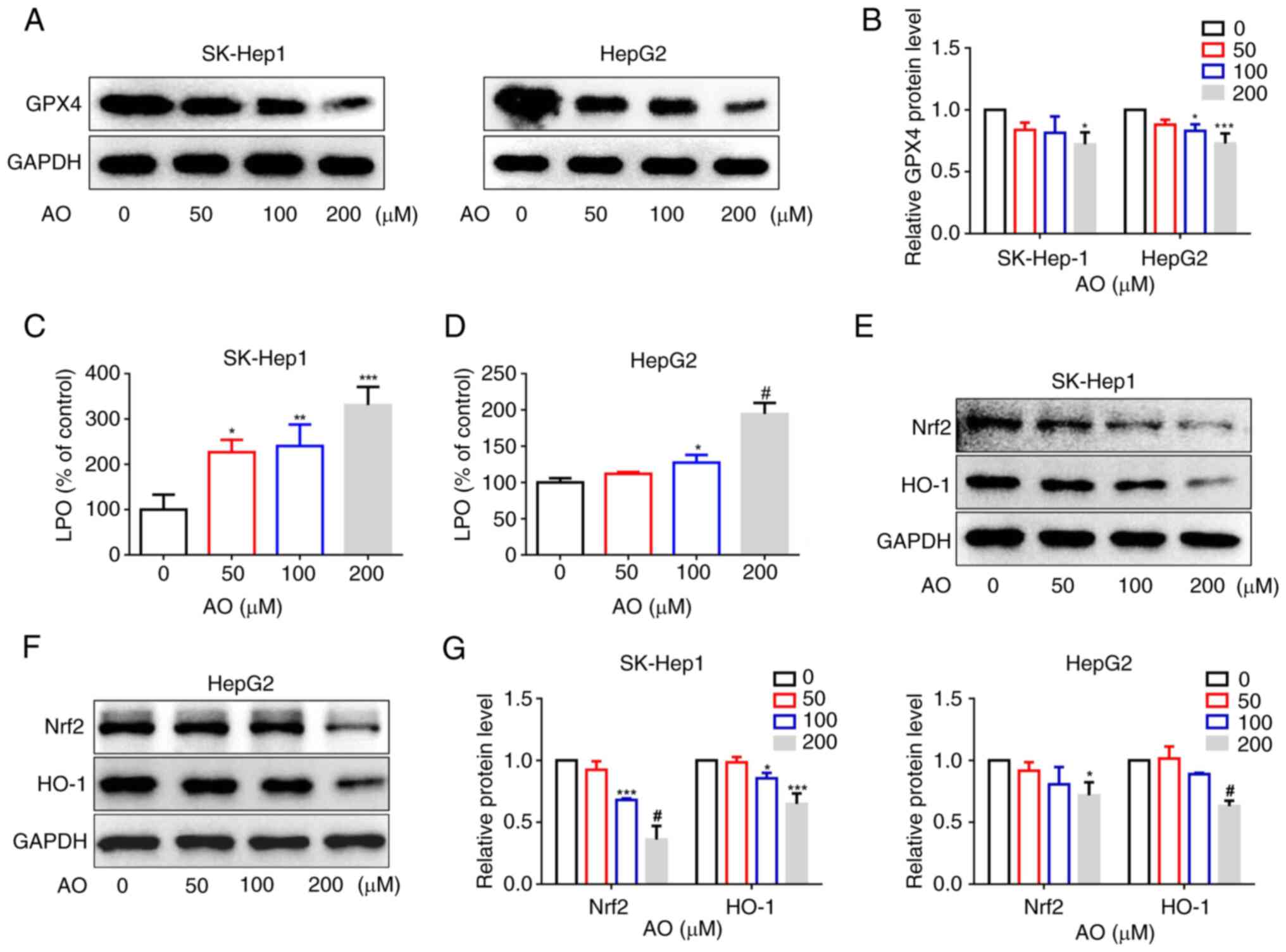 | Figure 4AO induces ferroptosis by
inactivating Nrf2/HO-1/GPX4 signaling. (A and B) SK-Hep1 and HepG2
cells were treated with AO (0, 50, 100 or 200 μM) for 24 h,
and the protein expression levels of GPX4 were measured by western
blotting. (C) SK-Hep1 and (D) HepG2 cells were treated with AO for
24 h and lipid peroxidation levels were detected. (E-G) SK-Hep1 and
HepG2 cells were treated with AO (0, 50, 100 or 200 μM) for
24 h, and the protein expression levels of Nrf2 and HO-1 were
measured by western blotting. *P<0.05,
**P<0.01, ***P<0.001,
#P<0.0001 vs. 0 μM. n=3. AO, aurantio-obtusin;
GPX4, glutathione peroxidase 4; HO-1, heme oxygenase 1; LPO, lipid
peroxide; Nrf2, nuclear factor erythroid 2-related factor 2. |
SCD1 expression levels are associated
with ferroptosis sensitivity in liver cancer cells
The present study investigated the sensitivity of
liver cancer cells to RSL3, a ferroptosis inducer, and observed
notable variations in the responsiveness of SK-Hep1 and HepG2 cells
to ferroptosis. As shown in Fig.
5A, treatment with 0.8 nM RSL3 led to a 90% inhibition of
SK-Hep1 cell viability, but had no noticeable effect on HepG2
cells. SCD1, an enzyme responsible for converting saturated fatty
acids into MUFAs, renders cancer cells sensitive to ferroptosis
when genetically or pharmacologically inhibited. The present study
assessed the baseline levels of SCD1 expression in SK-Hep1 and
HepG2 cells, and detected significantly lower SCD1 and GPX4
expression in SK-Hep1 cells and relatively higher levels in HepG2
cells (Fig. 5B). Additionally,
treatment with A939572, an inhibitor of SCD1 activity, enhanced the
susceptibility of cancer cells to ferroptosis induction and lipid
peroxidation compared with the control group (Fig. 5C-F). Subsequently, the preset
study explored the potential mechanisms underlying ferroptosis
sensitization by blocking SCD1. Transient silencing was performed
using siRNAs to modulate SCD1 expression levels in liver cancer
cells. Notably, SCD1 knockdown significantly reduced SCD1 protein
expression but had no effect on GPX4 protein expression, suggesting
that silencing SCD1 itself did not regulate GPX4 activity (Fig. 5G). Furthermore, silencing SCD1 or
pharmacologically inhibiting SCD1 using A939572 amplified
RSL3-reduced GPX4 protein levels (Fig. 5H and I). This finding suggested
that inhibiting SCD1 may enhance the sensitivity of liver cancer
cells to ferroptosis induction. In summary, these findings
indicated that SCD1 may have a crucial role in determining the
sensitivity of liver cancer cells to ferroptosis.
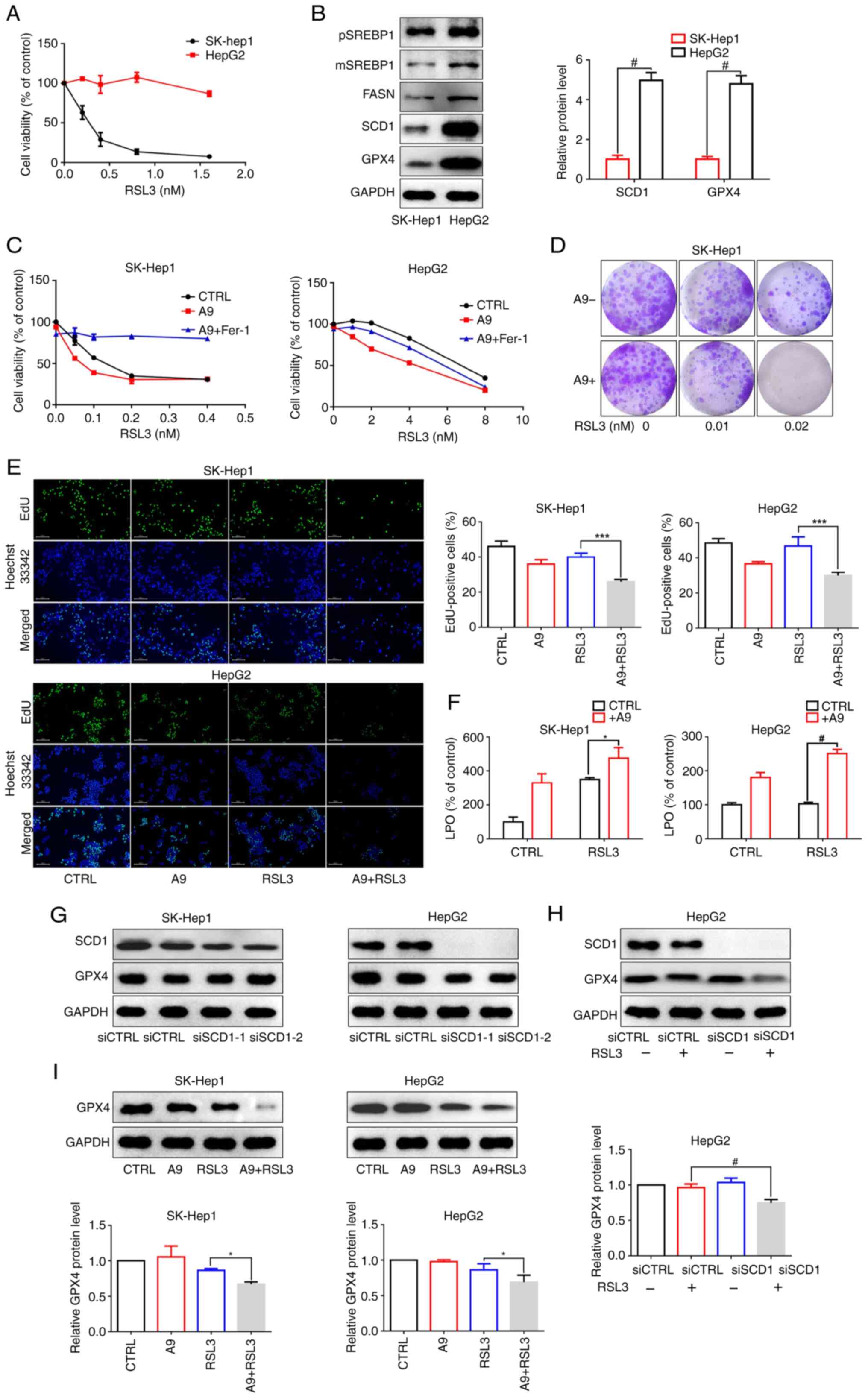 | Figure 5Relationship between SCD1 expression
and the sensitivity of liver cancer cells to RSL3. (A) Cell
Counting Kit-8 assay was used to evaluate the effects of RSL3 on
SK-Hep1 and HepG2 cell viability. (B) Western blot analysis was
performed to determine the basal levels of pSREBP1, mSREBP1, FASN,
SCD1 and GPX4 protein expression in SK-Hep1 and HepG2 cells. (C)
Cell viability was measured in SK-Hep1 and HepG2 cells treated with
different concentration of RSL3, or a combination of A939572 (8
μM) and Fer-1 (5 nM) for 48. (D) SK-Hep1 cells were treated
with different concentration of RSL3 combined with A939572 (8
μM) for 7 days, and colony formation was measured. (E and F)
SK-Hep1 and HepG2 cells were treated with A939572 (8 μM),
RSL3 (SK-Hep1: 0.01 nM, HepG2: 1 nM) or the combination for 24 h.
(E) Cell proliferation was assessed by EdU staining (magnification,
x200) and (F) lipid peroxidation levels were detected using the LPO
Content Assay Kit. (G) Expression levels of GPX4 were measured by
western blotting after transfection with siSCD1. (H) HepG2 cells
were transfected with siCTRL or siSCD1 and then treated with RSL3
for 24 h, followed by western blot analysis of SCD1 expression. (I)
Protein expression levels of GPX4 were detected in SK-Hep1 and
HepG2 cells treated with A939572 (8 μM), RSL3 (SK-Hep1: 0.01
nM, HepG2: 1 nM), or their combination for 24 h.
*P<0.05, ***P<0.001,
#P<0.0001. n=3. CTRL, control; FASN, fatty acid
synthase; Fer-1, ferrostatin-1; GPX4, glutathione peroxidase 4;
LPO, lipid peroxide; mSREBP1, mature SREBP1; pSREBP1, precursor
SREBP1; SCD1, stearoyl-CoA desaturase 1; si, small interfering;
SREBP1, sterol regulatory element-binding protein 1. |
AO sensitizes liver cancer cells to
RSL3-induced ferroptosis by suppressing SCD1 expression
As aforementioned, inhibiting SCD1 increased the
susceptibility of liver cancer cells to RSL3-induced ferroptosis.
SCD1 is a downstream target of SREBP1; therefore, the present study
investigated whether AO affects SCD1 expression in liver cancer
cells using western blot analysis. As shown in Fig. 6A, treatment with 200 μM AO
for 24 h significantly downregulated SCD1 expression in the liver
cancer cells. Moreover, the combination of AO and RSL3
significantly reduced cell viability and proliferation (Fig. 6B and D) and colony formation
(Fig. 6C) compared with RSL3 or
AO alone. These effects were reversed by Fer-1, an effective and
selective ferroptosis inhibitor that prevents membrane lipid damage
via a reductive mechanism, thereby reversing the inhibitory effect
on cell viability (Fig. 6B).
Concurrently, LPO levels were markedly increased and GPX4 levels
were markedly reduced when AO was combined with RSL3 compared with
the RSL3 treatment group (Fig. 6E and
F). In summary, these results suggested that AO enhanced
RSL3-induced ferroptosis in liver cancer cells by inhibiting SCD1
expression.
 | Figure 6AO sensitizes liver cancer cells to
ferroptosis by suppressing SCD1 expression. (A) Expression levels
of SCD1 protein in cells treated with AO for 24 h.
*P<0.05, **P<0.01 vs. 0 μM. (B)
Cell viability was measured in SK-Hep1 and HepG2 cells treated with
AO (50 μM), RSL3 (SK-Hep1: 0.01 nM, HepG2: 1 nM), or the
combination in the presence or absence of Fer-1 (5 nM) for 48 h.
(C) HepG2 cells were treated with different concentration of RSL3
combined with AO (50 μM) for 7 days, and colony formation
was measured. (D and E) SK-Hep1 and HepG2 cells treated with AO (50
μM), RSL3 (SK-Hep1: 0.01 nM, HepG2: 1 nM) or the combination
for 24 h. (D) Cell proliferation was assessed by EdU staining
(magnification, x200) and (E) lipid peroxidation levels were
detected using the Lipid Peroxide Content Assay Kit. (F) Protein
expression levels of GPX4 were detected in SK-Hep1 and HepG2 cells
treated with AO (50 μM), RSL3 (SK-Hep1: 0.01 nM, HepG2: 1
nM), or their combination for 24 h. *P<0.05,
**P<0.01, ***P<0.001. n=3. AO,
aurantio-obtusin; CTRL control; Fer-1, ferrostatin-1; GPX4,
glutathione peroxidase 4; LPO, lipid peroxide; SCD1, stearoyl-CoA
desaturase 1. |
Antitumor efficacy of AO and RSL3
combined therapy in a human liver cancer xenograft model
To further explore the potential anticancer
therapeutic benefits of simultaneous AO and RSL3 treatment in
vivo, experiments were conducted using a human liver cancer
xenograft model in nude mice. Mice subcutaneously injected with
HepG2 cells were treated with AO or RSL3, alone or in combination,
for 15 days. Tumor growth was significantly slower in the
combination treatment group than in the groups treated with AO or
RSL3 alone (Fig. 7A and B).
Furthermore, the tumor weight was markedly lower in the combination
treatment group than in the other groups (Fig. 7C). Throughout the course of the
animal study, total body weight was monitored as an indicator of
treatment-related toxicity. No significant weight loss was observed
in any of the treatment groups (Fig.
7D), indicating that the treatments were well-tolerated.
Additionally, a decrease in GPX4 expression was observed in the
tumors from the combination treatment group compared with the RSL3
group (Fig. 7E). Similarly, no
hepatic or renal toxicity was observed after AO treatment (Fig. 7F). In summary, these findings
highlight the therapeutic potential of combining AO with a
ferroptosis inducer in liver cancer treatment.
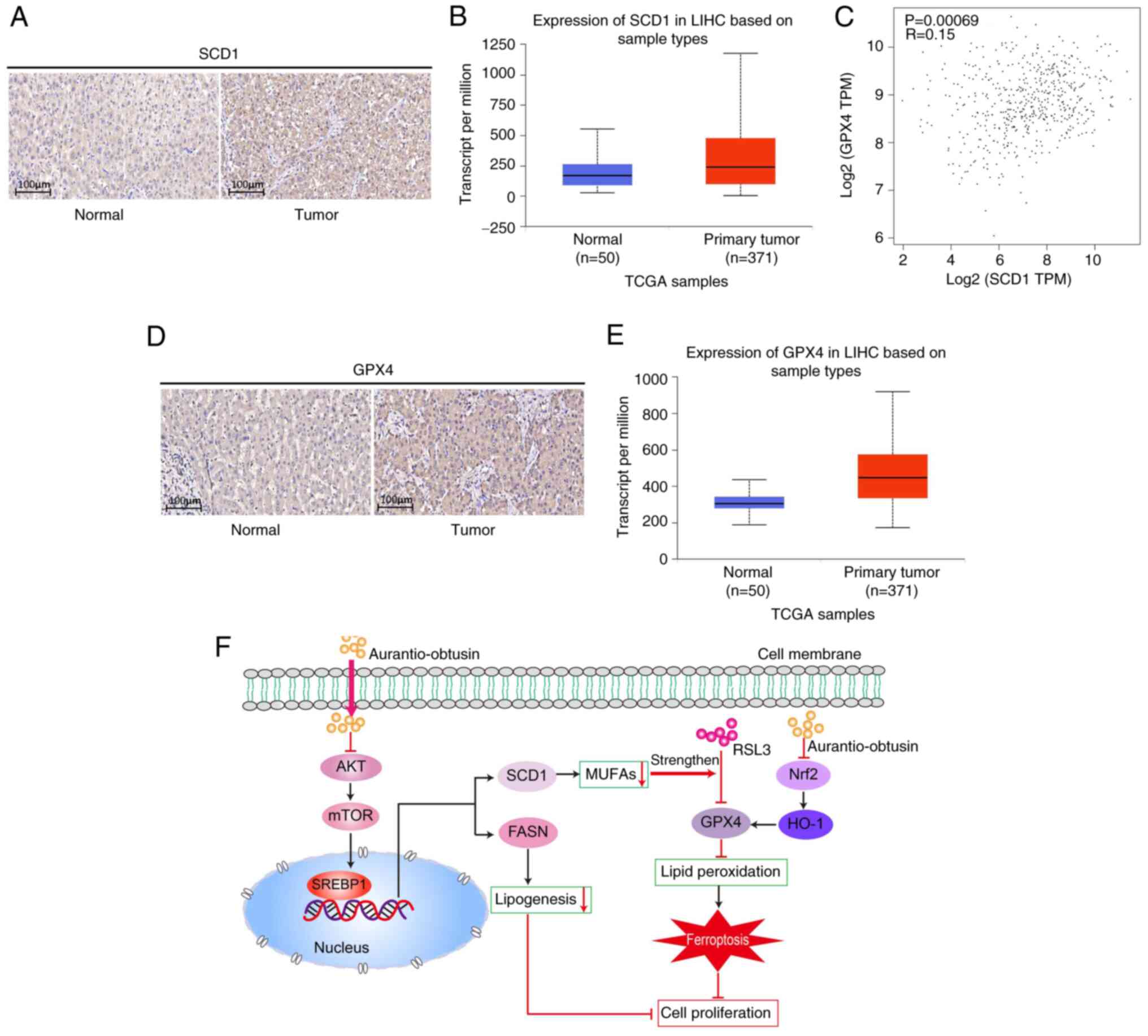
SCD1/GPX4 is differentially expressed in
specimens from patients with liver cancer
To investigate the relationship between SCD1 and
liver cancer progression, SCD1 expression w/as assessed in
specimens from patients with liver cancer. Immunohistochemistry
revealed a marked increase in SCD1 protein levels in liver cancer
tissues compared with those in normal liver tissues (Fig. 8A). Furthermore, liver cancer
samples from TCGA database, which included 371 primary liver tumor
samples and 50 normal liver tissue samples, were analyzed. The
results consistently demonstrated that SCD1 expression in primary
liver tumors was significantly elevated compared with that in
normal liver tissues (Fig. 8B).
Notably, the bioinformatics analysis utilizing data from TCGA
database also revealed a weak positive correlation between the
expression levels of SCD1 and GPX4 in liver cancer tissues
(Fig. 8C). Similarly, the
immunohistochemistry analyses confirmed a substantial increase in
GPX4 protein levels in liver cancer tissues compared with in normal
liver tissues, consistent with the findings from TCGA database
(Fig. 8D and E). These findings
collectively suggested that both SCD1 and GPX4 were significantly
upregulated in liver cancer tissues compared with in normal liver
tissues, and have the potential to serve as valuable biomarkers for
personalized treatment in patients with liver cancer.
Discussion
The present study conducted in vivo and in
vitro experiments to investigate the inhibitory effects of AO
on liver cancer cells. Lipid metabolism is frequently disrupted in
cancer and some reports have suggested that abnormal lipid
metabolism may serve an essential role in liver carcinogenesis
(6,12,48). Altered de novo lipogenesis
(DNL) is a pivotal deregulated metabolic event in cancer. SREBP1
controls the transcription of major enzymes involved in DNL,
including ACLY, ACACA, FASN and SCD1. Previous studies have
reported an increase in DNL in liver cancer samples (9,10,49,50). Multiple mechanisms, such as
activation of the AKT/mTOR pathway, can lead to increased SREBP1
induction and enhanced expression of ACLY, ACACA, FASN and SCD1. In
liver cancer, SCD1 has been reported to be upregulated and to be
associated with shorter disease-free survival (32,51). This finding is consistent with the
results of the present analysis using TCGA database. In addition,
in a combined proteomic and lipidomic profiling study, upregulated
hepatic SCD1 was identified as a reliable marker for liver cancer
diagnosis and progression (52).
A previous study demonstrated contrasting roles for AKT and AMPK in
fatty acid synthesis (46).
Mechanistically, SCD1 expression is induced by the AKT/mTOR
pathway, whereas AMPK suppresses its expression (53-56). The present study confirmed that AO
can inhibit the expression of SCD1 by suppressing the
AKT/mTOR/SREBP1 pathway, rather than the AMPK pathway.
Ferroptosis is a form of programmed cell death
triggered by metabolically regulated lipid peroxidation. Notably,
it can enhance the effectiveness of both targeted therapy and
chemotherapy in the treatment of cancer (57). The present study observed that
lower SCD1 expression led to increased susceptibility to
ferroptosis in SK-Hep1 and HepG2 cancer cell lines. SCD1-catalyzed
MUFAs effectively suppress ferroptosis by substituting
polyunsaturated fatty acids in the lipid membrane, thereby reducing
the accumulation of lipid ROS (58). SCD1 has been extensively studied
for a number of years in the context of metabolic diseases, such as
diabetes and obesity (59,60).
However, to the best of our knowledge, the exact role of SCD1 in
the development of cancer remains unclear. It has been indicated
that SCD1 has a crucial role in the regulation of liver
tumor-initiating cells and sorafenib resistance by modulating
endoplasmic reticulum stress-mediated differentiation (51). In the present study, it was
observed that the combination of A939572 and RSL3 led to a
significant inhibition of proliferation and a marked decrease in
colony formation in SK-Hep1 and HepG2 cancer cells. This suggests
that the combination of SCD1 inhibitors with ferroptosis inducers
may enhance the anti-liver cancer effects of ferroptosis
inducers.
In mammalian cells, the GSH-GPX4 axis serves a
pivotal role in scavenging LPO, thereby inhibiting ferroptosis
(61-63). Inhibition of the Nrf2/HO-1 pathway
could suppress the expression of GPX4 (47). Recent studies have demonstrated
that the downregulation of GPX4 expression induces the accumulation
of LPO, promoting ferroptosis and resulting in increased
sensitivity of resistant liver cancer cells to sorafenib (24,64). Consequently, triggering
ferroptosis is recognized as a valuable approach to resensitize
liver cancer cells to therapies. Recent research has revealed that
GPX4 is a downstream target of SCD1/FADS2 (65). SCD1/FADS2 act as a positive
regulator of GPX4 and modulates the GSH/GSSG ratio to prevent an
excessive accumulation of ROS, thereby mediating oxidative stress
and ferroptosis in ascites-derived ovarian cancer cells (65).
AO, an anthraquinone monomer compound derived from
cassia seeds, has been the subject of modern pharmacological
studies (38,40,41). These studies have demonstrated the
efficacy of AO in reducing blood lipid levels, mitigating NAFLD and
exhibiting anti-influenza properties (37,38,40). Despite its diverse effects, the
potential therapeutic advantages of AO in cancer treatment remain
unclear. In the present study, it was shown that AO treatment at
micromolar concentrations could effectively reduce cell
proliferation and migration, while impeding de novo fatty
acid synthesis in liver cancer cells.
The present study investigated the anti-liver cancer
effects of AO in cellular and animal models. The findings
demonstrated that AO could effectively inhibit the growth of human
liver cancer cells in a xenograft tumor mouse model. Furthermore,
the synergistic application of AO with the ferroptosis inducer RSL3
exhibited superior inhibitory effects compared with their
individual use. Mechanistic insights from cell experiments revealed
that AO attenuated proliferation and migration of liver cancer
cells, and this action was mediated through inhibition of the
AKT/mTOR/SREBP1 signaling pathway, downregulation of SCD1,
suppression of lipid synthesis and concurrent inhibition of GPX4 by
Nrf2/HO-1 pathway, ultimately leading to the induction of
ferroptosis and anti-liver cancer effects (Fig. 8F). Additionally, the enhanced
inhibition of SCD1 by AO further augmented the induction of
ferroptosis by the ferroptosis inducer RSL3 (Fig. 8F). These findings highlight the
potential therapeutic efficacy of AO in combating liver cancer and
underscore the importance of targeting ferroptosis as a promising
strategy for cancer therapy. Although the present findings
indicated that AO may induce ferroptosis in liver cancer cells by
inhibiting the Nrf2/HO-1/GPX4 signaling pathway, the investigation
of this mechanism remains insufficient. To confirm that this is the
sole pathway through which AO regulates ferroptosis, further
studies utilizing a GPX4-knockdown model are necessary.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LJT and HWD designed the study, confirmed the
authenticity of all the raw data and revised the manuscript. XJT
provided ethical oversight for the animal experiments and performed
the experiments. WL and JD analyzed the data and wrote the
manuscript. WL, JD, XDC, YP and XCQ performed the experiments. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
This study adhered to the guidelines for animal and
human research ethics. Animal experiments were conducted following
protocols approved by the Institutional Animal Care and Use
Committee at Hunan Normal University (approval no. D2021059). Human
study protocols were approved by the Ethics Committee of Hunan
Provincial People's Hospital, Changsha, China [approval no.
(2023)-178]. The patients provided informed consent for their
tissues to be used in future research at the time of initial
collection. Consequently, the requirement for informed consent was
waived by the same committee due to the retrospective nature of the
study and the use of anonymized data.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
AO
|
aurantio-obtusin
|
|
mTOR
|
mammalian target of rapamycin
|
|
GPX4
|
glutathione peroxidase 4
|
|
SCD1
|
stearoyl-CoA desaturase1
|
|
SREBP1
|
sterol regulatory element-binding
protein 1
|
|
FASN
|
fatty acid synthase
|
|
GSH
|
glutathione
|
|
MUFAs
|
monounsaturated fatty acids
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
HO-1
|
heme oxygenase-1
|
|
Fer-1
|
ferrostatin-1
|
|
FBS
|
fetal bovine serum
|
|
LPO
|
lipid peroxide
|
|
PA
|
palmitic acid
|
|
AMPK
|
AMP-activated protein kinase
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
TCGA
|
The Cancer Gene Atlas
|
Acknowledgements
The authors would like to thank Ms. Xin Ying
(School of Medicine of Hunan Normal University, Changsha, China)
for their assistance in editing the diagram.
Funding
This work was supported by the Hunan Provincial Natural Science
Foundation (grant nos. 2023JJ30348, 2021JJ8028 and 2022JJ80073),
the Hunan Health Commission High-level Talent Project (grant no.
20230609-1014), the Hunan Administration of Traditional Chinese
Medicine (grant no. 201922) and the Scientific Research Project of
the Changsha Science and Technology Department (grant no.
kq2208125).
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen D, Wang J, Li Y, Xu C, Fanzheng M,
Zhang P and Liu L: LncRNA NEAT1 suppresses cellular senescence in
hepatocellular carcinoma via KIF11-dependent repression of CDKN2A.
Clin Transl Med. 13:e14182023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang L, Liu BX and Long HY: Ablative
strategies for recurrent hepatocellular carcinoma. World J Hepatol.
15:515–524. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aravinthan AD, Bruni SG, Doyle AC, Thein
HH, Goldaracena N, Issachar A, Lilly LB, Selzner N, Bhat M,
Sreeharsha B, et al: Liver transplantation is a preferable
alternative to palliative therapy for selected patients with
advanced hepatocellular carcinoma. Ann Surg Oncol. 24:1843–1851.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shukla A, Patkar S, Sundaram S, Shah SR,
Ingle M, Gupta A, Gopan A, Kamat M, Mohanka R, Singh S, et al:
Clinical profile, patterns of care & adherence to guidelines in
patients with hepatocellular carcinoma: Prospective multi-center
study. J Clin Exp Hepatol. 12:1463–1473. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakagawa H, Hayata Y, Kawamura S, Yamada
T, Fujiwara N and Koike K: Lipid metabolic reprogramming in
hepatocellular carcinoma. Cancers (Basel). 10:4472018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balsano C, Porcu C, Sideri S and Tavolaro
S: Fat and hepatocellular carcinoma. Hepatoma Res. 4:382018.
View Article : Google Scholar
|
|
8
|
Scheinberg T, Mak B, Butler L, Selth L and
Horvath LG: Targeting lipid metabolism in metastatic prostate
cancer. Ther Adv Med Oncol. 15:175883592311528392023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fhu CW and Ali A: Fatty acid synthase: An
emerging target in cancer. Molecules. 25:39352020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jones SF and Infante JR: Molecular
pathways: Fatty acid synthase. Clin Cancer Res. 21:5434–5438. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koundouros N and Poulogiannis G:
Reprogramming of fatty acid metabolism in cancer. Br J Cancer.
122:4–22. 2020. View Article : Google Scholar :
|
|
12
|
Qin XY, Su T, Yu W and Kojima S: Lipid
desaturation-associated endoplasmic reticulum stress regulates MYCN
gene expression in hepatocellular carcinoma cells. Cell Death Dis.
11:662020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Zhou Y, Xu H, Wang X, Zhang Y,
Shang R, O'Farrell M, Roessler S, Sticht C, Stahl A, et al:
Therapeutic efficacy of FASN inhibition in preclinical models of
HCC. Hepatology. 76:951–966. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Liu Y, Han A, Tang C, Xu R, Feng
L, Yang Y, Chen L and Lin Z: The NQO1/p53/SREBP1 axis promotes
hepatocellular carcinoma progression and metastasis by regulating
Snail stability. Oncogene. 41:5107–5120. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li C, Yang W, Zhang J, Zheng X, Yao Y, Tu
K and Liu Q: SREBP-1 has a prognostic role and contributes to
invasion and metastasis in human hepatocellular carcinoma. Int J
Mol Sci. 15:7124–7138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shimano H and Sato R: SREBP-regulated
lipid metabolism: Convergent physiology-divergent pathophysiology.
Nat Rev Endocrinol. 13:710–730. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peterson TR, Sengupta SS, Harris TE,
Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter
AE, Finck BN and Sabatini DM: mTOR complex 1 regulates lipin 1
localization to control the SREBP pathway. Cell. 146:408–420. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bakan I and Laplante M: Connecting mTORC1
signaling to SREBP-1 activation. Curr Opin Lipidol. 23:226–234.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tao T, Su Q, Xu S, Deng J, Zhou S, Zhuang
Y, Huang Y, He C, He S, Peng M, et al: Down-regulation of PKM2
decreases FASN expression in bladder cancer cells through
AKT/mTOR/SREBP-1c axis. J Cell Physiol. 234:3088–3104. 2019.
View Article : Google Scholar
|
|
20
|
Li Y, Yang W, Zheng Y, Dai W, Ji J, Wu L,
Cheng Z, Zhang J, Li J, Xu X, et al: Targeting fatty acid synthase
modulates sensitivity of hepatocellular carcinoma to sorafenib via
ferroptosis. J Exp Clin Cancer Res. 42:62023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Z, Zhou C, Zhang Y, Tian X, Wang H,
Wu J and Jiang S: From synergy to resistance: Navigating the
complex relationship between sorafenib and ferroptosis in
hepatocellular carcinoma. Biomed Pharmacother. 170:1160742024.
View Article : Google Scholar
|
|
23
|
Dahiya M and Dureja H: Sorafenib for
hepatocellular carcinoma: Potential molecular targets and
resistance mechanisms. J Chemother. 34:286–301. 2022. View Article : Google Scholar
|
|
24
|
Wang C, Zheng C, Wang H, Shui S, Jin H,
Liu G, Xu F, Liu Z, Zhang L, Sun D and Xu P: Dual degradation
mechanism of GPX4 degrader in induction of ferroptosis exerting
anti-resistant tumor effect. Eur J Med Chem. 247:1150722023.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B,
Wu F, Wang Q, Wang S, Rong D, Reiter FP, et al: The mechanisms of
sorafenib resistance in hepatocellular carcinoma: Theoretical basis
and therapeutic aspects. Signal Transduct Target Ther. 5:872020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao Z, Wang D, Yang J, Li M, Ling C, Lv D,
Cao Y, Chen Z, Shi C, Shen H and Tang Y: Iron deficiency in
hepatocellular carcinoma cells induced sorafenib resistance by
upregulating HIF-1α to inhibit apoptosis. Biomed Pharmacother.
163:1147502023. View Article : Google Scholar
|
|
27
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Raeisi M, Hassanbeigi L, Khalili F,
Kharrati-Shishavan H, Yousefi M and Mehdizadeh A: Stearoyl-CoA
desaturase 1 as a therapeutic target for cancer: A focus on
hepatocellular carcinoma. Mol Biol Rep. 49:8871–8882. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo Z, Huo X, Li X, Jiang C and Xue L:
Advances in regulation and function of stearoyl-CoA desaturase 1 in
cancer, from bench to bed. Sci China Life Sci. 66:2773–2785. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sen U, Coleman C and Sen T: Stearoyl
coenzyme A desaturase-1: Multitasker in cancer, metabolism, and
ferroptosis. Trends Cancer. 9:480–489. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu HH, Xu Y, Li CJ, Hsu SJ, Lin XH, Zhang
R, Chen J, Chen J, Gao DM, Cui JF, et al: An SCD1-dependent
mechanoresponsive pathway promotes HCC invasion and metastasis
through lipid metabolic reprogramming. Mol Ther. 30:2554–2567.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bansal S, Berk M, Alkhouri N, Partrick DA,
Fung JJ and Feldstein A: Stearoyl-CoA desaturase plays an important
role in proliferation and chemoresistance in human hepatocellular
carcinoma. J Surg Res. 186:29–38. 2014. View Article : Google Scholar :
|
|
33
|
Wang C, Shi M, Ji J, Cai Q, Zhao Q, Jiang
J, Liu J, Zhang H, Zhu Z and Zhang J: Stearoyl-CoA desaturase 1
(SCD1) facilitates the growth and anti-ferroptosis of gastric
cancer cells and predicts poor prognosis of gastric cancer. Aging
(Albany NY). 12:15374–15391. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tesfay L, Paul BT, Konstorum A, Deng Z,
Cox AO, Lee J, Furdui CM, Hegde P, Torti FM and Torti SV:
Stearoyl-CoA desaturase 1 protects ovarian cancer cells from
ferroptotic cell death. Cancer Res. 79:5355–5366. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar :
|
|
36
|
Chen H, Qi Q, Wu N, Wang Y, Feng Q, Jin R
and Jiang L: Aspirin promotes RSL3-induced ferroptosis by
suppressing mTOR/SREBP-1/SCD1-mediated lipogenesis in PIK3CA-mutant
colorectal cancer. Redox Biol. 55:1024262022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim M, Lim SJ, Lee HJ and Nho CW: Cassia
tora seed extract and its active compound aurantio-obtusin inhibit
allergic responses in IgE-mediated mast cells and anaphylactic
models. J Agric Food Chem. 63:9037–9046. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kwon KS, Lee JH, So KS, Park BK, Lim H,
Choi JS and Kim HP: Aurantio-obtusin, an anthraquinone from cassiae
semen, ameliorates lung inflammatory responses. Phytother Res.
32:1537–1545. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin S, Zhang Y, Wang Z, Zhang S, Li Y, Fan
Y, Li D, Li S and Bai Y: Preparation of novel anthraquinone-based
aspirin derivatives with anti-cancer activity. Eur J Pharmacol.
900:1740202021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou F, Ding M, Gu Y, Fan G, Liu C, Li Y,
Sun R, Wu J, Li J, Xue X, et al: Aurantio-obtusin attenuates
non-alcoholic fatty liver disease through AMPK-mediated autophagy
and fatty acid oxidation pathways. Front Pharmacol. 12:8266282022.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu Y, Sun X, Hu X, Xu Y, Li T and Wu Z:
Pharmacological properties and underlying mechanisms of
aurantio-obtusin (review). Exp Ther Med. 26:3802023. View Article : Google Scholar
|
|
42
|
Li YJ, Wu RY, Liu RP, Wu KY, Ding MN, Sun
R, Gu YQ, Zhou F, Wu JZ, Zheng Q, et al: Aurantio-obtusin
ameliorates obesity by activating PPARα-dependent mitochondrial
thermogenesis in brown adipose tissues. Acta Pharmacol Sin.
44:1826–1840. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hu M, Zhong Y, Liu J, Zheng S, Lin L, Lin
X, Liang B, Huang Y, Xian H, Li Z, et al: An adverse outcome
pathway-based approach to assess aurantio-obtusin-induced
hepatotoxicity. Toxicology. 478:1532932022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chandrashekar DS, Karthikeyan SK, Korla
PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne
U, et al: UALCAN: An update to the integrated cancer data analysis
platform. Neoplasia. 25:18–27. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gu L, Zhu Y, Lin X, Tan X, Lu B and Li Y:
Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes
lipid metabolism and hepatocarcinogenesis. Oncogene. 39:2437–2449.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Oh JM and Chun S: Ginsenoside CK inhibits
the early stage of adipogenesis via the AMPK, MAPK, and AKT
signaling pathways. Antioxidants (Basel). 11:18902022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang R, Gao W, Wang Z, Jian H, Peng L, Yu
X, Xue P, Peng W, Li K and Zeng P: Polyphyllin I induced
ferroptosis to suppress the progression of hepatocellular carcinoma
through activation of the mitochondrial dysfunction via
Nrf2/HO-1/GPX4 axis. Phytomedicine. 122:1551352024. View Article : Google Scholar
|
|
48
|
Yamashita T, Honda M, Takatori H, Nishino
R, Minato H, Takamura H, Ohta T and Kaneko S: Activation of
lipogenic pathway correlates with cell proliferation and poor
prognosis in hepatocellular carcinoma. J Hepatol. 50:100–110. 2009.
View Article : Google Scholar
|
|
49
|
Zhou Y, Tao J, Calvisi DF and Chen X: Role
of lipogenesis rewiring in hepatocellular carcinoma. Semin Liver
Dis. 42:77–86. 2022. View Article : Google Scholar :
|
|
50
|
Li S, Liu R, Pan Q, Wang G, Cheng D, Yang
J, Chen H and Xu G: De novo lipogenesis is elicited dramatically in
human hepatocellular carcinoma especially in hepatitis C
virus-induced hepatocellular carcinoma. MedComm (2020). 1:178–187.
2020.PubMed/NCBI
|
|
51
|
Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY,
Cheng LKW, Ma S, Lin CH, Copland JA, Ding J, et al: Stearoyl-CoA
desaturase regulates sorafenib resistance via modulation of ER
stress-induced differentiation. J Hepatol. 67:979–990. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Muir K, Hazim A, He Y, Peyressatre M, Kim
DY, Song X and Beretta L: Proteomic and lipidomic signatures of
lipid metabolism in NASH-associated hepatocellular carcinoma.
Cancer Res. 73:4722–4731. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu G, Kuang S, Cao R, Wang J, Peng Q and
Sun C: Sorafenib kills liver cancer cells by disrupting
SCD1-mediated synthesis of monounsaturated fatty acids via the
ATP-AMPK-mTOR-SREBP1 signaling pathway. FASEB J. 33:10089–10103.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Igal RA: Stearoyl-CoA desaturase-1: A
novel key player in the mechanisms of cell proliferation,
programmed cell death and transformation to cancer. Carcinogenesis.
31:1509–1515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li L, Wang C, Calvisi DF, Evert M, Pilo
MG, Jiang L, Yuneva M and Chen X: SCD1 expression is dispensable
for hepatocarcinogenesis induced by AKT and Ras oncogenes in Mice.
PLoS One. 8:e751042013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z,
Cai K, Zhao Y and Luo Z: HCAR1/MCT1 regulates tumor ferroptosis
through the lactate-mediated AMPK-SCD1 activity and its therapeutic
implications. Cell Rep. 33:1084872020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang Y, Wu X, Ren Z, Li Y, Zou W, Chen J
and Wang H: Overcoming cancer chemotherapy resistance by the
induction of ferroptosis. Drug Resist Updat. 66:1009162023.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yi J, Zhu J, Wu J, Thompson CB and Jiang
X: Oncogenic activation of PI3K-AKT-mTOR signaling suppresses
ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA.
117:31189–31197. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tibori K, Orosz G, Zámbó V, Szelényi P,
Sarnyai F, Tamási V, Rónai Z, Mátyási J, Tóth B, Csala M and
Kereszturi É: Molecular mechanisms underlying the elevated
expression of a potentially type 2 diabetes mellitus associated
SCD1 variant. Int J Mol Sci. 23:62212022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Martín-Núñez GM, Cabrera-Mulero R,
Rojo-Martínez G, Gómez-Zumaquero JM, Chaves FJ, de Marco G,
Soriguer F, Castaño L and Morcillo S: Polymorphisms in the SCD1
gene are associated with indices of stearoyl CoA desaturase
activity and obesity: A prospective study. Mol Nutr Food Res.
57:2177–2184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhou J, Zhang L, Yan J, Hou A, Sui W and
Sun M: Curcumin induces ferroptosis in A549 CD133+ cells
through the GSH-GPX4 and FSP1-CoQ10-NAPH pathways. Discov Med.
35:251–263. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Xie Y, Kang R, Klionsky DJ and Tang D:
GPX4 in cell death, autophagy, and disease. Autophagy.
19:2621–2638. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang H, Wang C, Li B, Zheng C, Liu G, Liu
Z, Zhang L and Xu P: Discovery of ML210-based glutathione
peroxidase 4 (GPX4) degrader inducing ferroptosis of human cancer
cells. Eur J Med Chem. 254:1153432023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang
K and Tang N: GSTZ1 sensitizes hepatocellular carcinoma cells to
sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis.
Cell Death Dis. 12:4262021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Xuan Y, Wang H, Yung MM, Chen F, Chan WS,
Chan YS, Tsui SK, Ngan HY, Chan KK and Chan DW: SCD1/FADS2 fatty
acid desaturases equipoise lipid metabolic activity and
redox-driven ferroptosis in ascites-derived ovarian cancer cells.
Theranostics. 12:3534–3552. 2022. View Article : Google Scholar : PubMed/NCBI
|