Introduction
Acute myeloid leukemia (AML) is a type of tumor that
affects stem cell precursors of the myeloid lineage, including red
blood cells, platelets and white blood cells (1,2).
The development of AML is caused by genetic mutations such as
familial mutations in CEBPA, DDX4 and RUNX1 genes, resulting in
uncontrolled proliferation and hematopoietic stem cell
abnormalities (1,2). Among adults, AML is the second most
common form of leukemia worldwide and the 5-year survival rate is
<30% (3). Cytarabine is the
first-line drug used to treat AML. Although cytarabine-based
regimens can induce a complete response in most newly diagnosed
patients with AML, the overall clinical outcome remains
unsatisfactory due to drug resistance (4). Therefore, it is crucial to develop
strategies to overcome drug resistance.
Multiple mechanisms, including apoptosis, autophagy
and ferroptosis, have been suggested to be involved in drug
resistance (5-7). Ferroptosis is a distinct
iron-dependent form of cell death that differs from apoptosis,
necrosis and autophagy (8). It
has been reported that enhancing ferroptosis can reverse drug
resistance and improve cancer treatment (9). For example, it has been shown that
the use of ferroptosis inducers can synergistically sensitize
ovarian cancer cells to a PARP inhibitor (10). Liu et al (11) demonstrated that enhancing
ferroptosis by impairing STAT3-Nrf2-glutathione peroxidase 4 (GPX4)
signaling could increase the sensitivity of osteosarcoma cells to
cisplatin. Notably, it has also been suggested that inducing
ferroptosis can reverse the resistance of cytarabine in AML
(12); however, the underlying
mechanism has yet to be fully elucidated.
Sirtuin 1 (SIRT1) is a member of the class III
family of NAD(+)-dependent histone deacetylases, which serves an
important role in various cellular processes, such as cell
proliferation, apoptosis, inflammation, oxidation response and drug
resistance (13,14). Previous studies have shown that
SIRT1 contributes to drug resistance by deacetylating its
downstream targets, such as FOXO3 and WEE1 (15,16). In addition, SIRT1 upregulation by
cytarabine has been reported to be inhibited by Tenovin-6 in acute
lymphoblastic leukemia (17).
However, the mechanism by which SIRT1 regulates the drug resistance
of leukemia cells is currently unclear. High mobility group box-1
protein (HMGB1) is a nuclear DNA-binding protein involved in
nucleosome stabilization and gene transcription. In acute kidney
injury, cytoplasmic HMGB1 can induce ferroptosis by regulating
ACSL4 (18). The present study
aimed to explore the effects of SIRT1 knockdown on the induction of
ferroptosis via the HMGB1/ACSL4 pathway, which may reverse
cytarabine resistance in AML. The present findings demonstrated
that SIRT1 inhibition could be a promising strategy to overcome
cytarabine resistance.
Materials and methods
Cell culture and reagents
The leukemia cell lines HL60, K562 and Kasumi-1
(Kas-1) were purchased from Procell Life Science & Technology
Co., Ltd. HL60 and Kas-1 cells were isolated from AML, whereas K562
cells were isolated from chronic myeloid leukemia. Cells were
maintained in RPMI 1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.), 1% 100 U/ml penicillin G and 100 mg/ml
streptomycin. Cells were cultured at 37°C with 5% CO2.
Cytarabine was purchased from MilliporeSigma. Drug-resistant
leukemia cell lines were established as described previously
(19). Briefly, 105
cells were inoculated into a 6-well plate, and when cells reached
80% confluence, different concentrations (0, 1, 2, 5 and 10
μmol) of cytarabine were added. Subsequently, the medium was
changed every 3-4 days for 3-4 weeks. The IC50
concentrations of cytarabine in HL60, K562 and Kas-1 cells were 5
μmol, 100 nmol and 50 nmol, respectively.
Small interfering RNA (siRNA)
transfection
SIRT1, HMGB1, ACSL4 and negative control (NC) siRNA
sequences were obtained from Shanghai GenePharma Co., Ltd. siRNA
transfection was performed using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Briefly, 105
cytarabine-resistant HL60 (HL60/C) cells or HL60 cells were
inoculated into a 6-well plate and were grown to 40-50% confluence.
Subsequently, 50 nmol siRNA was diluted in Opti-MEM (Thermo Fisher
Scientific, Inc.), mixed with 2 μl Lipofectamine 3000, added
to the cell culture plate and cultured for 48 h at 37°C with 5%
CO2. The siRNA sequences were as follows: si-SIRT1,
forward 5′-GUGGCAGAUUGUUAUUAAUTT-3′, reverse
5′-AUUAAUAACAAUCUGCCACTT-3′; si-SIRT1-2, forward
5′-UUCUGAAAUAUUCAAUAUCAA-3′, reverse 5′-GAUAUUGAAUAUUUCAGAAAA-3′;
si-SIRT1-3, forward 5′-UUUUCCUUCCUUAUCUGACAA-3′, reverse
5′-GUCAGAUAAGGAAGGAAAACU-3′; si-HMGB1-1, forward
5′-GCUCAAGGAGAAUUUGUAATT-3′, reverse 5′-UUACAAAUUCUCCUUGAGCTT-3′;
si-HMGB1-2,forward 5′-ACAAAAAAUGCAUAUGAUGAC-3′,
reverse5′-CAUCAUAUGCAUUUUUUGUGC-3′; si-HMGB1-3, forward
5′-AGUUUCUUCGCAACAUCACCA-3′, reverse 5′-GUGAUGUUGCGAAGAAACUGG-3′;
si-ACSL4-1, forward 5′-GAUGGAUGCUUACAGAUUA-3′, reverse
5′-UAAUCUGUAAGCAUCCAUC-3′; si-ACSL4-2, forward
5′-AUAUUGUUAUUAACAAGUGGA-3′, reverse 5′-CACUUGUUAAUAACAAUAUAC-3′;
si-ACSL4-3,forward 5′-ACUGUAUAUUGUUAUUAACAA-3′, reverse
5′-GUUAAUAACAAUAUACAGUGC-3′; si-NC, forward
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′.
Co-immunoprecipitation (IP) assay
The possible protein interaction network of SIRT1
was constructed using the STRING database (https://string-db.org/).The SIRT1-HA and HMGB1-Flag
overexpression vectors were constructed using pcDNA3.1 plasmids
(Beyotime Institute of Biotechnology). Briefly, 1 μg
SIRT1-HA and HMGB1-Flag vectors were diluted in Opti-MEM, mixed
with 2 μl Lipofectamine 3000 and transfected into 293T
(Procell Life Science & Technology Co., Ltd.), HL60 and HL60/C
cells. After culturing at 37°C for 24 h, the cells were collected
and added to 100 μl RIPA lysis buffer (Beyotime Institute of
Biotechnology) on ice for 30 min, and the supernatant was collected
by centrifugation at 10,000 x g for 10 min at 4°C. Subsequently, 5
μg anti-Flag (cat. no. AE063; ABclonal Biotech Co., Ltd.)
and anti-HA (cat. no. AE105; ABclonal Biotech Co., Ltd.) antibodies
were added to the protein A/G agarose beads (Beyotime Institute of
Biotechnology) and incubated at 4°C overnight. Next, the 10
μl antibody-protein A/G agarose bead complex was added to
the cell lysate and slowly shaken at 4°C for 2-4 h to conjugate the
antibody to the protein A/G agarose beads. After the
immunoprecipitation reaction, the agarose beads were centrifuged at
1,500 x g at 4°C for 3 min and the precipitation was collected. The
agarose beads were washed with 1 ml RIPA lysis buffer (Beyotime
Institute of Biotechnology) 3-4 times. Finally, SDS loading buffer
was added for western blot analysis.
Cell viability analysis
The Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc.) was used to determine cell viability. AML cells
(5×104/well) were plated in 96-well plates and treated
with cytarabine (5 μmol) at 37°C for 24 h. Subsequently, 10
μl CCK-8 solution was added to each well and the cells were
incubated at 37°C for 2-3 h. The absorbance was measured at 450 nm
using a microplate reader.
EdU proliferation assay
Cells were transfected with si-NC or si-SIRT1 and
were treated with cytarabine (5 μmol) at 37°C for 24 h.
Cells (5×104 cells/well) were then plated in 24-well
plates, washed with PBS and incubated in serum-free medium
containing 10 μmol/l EdU (Guangzhou RiboBio Co., Ltd.) for 2
h at 37°C. Cells were fixed in 4% paraformaldehyde (Beyotime
Institute of Biotechnology) for 15 min at 4°C, after which, they
were stained with Apollo solution and DNA staining solution
(Beyotime Institute of Biotechnology) at room temperature. Images
of the cells were then captured using a fluorescence microscope
(Nikon Corporation).
Apoptosis analysis
AML cells (5×104/well) were plated in
24-well plates and treated with cytarabine (5 μmol) at 37°C
for 24 h. Flow cytometric analysis was subsequently performed using
the Annexin V-FITC kit (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. Apoptotic cells were
analyzed by flow cytometry (LSRFortessa; BD Biosciences) and
FlowJo-V10 software (FlowJo, LLC) was used to process the
experimental results.
Determination of reactive oxygen species
(ROS)
The intracellular ROS production in AML cells was
determined using the 2′,7′-dichlorofluorescein diacetate (DCFH-DA)
probe. Briefly, cells were harvested and washed with serum-free
medium, after which, they were incubated with 10 μM DCFH-DA
diluted in serum-free medium at 37°C for 30 min. Subsequently, the
cells were washed with serum-free medium to remove the unbound
DCFH-DA probe, and fluorescence-labeled cells were analyzed by flow
cytometry (LSRFortessa; BD Biosciences) and FlowJo-V10 software was
used to process the experimental results.
Reverse transcription-quantitative PCR
(RT-qPCR)
After treatment, total RNA was isolated from AML
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc) and was reverse transcribed into cDNA using
PrimeScript™ RT Master Mix (Takara Biotechnology Co., Ltd.)
according to the manufacturer's instructions. qPCR was performed
using the Fast SYBR Green Master Mix (Thermo Fisher Scientific,
Inc.) on a 7900 Real-Time PCR System (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The qPCR conditions were as follows: 95°C
for 10 min, followed by 35 cycles at 94°C for 30 sec, 60°C for 15
sec and 72°C for 30 sec. Relative mRNA expression was calculated
using the 2−ΔΔCq method (20). GAPDH was used as the internal
control. The primer sequences were as follows: HMGB1, forward
5′-TAACTAAACATGGGCAAAGGAG-3′, reverse 5′-TAGCAGACATGGTCTTCCAC-3′;
SIRT1, forward 5′-TAGCCTTGTCAGATAAGGAAGGA-3′, reverse
5′-ACAGCTTCACAGTCAACTTTGT-3′; GAPDH, forward
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse
5′-TGGTGAAGACGCCAGTGGA-3′.
Immunofluorescence staining
AML cells were fixed in 4% formaldehyde at 4°C for
20 min followed by permeabilization with 0.1% Triton X-100 at room
temperature for 10 min. The cells were then washed three times with
PBS, blocked with 5% bovine serum albumin (Beyotime Institute of
Biotechnology) for 30 min at room temperature, and incubated
overnight at 4°C with anti-GPX4 (1:1,000; cat. no. A11243),
anti-HMGB1 (1:1,000; cat. no. A2553) and anti-SIRT1 (1:1,000; cat.
no. A11267) (all from ABclonal Biotech Co., Ltd.), followed by
incubation with CoraLite 488-conjugated secondary antibody (1:200;
cat no. SA00013-2; Wuhan Sanying Biotechnology). The nuclei were
stained with DAPI at room temperature for 5 min. Cells were
visualized using a confocal microscope (Olympus Corporation).
Western blot analysis
After treatment, AML cells were collected and lysed
in RIPA lysis buffer (Beyotime Institute of Biotechnology). The
lysates were incubated on ice for 20 min and centrifuged at 10,000
x g for 30 min at 4°C, before the protein concentration was
determined using a BCA kit (Beyotime Institute of Biotechnology).
Protein samples (20 μg) were separated by SDS-PAGE on 10%
gels and were transferred onto polyvinylidene difluoride membranes
(MilliporeSigma). The membranes were blocked with 5% non-fat dry
milk diluted in Tris-buffered saline-0.5% Tween-20. Subsequently,
the membranes were incubated with anti-SIRT1 (1:1,000; cat. no.
A11267), anti-GPX4 (1:1,000, cat. no. A11243), anti-HMGB1 (1:1,000;
cat. no. A2553),anti-ACSL4 (1:1,000; cat. no. A20414) and
anti-GAPDH (1:5,000; cat. no. A19056) (all from ABclonal Biotech
Co., Ltd.) overnight at 4°C. After three washes, the membranes were
incubated with a HRP-conjugated secondary antibody (1:10,000; cat.
no. AS014; ABclonal Biotech Co., Ltd.) for 60 min at room
temperature. Protein bands were visualized using a
chemiluminescence imaging system (Tanon-4600; Tanon Science and
Technology Co., Ltd.) and ImageJ 1.8.0.345 software (National
Institutes of Health) was used for gray value analysis.
Measurements of superoxide dismutase
(SOD), glutathione (GSH) and malondialdehyde (MDA)
AML cells were collected and lysed in RIPA lysis
buffer (Beyotime Institute of Biotechnology). The MDA, GSH and SOD
levels in cell lysates were measured using MDA (cat. no. ab287797;
Abcam), GSH (cat. no. E-EL-0026; Wuhan Elabscience Biotechnology
Co., Ltd.) and SOD assay kits (cat. no. ab316899;Abcam) according
to the manufacturers' instructions.
Xenograft assay
A total of 15 nude mice (male; age, 7-8 weeks;
weight, 20-22 g) were purchased from the Guangzhou Ruige Biological
Technology Co., Ltd. The mice were raised under pathogen-free
conditions (temperature: 20-26°C; humidity: 40-70%) with a 12-h
light/dark cycle, and had free access to water and food.
For in vivo imaging, 9 nude mice were
randomly divided into three groups: Control, si-NC and si-SIRT1
(n=3). Briefly, 2×106 HL60/C cells transfected with
si-NC or si-SIRT1 were resuspended in 100 μl Matrigel (cat.
no. 356234; Corning, Inc.) and were injected into the dorsal flanks
right of the midline of nude mice. Meanwhile, the same operation
with 2×106 HL60 cells was performed in the control
group. The mice were observed every day. At day 7, mice were
intraperitoneally injected with cytarabine (20 mg/kg, three times a
week) for 2 weeks. The mice in all groups underwent a luciferase
activity assay. Briefly, anesthesia was induced by 2-3% isoflurane
inhalation and was maintained using 1.5-2% isoflurane. The mice
were intraperitoneally injected with luciferin potassium salt (150
mg/kg; Shanghai Yeasen Biotechnology Co., Ltd.)and luciferase was
detected after 30 min using a live animal imaging system
(AniView100; Guangzhou Boluteng Biotechnology Co., Ltd.).
Subsequently, these 9 mice were euthanized by cervical
dislocation.
In addition, another xenograft assay was conducted.
A total of 6 nude mice were randomly divided into two groups: si-NC
and si-SIRT1 (n=3); 2×106 HL60/C cells transfected with
si-NC or si-SIRT1 were resuspended in 100 μl Matrigel and
were injected into the dorsal flanks right of the midline of nude
mice (n=3). At day 7, mice were intraperitoneally injected with
cytarabine (20 mg/kg, three times a week) for 2 weeks. The tumor
growth was observed continuously for 21 days. Prior to sacrifice,
0.2 ml of blood was collected from the orbit of the mice, the blood
was left at room temperature for 2 h, centrifuged for 10,000 x g
for 5 min at 4°C and serum was collected; subsequently, these 6
mice were euthanized by cervical dislocation to collect tumor
tissue samples before reaching the humane endpoint: Tumor weight
did not exceed 10% of body weight.
The animal experiments were performed by Guangzhou
Seyotin Biotechnology Co., Ltd. and all experiments were approved
by the Animal Ethics Committee of Guangzhou Seyotin Biotechnology
Co., Ltd. (approval nos. SYT20203010 and SYT2024079).
Transmission electron microscopy
AML cells were harvested and fixed in 2.5%
glutaral-dehyde for 3 h at 4°C. After three washes with 0.1 M
phosphate buffer, the cells were fixed with 1% OsO4 at
room temperature for 2 h. The cells were washed again with 0.1 M
phosphate buffer, and were then dehydrated with graded acetone and
embedded in Epon resin for 12 h at 37°C, for 12 h at 45°C and for
24 h at 60°C, then sectioned at 50-70 nm with an ultramicrotome.
The sections were double stained with 3% uranyl acetate and lead
citrate (ddH2O, 30 ml; lead nitrate, 1.33 g; sodium
citrate, 1.76 g) for 30 min at room temperature and observed with a
transmission electron microscope (Hitachi, Ltd.).
Isolation of cytoplasmic proteins
After treatment, nuclear and cytoplasmic protein
fractions were extracted from HL60/C cells using a Nuclear and
Cytoplasmic Protein Extraction Kit (Beyotime Institute of
Biotechnology). The protein concentration was measured using a BCA
kit (MilliporeSigma). Equal amounts of cytoplasmic proteins were
loaded on gels for SDS-PAGE and were transferred to membranes.
Western blot analysis was performed with anti-HMGB1,anti-GAPDH and
anti-Histone (1:1,000; cat. no. AF0863; Affinity Biosciences).
Statistical analysis
The experiments were repeated three times and all
data are presented as the mean ± SD. GraphPad Prism 7 (Dotmatics)
was used to analyze and plot the data. Multigroup comparisons were
performed using one-way ANOVA followed by the Tukey post hoc
multiple comparisons test. Comparisons between two groups were
performed using an unpaired Student's t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
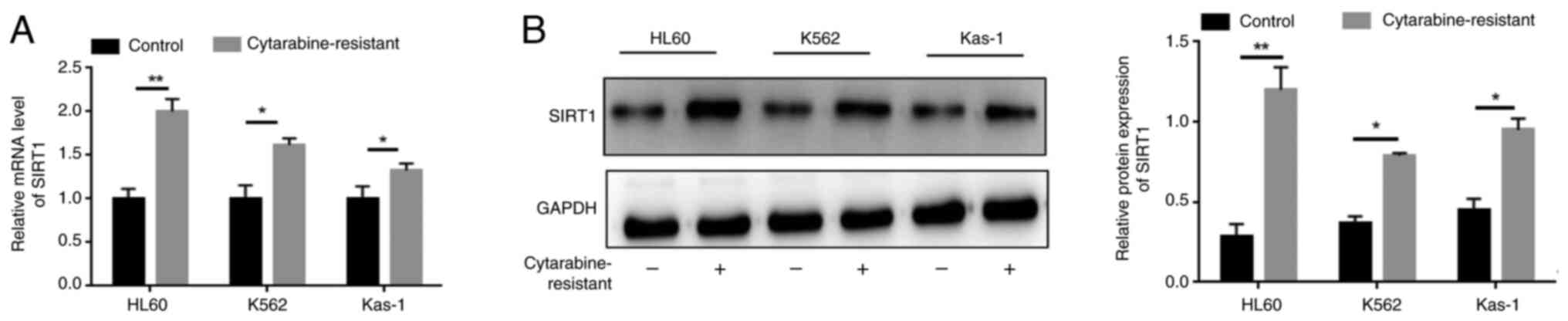
SIRT1 expression is significantly
upregulated in cytarabine-resistant leukemia cells
To address the involvement of SIRT1 in cytarabine
resistance, its expression levels in cytarabine-resistant leukemia
cells (HL-60, K562 and Kas-1) were detected. As shown in Figs. 1A and S1, the relative mRNA expression levels
of SIRT1, HMGB1and ACSL4 were significantly increased in
cytarabine-resistant leukemia cells compared with those in the
control group. Similarly, the protein expression levels of SIRT1 in
cytarabine-resistant leukemia cells were markedly increased
(Fig 1B). Taken together, these
findings indicated that SIRT1 may be elevated in
cytarabine-resistant leukemia cells.
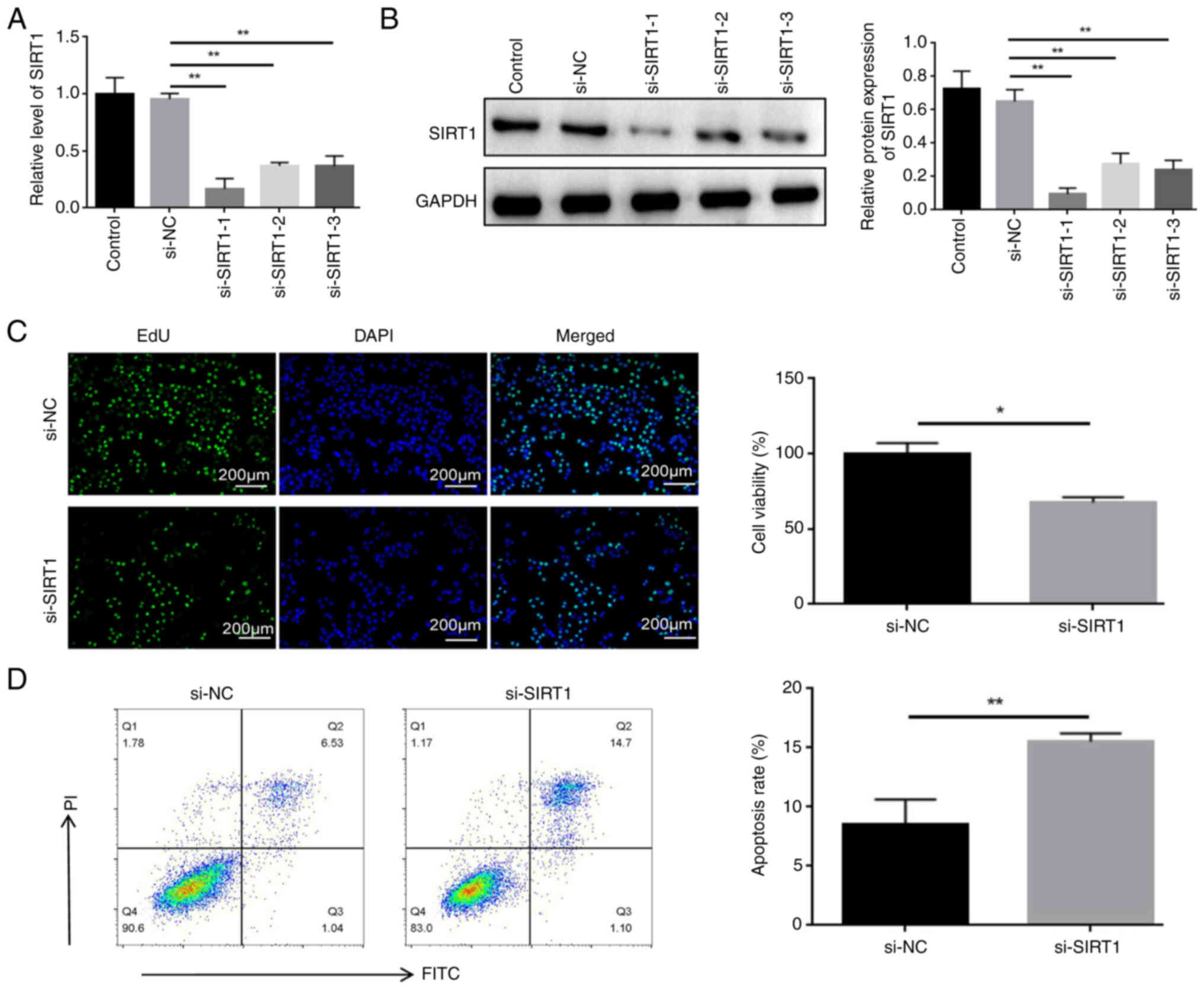
Knockdown of SIRT1 enhances apoptosis and
inhibits proliferation of HL60/C cells treated with cytarabine
To confirm the potential role of SIRT1 in
cytarabine-resistant leukemia cells, HL60/C cells were transfected
with si-SIRT1 sequences. The knockdown of SIRT1 was confirmed in
HL60/C through RT-qPCR (Fig. 2A)
and western blot analysis (Fig.
2B). Among the siRNAs, si-SIRT1-1 had the best knockdown effect
and was therefore used in subsequent experiments. Furthermore, cell
viability was significantly reduced in the si-SIRT1 group (Fig. 2C), and the IC50 value
was also significantly decreased (Fig. S2). In addition, transfection of
si-SIRT1 into HL60 cells was detected by RT-qPCR (Fig. S3A), and knockdown of SIRT1 did
not affect the viability of HL60 parental cells (Fig. S3B). The apoptosis rate was
significantly increased in the si-SIRT1 group (Fig. 2D)l; however, the apoptosis rate of
HL60 parental cells was the highest (Fig. S4). Regarding the rate of
apoptosis, Q2 represents late apoptosis and Q3 represents early
apoptosis.
Knockdown of SIRT1 enhances the
ferroptosis of HL60/C cells
To confirm the potential role of SIRT1 in
ferroptosis, HL60/C cells were transfected with si-SIRT1 sequences.
The results showed that SOD and glutathione (GSH) activities were
decreased, whereas MDA levels were elevated in the si-SIRT1 group
compared with those in the si-NC group (Fig. 3A-C). In addition, knockdown of
SIRT1 significantly enhanced ROS levels compared with those in the
si-NC group (Fig. 3D). It was
also observed that knockdown of SIRT1 reduced the expression levels
of GPX4 in HL60/C cells (Fig. 3E and
G). Moreover, mitochondrial damage is closely associated with
ferroptosis, and knockdown of SIRT1 elevated mitochondrial damage
in HL60/C cells compared with that in the si-NC group (Fig. 3F). These findings suggested that
SIRT1 may serve a critical role in ferroptosis in
cytarabine-resistant leukemia cells.
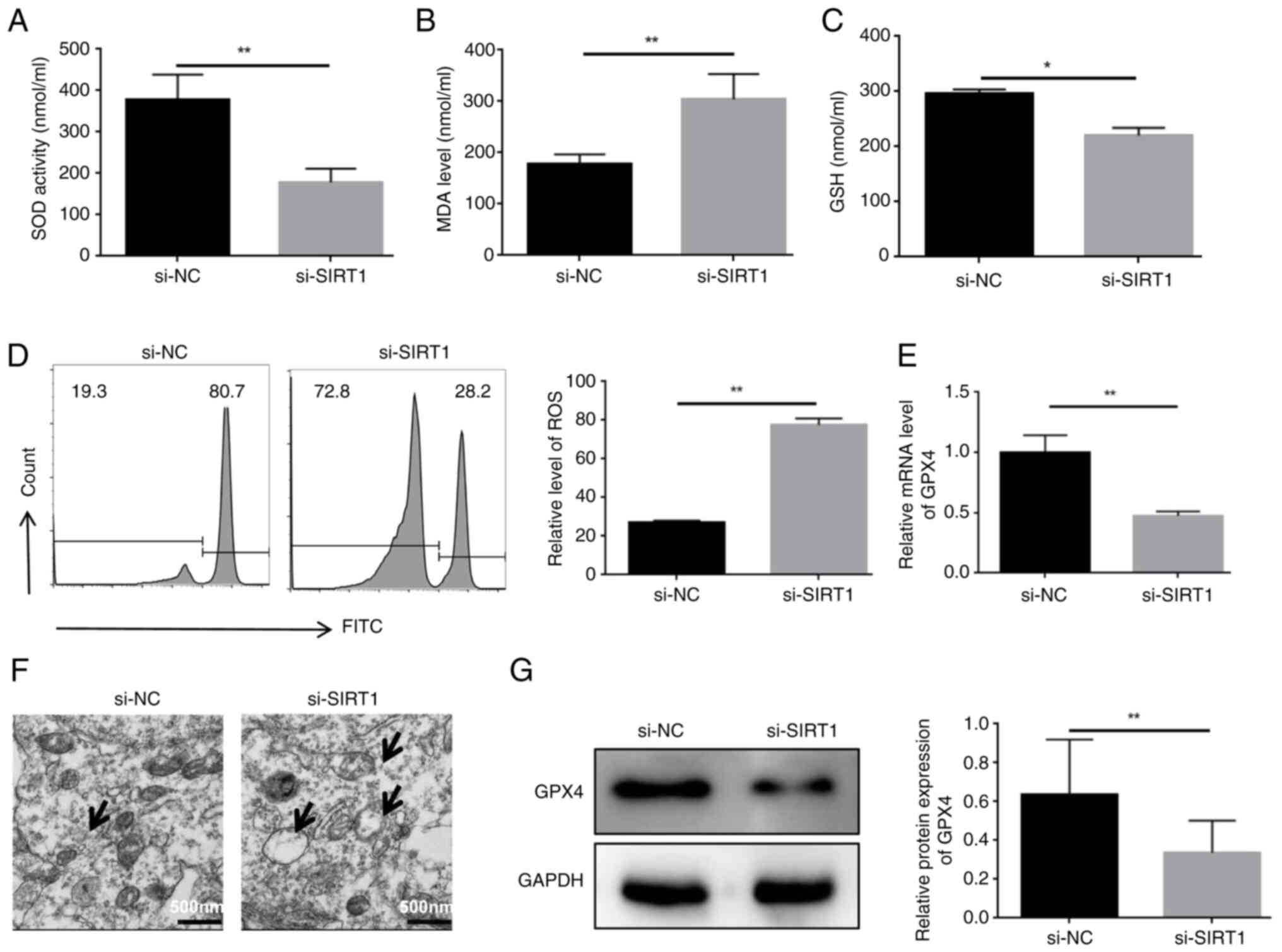 | Figure 3Knockdown of SIRT1 enhances the
ferroptosis of HL60/C cells. ELISAs were used to detect the effect
of SIRT1 knockdown on (A) SOD, (B) MDA and (C) GSH in HL60/C cells.
(D) Flow cytometry detected the impact of SITR1 knockdown on ROS
levels in HL60/C cells. (E) mRNA expression levels of GPX4 were
detected by reverse transcription-quantitative PCR. (F)
Transmission electron microscopy was used to observe mitochondrial
damage, as indicated by arrows. (G) Western blot analysis of the
effect of SIRT1 knockdown on GPX4 expression in HL60/C cells.
*P<0.05, **P<0.01. GPX4, glutathione
peroxidase 4; GSH, glutathione; HL60/C, cytarabine-resistant HL60;
MDA, malondialdehyde; NC, negative control; ROS, reactive oxygen
species; si, small interfering; SIRT1, sirtuin 1; SOD, superoxide
dismutase. |
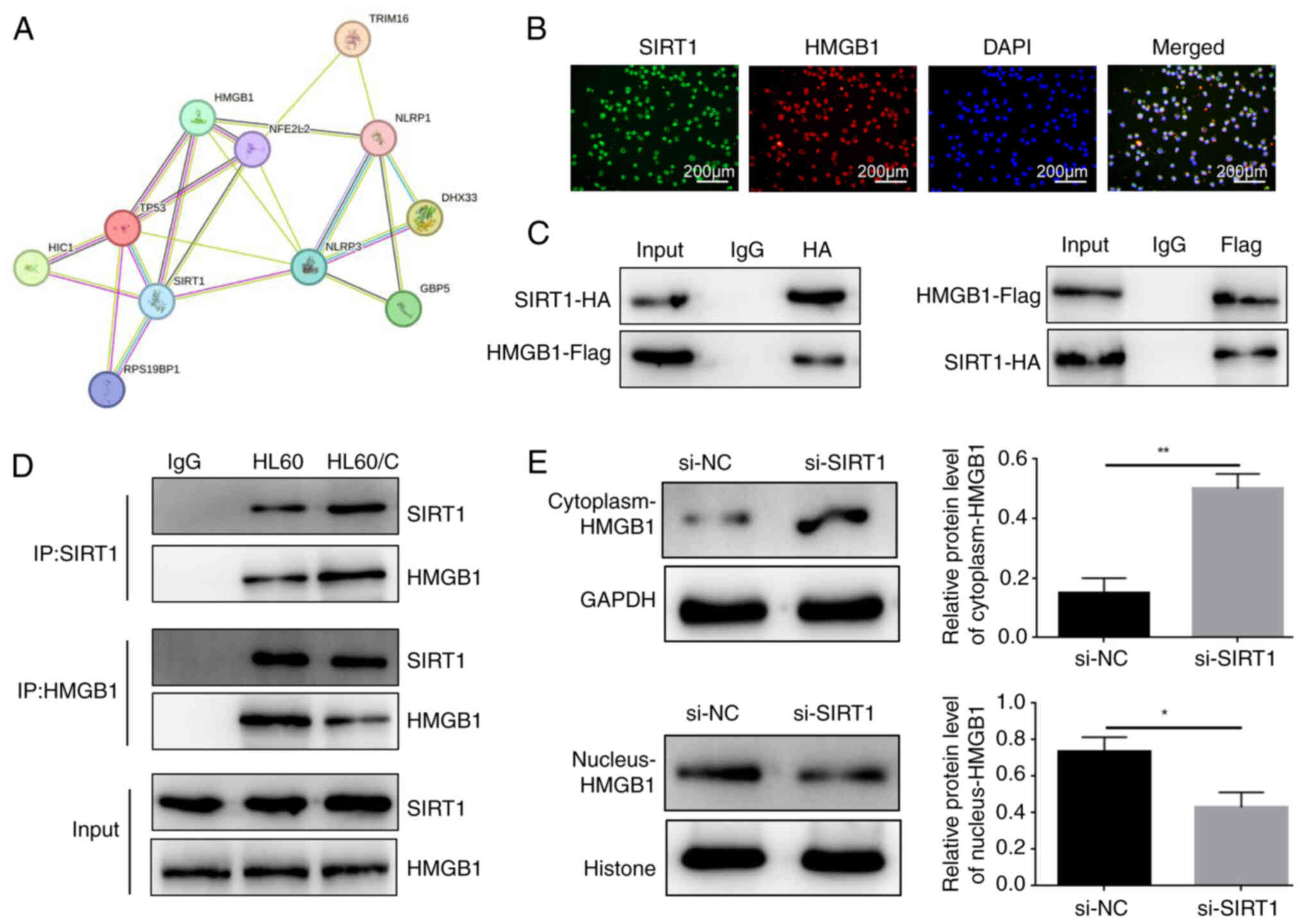
SIRT1 regulates HMGB1 expression and
inhibits the cytoplasmic translocation of HMGB1
The possible protein interaction network of SIRT1
was constructed using the STRING database (https://string-db.org/) and it was indicated that
SIRT1 may regulate HMGB1 expression (Fig. 4A). Notably, through double
immunofluorescence labelling, SIRT1 and HMGB1 were localized. The
results showed that SIRT1 and HMGB1 were expressed in the nucleus,
with obvious co-localization (Fig.
4B). Subsequently, the overexpression vectors SIRT1-HA and
HMGB1-Flag were transfected into 293T, HL60 and HL60/C cells, and
the mRNA expression levels of SIRT1 and HMGB1 were significantly
upregulated (Figs. S5 and S6).
Furthermore, the results of co-immunoprecipitation confirmed the
interaction between SIRT1 and HMGB1 in 293T cells (Fig. 4C). In addition, the same results
were detected in HL60 and HL60/C cells (Fig. 4D). Notably, it was also observed
that si-SIRT1 increased the expression levels of HMGB1 in the
cytoplasm and decreased the levels in the nucleus of HL60/C cells.
These findings indicated that SIRT1 may interact with HMGB1 and
inhibit its cytoplasmic translocation.
Knockdown of HMGB1 reduces the
ferroptosis of HL60/C cells
To confirm the potential role of HMGB1 in
ferroptosis, HL60/C cells were transfected with si-HMGB1 sequences.
The knockdown of HMGB1 was confirmed through RT-qPCR and western
blot analysis, and si-HMGB1-1 has the best knockdown effect
(Figs. 5A and S7). Subsequently, the results of the
EdU proliferation assay showed that the viability of
si-HMGB1-1-transfected cells was markedly higher than that in the
si-NC group (Fig. 5B). The
present study also examined the effects of HMGB1 knockdown on the
expression of ferroptosis-related markers. The results showed that
knockdown of HMGB1 elevated the activities of SOD, and the levels
of GSH and GPX4, compared with those in the si-NC group (Fig. 5C, F and G). By contrast, knockdown
of HMGB1 reversed the elevation of MDA and ROS in HL60/C cells
(Fig. 5D and E). Taken together,
these findings suggested that HMGB1 may induce the ferroptosis of
HL60/C cells.
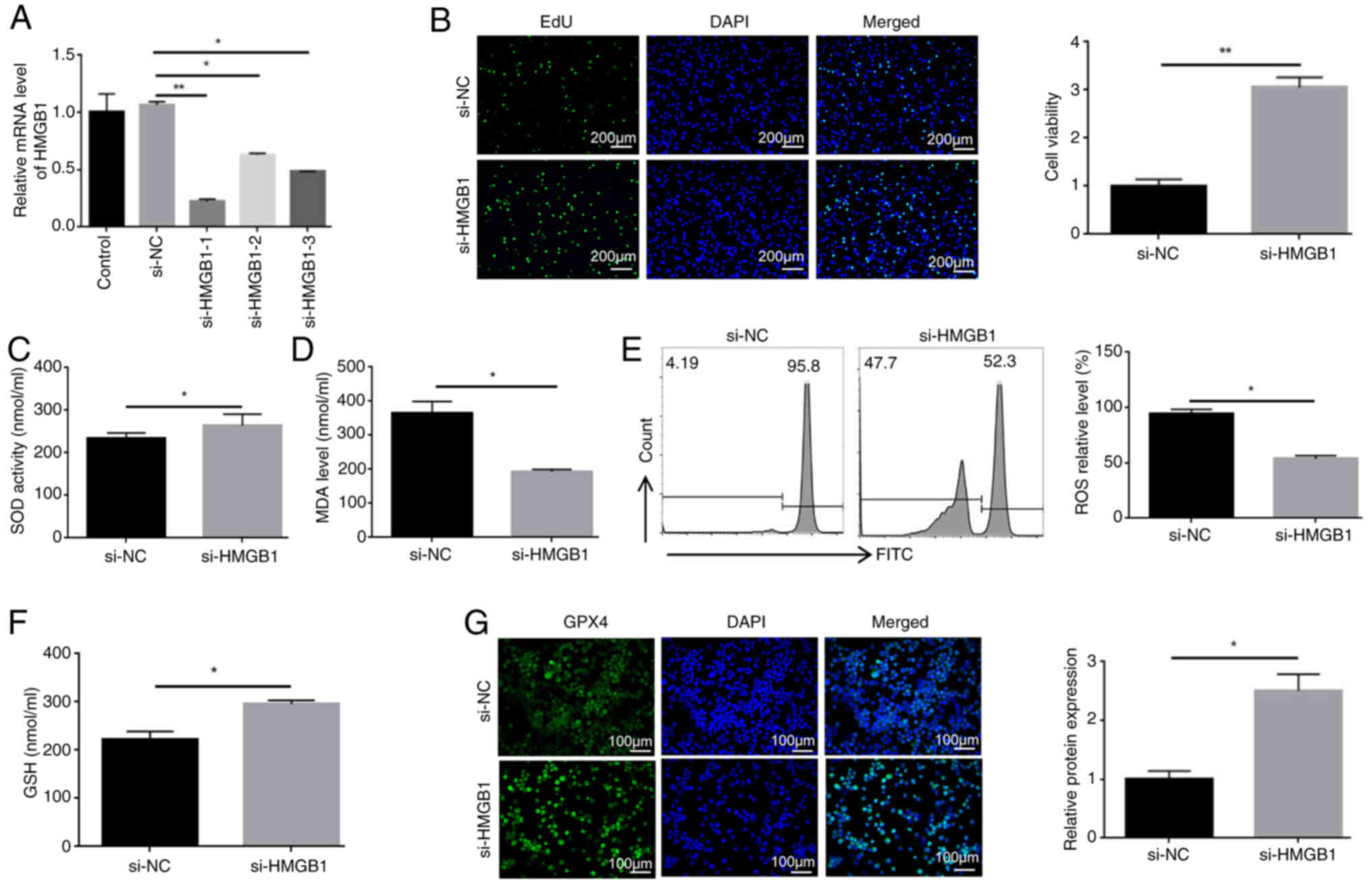 | Figure 5Knockdown of HMGB1 reduces the
ferroptosis of HL60/C cells. (A) Reverse transcription-quantitative
PCR analysis of the knockdown efficiency of si-HMGB1 sequences. (B)
EdU proliferation assay was used to detect the effect of HMGB1
knockdown on the viability of HL60/C cells. ELISAs were used to
determine the effect of HMGB1 knockdown on (C) SOD activities and
(D) MDA levels in HL60/C cells. (E) Flow cytometry detected the
impact of HMGB1 knockdown on ROS levels in HL60/C cells. (F) ELISA
was used to detect the levels of GSH in HL60/C cells. (G)
Immunofluorescence staining analyzing the effects of HMGB1
knockdown on GPX4 expression in HL60/C cells.
*P<0.05, **P<0.01. GPX4, glutathione
peroxidase 4; GSH, glutathione; HL60/C, cytarabine-resistant HL60;
HMGB1, high mobility group box-1 protein; MDA, malondialdehyde; NC,
negative control; ROS, reactive oxygen species; si, small
interfering; SOD, superoxide dismutase. |
Knockdown of HMGB1 reduces the expression
of ACSL4 in HL60/C cells
The present results suggested that, after knockdown
of HMGB1, the relative mRNA and protein expression levels of ACSL4
were significantly reduced in HL60/C cells (Fig. 6A and B). To validate the
relationship between SIRT1 and ASCL4, and the effect of this
pathway on ferroptosis, HL60/C cells were transfected with si-SIRT1
and/or si-ACSL4. As shown in Fig.
S8, si-ACSL4 significantly reduced the mRNA expression levels
of ACSL4 compared with those in the si-NC group. Notably, it was
observed that ROS and MDA levels were markedly increased in HL60/C
cells in response to si-SIRT1; however, this effect was
counteracted by knocking down ACSL4 (Fig. 6C and D). Similarly, knockdown of
SIRT1 decreased SOD activity and relative expression levels of GPX4
in HL60/R cells, but both were elevated in the si-SIRT1 + si-ACSL4
group (Fig. 6E and F). Taken
together, these findings suggested that knockdown of SIRT1 may
induce the cytoplasmic translocation of HMGB1 and increase
ferroptosis through the HMGB1/ACSL4 pathway in HL60/C cells.
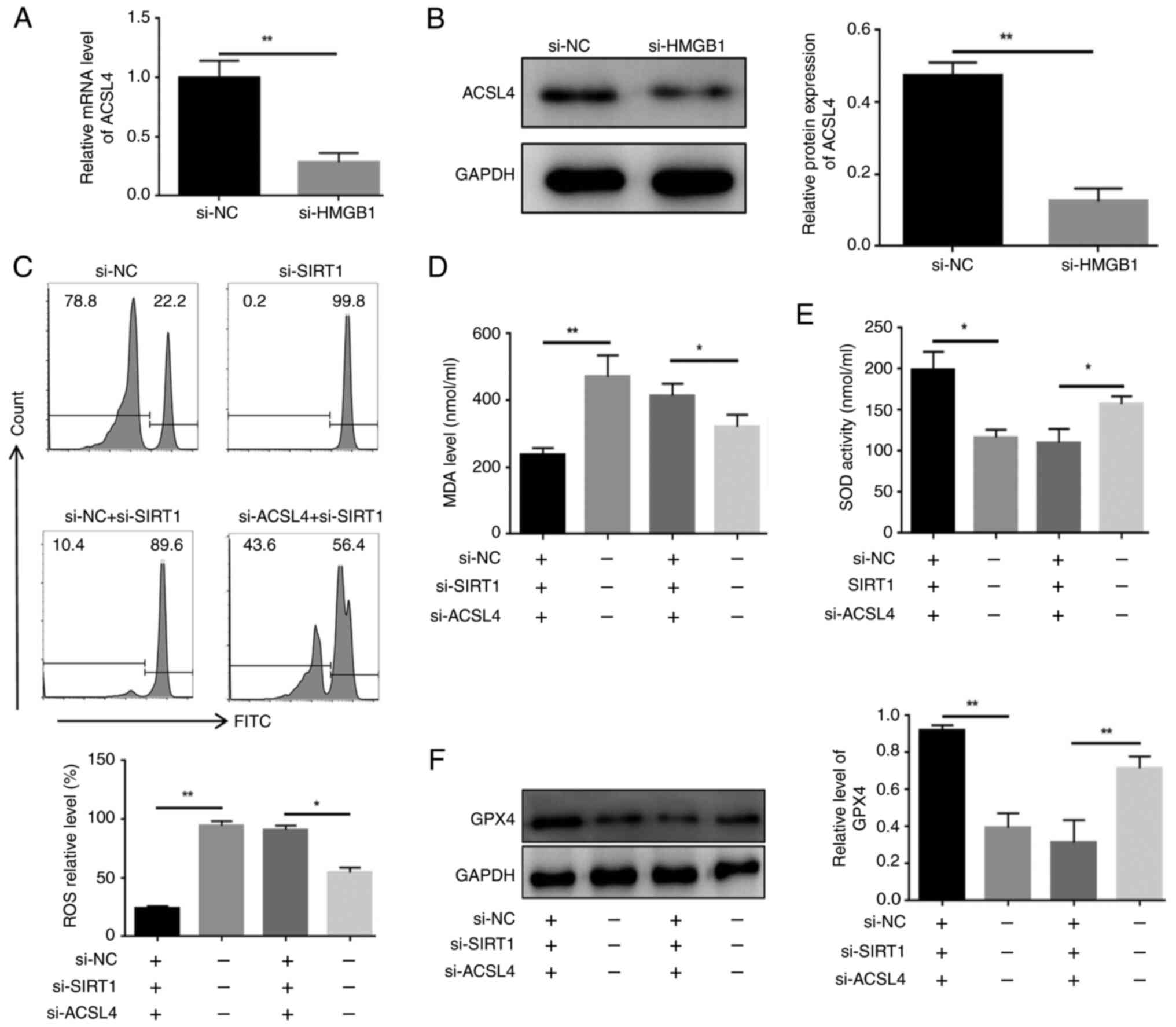 | Figure 6Knockdown of HMGB1 reduces the
expression of ACSL4 in HL60/C cells. (A) Reverse
transcription-quantitative PCR and (B) western blot analysis of the
expression levels of ACSL4 in HL60/C cells with HMGB1 knockdown.
(C) Flow cytometry detection of the impact of SIRT1 knockdown
and/or ACSL4 knockdown on ROS levels in HL60/C cells. ELISAs were
used to examine the effects of SIRT1 knockdown and/or ACSL4
knockdown on (D) MDA and (E) SOD in HL60/C cells. (F) Western blot
analysis of the expression levels of GPX4 in HL60/C cells after
SIRT1 knockdown and/or ACSL4 knockdown. *P<0.05,
**P<0.01. GPX4, glutathione peroxidase 4; HL60/C,
cytarabine-resistant HL60; HMGB1, high mobility group box-1
protein; MDA, malondialdehyde; NC, negative control; ROS, reactive
oxygen species; si, small interfering; SIRT1, sirtuin 1; SOD,
superoxide dismutase. |
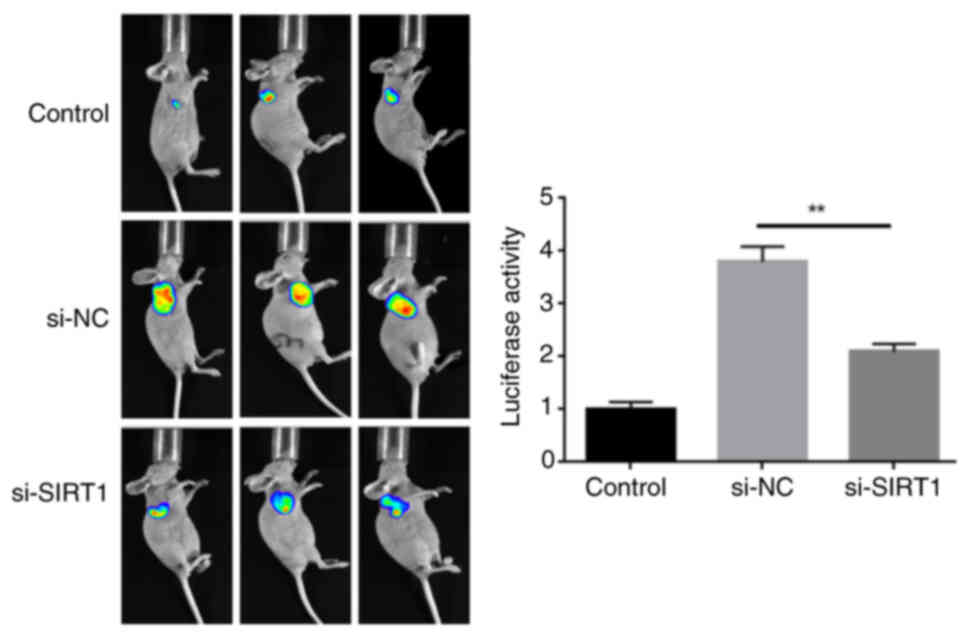
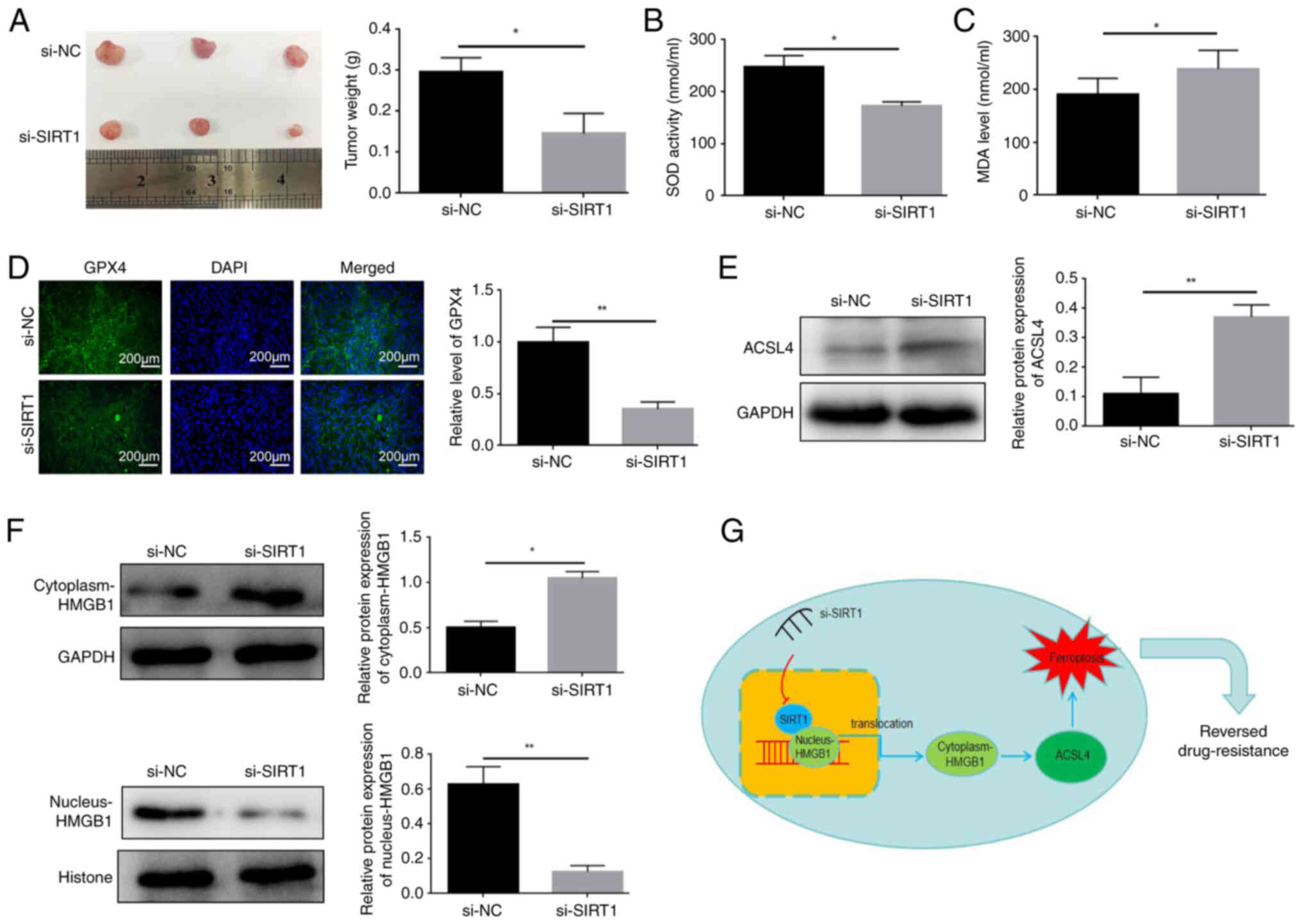
Knockdown of SIRT1 blocks the growth of
HL60/C cells in vivo
To evaluate whether knockdown of SIRT1 affects the
growth of HL60/C cells in vivo, in vivo imaging was
used to observe the size of the tumor. In vivo imaging
results showed that tumor growth was significantly inhibited in the
si-SIRT1 group compared with that in the si-NC group (Fig. 7).
In order to further explore the mechanism, a further
6 nude mice were randomly divided into two groups: si-SIRT1 or
si-NC (n=3); HL60/C cells transfected with si-SIRT1 or si-NC were
subcutaneously injected into nude mice. At day 7, mice were
intraperitoneally injected with cytarabine (20 mg/kg, three times a
week) for 2 weeks. Tumor growth was measured every other day. The
results showed that knockdown of SIRT1 significantly decreased the
weight of tumors derived from HL60/C cells in mice (Fig. 8A). Moreover, serum and tumor
tissue samples of 6 nude mice were collected, The si-SIRT1 group
exhibited decreased SOD activity and increased the MDA content in
serum compared with those in the si-NC group (Fig. 8B and C). Meanwhile, SIRT1
knockdown significantly inhibited GPX4 expression in tumor tissues
(Fig. 8D). Furthermore, it was
observed that the protein expression levels of ACSL4 in tumor
tissues were increased in the si-SIRT1 group of mice compared with
those in the si-NC group (Fig.
8E). In addition, compared with those in the si-NC group, the
protein expression levels of HMGB1 were increased in the cytoplasm
and were decreased in the nucleus of HL60/C cells after knockdown
of SIRT1 in mice tumor tissues (Fig.
8F). These findings suggested that knockdown of SIRT1 may
promote the translocation of HMGB1 from the nucleus to the
cytoplasm.
Discussion
The emergence of drug resistance is an important
problem in the treatment of AML. It has been reported that inducing
ferroptosis is an important strategy to reduce drug resistance;
however, the underlying mechanism remains unclear. The present
study revealed that SIRT1 expression was upregulated in
cytarabine-resistant leukemia cells. Knockdown of SIRT1 reversed
AML drug resistance by promoting ferroptosis. Mechanistically,
inhibition of SIRT1 expression could promote the translocation of
HMGB1 from the nucleus to the cytoplasm, and thereby enhance
ACSL4-mediated ferroptosis (Fig.
8G).
Ferroptosis is a form of cell death different from
apoptosis, necrosis and autophagy. It has been shown that
ferroptosis serves a key role in tumor suppression, thus providing
new opportunities for cancer treatment (9). The development of drug resistance to
cancer treatments remains a major challenge, and some preclinical
and clinical studies have focused on overcoming drug resistance
(3,4). Notably, ferroptosis has been shown
to be associated with cancer treatment resistance and inducing
ferroptosis may reverse resistance (21-23). Previous research has demonstrated
that erastin, a ferroptosis inducer, can improve the sensitivity of
leukemia cells to cytarabine (12); however, the underlying mechanism
remains unclear. GPX4 is a crucial regulator of ferroptosis, which
converts lipid hydroperoxides by reducing GSH, whereas SOD
functions as an antioxidant that removes excessive ROS and lipid
oxidation (24,25). Prior research has shown that as
ferroptosis progresses, lipid peroxidation generates elevated
levels of ROS and MDA (25). In
the present study, knockdown of SIRT1 could enhance GPX4 expression
and SOD activity, whereas it decreased ROS and MDA content in
HL60/C cells. These findings suggested that SIRT1 knockdown could
reverse cytarabine resistance in AML by enhancing ferroptosis.
A growing body of evidence has established that
HMGB1 serves a significant role in multiple drug resistance,
including resistance to thiopurines, 5-fluorouracil and cytarabine
(3). Upregulation of HMGB1
expression in erastin-treated HL60 cells expressing
NRASQ61L has previously been demonstrated (26). As expected, the present results
showed that silencing HMGB1 increased the levels of GPX4 and SOD
but decreased the levels of MDA and ROS in HL60/C cells, which was
consistent with previous findings (6). These results provide evidence
supporting the role of HMGB1 in the process of ferroptosis in
cytarabine-resistant leukemia cells. HMGB1 is normally localized in
the nucleus of cells, but is released extracellularly when the body
is exposed to stimuli or damage (27,28). HMGB1 can function as both a
nuclear DNA-binding molecule and a secreted protein. The
localization and biological function of HMGB1 are determined by its
post-translational modification (29). It has previously been shown that
treatment with chemotherapeutic drugs can promote the translocation
of HMGB1 from the nucleus to the cytoplasm (30). The present study observed an
increase in the cytoplasmic levels of HMGB1 and a decrease in its
nuclear level in the si-SIRT1 group compared with in the si-NC
group. While SIRT1 has been identified as serving an important role
in cytarabine resistance, the underlying mechanism remains
partially understood (17,31).
A previous study reported that SIRT1 could inhibit HMGB1
translocation to the cytoplasm and the expression of inflammatory
cytokines, attenuating bone cancer pain symptoms in mice (32). Furthermore, cytoplasmic
translocation of HMGB1 has been detected in high glucose-treated
mesangial cells, which can increase positive regulators of
ferroptosis, such as ACSL4, and enhance ferroptosis (33). In the present study, the results
showed that knockdown of SIRT1 elevated the translocation of HMGB1
from the nucleus to the cytoplasm, and increased the expression
levels of ACSL4 in vivo.
In conclusion, the present study demonstrated that
induction of ferroptosis by SIRT1 knockdown may alleviate
cytarabine resistance in AML by activating the HMGB1/ACSL4 pathway.
Therefore, targeting SIRT1 may hold promise as a strategy to
overcome cytarabine resistance in AML.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
QK and HC conceived and designed the experiments. QL
and YT performed the experiments. XL, XL, YC and YL analyzed and
interpreted the data. QK and HC confirm the authenticity of all the
raw data, QK wrote the paper. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All experiments were approved by the Animal Ethics
Committee of the Guangzhou Seyotin Biotechnology Co., Ltd.
(approval no. SYT20203010). The requirement to obtain additional
approval from the authors' own institution for the outsourcing of
the animal experiments was waived by the ethics committee of the
Third Affiliated Hospital of Sun Yat-sen University (Guangzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
AML
|
acute myeloid leukemia
|
|
HMGB1
|
high mobility group box-1 protein
|
|
CCK-8
|
Cell Counting Kit-8
|
|
ROS
|
reactive oxygen species
|
Acknowledgements
Not applicable.
Funding
This work was supported by the Doctoral Start-up Fund of Natural
Science Foundation of Guangdong Province under Grant (grant no.
2016A030310161) and the National Natural Science Foundation for
Young Scientists of China (grant no. 82100181).
References
|
1
|
Pelcovits A and Niroula R: Acute myeloid
leukemia: A review. R I Med J. 103:38–40. 2013.
|
|
2
|
Ganesan S, Mathews V and Vyas N:
Microenvironment and drug resistance in acute myeloid leukemia: Do
we know enough? Int J Cancer. 150:1401–1411. 2022. View Article : Google Scholar
|
|
3
|
Heuser M, Ofran Y, Boissel N, Brunet Mauri
S, Craddock C, Janssen J, Wierzbowska A and Buske C; ESMO
Guidelines Committee: Electronic address:
clinicalguidelines@esmo.org: Acute myeloid leukaemia in adult
patients: ESMO Clinical Practice Guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 31:697–712. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cros E, Jordheim L, Dumontet C and
Galmarini CM: Problems related to resistance to cytarabine in acute
myeloid leukemia. Leuk Lymphoma. 45:1123–1132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011. View Article : Google Scholar
|
|
6
|
Ye F, Chai W, Xie M, Yang M, Yu Y, Cao L
and Yang L: HMGB1 regulates erastin-induced ferroptosis via
RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am J
Cancer Res. 9:730–739. 2019.
|
|
7
|
Krynetskaia N, Xie H, Vucetic S, Obradovic
Z and Krynetskiy E: High mobility group protein B1 is an activator
of apoptotic response to antimetabolite drugs. Mol Pharmacol.
73:260–269. 2008. View Article : Google Scholar
|
|
8
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang C, Liu X, Jin S, Chen Y and Guo R:
Ferroptosis in cancer therapy: A novel approach to reversing drug
resistance. Mol Cancer. 21:472022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hong T, Lei G, Chen X, Li H, Zhang X, Wu
N, Zhao Y, Zhang Y and Wang J: PARP inhibition promotes ferroptosis
via repressing SLC7A11 and synergizes with ferroptosis inducers in
BRCA-proficient ovarian cancer. Redox Biol. 42:1019282021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Q and Wang K: The induction of
ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the
sensitivity of osteosarcoma cells to cisplatin. Cell Biol Int.
43:1245–1256. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze
MT, Zeh HJ, Kang R and Tang D: The ferroptosis inducer erastin
enhances sensitivity of acute myeloid leukemia cells to
chemotherapeutic agents. Mol Cell Oncol. 2:e10545492015. View Article : Google Scholar
|
|
13
|
Shen P, Deng X, Chen Z, Ba X, Qin K, Huang
Y, Li T, Yan J and Tu S: SIRT1: A potential therapeutic target in
autoimmune diseases. Front Immunol. 12:7791772021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim TH, Young SL, Sasaki T, Deaton JL,
Schammel DP, Palomino WA, Jeong J-W and Lessey BA: Role of SIRT1
and progesterone resistance in normal and abnormal endometrium. J
Clin Endocrinol Metab. 107:788–800. 2022. View Article : Google Scholar :
|
|
15
|
Liu Z, Li C, Yu C, Chen Z, Zhao C and Ye
L: TSPYL2 reduced gefitinib resistance and DNA damage repair via
suppressing SIRT1-mediated FOXO3 deacetylation. Future Med Chem.
14:407–419. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu X, Su Q, Xie H, Song L, Yang F, Zhang
D, Wang B, Lin S, Huang J, Wu M and Liu T: SIRT1 deacetylates WEE1
and sensitizes cancer cells to WEE1 inhibition. Nat Chem Biol.
19:585–595. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin Y, Cao Q, Chen C, Du X, Jin B and Pan
J: Tenovin-6-mediated inhibition of SIRT1/2 induces apoptosis in
acute lymphoblastic leukemia (ALL) cells and eliminates ALL
stem/progenitor cells. BMC Cancer. 15:2262015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao Z, Li G, Wang Y, Li Y, Xu H, Liu W,
Hao W, Yao Y and Zeng R: Cytoplasmic HMGB1 induces renal tubular
ferroptosis after ischemia/reperfusion. Int Immunopharmacol.
116:1097572023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gu L, Zhang G and Zhang Y: A novel method
to establish glucocorticoid resistant acute lymphoblastic leukemia
cell lines. J Exp Clin Cancer Res. 38:2692019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Mandke P and Vasquez KM: Interactions of
high mobility group box protein 1 (HMGB1) with nucleic acids:
Implications in DNA repair and immune responses. DNA Repair (Amst).
83:1027012019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Luo L, Wang S, Chen B, Zhong M, Du R, Wei
C, Huang F, Kou X, Xing Y and Tong G: Inhibition of inflammatory
liver injury by the HMGB1-A box through HMGB1/TLR-4/NF-kappaB
signaling in an acute liver failure mouse model. Front Pharmacol.
13:9900872022. View Article : Google Scholar
|
|
23
|
Ling VY, Straube J, Godfrey W, Haldar R,
Janardhanan Y, Cooper L, Bruedigam C, Cooper E, Shirazi PT,
Jacquelin S, et al: Targeting cell cycle and apoptosis to overcome
chemotherapy resistance in acute myeloid leukemia. Leukemia.
37:143–153. 2023. View Article : Google Scholar
|
|
24
|
Zhang H, Liu L, Chen L, Liu H, Ren S and
Tao Y: Long noncoding RNA DANCR confers cytarabine resistance in
acute myeloid leukemia by activating autophagy via the
miR-874-3P/ATG16L1 axis. Mol Oncol. 15:1203–1216. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chromik J, Safferthal C, Serve H and Fulda
S: Smac mimetic primes apoptosis-resistant acute myeloid leukaemia
cells for cytarabine-induced cell death by triggering necroptosis.
Cancer Lett. 344:101–109. 2014. View Article : Google Scholar
|
|
26
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar :
|
|
27
|
Wang X, Wang Z, Cao J, Dong Y and Chen Y:
Melatonin alleviates acute sleep deprivation-induced memory loss in
mice by suppressing hippocampal ferroptosis. Front Pharmacol.
12:7086452021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng H, Chen JN, Yu X, Jiang P, Yuan L,
Shen HS, Zhao LH, Chen PF and Yang M: HMGB1 enhances drug
resistance and promotes in vivo tumor growth of lung cancer cells.
DNA Cell Biol. 35:622–627. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Splichal I, Donovan SM, Jenistova V,
Splichalova I, Salmonova H, Vlkova E, Bunesova VN, Sinkora M,
Killer J, Skrivanova E and Splichalova A: High mobility group box 1
and TLR4 signaling pathway in gnotobiotic piglets
colonized/infected with L. amylovorus, L. mucosae, E. coli Nissle
1917 and S. Typhimurium. Int J Mol Sci. 20:62942019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li J, Zhou W, Mao Q, Gao D, Xiong L, Hu X,
Zheng Y and Xu X: HMGB1 promotes resistance to doxorubicin in human
hepatocellular carcinoma cells by inducing autophagy via the
AMPK/mTOR signaling pathway. Front Oncol. 11:7391452021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bhanot H, Weisberg EL, Reddy MM, Nonami A,
Neuberg D, Stone RM, Podar K, Salgia R, Griffin JD and Sattler M:
Acute myeloid leukemia cells require 6-phosphogluconate
dehydrogenase for cell growth and NADPH-dependent metabolic
reprogramming. Oncotarget. 8:67639–67650. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen X, Chen C, Fan S, Wu S, Yang F, Fang
Z, Fu H and Li Y: Omega-3 polyunsaturated fatty acid attenuates the
inflammatory response by modulating microglia polarization through
SIRT1-mediated deacetylation of the HMGB1/NF-kappaB pathway
following experimental traumatic brain injury. J Neuroinflammation.
15:1162018. View Article : Google Scholar
|
|
33
|
Wu Y, Zhao Y, Yang HZ, Wang YJ and Chen Y:
HMGB1 regulates ferroptosis through Nrf2 pathway in mesangial cells
in response to high glucose. Biosci Rep. 41:BSR202029242021.
View Article : Google Scholar : PubMed/NCBI
|