In recent years, developments in the medical space
have resulted in the establishment of several tumor treatment
methods. However, drug therapy remains to be a primary strategy in
the treatment for various types of cancers (1). Unfortunately, drug resistance has
become a huge obstacle for cancer therapy, often leading to relapse
and metastasis. Drug resistance refers to a state in which tumor
cells become insensitive to antitumor drugs, a significant factor
contributing to the failure of therapy and a pressing challenge in
cancer treatment (2). Tumor cells
can develop resistance to chemotherapeutic drugs through either
natural or acquired mechanisms. Natural drug resistance refers to
the natural resistance of tumor cells to chemotherapeutic drugs,
either because the drug within the cell does not reach the
concentration required to inactivate the target or because the
tumor cells fail to respond to the induction of apoptotic
mechanisms (3). Acquired drug
resistance develops gradually during the course of drug therapy:
Typically, drug resistance is acquired by tumor cells following
long-term treatment with small doses of cytotoxic drugs (4). While continuous chemotherapy can
inhibit tumor growth and extend patient survival, cancer cells
adapt to effects of drugs over time through mutations. This
adaptation reduces drug efficacy and may lead to the treatment
becoming ineffective, resulting in the development of drug
resistance (5).
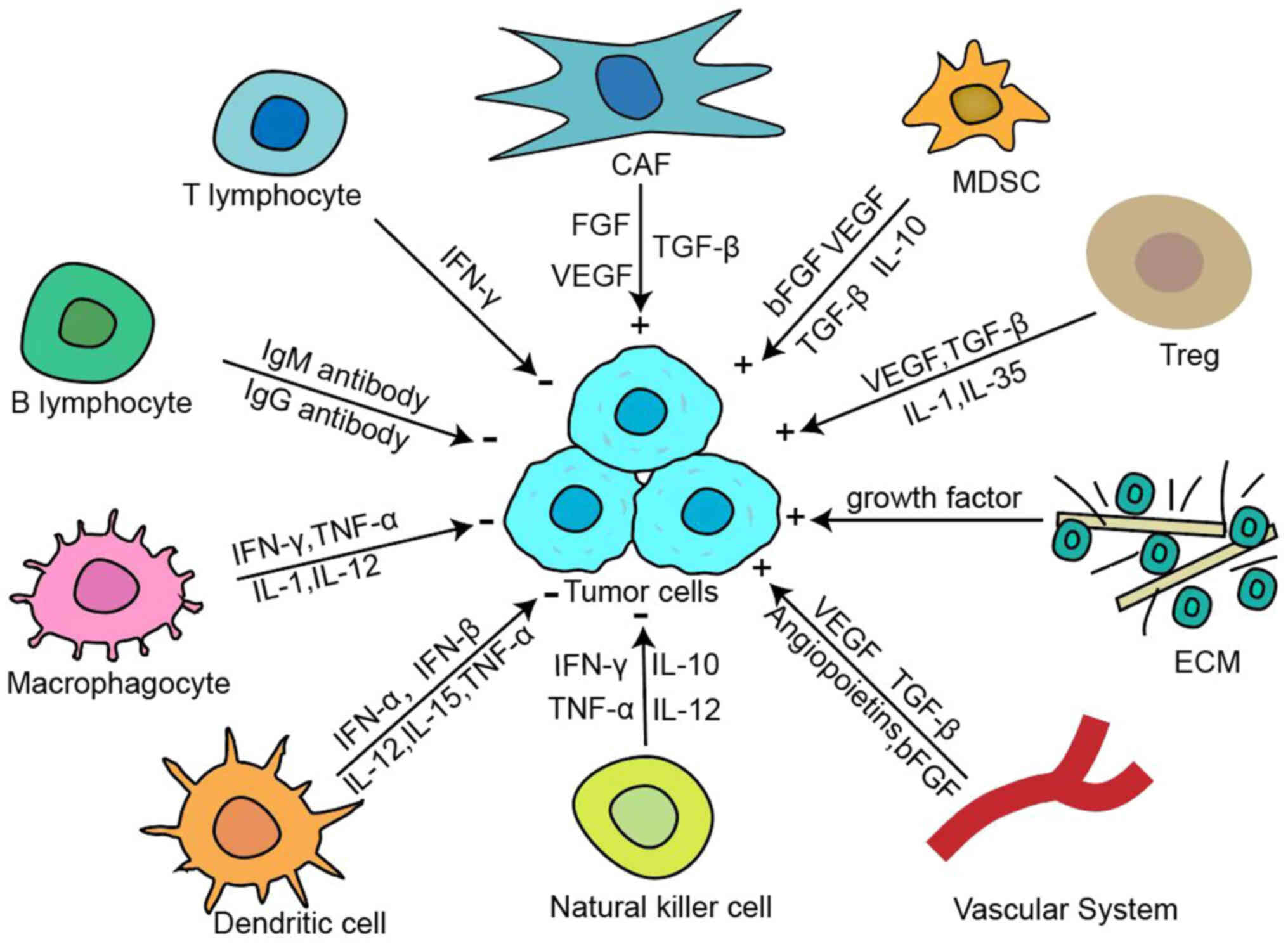
The tumor immune microenvironment (TME) is a complex
network of cells, molecules and physiological factors present at
the tumor, which together with tumor cells constitute the tumor
tissue ecosystem. Although the specific components of each TME are
differentiated and dependent on the type of tumor, the TME often
has common characteristics, such as the tumor cells themselves, the
surrounding blood vessels and cytokines, immune cells, stromal
cells and extracellular matrix (ECM) (6). The TME significantly influences
tumor initiation and progression, exhibiting substantial
differences from the microenvironment of normal tissues. Within the
TME, tumor cells interact with surrounding cells, altering their
function through interactions with the tumor-associated macrophages
(TAMs), cancer-associated fibroblasts (CAFs) and stromal cells.
These interactions facilitate tumor growth, invasion and metastasis
(7).
In addition, the TME significantly decreases the
efficacy of immunotherapy, and immune cells in the TME may be
altered by tumor cells, losing their immune monitoring effect on
the tumor, and even being transformed into suppressive cells by
tumor cells, thus creating an environment that favors tumor escape.
The immunosuppressive TME includes the immunosuppressive factors
and cells, and physical and mechanical barriers, where impaired
tumor antigen presentation process as well as metabolic alterations
can be considered as the main site of drug resistance. The typical
composition of the TME is demonstrated in Fig. 1.
Currently, the relationship between the TME and
tumor drug resistance is not fully understood. The present review
primarily discussed the relationship between the TME and tumor drug
resistance based on existing research, highlighting the challenges
and directions for future research. It also described the
mechanisms by which the TME interacts with the tumor and the
occurrence of tumor drug resistance, to showcase potential novel
treatment strategies and drug targets. New strategies for
overcoming tumor drug resistance and improving the effects of
cancer treatment are also highlighted.
Tumor antigens bind to MHC-I molecules and are
presented on the surface of tumor cells. This renders them
recognizable by immune effector cells. Therefore, tumor cells with
dysregulated presentation of antigens bound to MHC-I for
presentation are more likely to avoid detection by the immune
system (8). In general, tumor
cells can downregulate the antigen presentation by MHC-I via either
deletion of the MHC-Ⅰgene or inhibition of MHC-Ⅰgene transcription.
The human MHC genes encodes various molecules expressed on white
blood cells called human leucocyte antigen (HLA). The expression of
HLA-I antigen is downregulated in most malignant tumors, such as
melanoma, gastric cancer (GC), breast cancer and ovarian cancer.
Moreover, the degree of this downregulation is positively
correlated with the degree of malignancy and metastasis of
tumors.
The elimination of tumor cells by the immune system
relies on the reaction of immune effector cells to antigens present
on the surface of tumor cells. However, tumor cells often exhibit
reduced or absent expression of tumor antigens, inhibiting the
activation of T cells by dendritic cells (DCs) and evading
recognition and destruction by cytotoxic T lymphocytes (CTLs). It
has been reported that tumor cells will undergo antigen mutation
when co-cultured with monoclonal and polyclonal transgenic CTLs,
which specifically recognize the tumor antigen P1A, and thus, P1A
is not readily recognized by CTLs (9). In addition to generating antigenic
mutations, tumor cells can also evade recognition by the immune
system by shedding the antigens expressed on their surface. For
example, carcinoembryonic antigen can be shed from the surface of
tumor cells, which renders tumor cells unrecognizable by immune
effector cells. Similar to the loss of tumor antigens, tumor cells
can also evade recognition and attack by NK cells following the
release of natural killer cell group 2-member D (NKG2D) ligands
(10).
Activation of T cells requires the induction of the
first signal produced by the binding of T cell receptors to antigen
peptide-MHC complexes, and also the second signal provided by the
binding of costimulatory molecules (CMs) on antigen-presenting
cells or tumor cells to CM receptors on T cells. Tumor cells that
only express MHC-I antigens, but lack CMs participate in the
antigen presentation process, but cannot activate T cells and
elicit strong immune responses. Such tumor cells can lead to the
emergence of immune tolerance (11). The B7 family of molecules and
their receptors are the most important CM pairs participating in
the activation of T cells. The binding of each molecule to its
receptors promotes activation and proliferation of T cells. Studies
found that tumor cells downregulated the expression of B7-1 and
B7-2 molecules, meaning there is insufficient induction of T cell
activation signals such that T cells do not proliferate, whereas
the expression of B7-H1 and B7-H4 molecules is upregulated. Once
they bind to the receptors, these inhibitory CMs generate
inhibitory signals, induce the apoptosis of T cells, and inhibit
the antitumor immune response of the body (12,13).
The TME contains numerous immunosuppressive
cytokines, including TGF-β, IL-6 and IL-10, which impede antitumor
immune responses through direct or indirect mechanisms. TGF-β, for
example, hinders the proliferation of immune effector cells,
suppresses DC maturation, reduces CTL and NK cell activation,
reduces the levels of antitumor immune cytokines such as IFN-γ and
TNF-α, and inhibits MHC-II antigen expression induced by IFN-γ in
melanoma cells (14). IL-10 can
reduce the expression of CMs on DCs, inhibit tumor antigen
presentation, alter their phenotypes, inhibit the activity of T
cells, and block T cell-mediated attacks on tumor cells. It has
been shown that TNF can induce hemorrhagic necrosis of certain
tumor blood vessels, specifically eliminate tumor cells, and
modulate the immune functions of cells, while tumors can express
soluble TNF binding protein, which prevents the eliminating effect
of TNF (15). Additionally,
vascular endothelial growth factor (VEGF) has been implicated in
the development of tumor immune escape mechanisms. As a specific
endothelial cell stimulating factor, VEGF can accelerate tumor
neovascularization, increase the permeability of blood vessels, and
promote the infiltration and metastasis of tumor cells. It can also
suppress the maturation of DCs, affecting antigen presentation
function, and induce the programmed death ligand 1 (PD-L1)
expression in mature DCs, eventually stimulating the T cells and
the formation of CTLs (16,17).
The ECM in the TME supports the proliferation and
metastasis of tumor cells. The ECM is also an important component
of a hypoxic TME is composed of CAFs, TAMs, mesenchymal stem cells
(MSCs), inflammatory cells and endothelial cells (18). The ECM is an important
non-cellular component of the TME and plays a key role in
maintaining tissue structure and function. As a physical barrier of
the tumor, the ECM can dissolve drugs or delay drug delivery
(19). Fibronectin (FN),
hyaluronic acid, collagen (Col) and laminin are the main components
of the ECM, and can form ECM fibers. Specific enzymes, such as
lysyl oxidase cross-link these fibers, regulating tumor hardness
and promoting fibrosis (20).
Laminin has been revealed to be highly expressed in
multidrug-resistant cells (21,22). Alterations in the ECM can
facilitate tumor cell invasion and metastasis, influence the
penetration and distribution of chemotherapeutic drugs, and
diminish their effectiveness.
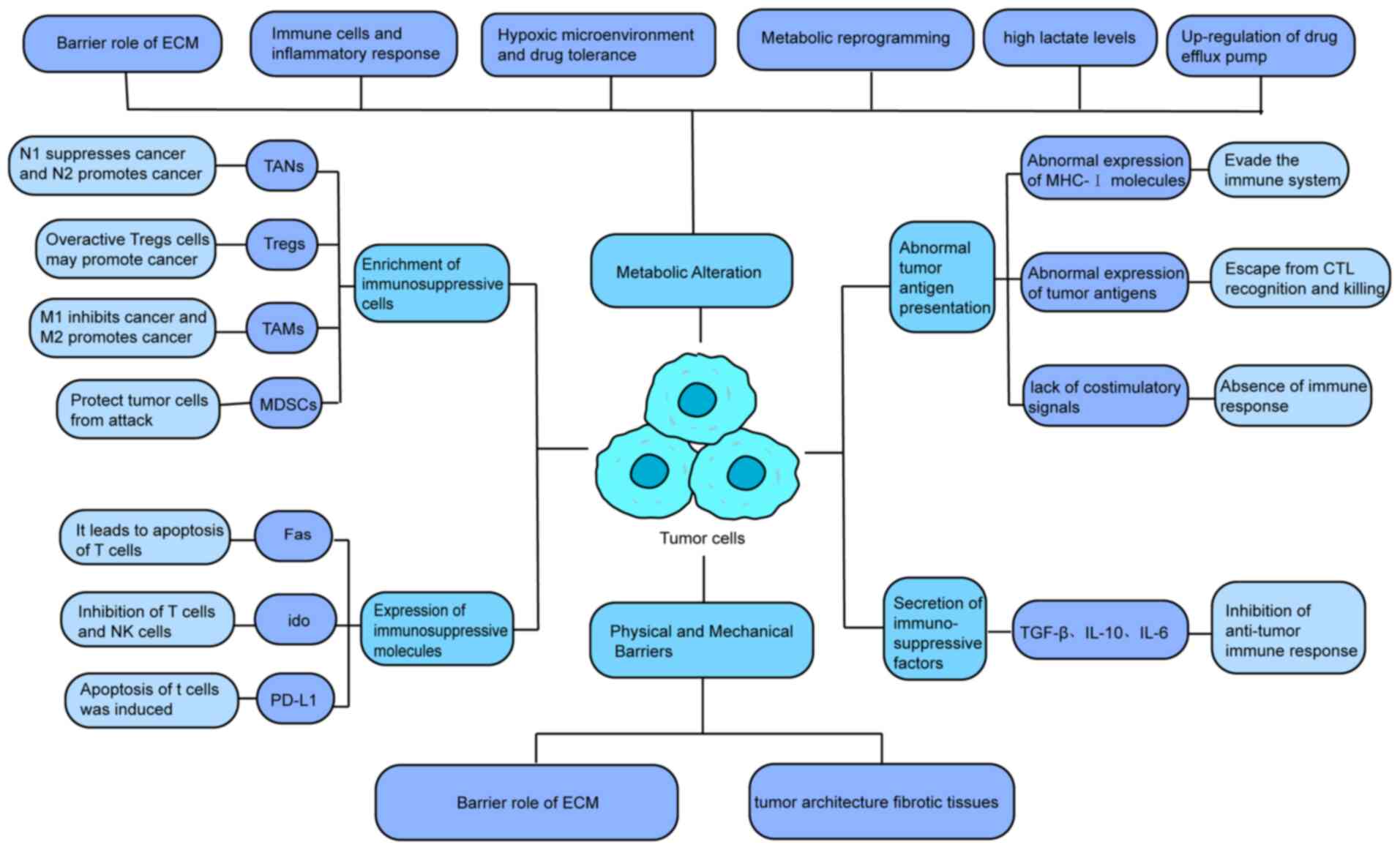
During tumor growth, immunosuppressive cells undergo
differentiation, proliferation and aggregation at the tumor site,
including TAMs, regulatory T cells (Tregs), tumor-associated
neutrophils (TANs), myeloid-derived suppressor cells (MDSCs) and
tumor-associated DCs (TADCs) (26). Several immunomodulatory factors
have been reported to date. In the present review, the main
components of the TME and their mechanisms of action were
introduced.
TAMs are a special class of macrophages derived from
monocytes in the peripheral blood and migrate to tumor tissues.
They are an important part of the TME, contributing to the
occurrence and progression of tumors by creating an
immunosuppressive microenvironment (27). TAMs typically differentiate into
one of two activation states: M1 macrophages, which have a
tumor-suppressive function, and M2 macrophages, which have a
tumor-promoting function (28).
M1 macrophages exert proinflammatory effects, eliminate tumor cells
and inhibit tumor growth. By contrast, M2 macrophages inhibit
inflammatory processes and modulate the immune cells thereby
promoting tumor growth, invasion, metastasis and angiogenesis
(29). M1 macrophages eliminate
cancer cells through antibody-dependent cell-mediated cytotoxicity
such as TNF, immune killer molecules and inflammatory cytokines
(30). In comparison, activated
M2 macrophages have been reported to accelerate the proliferation,
invasion and metastasis of tumor cells by enhancing angiogenesis
and inhibiting the antitumor function of immune cells (31). VEGF, IL-1, TNF-α, prostaglandin
E2, adrenomedullin and brain signaling protein 4D contribute to
angiogenesis (32).
By interacting with other cells, TAMs regulate the
composition and function of the TME and affect the behavior and
characteristics of tumor cells. Indeed, targeting TAMs is being
considered as a strategy for cancer treatment. M2 macrophages are
also closely associated with tumor angiogenesis and
lymphangiogenesis, known to directly modulate tumor proliferation
and metastasis, induce tumor drug resistance, and secrete cytokines
such as IL-10 and TGF-β, which inhibit immune responses (33). The secretion of PD-L1, cytotoxic T
lymphocyte-associat-edantigen-4 (CTLA-4) and other molecules may
enhance CTL apoptosis and inhibit CTL activation (34), resulting in immunosuppression.
Tregs constitute a subset of immune cells primarily
responsible for maintaining immune homeostasis and suppressing the
immune response against cancer (38). They achieve this by restraining
the activity of other immune cells, thereby preventing excessive
immune reactions and the development of autoimmune diseases. Tregs
are categorized into two primary types: Natural Tregs (nTregs),
which originate in the thymus, and induced Tregs (iTregs), which
differentiate in peripheral tissues (39,40). nTregs, also known as
thymus-derived Tregs, primarily control the inflammatory response
following infection and maintain normal immune tolerance. iTregs,
also known as peripherally-induced Tregs (41), can generate immune factors and
thereby decrease the activity of immune cells or directly bind to
immune cells to inhibit their activity. Tregs are a double-edged
sword. They can regulate the strength and duration of the immune
response by inhibiting the activity and function of other immune
cells to ensure the normal response of the immune system to
external antigens and prevent the occurrence of autoimmune
diseases. Within the TME, hyperactive Tregs cells can inhibit
immune cell assault on tumors, consequently allowing tumor evasion
and proliferation (42).
Tregs participate in immune regulation, autoimmune
diseases and tumor immune escape among other processes. Further
studies into the function and regulatory mechanism of Tregs are
required to improve our understanding of the immune balance,
occurrence and development of immune-related diseases. This is
essential for developing new immunoregulatory therapies. Tregs have
been reported to inhibit DCs by expressing IL-10, TGF-β and IL-35,
and secrete granzyme and perforin to eliminate effector cells
directly, and inhibit the proliferation of effector cells. In
addition, Tregs highly express CD39 and CD73 molecules, which
increase adenosine production, and bind to adenosine receptors on
the surface of effector cells to exert inhibitory effects (43).
TANs are a class of neutrophils that are present in
the TME. As an important type of cell of the immune system, TANs
participate in tumor development and progression. They are
functionally divided into tumor-suppressive N1 cells and
tumor-promoting N2 cells (47).
TGF-β induces N1 to N2 polarization of TANs (48). N1 TANs can activate the effector T
cells to prevent tumor development, promote tumor cell apoptosis
via tumor necrosis factor-associated apoptosis-inducing ligand, and
promote the release of matrix metalloproteinase 8 (MMP8) to degrade
the ECM, which favors tumor metastasis (49). Conversely, N2 TANs enhance
angiogenesis by stimulating VEGF, facilitate tumor advancement and
metastasis through cathepsin G and reactive oxygen species (ROS)
(50), and affect tumor
aggressiveness and prognosis via various mechanisms. TANs form a
complex network alongside tumor cells and other immune cells. They
regulate the inflammatory milieu, modulate immune responses, and
mediate cell-to-cell interactions within the TME, thereby
influencing tumor progression. Investigating the functions and
mechanisms of TANs is instrumental in identifying novel therapeutic
targets and devising innovative strategies for cancer
treatment.
MDSCs, as one of the most important types of stromal
cells of the TME, are multifunctional cells derived from the bone
marrow, and they exhibit immunosuppressive effects in pathological
conditions to protect tumor cells from attack by the host's immune
system via the production of ROS to inhibit T cell function
(54,55). Their morphology is very similar to
that of granulocytes or monocytes. Therefore, the two major
populations of MDSCs are called polymorphonuclear neutrophil MDSCs
(PMN-MDSCs) and monocyte MDSCs (M-MDSCs). Both types can inhibit
the activity of the TME, in which M-MDSC shows significant
plasticity and differentiation direction regulated by the TME
(56). M-MDSCs may exhibit
non-specific inhibition via various mechanisms and secrete various
immunosuppressive factors, such as nitrous oxide,
granulocyte-macrophage colony-stimulating factor, IL-10 and IL-6,
among other factors, which can directly or indirectly inhibit the
activity of T cells (57). By
contrast, PMN-MDSCs exhibit antigen-specific T cell tolerance and
function as non-specific suppressor cells capable of producing
cytokines that promote tumor angiogenesis. MDSCs are important
regulatory cells in the TME, and their immunosuppressive function
influences tumor growth and response to treatment. In-depth studies
on MDSCs are required to determine the mechanisms of tumor immune
escape and provide new ideas and strategies for individualized and
precise treatment. Aggregation of MDSCs can directly stimulate
tumor angiogenesis by releasing various pro-angiogenic factors.
Moreover, MDSCs can enhance the expression of immunosuppressive
factors, including arginase 1, ROS and inducible nitric oxide
synthase to induce apoptosis of activated T cells (58-60).
There is strong evidence indicating that tumors can
interfere with the differentiation of monocytes into normal DCs and
induce their differentiation into other types of monocytes of the
same lineage, thus acting as immunosuppressive cells in the TME.
Such cells are called TADCs. The expression of the surface antigen
presentation related molecules MHC-I, MHC-II and activation
molecules CD80 and CD86, are decreased on TADCs. This decreases its
antigen processing and presentation ability. Moreover, TADCs
exhibit upregulated expression of signal transducers and activators
of transcription protein3 (STAT3), IL-12 transcription, suppression
of DC maturation and the activation of T cells (63,64). Bregs, a special subtype of B
lymphocytes, can secrete IL-10, TGF-β and IL-35, and promote Treg
generation, which also exerts immunosuppressive effects (65).
Factor-associated suicide (Fas) is a crucial death
receptor known to trigger apoptosis. Under physiological
conditions, T cells can induce apoptosis in Fas-positive target
cells via Fas/Fas ligand (FasL)-mediated pathways. However, tumor
cells possess mechanisms to evade T cell attacks; They can
downregulate Fas expression or harbor Fas mutations, rendering
themselves resistant to T cell-mediated apoptosis. Moreover, tumor
cells often exhibit upregulated expression of FasL, which binds to
Fas receptors on T cells, leading to T cell apoptosis (66). In addition to aberrant expression
of Fas/FasL molecules and the development of resistance to
apoptosis, tumor cells also exhibit upregulated expression of
certain other immunosuppressive molecules and induce apoptosis of
immune effector cells. For example, indoleamine2, 3-dioxygenase
(IDO), which is upregulated in tumor cells, promotes tryptophan
degradation. On the one hand, tryptophan deficiency can result in
the arrest of T cells in the G1 phase of the cell cycle and
suppress the proliferation of T cells. On the other hand,
tryptophan degradation generates metabolites with cytotoxic and
pro-apoptotic effects, which inhibit apoptosis-inducing effects on
both T cells and NK cells (67).
In addition, upregulation of B7-H1 molecule (also known as PD-L1),
which is present on the surface of tumor cells, when combined with
the T-cell inhibitory receptor PD-1, can induce T cell tolerance.
Furthermore, it stimulates the secretion of IL-10 and induces
apoptosis of T cells (68,69).
A large quantity of immune cells is present in the
TME, such as TAMs, MDSCs and Tregs (70), which regulate tumor growth and
drug response by secreting cytokines, chemokines and growth
factors. For example, TAMs can suppress the antitumor immune
response by secreting immunosuppressive factors such as IL-10 and
TGF-β, thereby reducing the effectiveness of immunotherapy
(71).
Hypoxia occurs in tumor tissues due to rapid growth
which is not matched by an increase in blood and oxygen supply, and
a hypoxic environment induces activation of various pro-survival
signaling pathways, such as hypoxia inducible factor-1α (HIF-1α)
pathway (72). HIF-1α promotes
tumor cell survival and adaptation by regulating the expression of
multiple genes, such as VEGF and glucose transporter (GLUT) 1,
thereby improving tumor cell tolerance to chemotherapeutic drugs
(73). Hypoxia also enhances the
stability of HIF-1α which in turn modulates the hypoxia-induced
gene expression and metabolism (74), including genes related to
angiogenesis, erythropoiesis, glucose uptake and anaerobic
metabolism (75) and multiple
drug resistance genes including multi-drug resistance gene (MDR1)
and the alkalized ECM protein (76). Studies have demonstrated that its
upregulation increases the aggressiveness of tumor cells and
emergence of drug-resistant phenotypes in high-grade gliomas
(77). For example, in treating
glioblastoma (GBM), hypoxia is a common feature of rapidly growing
tumors such as GBM because they quickly outpace the vascular system
and the nutrient supply (78).
Mitochondria are initiators of oxidative phosphorylation channels
which support hypoxic utilization. This suggests that inhibiting
mitochondrial oxidative phosphorylation channels and blocking the
TME feeding cycle may be an effective therapeutic strategy to
improve tumor drug resistance (79).
Tumor cells adapt to changes in the TME through
metabolic reprogramming, such as by enhancing the use of the
glycolytic pathway through the Warburg effect, which not only
supports the rapid proliferation of tumor cells but also affects
the metabolism and effects of drugs (80). Metabolic changes in tumor cells
can lead to a reduction in the active form of drugs within the
cell, or affect drug uptake and efflux by altering the permeability
of the cell membranes (81).
Shigeta et al (82) found
that gemcitabine-resistant urothelial cancer cells have metabolic
reprogramming, which promotes pyrimidine biosynthesis by increasing
the metabolic stimulation of aerobic glycolysis and pentose
phosphate pathways, thereby reducing the therapeutic effect of
gemcitabine. Li et al (83) revealed that the downregulation of
retinoic acid receptor responder 2 promotes breast cancer brain
metastasis (BCBrM) through lipid metabolic reprogramming, which can
be used as a potential target for BCBrM treatment (83).
Tumor tissues often create an acidic
microenvironment by generating acidic metabolites. This acidity can
reduce the effectiveness of chemotherapy drugs by lowering their
concentrations both inside and outside the cells (84). High lactate levels in the TME
contribute to immunosuppression by inhibiting the function of
immune cells such as T cells and natural killer (NK) cells
(85). Lactate promotes tumor
progression by stabilizing HIF-1α, enhancing angiogenesis, and
altering the behavior of CAFs (86). For example, in GBM treatment,
byproducts of cell fermentation such as lactic acid, glutamic acid
and carbon dioxide contribute to the acidification of the
microenvironment, promoting drug resistance, increased invasiveness
and higher rates of tumor recurrence (87). Li et al (88) found that lactate accumulates in
the tumor environment of metastatic CRC and acts as a substrate for
histone acidification, which is further induced by enhanced
cellular glycolysis under hypoxia (88). Wang et al (89) found that Proprotein convertase
subtilisin/kexin type 9 (PCSK9) mediated by macrophage migration
inhibitory factor and lactate levels play an important role in
macrophage phenotypes polarization, targeting PCSK9 expression or
activity can be used to effectively control colon cancer (89).
Cytokines and growth factors in the TME can induce
tumor cells to upregulate MDR proteins such as P-glycoprotein
(P-gp) and MDR-associated protein, which are able to actively pump
drugs out of tumor cells, thereby reducing the intracellular
accumulation of drugs and leading to resistance (90).
The ECM acts as structural support in the TME while
also forming a physical barrier to drug penetration. The components
of the ECM include Col, elastin and glycosaminoglycans, which limit
the spread of drugs within tumor tissues (91). In addition, the high-density fiber
web in the ECM may trap and sequester a drug, reducing its
effective concentration in tumor cells (92).
The TME not only affects tumor cells through
biochemical signals, but also affects drug delivery and efficacy
through physical and mechanical barriers. Tumor cells often exhibit
different mechanical properties compared with normal cells, such as
higher rigidity and the ability to deform (93). These mechanical properties enable
tumor cells to survive and move through the high-density ECM, thus
promoting tumor invasion and metastasis (94). Abnormal accumulation and
recombination of the ECM in tumor tissues can lead to the increased
density and rigidity of the matrix, thus forming a physical barrier
that limits the spread and penetration of drugs in tumor tissues
(95). This dense and rigid ECM
prevents the passage of drug molecules, making it difficult for
drugs to reach deep inside the tumor, thus reducing the
effectiveness of treatment (96).
Tumor blood vessels often exhibit abnormal structures, such as
irregularity, distortion and increased permeability, which can lead
to inefficient drug delivery. On the one hand, increased vascular
permeability leads to the rapid delivery of drugs into the tumor
stroma (97). On the other hand,
the abnormal structure of blood vessels can lead to uneven drug
distribution, such that certain tumor areas do not receive
sufficient quantities of a drug, thus affecting the overall
therapeutic effect of said drug (98). Rapid tumor growth can lead to
increased interstitial pressure, which is primarily caused by tumor
cell proliferation, ECM accumulation and abnormal function of new
blood vessels. High interstitial pressure compresses the blood
vessels inside the tumor, further reducing the ability of the drug
to enter the tumor tissue through the blood vessels (99).
Cytoskeletal proteins have long been recognized as
structural proteins that provide the necessary mechanical structure
for cell development and tissue balance. As the cancer genome
project completed, scientists are surprised to find that a large
number of mutations was annotated as cytoskeleton or related
proteins (100). Cells respond
to environmental conditions, both chemical stimuli and physical
stimuli such as mechanical stress. For example, cells can sense the
hardness of the surrounding material through transmembrane proteins
such as integrin, and further trigger the structural changes of
cytoskeletal proteins such as actin and myosin, and the shape of
the cell will change accordingly (101). It was found that the actin
cytoskeleton can form a chelate with tripartite motif
(TRIM)-containing protein (TRIM21), in cells under the condition of
high tension; consequently, TRIM21 loses the 'freedom'. Thus, the
phosphofructokinase activity was maintained and glycolysis was
promoted (102). The mechanisms
underlying tumor suppression by immune cells are shown in Fig. 2.
Administration of chemotherapeutic drugs such as
paclitaxel promotes the secretion of CSF1 and IL-34 from breast
cancer cells, thereby attracting macrophages to infiltrate the TME.
Subsequently, this inhibits the activation and proliferation of
CTLs, thereby diminishing their antitumor immune capacity. It also
inhibits the antitumor effect of cytotoxic drugs (103). Conversely, neutralizing
antibodies targeting CSF1 can inhibit TAM infiltration in breast
cancer, thereby enhancing the antitumor immune response of CTLs to
a certain extent and improving the efficacy of chemotherapy. In
lung cancer, the cytotoxic drug doxorubicin (DOX) induces the
production of IL-34 from tumor cells through the activation of the
NF-κB pathway. IL-34, regulated by EBPβ, augments the
immunosuppressive and tumor-promoting functions of TAMs. This
prevents CTL responses and promotes regulatory T lymphocyte
responses, thereby facilitating tumor immune evasion, maintaining
TME homeostasis during chemotherapy, and reducing chemotherapeutic
efficacy (104).
Tumor treatments act by improving the CTL response
and achieving immune checkpoint blockade; however, certain patients
do not respond to programmed death-1/ligand-1 (PD-1/-L1) antibody
treatment. TAMs express PD-L1 and CTLA-4 ligands. By binding to
PD-1 and CTLA-4 on T cells, they inhibit the immune response of T
lymphocytes directly and decrease the efficacy of immunotherapy
(105).
IL-6, secreted by TAMs can decrease the levels of
the tumor suppressor miR-204-5p by activating STAT3, thereby
enhancing the anti-apoptotic ability of CRC cells and the
development of resistance to 5-fluorouracil (5-FU) and oxaliplatin
(OXA) (106). It has been
demonstrated that M2-type TAMs in breast cancer tissues and tumor
cells promote the development of tumor resistance to DOX via the
paracrine circuit of IL-6 (107). IL-10 is primarily activated and
released by M2-type TAMs, which stimulates STAT3 to upregulate the
expression of the protooncogene Bcl-2, thereby reducing the
apoptotic effect of paclitaxel on breast cancer (108). CCL22, secreted by M2-type TAMs
in colorectal tumors can promote epithelial-mesenchymal transition
(EMT) of tumor cells and inhibit caspase-mediated cell apoptosis.
It can also promote resistance of tumor cells to 5-FU (109).
The hypoxic conditions within GC lesions activate
the HIF-1α, thereby increasing the expression of HMGB1. This
elevation in HMGB1 levels can facilitate the migration of
macrophages to a tumor and stimulate the production of growth
differentiation factor 15. Consequently, this process accelerates
the oxidation of fatty acid β in GC cells, ultimately promoting
tumor resistance (110). It has
been revealed that M2-type TAMs induce gemcitabine resistance in
pancreatic ductal adenocarcinoma (PDAC) via the insulin-like growth
factor (IGF) (111). Cathepsins,
a type of cysteine proteinase, participate in the development of
cancer drug resistance. Upon activation by IL-4, cathepsins B and S
within TAMs are implicated in drug resistance in lung, colon and
breast cancer. This mechanism may involve the degradation of drug
target proteins by cathepsins.
Macrophages in breast cancer tissues inhibit tumor
cell apoptosis and induce autophagy by activating PI3K/AKT/survivin
signaling pathway, leading to a reduction in sensitivity of breast
cancer cells to DOX (112). TAMs
can enhance the anti-apoptotic ability of breast adenocarcinoma
cells via the CCL2/PI3K/AKT/mammalian target of rapamycin (mTOR)
signaling pathway. This further leads to the development of
resistance to tamoxifen (113).
In GC, conventional chemotherapeutic drugs such as 5-FU induces
GLUT3-dependent glycolysis in M2-type TAMs, which activates the
CCL8/Janus kinase1 (JAK1)/STAT3 signaling pathway. It also enhances
the tolerance of tumor cells to 5-FU (114). In addition, studies it has been
indicated that TAMs in GC can promote drug resistance in tumor
cells by activating the HIF1α/leukemia inhibitory factor/STAT3
signaling pathway (115).
It has been indicated that TAMs in malignant tumors
contribute to ECM degradation and remodeling by producing MMPs. In
addition, TAMs regulate the synthesis and release of pro-angiogenic
factors, thereby facilitating tumor blood vessel formation.
Stockmann et al (116)
found that VEGF derived from TAMs induces tumor resistance to
cyclophosphamide by promoting the formation of an abnormal tumor
vascular network characterized by low vascular density, low
tortuosity and low adventitia cell coverage. In line with this,
TAMs induce the formation of abnormal blood vessels with low
perfusion by secreting VEGF, limiting the entry of chemotherapeutic
drugs into the tumor to exert antitumor effects, and significantly
reducing the effect of chemotherapy (117). In lung adenocarcinoma, M2-type
TAMs can upregulate VEGF-C expression and its receptor VEGFR3,
promoting tumor growth and downregulation of the tumor suppressor
genes p53 and PTEN, thereby inhibiting tumor cell apoptosis and
reducing their sensitivity to chemotherapeutic drugs (118). In ovarian cancer treatment with
anti-VEGF drugs, the expression of VEGFR1 and VEGFR3 in TAMs may
decrease. Despite this reduction, TAMs retain their capacity to
facilitate tumor cell tolerance to anti-VEGF drugs by activating
alternative angiogenic pathways (119).
CSCs, a subset of cancer cells, are multipotent
cells with self-renewal and tumor initiation properties. These
features contribute to the occurrence, development and drug
resistance of tumors. It has been found that the M2-type TAMs can
induce tumor cells to acquire stem cell characteristics, thereby
promoting the malignant progression of tumors (120). IL-17 in ovarian cancer tissues
is primarily derived from TAMs and CD4+ T lymphocytes,
which bind to the IL-17 receptor expressed on tumor cells with a
stem cell phenotype. It can activate the NF-κB/p38 mitogen
activated protein kinase (MAPK) signaling pathway, and hence the
stem cell characteristics of tumor cells and the promotion of tumor
progression and drug resistance (121). Milk fat globulin epidermal
growth factor8 and IL-6 secreted by M2 TAMs synergistically
activate the transcription factor STAT3 which enhances the stem
cell properties of pancreatic cancer cells. They inhibit a CTL
immune response, thereby promoting tumor drug resistance, while
reducing the infiltration of TAMs and the stem cell characteristics
of tumor cells (122).
Similarly, TAM-derived IL-10 can also significantly enhance the
stem cell characteristics of small cell lung cancer cells by
activating the JAK1/STAT1/NF-κB/Notch1 signaling pathway (123).
TAMs can regulate the expression of drug metabolism
enzymes intra and extracellularly and alter the metabolic processes
mediated by drugs in tumor cells. For example, cytokines secreted
by TAMs, such as IL-10 and TGF-β, can upregulate the CYP450 enzyme
system in tumor cells, thereby altering the metabolic rate of
chemotherapeutic drugs and affecting the activity and half-life of
drugs (124). TAMs inhibit the
activity of effector T cells and NK cells, thereby weakening the
immune system's attack on the tumor. Through these immunomodulatory
effects, TAMs indirectly affect the efficacy of antitumor drugs,
increasing the likelihood of tumor cell survival and proliferation
(125). TAMs promote the
degradation and remodeling of ECM by secreting a variety of
stroma-degrading enzymes, change the physical and chemical
characteristics of the TME, and affect the distribution and
metabolism of drugs in tumor tissues (126).
TAMs can promote the repair of DNA damage in tumor
cells induced by chemotherapy, promote the proliferation of
endothelial cells by secreting certain cytokines such as VEGF and
1L-1, promote angiogenesis, and provide nutrients and oxygen for
tissue repair (127). TAMs
degrade and reshape the ECM by secreting matrix remodeling enzymes
such as MMPs to provide spatial and structural support for the
growth and migration of new tissues (128). This promotes cell survival and
tissue repair by activating multiple signaling pathways. For
example, TAMs can promote the proliferation and survival of tumor
cells by secreting epidermal growth factor (EGF) and activating the
EGF receptor signaling pathway (129). After chemotherapy, TAMs promote
tissue repair by secreting anti-inflammatory factors and
immunosuppressive factors that regulate the local immune
environment and reduce inflammatory response and immune attack. For
example, TAMs reduce inflammatory damage by secreting IL-10 and
TGF-β, inhibiting the activity of effector T cells and NK cells
(130). Several studies have
shown that TAMs can support the survival and proliferation of CSCs
by secreting specific cytokines, such as IL-6 and CSF-1. These
cytokines can activate key signaling pathways in CSCs, such as
JAK/STAT3 and PI3K/AKT, and enhance drug resistance and repair
ability of CSCs (131).
Interestingly, Lurbinectedin (PM01183) enhanced the
antitumor of gemcitabine in PDAC by exhibiting a specific depletion
of TAMs that downregulated cytidine deaminase expression and
increased gemcitabine-mediated DNA damage (132). Radioresistance of tumor cells
could potentially be one of the causes for local recurrence post
treatment. HMGB1 was shown to play a role in bladder cancer
radioresistance through its intracellular functions in promoting
DNA damage repair and autophagy. Moreover, combining radiation and
HMGB1 inhibition significantly impaired tumor infiltrating MDSCs
and TAMs-but not Tregs- and shifted the overall tumor immune
balance towards antitumor response (133). Hong et al (134) experimentally verified that TAMs
enhanced WTAP-mediated N6-methyladenosine RNA methylation through a
CXCL16/CXCR6 axis and promoted cisplatin resistance in ovarian
cancer cells.
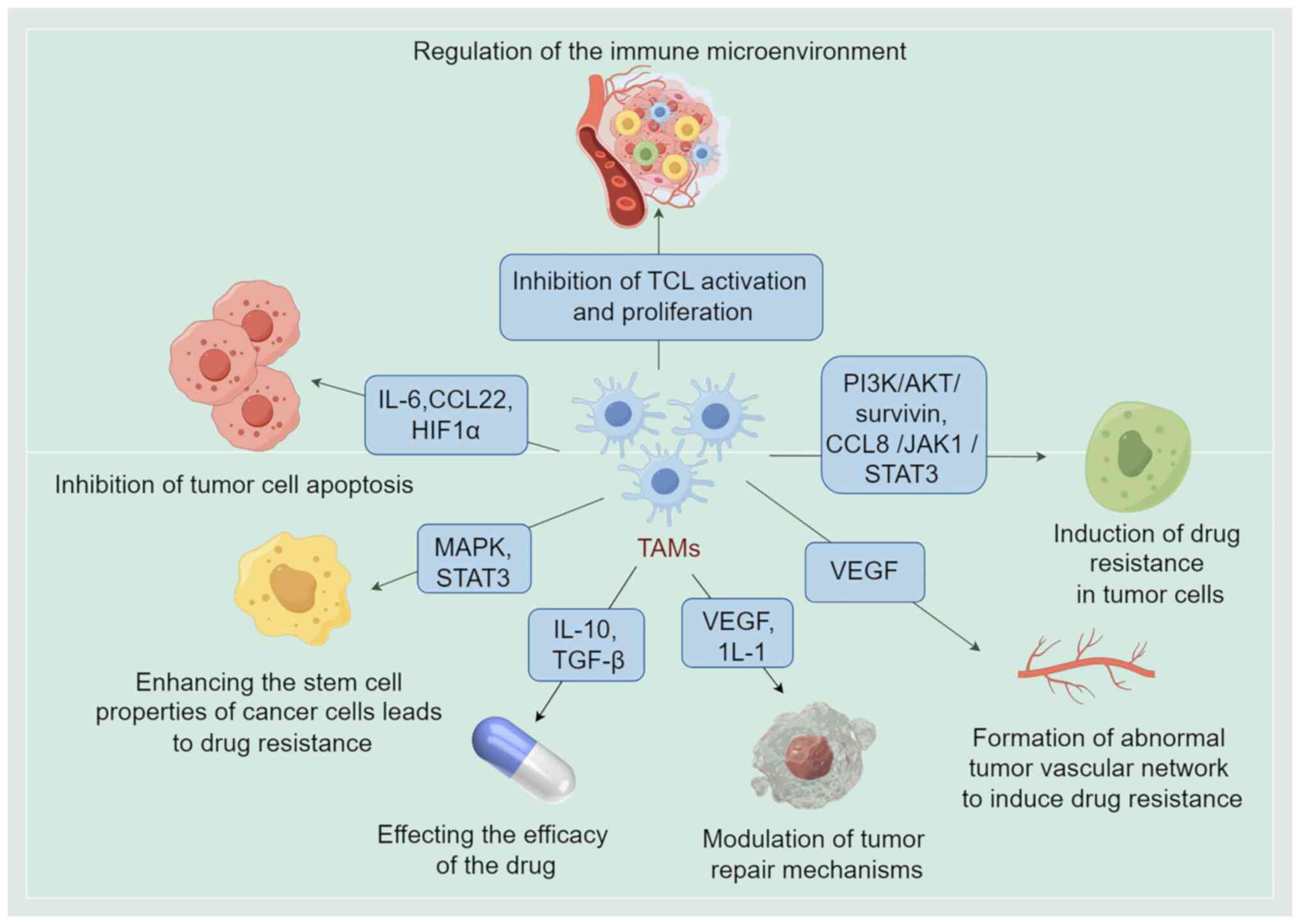
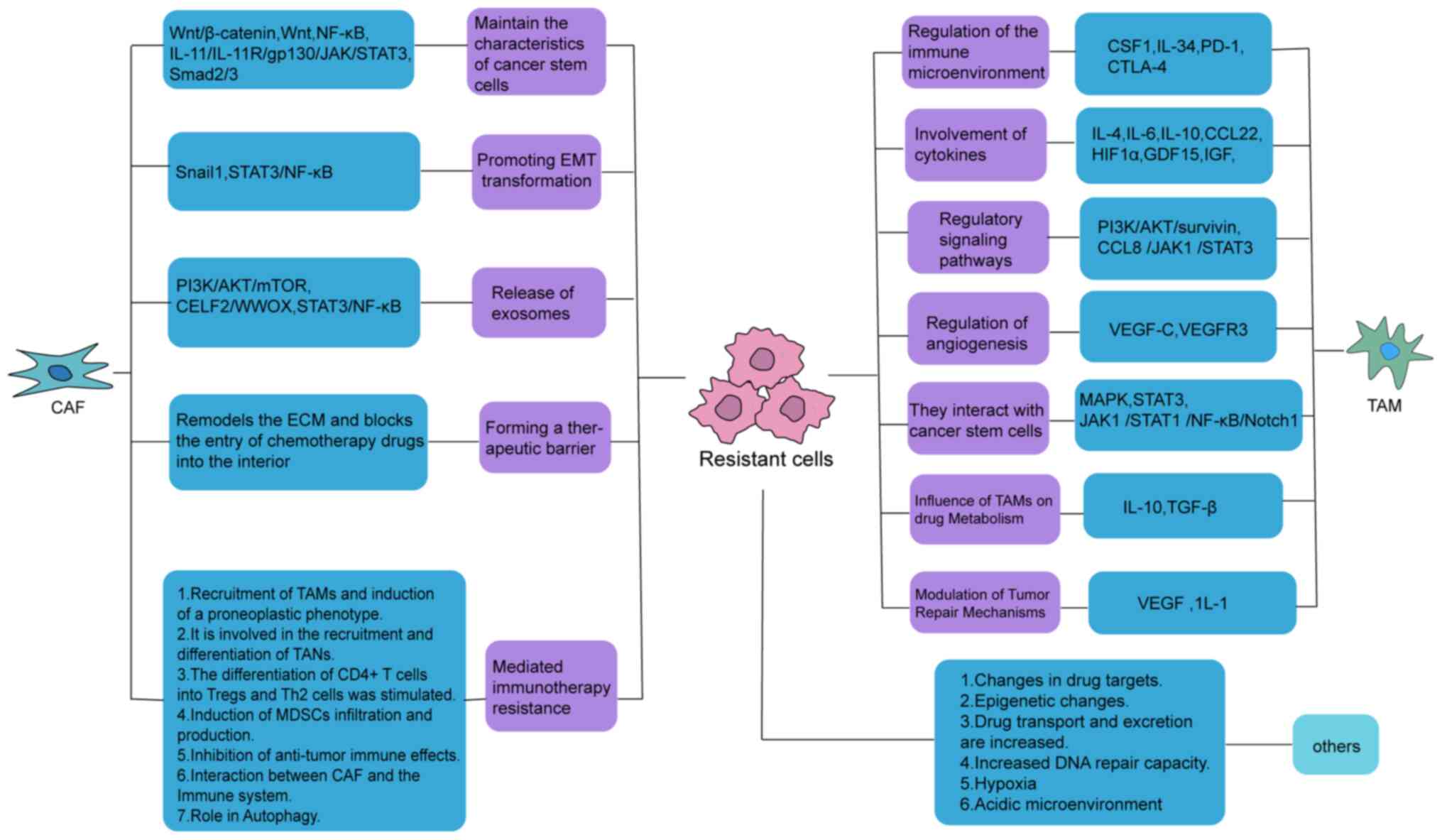
Therefore, TAMs promote tumor drug resistance by
modulating the immune microenvironment, cytokines, signaling
pathways, angiogenesis and CSCs (Fig.
3).
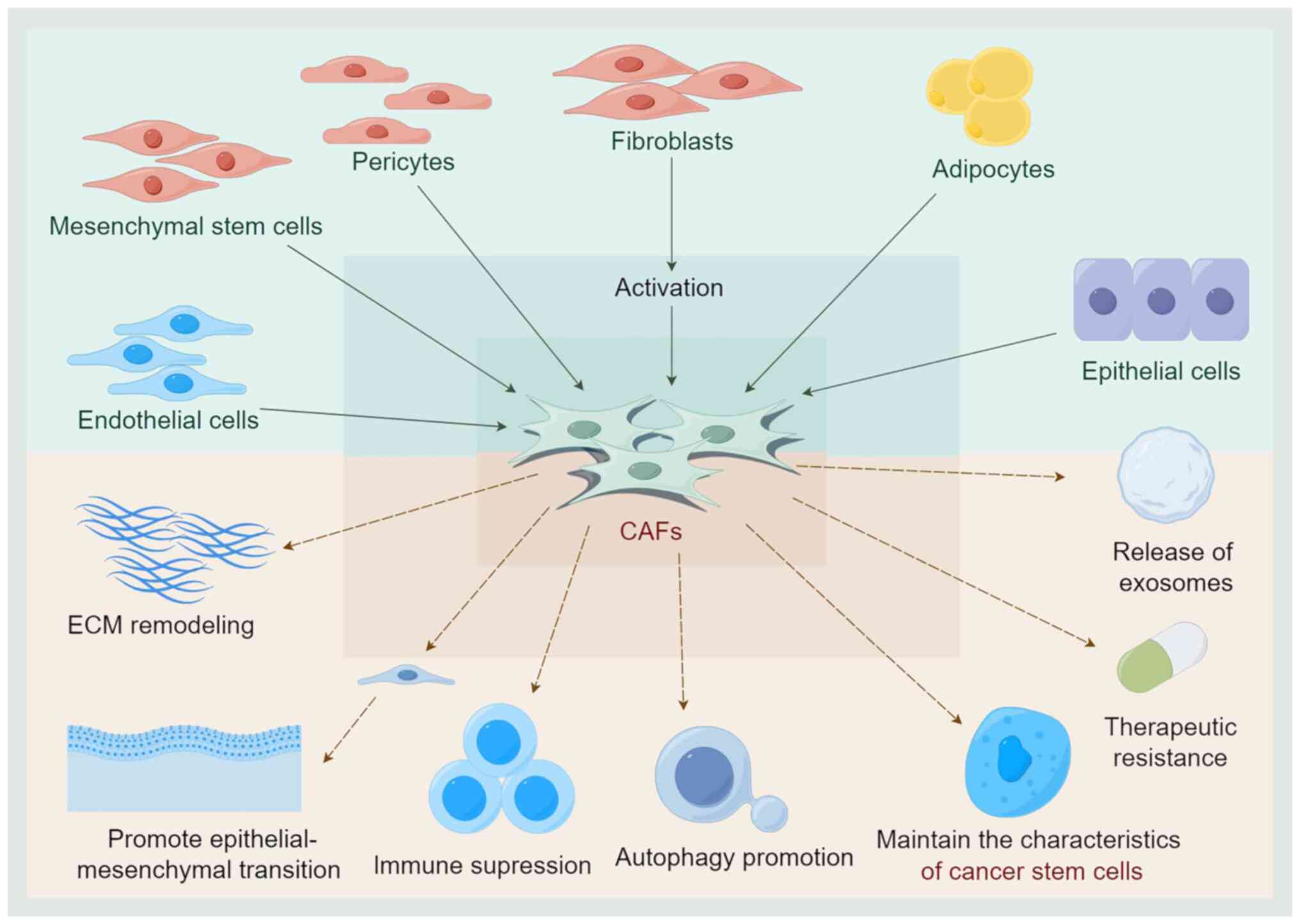
CAFs are generated by various cellular sources.
Currently, researchers have identified that CAFs are primarily
activated by fibroblasts in normal tissues. Various cytokines
secreted by tumor cells participate in this process, including
TGF-β, EGF, platelet-derived growth factor (PDGF) and fibroblast
growth factor 2 (135,136). In addition to fibroblasts, CAFs
can also be produced by the transformation of MSCs, endothelial
cells and epithelial cells. Furthermore, the transformation of
adipocytes or pericytes into CAFs through trans-differentiation is
also a mechanism of CAF generation (137).
In CRC, CAFs enhance the secretion of lncRNA H19 by
activating the Wnt/β-catenin pathway. This enhances the stemness of
CSCs, promoting resistance to OXA in CRC cells (138). Moreover, CAFs induce the
expression of Wnt signaling-related genes in CRC cells, such as T
lymphoma invasion and metastasis inducing protein-1, mediating
resistance to 5-FU (139). In
addition, activation of the NF-κB pathway can regulate the
self-renewal of CSCs, also contributing to drug resistance
(140). In GC, CAFs activate the
NF-κB pathway by secreting the neuroregulatory factor 1, and this
process is closely related to GC drug resistance and prognosis
(141). In addition, CAFs may
activate the IL-11/IL-11R/gp130/JAK/STAT3 anti-apoptotic signaling
pathway in GC cells by secreting IL-11, to promote the maintenance
of chemotherapeutic resistance of CSCs (142). There is evidence that CAFs can
secrete TGF-β1 through the Smad2/3 pathway and upregulate the
expression of STAT4 in PDAC tumor cells, thereby maintaining the
characteristics of CSCs and promoting the resistance of tumor cells
to gemcitabine (143).
EMT, regulated by transcription factors such as ZEB
and Snail, is a key process that modulates cancer progression. In
CRC, the expression of Snail1 in the tumor stroma enhances
chemoresistance in tumor cells. As a result, CAFs facilitate the
emergence of resistance in CRC to drugs such as 5-FU and paclitaxel
by increasing Snail1 expression (144). In addition, CAFs induce EMT of
tumor cells by upregulating the expression of cyclooxygenase-2 and
prostaglandin E2 synthesis in CRC cells, thereby reducing the
sensitivity of CRC cells to OXA (145). In esophageal squamous cell
carcinoma (ESCC), IL6, secreted by CAFs, upregulates the expression
of CXCR7 in tumor cells and promotes the process of EMT via the
STAT3/NF-κB pathway. This suggests that CAFs may affect cisplatin
resistance by secreting IL-6 (146).
Exosomes secreted by CAFs promote the occurrence and
development of gastrointestinal tumors and chemotherapeutic
resistance. The mechanism by which CAFs promote chemotherapeutic
resistance is as follows: i) They can deliver molecules that may
induce drug resistance: CAF-derived exosomes carry microRNAs
(miRNAs or miRs), which can alter the gene expression profile of
tumor cells and promote drug resistance by downregulating tumor
suppressor genes or upregulating drug-resistance-related genes
(147). Proteins contained in
exosomes (such as MMPs and TGF-β) can activate signaling pathways
within tumor cells, enhancing their viability and drug resistance
(148). ii) Regulation of the
TME: CAF-derived exosomes create a microenvironment conducive to
tumor growth and drug resistance by regulating immune cells, such
as inhibiting T cell activity and promoting the accumulation of
immunosuppressive cells (149).
Factors in exosomes can promote the formation of new blood vessels,
provide more nutrients and oxygen to tumor cells, and also affect
the distribution and effectiveness of chemotherapeutic drugs by
altering the permeability of blood vessels (150). iii) Enhance autophagy of tumor
cells: MiRNAs and proteins in exosomes can activate
autophagy-related signaling pathways (such as MTOR and AMPK),
enhance the survival capacity of tumor cells under chemotherapeutic
pressure, and reduce the cytotoxicity of chemotherapy drugs
(151).
It has been revealed that cricN4BP2L2 in exosomes
secreted by CAFs activates the PI3K/AKT/mTOR pathway by binding to
EIF4A3, which then promotes the stemness and OXA resistance of CRC
cells (155). In addition,
miR-625-3p in exosomes secreted by CAFs may enhance the invasive
capacity of CRC cells and promote chemotherapeutic resistance of
CRC cells by inhibiting the CELF2/WWOX pathway (156). In PDAC, once taken up by
adjacent tumor cells, miR-106b in exosomes secreted by CAFs
directly targets tumor protein 53 and induces nucleoprotein 1 to
promote tumor cell survival and GEM resistance (157). In addition, in ESCC, IL-6
secreted by CAFs and miR-21 packaged in exosomes upregulates STAT3
expression in tumor cells, promotes the generation of MDSCs of
monocytes, and finally enhances the resistance of ESCC cells to
cisplatin (158). In HCC,
CAF-derived exosomes deliver circ-ZFR to tumor cells and promote
cisplatin resistance of tumor cells by inhibiting the STAT3/NF-κB
pathway (159).
Emerging evidence indicates that CAFs participate in
ECM remodeling through a several pathways, which increases the
density and stiffness of tumor tissues compared with normal
tissues. The remodeled ECM prevents the entry of chemotherapeutic
agents into tumor cells, limiting their therapeutic efficacy. In
CRC, CAFs are particularly active in synthesizing ECM, which
substantially impedes the penetration of the majority of antitumor
drugs into solid tumors (160).
In addition, increased adhesion between tumor cells and the ECM can
arrest the cell cycle induced by the quiescent state, which
promotes the development of drug resistance. Taken together, these
findings suggest that the influence of ECM and CAFs on the response
to chemotherapy is multifaceted, not only forming a protective
barrier to block drug diffusion, but also enhancing anti-apoptotic
effects through the integrin and focal adhesion kinase (FAK)
signaling pathways, as well as forming a hypoxic environment and
increased metabolic stress promoting tumor growth, acquisition of a
CSC phenotype and treatment resistance (161).
CAFs induce the recruitment and differentiation of
monocytes into tumor-promoting macrophage subsets through the
secretion of abundant soluble mediators. This process inhibits T
cell proliferation and enhances immunosuppression. For example, in
triple-negative breast cancer, CAFs regulate the CXCL12-CXCR4 axis
to redirect monocytes into immunosuppressive lipid-associated
macrophages (LAMs), thereby inhibiting T cell activation and
proliferation. Depletion of genes in this subset of LAMs prevents
tumor growth (162). This
finding suggests a potential targeting strategy for reducing immune
suppression in breast cancer. For example, IGFBP7 secreted by CAFs
promotes the polarization of macrophages towards an M2/TAM
phenotype by regulating the FGF2/FGFR1/PI3K/AKT axis. IGFBP7 is
upregulated in various tumors, and its complex biological role and
molecular mechanism in tumorigenesis remains the focus of current
attention. Similarly, M2/TAMs can influence the activation of CAFs
(163). Activated CAFs further
enhances TAM activity, which reinforces the cycle of
immunosuppression.
It has been demonstrated that CAF-derived CLCF1
promotes the secretion of CXCL6 and TGF-β in tumor cells, enhancing
TAN infiltration and polarization in a paracrine manner in HCC.
Conversely, CXCL6 and TGF-β secreted by tumor cells activate the
ERK1/2 signaling pathway in CAFs to promote the production of
CLCF1, thereby forming a positive feedback loop to accelerate the
induction of liver cancer cell stemness and TANs 'N2' polarization.
This suggests that CLCF1 holds promise as a prognostic biomarker
for HCC. Moreover, it has been proposed that targeted inhibition of
CLCF1 or ERK1/2 signaling may represent a viable therapeutic
strategy for patients with HCC (164). In another study on HCC,
peripheral blood neutrophils were recruited by CAFs to HCC via the
SDF1a/CXCR4 pathway, and IL6 were released by CAF-induced TANs to
differentiate into PDL1+TANs via the JAK-STAT3 signaling
pathway, thereby inhibiting the activity of T cells (165). Therefore, CAF-mediated
activation of TANs provides a novel mechanism for tumor
immunotherapy.
Previous evidence has demonstrated that IL-1/NF-κB
and TGF-β signaling pathways modulate the differentiation of normal
mesothelial cells into antigen-presenting CAFs (apCAFs), which in
turn promotes the differentiation of CD4+T cells into
FoxP3 Tregs (166). It was also
identified that mesothelial cells are the origin of apCAFs.
Therefore, further research is required to expand our understanding
of the transformation process of mesothelial cells into apCAFs and
the function of apCAFs may provide a novel direction for designing
novel immunotherapies. CAFs can promote the transformation of Th
cells into Th2 cells, exert immunosuppressive effects, and mediate
tumor immune escape. In pancreatic cancer, tumor cells promote the
secretion of IL-1β by releasing inflammasome adaptor proteins,
leading to the activation of CAFs. The activated CAFs secrete
thymic stromal lymphopoietin, produce chemokines that attract Th2,
promote Th2 polarization, and promote tumor growth (167). Chen et al (168) constructed salvianolic acid B
pegylated liposomes and found that they interfered with the
activation of CAFs by inhibiting TGF-β1/Smad signaling. Moreover,
it was observed that the levels of Th2 cytokines and the chemokine
CXCL13 were decreased in tumor tissues, and this was correlated
with the inactivation of CAFs. Thus, it has been suggested that
inactivating CAFs and inhibiting the differentiation of Th2 cells
may be an effective approach to immunotherapy.
CAFs facilitate the migration and induction of
MDSCs through the secretion of a variety of cytokines and
chemokines, thereby exerting inhibitory effects on both adaptive
and innate immunity. It has been suggested that CAFs derived from
lung squamous cell carcinoma promote the migration of
CD14+CCR2+ monocytes to the local TME by
releasing CCL2. Subsequently, these monocytes are reprogrammed into
MDSCs. MDSCs, in turn, enhance the production of ROS by
upregulating the expression of NOX2 and IDO1. Moreover, it was
found that the proliferation of CD8+T cells was
decreased, causing immune tolerance (169). This also suggests that efficient
removal of excess ROS may improve T cell responses. A recent study
in ESCC demonstrated that IL-6 and exosomal miR-21 secreted by CAFs
induced the production of M-MDSCs by activating the STAT3 signaling
pathway, while abrogating the differentiation towards DCs and
macrophages. It cannot effectively activate antitumor immunity and
promote drug resistance of tumor cells (139). These data suggest that targeting
the IL-6/miR-21-STAT3 axis may be a potential approach for
reversing drug resistance.
CAFs produce excess metabolites through aerobic
glycolysis thereby enhancing the anabolic demand of adjacent cancer
cells, and this is termed the 'reverse Warburg effect' (170). In a study on prostate cancer,
lactate released by CAF glycolysis reduced Th1 through sirtuin 1
(SIRT1)-mediated deacetylation of T-bet, and activated NF-κB to
upregulate the expression of FoxP3, thereby stimulating the
polarization of CD4+T cells to Tregs (171). CAFs also induce immune tolerance
by increasing IDO and arginase 2 (ARG2) activity. IDO1-induced
tryptophan degradation can inhibit the proliferation of T cells and
promote the differentiation of Tregs. Tryptophan metabolites,
including kynurenine and 3-hydroxykynurenine, can induce apoptosis
of activated T cells and NK cells. ARG2 inhibits cancer immune
responses by depleting arginine, thereby inducing T cell anergy and
promoting TAM reprogramming to an immunosuppressive phenotype
(172). Therefore, CAFs regulate
immune cells via metabolites generated by metabolic reprogramming
to achieve antitumor immunosuppression. These findings suggest that
excessive production of metabolites by CAFs is involved in the
process of immunosuppression.
CAFs inhibit the T-cell priming process. DCs are
the most important antigen-presenting cells involved in the
activation of T lymphocytes. CAFs inhibit the differentiation and
maturation of DCs by secreting TGF-β and downregulating the
expression of MHC-II molecules and the CMs, CD40, CD80 and CD86
expressed on the surface of DCs (173). CAFs suppress the infiltration of
CTLs into the tumor. In a previous study employing paired syngeneic
mouse models of breast cancer featuring varying levels of CAFs, it
was observed that an increased abundance of CAFs was associated
with lower levels of CTLs. Models rich in CAFs displayed a
phenotype characterized by CTL exclusion. Targeting a specific
subset of CAFs by genetically knocking out the
myofibroblastic-restricted receptor Endo180 (Mrc2) in mice
increased the infiltration of CTLs and responsiveness to immune
checkpoint inhibitors (174).
CAFs can also directly or indirectly inhibit the
antitumor immunity of NK cells via several mechanisms. One such
mechanism is that CAFs reduce NK cell activity and function by
downregulating the expression of their activating
receptors/receptor-associated ligands. The poliovirus receptor
(PVR/CD155), is a ligand that activates the NK receptor DNAM-1, and
CAFs can suppress PVR expression on the cell surface, thereby
inhibiting the eliminating activity of NK cells (175). Second, CAFs inhibit NK
cell-mediated antitumor elimination by secreting inhibitory factors
that bind to inhibitory receptors on the surface of NK cells. In
PDAC, NetG1+CAFs were reported to induce
immunosuppression, and its deletion reprograms them to an antitumor
phenotype that allows NK cells to eliminate cancer cells. The
mechanism involved depends, in part, on its ability to limit the
secretion of immunosuppressive cytokines and increase the
expression of IL-15 (176).
Notably, it has been found that TGF-β is a key factor that links
CAFs to NK cells. TGF-β secreted by CAFs can significantly inhibit
the activation and cytotoxic activity of NK cells by suppressing
the activation of receptors of NK cells, reducing the production of
interferon-γ, and blocking the metabolic signal transduction
downstream of stimulatory cytokines (177). Although some progress has been
made in understanding the mechanism by which CAFs inhibit NK cells,
the detailed mechanisms by which CAFs function are complex and
require further study.
In CRC, CAFs upregulate the expression levels of
immune checkpoint molecules TIM3, LAG3 and CD39 and downregulate
that of CD137 through cathepsin S-dependent tumor antigen
cross-presentation, which decreases T cell toxicity (178). Conversely, CAFs can also alter T
cell immunity by upregulating the expression of immune checkpoint
ligands. α-SMA+CAFs upregulate PD-L1 expression in lung
adenocarcinoma cells by secreting CXCL2, and the interaction
between PD-L1 and PD-1 contributes to the immune escape of tumor
cells (179).
The interaction between CAFs and the immune system
plays a crucial role in the TME. CAFs affect the function of immune
cells through multiple mechanisms, thereby promoting tumor growth
and immune escape. In the present review, a few key aspects of the
interaction between CAFs and the immune system were described: i)
Establishment of an immunosuppressive environment: CAFs inhibit
antitumor immune responses by secreting a variety of
immunosuppressive factors, such as TGF-β, IL-6 and IL-10. These
factors can inhibit effector T cells and NK cells, reducing their
capacity to eliminate cells (180). ii) Promoting immune escape: CAFs
downregulate the expression of MHC-I and reduce the possibility of
tumor cells being recognized by immune cells and thus reduce the
likelihood of being cleared. Secretion of CXCL12 can direct immune
cells away from the tumor area, thereby reducing immune
cell-mediated attacks on the tumor cells (181). iii) Influencing the infiltration
of antitumor immune cells: CAFs promote angiogenesis by secreting
angiogenic factors (such as VEGF), but the new blood vessels are
typically structurally abnormal, making it difficult for immune
cells to pass through (182).
CAFs change the structure and composition of the ECM by secreting
MMPs and other matrix remodeling enzymes, thus limiting the
migration of immune cells (183). iv) Directly interacting with
immune cells: CAFs interact with DCs to inhibit their maturation
and antigen-presenting function, reducing the effectiveness of the
antitumor immune response. Interactions with macrophages promote
their maturation towards an M2 phenotype, which has a pro-tumor and
immunosuppressive function (184).
Promotion of autophagy in tumor cells: Autophagy
allows tumor cells to survive stresses induced by chemotherapy, and
CAFs can influence this process through direct cell-cell
interactions or paracrine signaling. CAFs promote autophagy in
tumor cells by secreting autophagy-related factors, such as TGF-β,
IL-6 and CXCL12. These factors enhance the autophagic activity of
tumor cells by activating downstream signaling pathways such as the
PI3K/AKT, AMPK and mTOR pathways (185). Metabolic interactions between
CAFs and tumor cells, such as the exchange of lactic acid, amino
acids and lipids, can regulate the autophagic activity of tumor
cells (186).
As a support cell for autophagy: CAFs participate
in ECM remodeling by secreting matrix degrading enzymes such as
MMPs. This process may indirectly affect the autophagic activity of
tumor cells by regulating the composition and structure of the ECM
(187). CAFs can enhance the
resistance of tumor cells to chemotherapeutic drugs by promoting
autophagy in tumor cells. The specific mechanism may involve
autophagy-mediated cell survival signaling and anti-apoptotic
effects (188). It has been
shown that CAFs in breast cancer enhance the autophagy activity of
breast cancer cells by secreting IL-6 and TGF-β, thus promoting the
survival of tumor cells and chemotherapeutic resistance (189). In pancreatic cancer, CAFs
facilitate tumor cell survival in nutrient deficient and hypoxic
environments by promoting autophagy (190). As the most abundant stromal
cells in the TME, CAFs influence the emergence of tumor resistance
(Fig. 4).
Taken together, cellular and molecular factors in
the TME can influence the occurrence of drug resistance of tumor
cells, including through immune escape mechanisms, creating a
hypoxic environment, acidic microenvironment and regulating
extracellular mechanism among other means, which may affect the
sensitivity of tumor cells by regulating the environment and
metabolic state of tumor cells (Fig.
5).
The development of drug resistance related to the
TME is a major factor that reduces the effectiveness of several
treatments, which decreases patient survival as well as the quality
of life of patients. Thus, therapeutic potential of targeting TME
especially the TAMs and CAFs, cannot be underestimated for the
treatment of a wide range of cancer. A more thorough investigation
of pharmacological blockade for drug-resistant cancer therapy has
been augmented with the scientific rigor and depth of the
content.
Considering the therapeutic effects of TAMs, they
are not being investigated for potential alleviation/prevention of
tumor drug resistance and malignant progression. The combination of
TAMs with commonly used chemotherapeutic drugs, targeted
therapeutic drugs, or immunotherapeutic drugs can reduce tumor drug
resistance and enhance antitumor efficacy. Current antitumor
strategies targeting TAMs include: i) Inhibition of macrophage
recruitment in the TME; ii) removal of M2 TAMs from the TME; iii)
repolarization of M2 TAMs into M1 TAMs; and iv) delivery of
antitumor drugs.
Chemokine-chemokine receptor signaling pathways,
such as CCL2-CCR2 and CCL5-CCR5, act as the primary mechanisms for
recruiting macrophages into the TME. Interventions aimed at
blocking this signaling to impede macrophage infiltration into the
TME have been investigated in both preclinical models and clinical
trials. CCR2 antagonists such as PF-04136309 and MLN1202 have
demonstrated efficacy in reducing macrophage infiltration in the
TME in preclinical animal studies. Moreover, combining CCR2
antagonists with the chemotherapeutic regimen FOLFIRINOX has been
shown to augment CTL immune responses in pancreatic tumors, thereby
enhancing the efficacy of chemotherapy and inhibiting tumor
progression (191). It has been
demonstrated that inhibition of CCL5-CCR5 signaling can enhance the
therapeutic effect of tumors such as breast cancer, GC, CRC and
pancreatic cancer, and CCR5 antagonists can reduce the tumor burden
of GC and inhibit its peritoneal metastasis, ultimately prolonging
the survival of patients (192-195). In vitro cell experiments
have indicated that CCR5 antagonists can suppress the proliferation
of pancreatic cancer cells and cell apoptosis. Moreover, animal
transplantation tumor models have also revealed that CCR5
antagonists can alleviate liver metastasis of pancreatic cancer
(196). Therefore, CCR2 and CCR5
antagonists combined with conventional chemotherapeutic drugs are
promising treatments for cancer.
Targeted depletion of M2 TAMs can promote antitumor
immune responses and inhibit tumor growth. Lee et al
(197) found that the hybrid
peptide MEL-dKLA could target M2 TAMs and induce mitochondrial
apoptosis, and had a low affinity for M1 TAMs, T lymphocytes, DCs
and other immune cells. Moreover, in vivo experiments have
validated its capacity to suppress tumor angiogenesis and decrease
tumor burden. CSF-1 plays a pivotal role in the polarization of M2
TAMs. Anti-CSF-1 receptor antibodies AFS98 and M279 have
demonstrated efficacy in eliminating M2 TAMs in breast
adenocarcinoma by intercepting CSF-1 signal transduction. This
intervention not only inhibits tumor cell proliferation but also
extends the survival of animal models (198).
Corosolic acid (CA) can inhibit the transcription
factor STAT3. Liposome-packaged CA combined with anti-CD163
antibody can target TAMs, downregulate the expression of IL-10 and
upregulate the expression of TNF-α, as well as promote the
transformation of TAMs into M1 (40). Rodell et al (199) reported that Toll-like receptor
7/8 (TLR7/8) antagonist R848 induced the transformation of
macrophages into M1. R848 carried by nanoparticles can repolarize
M2 TAMs into M1 TAMs in a variety of tumor animal models, and its
combination with anti-PD-1 antibody can significantly improve its
antitumor immune ability. Immune-response gene 1 (IRG1) is
expressed as TAMs in both human and mouse tumors. It has been
demonstrated that deletion of IRG1 in mice can inhibit the growth
of multiple tumor types, and IRG1-deficient macrophages represent
an effective cell-based therapeutic strategy for cancer treatment
(200). N-methyl-D-aspartate
receptor (NMDAR) is an ion channel expressed on macrophages, and it
was found that blocking the TAM phenotype of NMDAR functional and
metabolic changes can improve promotion of antitumor immunity
mediated by T cells and NK cells (201).
Due to the strong phagocytic ability of
macrophages, their efficient migration to tumor lesions following
stimulation by chemokines, TAM-mediated antitumor drug delivery has
become an important research focus. In previous studies,
macrophages were incubated in the peritoneal cavity containing
antitumor drugs in the form of nanoparticles or liposome (LPO), and
then injected back into animal models. This caused a significant
prolongation of the circulating half-life of drugs, enhanced their
antitumor efficacy, and reduced drug toxicity (202). In an animal model of lung cancer
transplantation tumor, LPO-DOX delivered by TAMs exhibited higher
antitumor efficacy with the advantage of low toxicity (203).
A study of two distinct cohorts comprising over 160
PDAC samples showed that FAK activity was increased in CAFs within
PDAC compared with fibroblasts from healthy pancreatic tissue.
Moreover, elevated FAK activity in PDAC-associated fibroblasts
emerged as an independent prognostic indicator for both
disease-free and overall survival in multivariate analysis. This
highlights the FAK activity in fibroblasts as a pivotal regulator
of PDAC advancement and an independent prognostic marker for PDAC
progression (204). In a
transcriptomic analysis of OSCC xenografts, four genetic indices
including FN1, TGFB2, TGFBR2 and TGFBI were detected as CAF indices
and were reported to be strong predictors of survival in binary
analysis of different subtypes of patients. Moreover, the CAF index
is more powerful than the EMT score in predicting survival outcomes
(205). In addition, using
independent CAF-related prognostic genes, a model for predicting
the prognosis of HCC was established. Furthermore, the immune
infiltration characteristics, chemosensitivity and
immunotherapeutic response of TME were analyzed. The prognostic
value the of CAF-related genes was also determined (206). CAFs can resist the
immunosuppressive effects and related mechanisms of PD-1/PD-L1
immunotherapy. Researchers have identified several key molecules,
such as TGF-β, Ln-γ2, Wnt2 and exosomes. Therapeutic strategies
targeting CAF biomarkers are currently in clinical trials with the
aim of improving patient survival (207). LncRNA Disheveled binding
antagonist of beta catenin3 antisense1 (DACT3-AS1), a CAF
derivative, targets miR-181a-5p/SIRT1. OXA resistance-induced
downregulation of DACT3-AS1 in CAFs-derived exosomes enhanced OXA
resistance through miR-181a-5p/SIRT1 signaling pathway in GC
(208).
Tumor response to drug exposure is largely affected
by TME, which can reduce the efficiency of chemotherapy drugs and
limit the efficacy of immunotherapy. Some clinical findings have
shown that targeting TME can enhance the efficacy of
chemotherapeutic drugs and immunotherapy. Emerging evidence
demonstrated that targeting CAFs can improve cancer chemotherapy by
increasing intratumoral drug uptake. Loeffler, a kind of fibroblast
activation protein of the oral DNA vaccine, can reduce the
expression of type 1 Col, increasing the absorption of the tumor
cells to chemotherapy drugs by 70%. Compared with the control
group, the life span of the vaccinated mice was 3-fold longer, and
the tumor growth was significantly inhibited (209). A pooled analysis of 3,771
patients treated with neoadjuvant therapy found that the
pathological complete response (PCR) of breast cancer was 31%, and
lymphocyte infiltration of the PCR was only 2% of breast cancer.
The presence of CD3 or CD20 lymphocyte infiltration and a low
proportion of CD146+CAFs indicate higher chemotherapy
sensitivity (210).
Among 1,201 patients with NSCLC treated with
PD-(L)1 blockade, acquired resistance is common, occurring in
>60% of initial responders. Memon et al (211) found that the relapse of tumor
can be raised by IFN-γ reaction gene or stable expression to
separate. Persistent inflammation of the acquired resistant TME,
rather than elimination or abandonment, may provide a new strategy
for reversing drug resistance (211).
Ferroptosis is an iron-dependent mode of programmed
cell death characterized by the accumulation of lipid ROS to
manipulate cancer cell mutation and drug resistance through the
lipid metabolic pathway that aggregates on phospholipid glutathione
peroxidase. Zhu et al (212) found that ferroptosis achieves
lethal levels by accumulating ROS and LPO products in the TME, and
ferroptosis-driven nanotherapeutics can reverse drug resistance,
showing superior efficacy over traditional methods. Preparation of
paclitaxel-loaded ginsenoside Rg3 liposomes has been shown to
inhibit the 1L-/STAT3/p-STAT3 pathway for tumor cells and the
double effect of TME remodeling, realizing the high tumor
inhibition rate of 90.3% (213).
Numerous studies have demonstrated that the TME
influences the formation of tumor drug resistance. The infiltration
and activation of immune cells affect the efficacy of tumor
treatment, but may also be utilized by tumor cells to escape immune
attack. Tumor cells can increase resistance to treatment via
various mechanisms. Although current immunotherapeutic strategies
have been successful, there are several limitations, such as
individual differences in patients, immune escape and drug
resistance, among others, which need to be further investigated and
accounted for. Future studies should focus on the complexity of the
TME, including the interaction of different immune cell subsets and
the communication between immune cells and tumor cells. Scientists
should explore novel therapeutic strategies, such as the combined
application of immune checkpoint inhibitors and the development of
personalized immunotherapeutic strategies, to address the
challenges of tumor resistance. Future investigations should aim to
enhance the synergy between basic bench research and clinical
applications, facilitating the translation of laboratory
discoveries into practical treatments. Moreover, efforts should be
directed towards refining current therapeutic strategies. In
summary, investigating the TME and drug resistance holds
theoretical and clinical importance. It is anticipated that future
studies will uncover novel breakthroughs for cancer treatment.
The data generated in the present study may be
requested from the corresponding author.
YL, JL, YZ and QG made substantial contributions to
this paper, both in terms of conception and design analysis. All
authors participated in the drafting and revision of the article,
finalized the version to be published and agreed to take
responsibility for all aspects of the work. All authors read and
approved the final version of the manuscript. Data authentication
is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the National Natural Science
Foundation of China (grant no. 82204433).
|
1
|
Maia A, Schöllhorn A, Schuhmacher J and
Gouttefangeas C: CAF-immune cell crosstalk and its impact in
immunotherapy. Semin Immunopathol. 45:203–214. 2023. View Article : Google Scholar :
|
|
2
|
Yan CY, Zhao ML, Wei YN and Zhao XH:
Mechanisms of drug resistance in breast cancer liver metastases:
Dilemmas and opportunities. Mol Ther Oncolytics. 28:212–229. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vesely MD, Zhang T and Chen L: Resistance
mechanisms to Anti-PD cancer immunotherapy. Annu Rev Immunol.
40:45–74. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fu T, Dai LJ, Wu SY, Xiao Y, Ma D, Jiang
YZ and Shao ZM: Spatial architecture of the immune microenvironment
orchestrates tumor immunity and therapeutic response. J Hematol
Oncol. 14:982021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tang Y, Zang H, Wen Q and Fan S: AXL in
cancer: A modulator of drug resistance and therapeutic target. J
Exp Clin Cancer Res. 42:1482023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang Y, Zhang H, Wang J, Liu Y, Luo T and
Hua H: Targeting extracellular matrix stiffness and
mechanotransducers to improve cancer therapy. J Hematol Oncol.
15:342022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
La Rocca A, De Gregorio V, Lagreca E,
Vecchione R, Netti PA and Imparato G: Colorectal cancer
bioengineered microtissues as a model to replicate Tumor-ECM
crosstalk and assess drug delivery systems in vitro. Int J Mol Sci.
24:56782023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y and Brekken RA: Direct and
indirect regulation of the tumor immune microenvironment by VEGF. J
Leukoc Biol. 111:1269–1286. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dong W, Xie Y and Huang H: Prognostic
value of Cancer-associated fibroblast-related gene signatures in
hepatocellular carcinoma. Front Endocrinol (Lausanne).
13:8847772022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu X, Liu Y, Qi Y, Huang Y, Hu F, Dong F,
Shu K and Lei T: Signal pathways involved in the interaction
between tumor-associated macrophages/TAMs and Glioblastoma cells.
Front Oncol. 12:8220852022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Larmonier N, Marron M, Zeng Y, Cantrell J,
Romanoski A, Sepassi M, Thompson S, Chen X, Andreansky S and
Katsanis E: Tumor-derived CD4(+)CD25(+) regulatory T cell
suppression of dendritic cell function involves TGF-beta and IL-10.
Cancer Immunol Immunother. 56:48–59. 2007. View Article : Google Scholar
|
|
12
|
Haque A, Banik NL and Ray SK: Emerging
role of combination of all-trans retinoic acid and interferon-gamma
as chemoimmunotherapy in the management of human glioblastoma.
Neurochem Res. 32:2203–2209. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Downs-Canner SM, Meier J, Vincent BG and
Serody JS: B cell function in the tumor microenvironment. Annu Rev
Immunol. 40:169–193. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rabinovich GA, Gabrilovich D and Sotomayor
EM: Immunosuppressive strategies that are mediated by tumor cells.
Annu Rev Immunol. 25:267–296. 2007. View Article : Google Scholar
|
|
15
|
Zulfiqar B, Mahroo A, Nasir K, Farooq RK,
Jalal N, Rashid MU and Asghar K: Nanomedicine and cancer
immunotherapy: Focus on indoleamine 2, 3-dioxygenase inhibitors.
Onco Targets Ther. 10:463–476. 2017. View Article : Google Scholar :
|
|
16
|
Makkouk A and Weiner GJ: Cancer
immunotherapy and breaking immune tolerance: new approaches to an
old challenge. Cancer Res. 75:5–10. 2015. View Article : Google Scholar
|
|
17
|
Vimalraj S: A concise review of VEGF,
PDGF, FGF, Notch, angiopoietin, and HGF signalling in tumor
angiogenesis with a focus on alternative approaches and future
directions. Int J Biol Macromol. 221:1428–1438. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang J, Zhang L, Wan D, Zhou L, Zheng S,
Lin S and Qiao Y: Extracellular matrix and its therapeutic
potential for cancer treatment. Signal Transduct Target Ther.
6:1532021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bigos KJ, Quiles CG, Lunj S, Smith DJ,
Krause M, Troost EG, West CM, Hoskin P and Choudhury A: Tumour
response to hypoxia: Understanding the hypoxic tumour
microenvironment to improve treatment outcome in solid tumours.
Front Oncol. 14:13313552024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rømer AMA, Thorseth ML and Madsen DH:
Immune modulatory properties of collagen in cancer. Front Immunol.
12:7914532021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Govaere O, Wouters J, Petz M, Vandewynckel
YP, Van den Eynde K, Van den Broeck A, Verhulst S, Dollé L,
Gremeaux L, Ceulemans A, et al: Laminin-332 sustains
chemoresistance and quiescence as part of the human hepatic cancer
stem cell niche. J Hepatol. 64:609–617. 2016. View Article : Google Scholar
|
|
22
|
Fukazawa S, Shinto E, Tsuda H, Ueno H,
Shikina A, Kajiwara Y, Yamamoto J and Hase K: Laminin β3 expression
as a prognostic factor and a predictive marker of chemoresistance
in colorectal cancer. Jpn J Clin Oncol. 45:533–540. 2015.PubMed/NCBI
|
|
23
|
Di Martino JS, Nobre AR, Mondal C, Taha I,
Farias EF, Fertig EJ, Naba A, Aguirre-Ghiso JA and Bravo-Cordero
JJ: A tumor-derived type III collagen-rich ECM niche regulates
tumor cell dormancy. Nat Cancer. 3:90–107. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Puttock EH, Tyler EJ, Manni M, Maniati E,
Butterworth C, Burger Ramos M, Peerani E, Hirani P, Gauthier V, Liu
Y, et al: Extracellular matrix educates an immunoregulatory tumor
macrophage phenotype found in ovarian cancer metastasis. Nat
Commun. 14:25142023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang L, Li C, Wang J, Yang G, Lv Y, Fu B,
Jian L, Ma J, Yu J, Yang Z, et al: Transformable ECM deprivation
system effectively suppresses renal cell carcinoma by reversing
anoikis resistance and increasing chemotherapy sensitivity. Adv
Mater. 34:e22035182022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tie Y, Tang F, Wei YQ and Wei XW:
Immunosuppressive cells in cancer: Mechanisms and potential
therapeutic targets. J Hematol Oncol. 15:612022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Y, Li C, Lu Y, Liu C and Yang W: Tumor
microenvironment-mediated immune tolerance in development and
treatment of gastric cancer. Front Immunol. 13:10168172022.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Labani-Motlagh A, Ashja-Mahdavi M and
Loskog A: The tumor microenvironment: A milieu hindering and
obstructing antitumor immune responses. Front Immunol. 11:9402020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang H, Tian T and Zhang J:
Tumor-associated macrophages (TAMs) in colorectal cancer (CRC):
From mechanism to therapy and prognosis. Int J Mol Sci.
22:84702021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiao M, He J, Yin L, Chen X, Zu X and Shen
Y: Tumor-associated macrophages: Critical players in drug
resistance of breast cancer. Front Immunol. 12:7994282021.
View Article : Google Scholar
|
|
31
|
Zaghdoudi S, Decaup E, Belhabib I, Samain
R, Cassant-Sourdy S, Rochotte J, Brunel A, Schlaepfer D, Cros J,
Neuzillet C, et al: FAK activity in cancer-associated fibroblasts
is a prognostic marker and a druggable key metastatic player in
pancreatic cancer. EMBO Mol Med. 12:e120102020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yin Y, Yao S, Hu Y, Feng Y, Li M, Bian Z,
Zhang J, Qin Y, Qi X, Zhou L, et al: The Immune-microenvironment
Confers Chemoresistance of colorectal cancer through
macrophage-derived IL6. Clin Cancer Res. 23:7375–7387. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li J, He K, Liu P and Xu LX: Iron
participated in breast cancer chemoresistance by reinforcing IL-6
paracrine loop. Biochem Biophys Res Commun. 475:154–160. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang H, Dai Z, Wu W, Wang Z, Zhang N,
Zhang L, Zeng WJ, Liu Z and Cheng Q: Regulatory mechanisms of
immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer
Res. 40:1842021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen S, Wang M, Lu T, Liu Y, Hong W, He X,
Cheng Y, Liu J, Wei Y and Wei X: JMJD6 in tumor-associated
macrophage regulates macrophage polarization and cancer progression
via STAT3/IL-10 axis. Oncogene. 42:2737–2750. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang B, Zhu J, Wang Y, Chen W, Fang S, Mao
W, Xu Z, Yang Y, Weng Q, Zhao Z, et al: Targeted xCT-mediated
Ferroptosis and Protumoral polarization of macrophages is effective
against HCC and enhances the efficacy of the Anti-PD-1/L1 response.
Adv Sci (Weinh). 10:e22039732023. View Article : Google Scholar
|
|
37
|
Li Y, Shen Z, Chai Z, Zhan Y, Zhang Y, Liu
Z, Liu Y, Li Z, Lin M, Zhang Z, et al: Targeting MS4A4A on
tumour-associated macrophages restores CD8+ T-cell-mediated
antitumour immunity. Gut. 72:2307–2320. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tanei T, Leonard F, Liu X, Alexander JF,
Saito Y, Ferrari M, Godin B and Yokoi K: Redirecting transport of
nanoparticle albumin-bound paclitaxel to macrophages enhances
therapeutic efficacy against liver metastases. Cancer Res.
76:429–439. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rodell CB, Arlauckas SP, Cuccarese MF,
Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ and Weissleder R:
TLR7/8-agonist-loaded nanoparticles promote the polarization of
tumour-associated macrophages to enhance cancer immunotherapy. Nat
Biomed Eng. 2:578–588. 2018. View Article : Google Scholar :
|
|
40
|
Andersen MN, Etzerodt A, Graversen JH,
Holthof LC, Moestrup SK, Hokland M and Møller HJ: STAT3 inhibition
specifically in human monocytes and macrophages by CD163-targeted
corosolic acid-containing liposomes. Cancer Immunol Immunother.
68:489–502. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Candido JB, Morton JP, Bailey P, Campbell
AD, Karim SA, Jamieson T, Lapienyte L, Gopinathan A, Clark W,
McGhee EJ, et al: CSF1R+ macrophages sustain pancreatic tumor
growth through T cell suppression and maintenance of key gene
programs that define the squamous subtype. Cell Rep. 23:1448–1460.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Larionova I, Cherdyntseva N, Liu T,
Patysheva M, Rakina M and Kzhyshkowska J: Interaction of
tumor-associated macrophages and cancer chemotherapy.
Oncoimmunology. 8:15960042019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xia C, Yin S, To KKW and Fu L:
CD39/CD73/A2AR pathway and cancer immunotherapy. Mol Cancer.
22:442023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang L, Dou X, Zheng Z, Ye C, Lu TX,
Liang HL, Wang L, Weichselbaum RR and He C: YTHDF2/m6 A/NF-κB axis
controls anti-tumor immunity by regulating intratumoral Tregs. EMBO
J. 42:e1131262023. View Article : Google Scholar
|
|
45
|
Wen Z, Liu T, Zhang Y, Yue Q, Meng H, He
Y, Yang Y, Li M, Zheng J and Lin W: Salidroside regulates tumor
microenvironment of non-small cell lung cancer via
Hsp70/Stub1/Foxp3 pathway in Tregs. BMC Cancer. 23:7172023.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shiri AM, Zhang T, Bedke T, Zazara DE,
Zhao L, Lücke J, Sabihi M, Fazio A, Zhang S, Tauriello DVF, et al:
IL-10 dampens antitumor immunity and promotes liver metastasis via
PD-L1 induction. J Hepatol. 80:634–644. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hume DA and MacDonald KP: Therapeutic
applications of macrophage colony-stimulating factor-1 (CSF-1) and
antagonists of CSF-1 receptor (CSF-1R) signaling. Blood.
119:1810–1820. 2012. View Article : Google Scholar
|
|
48
|
Lee C, Jeong H, Bae Y, Shin K, Kang S, Kim
H, Oh J and Bae H: Targeting of M2-like tumor-associated
macrophages with a melittin-based pro-apoptotic peptide. J
Immunother Cancer. 7:1472019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang H, Zepp M, Georges RB, Jarahian M,
Kazemi M, Eyol E and Berger MR: The CCR5 antagonist maraviroc
causes remission of pancreatic cancer liver metastasis in nude rats
based on cell cycle inhibition and apoptosis induction. Cancer
Lett. 474:82–93. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Aldinucci D and Casagrande N: Inhibition
of the CCL5/CCR5 Axis against the Progression of Gastric Cancer.
Int J Mol Sci. 19:14772018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hu Q, Wang R, Zhang J, Xue Q and Ding B:
Tumor-associated neutrophils upregulate PANoptosis to foster an
immunosuppressive microenvironment of non-small cell lung cancer.
Cancer Immunol Immunother. 72:4293–4308. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sheng Y, Peng W, Huang Y, Cheng L, Meng Y,
Kwantwi LB, Yang J, Xu J, Xiao H, Kzhyshkowska J, et al:
Tumor-activated neutrophils promote metastasis in breast cancer via
the G-CSF-RLN2-MMP-9 axis. J Leukoc Biol. 113:383–399. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chan YT, Tan HY, Lu Y, Zhang C, Cheng CS,
Wu J, Wang N and Feng Y: Pancreatic melatonin enhances anti-tumor
immunity in pancreatic adenocarcinoma through regulating
tumor-associated neutrophils infiltration and NETosis. Acta Pharm
Sin B. 13:1554–1567. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lv B, Wang Y, Ma D, Cheng W, Liu J, Yong
T, Chen H and Wang C: Immunotherapy: Reshape the tumor immune
microenvironment. Front Immunol. 13:8441422022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang W, Li S, Li C, Li T and Huang Y:
Remodeling tumor microenvironment with natural products to overcome
drug resistance. Front Immunol. 13:10519982022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Halama N, Zoernig I, Berthel A, Kahlert C,
Klupp F, Suarez-Carmona M, Suetterlin T, Brand K, Krauss J,
Lasitschka F, et al: Tumoral immune cell exploitation in colorectal
cancer metastases can be targeted effectively by Anti-CCR5 therapy
in cancer patients. Cancer Cell. 29:587–601. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Joyce JA and Fearon DT: T cell exclusion,
immune privilege, and the tumor microenvironment. Science.
348:74–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
DeNardo DG, Brennan DJ, Rexhepaj E,
Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD,
Junaid SA, et al: Leukocyte complexity predicts breast cancer
survival and functionally regulates response to chemotherapy.
Cancer Discov. 1:54–67. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Baghdadi M, Wada H, Nakanishi S, Abe H,
Han N, Putra WE, Endo D, Watari H, Sakuragi N, Hida Y, et al:
Chemotherapy-Induced IL34 enhances immunosuppression by
tumor-associated macrophages and mediates survival of
Chemoresistant lung cancer cells. Cancer Res. 76:6030–6042. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yang C, He L, He P, Liu Y, Wang W, He Y,
Du Y and Gao F: Increased drug resistance in breast cancer by
tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling
pathway. Med Oncol. 32:3522015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang R, Dong M, Tu J, Li F, Deng Q, Xu J,
He X, Ding J, Xia J, Sheng D, et al: PMN-MDSCs modulated by CCL20
from cancer cells promoted breast cancer cell stemness through
CXCL2-CXCR2 pathway. Signal Transduct Target Ther. 8:972023.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mei Y, Zhu Y, Yong KSM, Hanafi ZB, Gong H,
Liu Y, Teo HY, Hussain M, Song Y, Chen Q, et al: IL-37 dampens
immunosuppressive functions of MDSCs via metabolic reprogramming in
the tumor microenvironment. Cell Rep. 43:1138352024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wei C, Yang C, Wang S, Shi D, Zhang C, Lin
X and Xiong B: M2 macrophages confer resistance to 5-fluorouracil
in colorectal cancer through the activation of CCL22/PI3K/AKT
signaling. Onco Targets Ther. 12:3051–3063. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu S, Li Q, Yu Y, Cui Y, Li W, Liu T and
Liu F: Activated HIF1α of tumor cells promotes chemoresistance
development via recruiting GDF15-producing tumor-associated
macrophages in gastric cancer. Cancer Immunol Immunother.
69:1973–1987. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kullberg M, Martinson H, Mann K and
Anchordoquy TJ: Complement C3 mediated targeting of liposomes to
granulocytic myeloid derived suppressor cells. Nanomedicine.
11:1355–1363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ostrand-Rosenberg S, Lamb TJ and Pawelec
G: Here, There, and everywhere: Myeloid-derived suppressor cells in
immunology. J Immunol. 210:1183–1197. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Plesca I, Müller L, Böttcher JP, Medyouf
H, Wehner R and Schmitz M: Tumor-associated human dendritic cell
subsets: Phenotype, functional orientation, and clinical relevance.
Eur J Immunol. 52:1750–1758. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Dong Y, Chen J, Chen Y and Liu S:
Targeting the STAT3 oncogenic pathway: Cancer immunotherapy and
drug repurposing. Biomed Pharmacother. 167:1155132023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Anderson NM and Simon MC: The tumor
microenvironment. Curr Biol. 30:R921–R925. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Arner EN and Rathmell JC: Metabolic
programming and immune suppression in the tumor microenvironment.
Cancer Cell. 41:421–433. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jing X, Yang F, Shao C, Wei K, Xie M, Shen
H and Shu Y: Role of hypoxia in cancer therapy by regulating the
tumor microenvironment. Mol Cancer. 18:1572019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wu Q, You L, Nepovimova E, Heger Z, Wu W,
Kuca K and Adam V: Hypoxia-inducible factors: Master regulators of
hypoxic tumor immune escape. J Hematol Oncol. 15:772022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lian X, Yang K, Li R, Li M, Zuo J, Zheng
B, Wang W, Wang P and Zhou S: Immunometabolic rewiring in
tumorigenesis and anti-tumor immunotherapy. Mol Cancer. 21:272022.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Liu Z, Zou H, Dang Q, Xu H, Liu L, Zhang
Y, Lv J, Li H, Zhou Z and Han X: Biological and pharmacological
roles of m6A modifications in cancer drug resistance. Mol Cancer.
21:2202022. View Article : Google Scholar
|
|
75
|
Xu H, Jiao D, Liu A and Wu K: Tumor
organoids: Applications in cancer modeling and potentials in
precision medicine. J Hematol Oncol. 15:582022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yang L, Dong Y, Li Y, Wang D, Liu S, Wang
D, Gao Q, Ji S, Chen X, Lei Q, et al: IL-10 derived from M2
macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1
pathway in non-small cell lung cancer. Int J Cancer. 145:1099–1110.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yang D, Liu J, Qian H and Zhuang Q:
Cancer-associated fibroblasts: From basic science to anticancer
therapy. Exp Mol Med. 55:1322–1332. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhao Z, Mei Y, Wang Z and He W: The effect
of oxidative phosphorylation on cancer drug resistance. Cancers
(Basel). 15:622022. View Article : Google Scholar
|
|
79
|
Zhang H, Yue X, Chen Z, Liu C, Wu W, Zhang
N, Liu Z, Yang L, Jiang Q, Cheng Q, et al: Define cancer-associated
fibroblasts (CAFs) in the tumor microenvironment: New opportunities
in cancer immunotherapy and advances in clinical trials. Mol
Cancer. 22:1592023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Dey P, Kimmelman AC and DePinho RA:
Metabolic Codependencies in the tumor microenvironment. Cancer
Discov. 11:1067–1081. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Seebacher NA, Krchniakova M, Stacy AE,
Skoda J and Jansson PJ: Tumour microenvironment stress promotes the
development of drug resistance. Antioxidants (Basel). 10:18012021.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Shigeta K, Hasegawa M, Hishiki T, Naito Y,
Baba Y, Mikami S, Matsumoto K, Mizuno R, Miyajima A, Kikuchi E, et
al: IDH2 stabilizes HIF-1α-induced metabolic reprogramming and
promotes chemoresistance in urothelial cancer. EMBO J.
42:e1106202023. View Article : Google Scholar
|
|
83
|
Li YQ, Sun FZ, Li CX, Mo HN, Zhou YT, Lv
D, Zhai JT, Qian HL and Ma F: RARRES2 regulates lipid metabolic
reprogramming to mediate the development of brain metastasis in
triple negative breast cancer. Mil Med Res. 10:342023.PubMed/NCBI
|
|
84
|
Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X,
Shao Q, Zhou B, Zhou H, Wei S, et al: Tumor metabolite lactate
promotes tumorigenesis by modulating MOESIN lactylation and
enhancing TGF-β signaling in regulatory T cells. Cell Rep.
39:1109862022. View Article : Google Scholar
|
|
85
|
Linares JF, Cid-Diaz T, Duran A, Osrodek
M, Martinez-Ordoñez A, Reina-Campos M, Kuo HH, Elemento O, Martin
ML, Cordes T, et al: The lactate-NAD+ axis activates
cancer-associated fibroblasts by downregulating p62. Cell Rep.
39:1107922022. View Article : Google Scholar :
|
|
86
|
Mazurkiewicz J, Simiczyjew A, Dratkiewicz
E, Pietraszek-Gremplewicz K, Majkowski M, Kot M, Ziętek M,
Matkowski R and Nowak D: Melanoma cells with diverse invasive
potential differentially induce the activation of normal human
fibroblasts. Cell Commun Signal. 20:632022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen
S, Wang Y, Wang T and Hou Y: Carcinoma-associated fibroblasts
promote the stemness and chemoresistance of colorectal cancer by
transferring exosomal lncRNA H19. Theranostics. 8:3932–3948. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Li W, Zhou C, Yu L, Hou Z, Liu H, Kong L,
Xu Y, He J, Lan J, Ou Q, et al: Tumor-derived lactate promotes
resistance to bevacizumab treatment by facilitating autophagy
enhancer protein RUBCNL expression through histone H3 lysine 18
lactylation (H3K18la) in colorectal cancer. Autophagy. 20:114–130.
2024. View Article : Google Scholar :
|
|
89
|
Wang L, Li S, Luo H, Lu Q and Yu S: PCSK9
promotes the progression and metastasis of colon cancer cells
through regulation of EMT and PI3K/AKT signaling in tumor cells and
phenotypic polarization of macrophages. J Exp Clin Cancer Res.
41:3032022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ivey JW, Bonakdar M, Kanitkar A, Davalos
RV and Verbridge SS: Improving cancer therapies by targeting the
physical and chemical hallmarks of the tumor microenvironment.
Cancer Lett. 380:330–339. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wu P, Gao W, Su M, Nice EC, Zhang W, Lin J
and Xie N: Adaptive mechanisms of tumor therapy resistance driven
by tumor microenvironment. Front Cell Dev Biol. 9:6414692021.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Cheng J, Yan J, Liu Y, Shi J, Wang H, Zhou
H, Zhou Y, Zhang T, Zhao L, Meng X, et al: Cancer-cell-derived
fumarate suppresses the anti-tumor capacity of CD8+ T cells in the
tumor microenvironment. Cell Metab. 35:961–978.e10. 2023.
View Article : Google Scholar
|
|
93
|
Rahmanian M, Seyfoori A, Ghasemi M, Shamsi
M, Kolahchi AR, Modarres HP, Sanati-Nezhad A and Majidzadeh-A K:
In-vitro tumor microenvironment models containing physical and
biological barriers for modelling multidrug resistance mechanisms
and multidrug delivery strategies. J Control Release. 334:164–177.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Tajaldini M, Poorkhani A, Amiriani T,
Amiriani A, Javid H, Aref P, Ahmadi F, Sadani S and Khori V:
Strategy of targeting the tumor microenvironment via inhibition of
fibroblast/fibrosis remodeling new era to cancer
chemo-immunotherapy resistance. Eur J Pharmacol. 957:1759912023.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lopez-Crapez E, Costa L, Tosato G, Ramos
J, Mazard T, Guiramand J, Thierry A, Colinge J, Milhiet PE and
Bénistant C: Mechanical signatures of human colon cancers. Sci Rep.
12:124752022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhao Q, Chen J, Zhang Z, Xiao C, Zeng H,
Xu C, Yang X and Li Z: Modulating tumor mechanics with nanomedicine
for cancer therapy. Biomater Sci. 11:4471–4489. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zanotelli MR and Reinhart-King CA:
Mechanical forces in tumor angiogenesis. Adv Exp Med Biol.
1092:91–112. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Nicolas-Boluda A, Silva AKA, Fournel S and
Gazeau F: Physical oncology: New targets for nanomedicine.
Biomaterials. 150:87–99. 2018. View Article : Google Scholar
|
|
99
|
Arora I, Li S, Crowley MR, Li Y and
Tollefsbol TO: Genome-wide analysis on transcriptome and methylome
in prevention of mammary tumor induced by early life combined
botanicals. Cells. 12:142022. View Article : Google Scholar
|
|
100
|
Wang EJ, Chen IH, Kuo BY, Yu CC, Lai MT,
Lin JT, Lin LY, Chen CM, Hwang T and Sheu JJ: Alterations of
cytoskeleton networks in cell fate determination and cancer
development. Biomolecules. 12:18622022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Geiger B, Bershadsky A, Pankov R and
Yamada KM: Transmembrane crosstalk between the extracellular
matrix-cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2:793–805.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Park JS, Burckhardt CJ, Lazcano R, Solis
LM, Isogai T, Li L, Chen CS, Gao B, Minna JD, Bachoo R, et al:
Mechanical regulation of glycolysis via cytoskeleton architecture.
Nature. 578:621–626. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yu S, Li Q, Wang Y, Cui Y, Yu Y, Li W, Liu
F and Liu T: Tumor-derived LIF promotes chemoresistance via
activating tumor-associated macrophages in gastric cancers. Exp
Cell Res. 406:1127342021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Li J, Wang S, Wang N, Zheng Y, Yang B,
Wang X, Zhang J, Pan B and Wang Z: Aiduqing formula inhibits breast
cancer metastasis by suppressing TAM/CXCL1-induced Treg
differentiation and infiltration. Cell Commun Signal. 19:892021.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Gao D, Cazares LH and Fish EN: CCL5-CCR5
interactions modulate metabolic events during tumor onset to
promote tumorigenesis. BMC Cancer. 17:8342017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Yuan MX, Ji CY, Gao HQ, Sheng XY, Xie WX
and Yin Q: lncRNA TUG1 regulates angiogenesis via the
miR-204-5p/JAK2/STAT3 axis in hepatoblastoma. Mol Med Rep.
24:5532021. View Article : Google Scholar
|
|
107
|
Nywening TM, Belt BA, Cullinan DR, Panni
RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE,
et al: Targeting both tumour-associated CXCR2+ neutrophils and
CCR2+ macrophages disrupts myeloid recruitment and improves
chemotherapeutic responses in pancreatic ductal adenocarcinoma.
Gut. 67:1112–1123. 2018. View Article : Google Scholar
|
|
108
|
Inoue C, Miki Y, Saito R, Hata S, Abe J,
Sato I, Okada Y and Sasano H: PD-L1 induction by cancer-associated
fibroblast-derived factors in lung adenocarcinoma cells. Cancers
(Basel). 11:12572019. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Harryvan TJ, Visser M, de Bruin L, Plug L,
Griffioen L, Mulder A, van Veelen PA, van der Heden van Noort GJ,
Jongsma ML, Meeuwsen MH, et al: Enhanced antigen cross-presentation
in human colorectal cancer-associated fibroblasts through
upregulation of the lysosomal protease cathepsin S. J Immunother
Cancer. 10:e0035912022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Souza-Fonseca-Guimaraes F, Rossi GR,
Dagley LF, Foroutan M, McCulloch TR, Yousef J, Park HY, Gunter JH,
Beavis PA, Lin CY, et al: TGFβ and CIS inhibition overcomes NK-cell
suppression to restore antitumor immunity. Cancer Immunol Res.
10:1047–1054. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Francescone R, Barbosa Vendramini-Costa D,
Franco-Barraza J, Wagner J, Muir A, Lau AN, Gabitova L, Pazina T,
Gupta S, Luong T, et al: Netrin G1 promotes pancreatic
tumorigenesis through cancer-associated fibroblast-driven
nutritional support and immunosuppression. Cancer Discov.
11:446–479. 2021. View Article : Google Scholar
|
|
112
|
Huang KF, Zhang GD, Huang YQ and Diao Y:
Wogonin induces apoptosis and down-regulates survivin in human
breast cancer MCF-7 cells by modulating PI3K-AKT pathway. Int
Immunopharmacol. 12:334–41. 2012. View Article : Google Scholar
|
|
113
|
Ali SR, Jordan M, Nagarajan P and Amit M:
Nerve density and neuronal biomarkers in cancer. Cancers (Basel).
14:48172022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mhaidly R and Mechta-Grigoriou F: Role of
cancer-associated fibroblast subpopulations in immune infiltration,
as a new means of treatment in cancer. Immunol Rev. 302:259–272.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Timosenko E, Hadjinicolaou AV and
Cerundolo V: Modulation of cancer-specific immune responses by
amino acid degrading enzymes. Immunotherapy. 9:83–97. 2017.
View Article : Google Scholar
|
|
116
|
Stockmann C, Doedens A, Weidemann A, Zhang
N, Takeda N, Greenberg JI, Cheresh DA and Johnson RS: Deletion of
vascular endothelial growth factor in myeloid cells accelerates
tumorigenesis. Nature. 456:814–818. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wang S, Liu G, Li Y and Pan Y: Metabolic
reprogramming induces macrophage polarization in the tumor
microenvironment. Front Immunol. 13:8400292022. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhang M, Zhang H, Tang F, Wang Y, Mo Z,
Lei X and Tang S: Doxorubicin resistance mediated by cytoplasmic
macrophage colony-stimulating factor is associated with switch from
apoptosis to autophagic cell death in MCF-7 breast cancer cells.
Exp Biol Med (Maywood). 241:2086–2093. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Mehta AK, Kadel S, Townsend MG, Oliwa M
and Guerriero JL: Macrophage biology and mechanisms of immune
suppression in breast cancer. Front Immunol. 12:6437712021.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Li Y, Weng Y, Zhong L, Chong H, Chen S,
Sun Y, Li W and Shi Q: VEGFR3 inhibition chemosensitizes lung
adenocarcinoma A549 cells in the tumor-associated macrophage
microenvironment through upregulation of p53 and PTEN. Oncol Rep.
38:2761–2773. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Dalton HJ, Pradeep S, McGuire M,
Hailemichael Y, Ma S, Lyons Y, Armaiz-Pena GN, Previs RA, Hansen
JM, Rupaimoole R, et al: Macrophages facilitate resistance to
Anti-VEGF therapy by Altered VEGFR expression. Clin Cancer Res.
23:7034–7046. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Vahidian F, Duijf PHG, Safarzadeh E,
Derakhshani A, Baghbanzadeh A and Baradaran B: Interactions between
cancer stem cells, immune system and some environmental components:
Friends or foes? Immunol Lett. 208:19–29. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Pu Y and Ji Q: Tumor-associated
macrophages regulate PD-1/PD-L1 immunosuppression. Front Immunol.
13:8745892022. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Binnewies M, Pollack JL, Rudolph J, Dash
S, Abushawish M, Lee T, Jahchan NS, Canaday P, Lu E, Norng M, et
al: Targeting TREM2 on tumor-associated macrophages enhances
immunotherapy. Cell Rep. 37:1098442021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Chen D, Zhang X, Li Z and Zhu B: Metabolic
regulatory crosstalk between tumor microenvironment and
tumor-associated macrophages. Theranostics. 11:1016–1030. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Cassetta L and Pollard JW: A timeline of
tumour-associated macrophage biology. Nat Rev Cancer. 23:238–257.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Pan Y, Yu Y, Wang X and Zhang T:
Tumor-associated macrophages in tumor immunity. Front Immunol.
11:5830842020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Gao J, Liang Y and Wang L: Shaping
polarization of tumor-associated macrophages in cancer
immunotherapy. Front Immunol. 13:8887132022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Li C, Xu X, Wei S, Jiang P, Xue L and Wang
J: Tumor-associated macrophages: Potential therapeutic strategies
and future prospects in cancer. J Immunother Cancer. 9:e0013412021.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Basak U, Sarkar T, Mukherjee S,
Chakraborty S, Dutta A, Dutta S, Nayak D, Kaushik S, Das T and Sa
G: Tumor-associated macrophages: An effective player of the tumor
microenvironment. Front Immunol. 14:12952572023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Munir MT, Kay MK, Kang MH, Rahman MM,
Al-Harrasi A, Choudhury M, Moustaid-Moussa N, Hussain F and Rahman
SM: Tumor-associated macrophages as multifaceted regulators of
breast tumor growth. Int J Mol Sci. 22:65262021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Céspedes MV, Guillén MJ, López-Casas PP,
Sarno F, Gallardo A, Álamo P, Cuevas C, Hidalgo M, Galmarini CM,
Allavena P, et al: Lurbinectedin induces depletion of
tumor-associated macrophages, an essential component of its in vivo
synergism with gemcitabine, in pancreatic adenocarcinoma mouse
models. Dis Model Mech. 9:1461–1471. 2016.PubMed/NCBI
|
|
133
|
Ayoub M, Shinde-Jadhav S, Mansure JJ,
Alvarez F, Connell T, Seuntjens J, Piccirillo CA and Kassouf W: The
immune mediated role of extracellular HMGB1 in a heterotopic model
of bladder cancer radioresistance. Sci Rep. 9:63482019. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Hong L, Wang X, Zheng L, Wang S and Zhu G:
Tumor-associated macrophages promote cisplatin resistance in
ovarian cancer cells by enhancing WTAP-mediated N6-methyladenosine
RNA methylation via the CXCL16/CXCR6 axis. Cancer Chemother
Pharmacol. 92:71–81. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Kobayashi H, Gieniec KA, Lannagan TRM,
Wang T, Asai N, Mizutani Y, Iida T, Ando R, Thomas EM, Sakai A, et
al: The origin and contribution of cancer-associated fibroblasts in
colorectal carcinogenesis. Gastroenterology. 162:890–906. 2022.
View Article : Google Scholar
|
|
136
|
Hosomi S, Grootjans J, Huang YH, Kaser A
and Blumberg RS: New insights into the regulation of natural-killer
group 2 Member D (NKG2D) and NKG2D-ligands: Endoplasmic reticulum
stress and CEA-related cell adhesion molecule 1. Front Immunol.
9:13242018. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Comito G, Iscaro A, Bacci M, Morandi A,
Ippolito L, Parri M, Montagnani I, Raspollini MR, Serni S, Simeoni
L, et al: Lactate modulates CD4+ T-cell polarization and induces an
immunosuppressive environment, which sustains prostate carcinoma
progression via TLR8/miR21 axis. Oncogene. 38:3681–3695. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liang L, Li W, Li X, Jin X, Liao Q, Li Y
and Zhou Y: 'Reverse Warburg effect' of cancer associated
fibroblasts (Review). Int J Oncol. 60:672022. View Article : Google Scholar
|
|
139
|
Zhao Q, Huang L, Qin G, Qiao Y, Ren F,
Shen C, Wang S, Liu S, Lian J, Wang D, et al: Cancer-associated
fibroblasts induce monocytic myeloid-derived suppressor cell
generation via IL-6/exosomal miR-21-activated STAT3 signaling to
promote cisplatin resistance in esophageal squamous cell carcinoma.
Cancer Lett. 518:35–48. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Xiang H, Ramil CP, Hai J, Zhang C, Wang H,
Watkins AA, Afshar R, Georgiev P, Sze MA, Song XS, et al:
Cancer-associated fibroblasts promote immunosuppression by inducing
ROS-generating monocytic MDSCs in lung squamous cell carcinoma.
Cancer Immunol Res. 8:436–450. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Lin SC, Liao YC, Chen PM, Yang YY, Wang
YH, Tung SL, Chuang CM, Sung YW, Jang TH, Chuang SE, et al:
Periostin promotes ovarian cancer metastasis by enhancing M2
macrophages and cancer-associated fibroblasts via integrin-mediated
NF-κB and TGF-β2 signaling. J Biomed Sci. 29:1092022. View Article : Google Scholar
|
|
142
|
Chen X, Zhang W, Yang W, Zhou M and Liu F:
Acquired resistance for immune checkpoint inhibitors in cancer
immunotherapy: Challenges and prospects. Aging (Albany NY).
14:1048–1064. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Baik AH: Hypoxia signaling and oxygen
metabolism in cardio-oncology. J Mol Cell Cardiol. 165:64–75. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Dzobo K, Senthebane DA and Dandara C: The
tumor microenvironment in tumorigenesis and therapy resistance
revisited. Cancers (Basel). 15:3762023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Harris B, Saleem S, Cook N and Searle E:
Targeting hypoxia in solid and haematological malignancies. J Exp
Clin Cancer Res. 41:3182022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhang H, Deng T, Liu R, Ning T, Yang H,
Liu D, Zhang Q, Lin D, Ge S, Bai M, et al: CAF secreted miR-522
suppresses ferroptosis and promotes acquired chemo-resistance in
gastric cancer. Mol Cancer. 19:432020. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS,
Yan YR, Song FY, Wang FF, Zhu XH, Liao WJ, et al: CAFs secreted
exosomes promote metastasis and chemotherapy resistance by
enhancing cell stemness and epithelial-mesenchymal transition in
colorectal cancer. Mol Cancer. 18:912019. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Liu T, Han C, Fang P, Ma Z, Wang X, Chen
H, Wang S, Meng F, Wang C, Zhang E, et al: Cancer-associated
fibroblast-specific lncRNA LINC01614 enhances glutamine uptake in
lung adenocarcinoma. J Hematol Oncol. 15:1412022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Patil N, Allgayer H and Leupold JH:
MicroRNAs in the tumor microenvironment. Adv Exp Med Biol.
1277:1–31. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Zhu S, Mao J, Zhang X, Wang P, Zhou Y,
Tong J, Peng H, Yang B and Fu Q: CAF-derived exosomal lncRNA FAL1
promotes chemoresistance to oxaliplatin by regulating autophagy in
colorectal cancer. Dig Liver Dis. 56:330–342. 2024. View Article : Google Scholar
|
|
151
|
Meng Q, Deng Y, Lu Y, Wu C and Tang S:
Tumor-derived miRNAs as tumor microenvironment regulators for
synergistic therapeutic options. J Cancer Res Clin Oncol.
149:423–439. 2023. View Article : Google Scholar
|
|
152
|
Zhang P, Wang Q, Lu W, Zhang F, Wu D and
Sun J: NNT-AS1 in CAFs-derived exosomes promotes progression and
glucose metabolism through miR-889-3p/HIF-1α in pancreatic
adenocarcinoma. Sci Rep. 14:69792024. View Article : Google Scholar
|
|
153
|
Wang WZ, Cao X, Bian L, Gao Y, Yu M, Li
YT, Xu JG, Wang YH, Yang HF, You DY, et al: Analysis of mRNA-miRNA
interaction network reveals the role of CAFs-derived exosomes in
the immune regulation of oral squamous cell carcinoma. BMC Cancer.
23:5912023. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Miaomiao S, Xiaoqian W, Yuwei S, Chao C,
Chenbo Y, Yinghao L, Yichen H, Jiao S and Kuisheng C:
Cancer-associated fibroblast-derived exosome microRNA-21 promotes
angiogenesis in multiple myeloma. Sci Rep. 13:96712023. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Zhang Z, Shang J, Yang Q, Dai Z, Liang Y,
Lai C, Feng T, Zhong D, Zou H, Sun L, et al: Exosomes derived from
human adipose mesenchymal stem cells ameliorate hepatic fibrosis by
inhibiting PI3K/Akt/mTOR pathway and remodeling choline metabolism.
J Nanobiotechnology. 21:292023. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Zhang Y, Yin C, Wei C, Xia S, Qiao Z,
Zhang XW, Yu B, Zhou J and Wang R: Exosomal miR-625-3p secreted by
cancer-associated fibroblasts in colorectal cancer promotes EMT and
chemotherapeutic resistance by blocking the CELF2/WWOX pathway.
Pharmacol Res. 186:1065342022. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Huang H, Wang Z, Zhang Y, Pradhan RN,
Ganguly D, Chandra R, Murimwa G, Wright S, Gu X, Maddipati R, et
al: Mesothelial cell-derived antigen-presenting cancer-associated
fibroblasts induce expansion of regulatory T cells in pancreatic
cancer. Cancer Cell. 40:656–673.e7. 2022. View Article : Google Scholar :
|
|
158
|
Cheng Y, Li H, Deng Y, Tai Y, Zeng K,
Zhang Y, Liu W, Zhang Q and Yang Y: Cancer-associated fibroblasts
induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster
immune suppression in hepatocellular carcinoma. Cell Death Dis.
9:4222018. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Song M, He J, Pan QZ, Yang J, Zhao J,
Zhang YJ, Huang Y, Tang Y, Wang Q, He J, et al: Cancer-associated
fibroblast-mediated cellular crosstalk supports hepatocellular
carcinoma progression. Hepatology. 73:1717–1735. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Najafi M, Farhood B and Mortezaee K:
Extracellular matrix (ECM) stiffness and degradation as cancer
drivers. J Cell Biochem. 120:2782–2790. 2019. View Article : Google Scholar
|
|
161
|
Henke E, Nandigama R and Ergün S:
Extracellular matrix in the tumor microenvironment and its impact
on cancer therapy. Front Mol Biosci. 6:1602020. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Timperi E, Gueguen P, Molgora M, Magagna
I, Kieffer Y, Lopez-Lastra S, Sirven P, Baudrin LG, Baulande S,
Nicolas A, et al: Lipid-associated macrophages are induced by
cancer-associated fibroblasts and mediate immune suppression in
breast cancer. Cancer Res. 82:3291–3306. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Li D, Xia L, Huang P, Wang Z, Guo Q, Huang
C, Leng W and Qin S: Cancer-associated fibroblast-secreted IGFBP7
promotes gastric cancer by enhancing tumor associated macrophage
infiltration via FGF2/FGFR1/PI3K/AKT axis. Cell Death Discov.
9:172023. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Ueshima E, Fujimori M, Kodama H, Felsen D,
Chen J, Durack JC, Solomon SB, Coleman JA and Srimathveeravalli G:
Macrophage-secreted TGF-β1 contributes to fibroblast activation and
ureteral stricture after ablation injury. Am J Physiol Renal
Physiol. 317:F52–F64. 2019. View Article : Google Scholar
|
|
165
|
Deng Y, Cheng J, Fu B, Liu W, Chen G,
Zhang Q and Yang Y: Hepatic carcinoma-associated fibroblasts
enhance immune suppression by facilitating the generation of
myeloid-derived suppressor cells. Oncogene. 36:1090–1101. 2017.
View Article : Google Scholar
|
|
166
|
Zhou Y, Tang W, Zhuo H, Zhu D, Rong D, Sun
J and Song J: Cancer-associated fibroblast exosomes promote
chemoresistance to cisplatin in hepatocellular carcinoma through
circZFR targeting signal transducers and activators of
transcription (STAT3)/nuclear factor-kappa B (NF-κB) pathway.
Bioengineered. 13:4786–4797. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Chen Y, McAndrews KM and Kalluri R:
Clinical and therapeutic relevance of cancer-associated
fibroblasts. Nat Rev Clin Oncol. 18:792–804. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Chen Y, Hu M, Wang S, Wang Q, Lu H, Wang
F, Wang L, Peng D and Chen W: Nano-delivery of salvianolic acid B
induces the quiescence of tumor-associated fibroblasts via
interfering with TGF-β1/Smad signaling to facilitate chemo- and
immunotherapy in desmoplastic tumor. Int J Pharm. 623:1219532022.
View Article : Google Scholar
|
|
169
|
Xiang H, Ramil CP, Hai J, Zhang C, Wang H,
Watkins AA, Afshar R, Georgiev P, Sze MA, Song XS, et al:
Cancer-associated fibroblasts promote immunosuppression by inducing
ROS-generating monocytic MDSCs in lung squamous cell carcinoma.
Cancer Immunol Res. 8:436–450. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Bai XF, Liu J, Li O, Zheng P and Liu Y:
Antigenic drift as a mechanism for tumor evasion of destruction by
cytolytic T lymphocytes. J Clin Invest. 111:1487–1496. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Cheng C, Qu QX, Shen Y, Lv YT, Zhu YB,
Zhang XG and Huang JA: Overexpression of B7-H4 in tumor infiltrated
dendritic cells. J Immunoassay Immunochem. 32:353–364. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Blank C, Kuball J, Voelkl S, Wiendl H,
Becker B, Walter B, Majdic O, Gajewski TF, Theobald M, Andreesen R,
et al: Blockade of PD-L1 (B7-H1) augments human tumor-specific T
cell responses in vitro. Int J Cancer. 119:317–327. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Zheng Y, Tang L, Mabardi L, Kumari S and
Irvine DJ: Enhancing adoptive cell therapy of cancer through
targeted delivery of small-molecule immunomodulators to
internalizing or noninternalizing receptors. ACS Nano.
11:3089–3100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Farhood B, Najafi M and Mortezaee K: CD8+
cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell
Physiol. 234:8509–8521. 2019. View Article : Google Scholar
|
|
175
|
Inoue T, Adachi K, Kawana K, Taguchi A,
Nagamatsu T, Fujimoto A, Tomio K, Yamashita A, Eguchi S, Nishida H,
et al: Cancer-associated fibroblast suppresses killing activity of
natural killer cells through downregulation of poliovirus receptor
(PVR/CD155), a ligand of activating NK receptor. Int J Oncol.
49:1297–1304. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Van den Eynde A, Gehrcken L, Verhezen T,
Lau HW, Hermans C, Lambrechts H, Flieswasser T, Quatannens D, Roex
G, Zwaenepoel K, et al: IL-15-secreting CAR natural killer cells
directed toward the pan-cancer target CD70 eliminate both cancer
cells and cancer-associated fibroblasts. J Hematol Oncol. 17:82024.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q,
Deng S and Zhou H: Signaling pathways in cancer-associated
fibroblasts and targeted therapy for cancer. Signal Transduct
Target Ther. 6:2182021. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Yu W, Lei Q, Yang L, Qin G, Liu S, Wang D,
Ping Y and Zhang Y: Contradictory roles of lipid metabolism in
immune response within the tumor microenvironment. J Hematol Oncol.
14:1872021. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Kennel KB, Bozlar M, De Valk AF and Greten
FR: Cancer-associated fibroblasts in inflammation and antitumor
immunity. Clin Cancer Res. 29:1009–1016. 2023. View Article : Google Scholar :
|
|
180
|
Li X, Sun Z, Peng G, Xiao Y, Guo J, Wu B,
Li X, Zhou W, Li J, Li Z, et al: Single-cell RNA sequencing reveals
a pro-invasive cancer-associated fibroblast subgroup associated
with poor clinical outcomes in patients with gastric cancer.
Theranostics. 12:620–638. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Galbo PM Jr, Zang X and Zheng D: Molecular
features of Cancer-associated fibroblast subtypes and their
implication on cancer pathogenesis, prognosis, and immunotherapy
resistance. Clin Cancer Res. 27:2636–2647. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Glabman RA, Choyke PL and Sato N:
Cancer-associated fibroblasts: Tumorigenicity and targeting for
cancer therapy. Cancers (Basel). 14:39062022. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Bhattacharjee S, Hamberger F, Ravichandra
A, Miller M, Nair A, Affo S, Filliol A, Chin L, Savage TM, Yin D,
et al: Tumor restriction by type I collagen opposes tumor-promoting
effects of cancer-associated fibroblasts. J Clin Invest.
131:e1469872021. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Dong D, Yao Y, Song J, Sun L and Zhang G:
Cancer-associated fibroblasts regulate bladder cancer invasion and
metabolic phenotypes through autophagy. Dis Markers.
2021:66452202021. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Strickaert A, Corbet C, Spinette SA,
Craciun L, Dom G, Andry G, Larsimont D, Wattiez R, Dumont JE, Feron
O, et al: Reprogramming of energy metabolism: Increased expression
and roles of pyruvate carboxylase in papillary thyroid cancer.
Thyroid. 29:845–857. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Zhu Y, Li X, Wang L, Hong X and Yang J:
Metabolic reprogramming and crosstalk of cancer-related fibroblasts
and immune cells in the tumor microenvironment. Front Endocrinol
(Lausanne). 13:9882952022. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Xia H, Green DR and Zou W: Autophagy in
tumour immunity and therapy. Nat Rev Cancer. 21:281–297. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Liu L, Liu S, Luo H, Chen C, Zhang X, He L
and Tu G: GPR30-mediated HMGB1 upregulation in CAFs induces
autophagy and tamoxifen resistance in ERα-positive breast cancer
cells. Aging (Albany NY). 13:16178–16197. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Zeng Z, Hu P, Tang X, Zhang H, Du Y, Wen S
and Liu M: Dectection and analysis of miRNA expression in breast
cancer-associated fibroblasts. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi.
30:1071–1075. 2014.In Chinese. PubMed/NCBI
|
|
190
|
Izumi D, Toden S, Ureta E, Ishimoto T,
Baba H and Goel A: TIAM1 promotes chemoresistance and tumor
invasiveness in colorectal cancer. Cell Death Dis. 10:2672019.
View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Nywening TM, Wang-Gillam A, Sanford DE,
Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA,
Yano M, et al: Targeting tumour-associated macrophages with CCR2
inhibition in combination with FOLFIRINOX in patients with
borderline resectable and locally advanced pancreatic cancer: A
single-centre, open-label, dose-finding, non-randomised, phase 1b
trial. Lancet Oncol. 17:651–662. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Ma J, Song X, Xu X and Mou Y:
Cancer-associated fibroblasts promote the Chemo-resistance in
gastric cancer through secreting IL-11 targeting JAK/STAT3/Bcl2
pathway. Cancer Res Treat. 51:194–210. 2019. View Article : Google Scholar
|
|
193
|
Wei L, Lin Q, Lu Y, Li G, Huang L, Fu Z,
Chen R and Zhou Q: Cancer-associated fibroblasts-mediated ATF4
expression promotes malignancy and gemcitabine resistance in
pancreatic cancer via the TGF-β1/SMAD2/3 pathway and ABCC1
transactivation. Cell Death Dis. 12:3342021. View Article : Google Scholar
|
|
194
|
Li Z, Chan K, Qi Y, Lu L, Ning F, Wu M,
Wang H, Wang Y, Cai S and Du J: Participation of CCL1 in
Snail-positive fibroblasts in colorectal cancer contribute to
5-Fluorouracil/Paclitaxel Chemoresistance. Cancer Res Treat.
50:894–907. 2018. View Article : Google Scholar :
|
|
195
|
Saw PE, Chen J and Song E: Targeting CAFs
to overcome anticancer therapeutic resistance. Trends Cancer.
8:527–555. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Singh SK, Mishra MK, Eltoum IA, Bae S,
Lillard JW Jr and Singh R: CCR5/CCL5 axis interaction promotes
migratory and invasiveness of pancreatic cancer cells. Sci Rep.
8:13232018. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Lee C, Lee H, Cho H, Kim S, Choi I, Hwang
YS, Jeong H, Jang H, Pak S, Hwang DS, et al: Combination of
anti-PD-L1 antibody with peptide MEL-dKLA targeting M2
tumor-associated macrophages suppresses breast cancer metastasis.
Cancer Commun (Lond). 42:345–349. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Siewe N and Friedman A: Cancer therapy
with immune checkpoint inhibitor and CSF-1 blockade: A mathematical
model. J Theor Biol. 556:1112972023. View Article : Google Scholar
|
|
199
|
Rodell CB, Ahmed MS, Garris CS, Pittet MJ
and Weissleder R: Development of Adamantane-conjugated TLR7/8
agonists for supramolecular delivery and cancer immunotherapy.
Theranostics. 9:8426–8436. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Chen YJ, Li GN, Li XJ, Wei LX, Fu MJ,
Cheng ZL, Yang Z, Zhu GQ, Wang XD, Zhang C, et al: Targeting IRG1
reverses the immunosuppressive function of tumor-associated
macrophages and enhances cancer immunotherapy. Sci Adv.
9:eadg06542023. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Yuan D, Hu J, Ju X, Putz EM, Zheng S, Koda
S, Sun G, Deng X, Xu Z, Nie W, et al: NMDAR antagonists suppress
tumor progression by regulating tumor-associated macrophages. Proc
Natl Acad Sci USA. 120:e23021261202023. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Li M, Yang Y, Xiong L, Jiang P, Wang J and
Li C: Metabolism, metabolites, and macrophages in cancer. J Hematol
Oncol. 16:802023. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Khalaf K, Hana D, Chou JT, Singh C,
Mackiewicz A and Kaczmarek M: Aspects of the tumor microenvironment
involved in immune resistance and drug resistance. Front Immunol.
12:6563642021. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Begum A, McMillan RH, Chang YT, Penchev
VR, Rajeshkumar NV, Maitra A, Goggins MG, Eshelman JR, Wolfgang CL,
Rasheed ZA, et al: Direct interactions with cancer-associated
fibroblasts lead to enhanced pancreatic cancer stem cell function.
Pancreas. 48:329–334. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Ko YC, Lai TY, Hsu SC, Wang FH, Su SY,
Chen YL, Tsai ML, Wu CC, Hsiao JR, Chang JY, et al: Index of
Cancer-associated fibroblasts is superior to the
epithelial-mesenchymal transition score in prognosis prediction.
Cancers (Basel). 12:17182020. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Liu Y, Xun Z, Ma K, Liang S, Li X, Zhou S,
Sun L, Liu Y, Du Y, Guo X, et al: Identification of a tumour immune
barrier in the HCC microenvironment that determines the efficacy of
immunotherapy. J Hepatol. 78:770–782. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Yamamoto Y, Kasashima H, Fukui Y, Tsujio
G, Yashiro M and Maeda K: The heterogeneity of cancer-associated
fibroblast subpopulations: Their origins, biomarkers, and roles in
the tumor microenvironment. Cancer Sci. 114:16–24. 2023. View Article : Google Scholar
|
|
208
|
Qu X, Liu B, Wang L, Liu L, Zhao W, Liu C,
Ding J, Zhao S, Xu B, Yu H, et al: Loss of cancer-associated
fibroblast-derived exosomal DACT3-AS1 promotes malignant
transformation and ferroptosis-mediated oxaliplatin resistance in
gastric cancer. Drug Resist Updat. 68:1009362023. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Loeffler M, Krüger JA, Niethammer AG and
Reisfeld RA: Targeting tumor-associated fibroblasts improves cancer
chemotherapy by increasing intratumoral drug uptake. J Clin Invest.
116:1955–1962. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Denkert C, von Minckwitz G, Darb-Esfahani
S, Lederer B, Heppner BI, Weber KE, Budczies J, Huober J, Klauschen
F, Furlanetto J, et al: Tumour-infiltrating lymphocytes and
prognosis in different subtypes of breast cancer: A pooled analysis
of 3771 patients treated with neoadjuvant therapy. Lancet Oncol.
19:40–50. 2018. View Article : Google Scholar
|
|
211
|
Memon D, Schoenfeld AJ, Ye D, Fromm G,
Rizvi H, Zhang X, Keddar MR, Mathew D, Yoo KJ, Qiu J, et al:
Clinical and molecular features of acquired resistance to
immunotherapy in non-small cell lung cancer. Cancer Cell.
42:209–224.e9. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Zhu L, Meng D, Wang X and Chen X:
Ferroptosis-driven Nanotherapeutics to reverse drug resistance in
tumor microenvironment. ACS Appl Bio Mater. 5:2481–2506. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Zhu Y, Wang A, Zhang S, Kim J, Xia J,
Zhang F, Wang D, Wang Q and Wang J: Paclitaxel-loaded ginsenoside
Rg3 liposomes for drug-resistant cancer therapy by dual targeting
of the tumor microenvironment and cancer cells. J Adv Res.
49:159–173. 2023. View Article : Google Scholar :
|