Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common type of cancer, with an annual incidence of
~600,000 new cases globally (1,2).
Despite advances in surgery and radiotherapy, as well as the
incorporation of chemotherapy into treatment modalities, the 5-year
survival remains ~50% and has barely improved over the past decades
(3). The majority of HNSCC cases
are tobacco- and alcohol-associated, and in oropharyngeal cancer,
infection with high-risk human papillomavirus (HPV) is an oncogenic
factor (4). HPV-positive HNSCC is
biologically and clinically distinct from HPV-negative HNSCC
(5). Notably, most patients with
a HPV-positive tumor have a more favorable prognosis, with a 30-40%
higher 5-year survival rate than patients with a HPV-negative tumor
(6). However, the HPV status of
the tumor is not predictive for individual patient outcome. A
subgroup of patients with HPV-positive tumors has a worse
prognosis, characterized by a higher risk of disease recurrence
and/or secondary primary tumors. Although the underlying reason for
this less favorable prognosis is not completely understood,
exposure to additional risk factors, such as tobacco smoking, may
play a role (7). Treatment with
radiation or surgery alone is typically indicated for early-stage
disease, whereas combined approaches, including surgery,
radiotherapy and chemotherapy is generally applied for
locoregionally advanced disease (8,9).
For inoperable, locally advanced HNSCC, a combination treatment
with radiotherapy with cisplatin remains the standard of care. For
patients that are unfit for cisplatin, treatment with cetuximab, a
monoclonal antibody directed against epidermal growth factor
receptor, could be an alternative to chemotherapy. Novel
immunotherapies, including programmed cell death protein-1
checkpoint inhibitors such as nivolumab and pembrolizumab, could be
considered as treatment option for patients with recurrent or
metastatic HNSCC, irrespective of HPV-status (9). These immunotherapies have shown
durable responses, but this benefit was only observed in a limited
number of patients (9).
Furthermore, the currently used treatment modalities often result
in severe side effects and a reduction in quality of life, which is
particularly important for patients with an unfavorable prognosis.
Therefore, new agents that can improve survival rates of patients
with HPV-negative and HPV-positive HNSCC, without causing severe
side effects, are urgently needed.
Recent whole-exome sequencing studies have revealed
a wide spectrum of genetic aberrations and molecular diversity in
HNSCC (5,10). Frequently deregulated cellular
pathways include the cell cycle and the phosphatidylinositol
3-kinase (PI3K) signaling pathway, which regulate cell
proliferation, survival and apoptosis (11,12). Genetic alterations associated with
deregulation of the cell cycle machinery are detected in nearly all
cases of HNSCC (13).
Retinoblastoma (Rb1) tumor suppressor protein plays a critical role
in regulating cellular proliferation. Cyclin D-cyclin-dependent
kinase 4/6 (CDK4/6) may phosphorylate and inactivate Rb1, leading
to the release and activation of E2F transcription factors
necessary for G1-S phase cell cycle progression (14). In HPV-positive HNSCC, viral
oncoprotein E7 drives unrestrained proliferation by promoting Rb1
degradation, which also leads to p16 upregulation (14). In HPV-negative HNSCC, Rb1
inactivation occurs through hyperactivation of the Rb1 inhibitory
complex CDK4/6-Cyclin D. The CCND1 gene (encoding Cyclin D1,
the regulatory subunit of the complex) is amplified, and/or the
CDK4/6 inhibitor, p16, is inactivated in nearly all of these cancer
types, preventing the phosphorylation of Rb1 (15-17).
The Cancer Genome Atlas (TCGA) data demonstrates
that >50% of HPV-positive and HPV-negative HNSCC cases harbor
activated PI3K (and related pathways) signaling, mainly due to
mutations in or amplifications of PIK3CA, loss of
PTEN or activation of receptor tyrosine kinases (13). PI3K activation leads to synthesis
of phosphatidylinositol 3,4,5-trisphosphate (PIP3) at the plasma
membrane, resulting in the recruitment of pleckstrin homology
domain-containing proteins phosphoinositide dependent protein
kinase-1 (PDK1) and Akt. Akt is phosphorylated by PDK1 at Thr308,
resulting in activation of downstream proteins, including mammalian
target of rapamycin (mTOR) complex 1. The activation of the PI3K
pathway is associated with resistance to chemotherapy and other
targeted therapies and plays a crucial role in cell energy
metabolism (18). Therefore,
inhibition of the PI3K pathway may be an important step and one of
the most promising targets in anticancer therapy, including for
HNSCC (19,20).
There are several CDK4/6 inhibitors (CDKi) and
PI3K/Akt/mTOR pathway inhibitors (PI3Ki) available. Palbociclib
(PD-0332991; Pfizer) is a selective CDKi that was first approved
for the treatment of breast cancer (21). Ribociclib (LEE011; Novartis) is a
selective orally bioavailable CDKi that received Food and Drug
Administration (FDA) approval in March, 2017 (22). Both drugs bind to the ATP cleft of
CDK4 and CDK6. Alpelisib (BYL719; Novartis) is an oral selective
PI3K p110α isoform inhibitor (23,24) that has been approved by the FDA in
combination with fulvestrant for the treatment of hormone
receptor-positive, human epidermal growth factor receptor 2
(HER2)-negative, PIK3CA-mutated metastatic breast cancer.
This drug inhibits wild-type PI3K p110α and mutated PI3K p110α (as
a result of PIK3CA mutations). Buparlisib (BKM120; Novartis)
is a 2,6-dimorpholino pyrimidine derivative that significantly
inhibits wild-type and mutant PI3K catalytic subunit p110 (α, β, δ
and γ) (25). Gedatolisib
(PF-05212384; Pfizer) is a highly potent dual inhibitor of PI3K (α,
β, δ and γ) and mTOR (TORC1 and TORC2). In vitro,
gedatolisib potently inhibits class I PI3Ks, PI3K-α mutants and
mTOR (26).
The aim of the present study was to investigate the
in vitro antiproliferative effects of several CDKi
(palbociclib and ribociclib) and PI3Ki (gedatolisib, buparlisib and
alpelisib) in HPV-positive and -negative HNSCC cell lines. We
hypothesized that CDKi are effective inhibitors in HPV-negative
HNSCC cell lines, associated with hyperactivation of the Rb1
inhibitory complex, CDK4/6-Cyclin D. In addition, PI3Ki are
expected to be effective in both HPV-negative and -positive HNSCC
cell lines related to active PI3K/Akt/mTOR signaling.
Materials and methods
Cell lines and culture conditions
In total, five HPV16-positive HNSCC cell lines:
UD-SCC-2 (kindly provided by Thomas Hoffmann, University of Ulm,
Germany), 93-VU-147T (kindly provided by Johan P. De Winter, VU
Medical Center, Amsterdam, The Netherlands), UM-SCC-47 and
UM-SCC-104 (both kindly provided by Thomas E. Carey, University of
Michigan, USA) and UPCI-SCC-090 (kindly provided by Susanne M.
Gollin, University of Pittsburgh, USA) were used in the present
study. In addition, three HPV16-negative HNSCC cell lines:
UPCI-SCC-72 and UPCI-SCC-003 (both kindly provided by Susanne M.
Gollin) and UT-SCC-33 (kindly provided by R.A. Grenman, Turku
University, Finland) were used. The MCF-7, HeLa, CaSki and SiHa
cell lines were purchased from the American Type Culture Collection
and the HaCaT cell line was purchased from CLS Cell Line Service
GmbH. The normal oral keratinocyte (NOK) cell line was prepared
from gingival tissues obtained from oral surgeries and immortalized
by activation of h-TERT, as described previously (27-29). The NOK cell line was immortalized
and kindly provided by Karl Munger, Tufts University Medical
School, USA.
Cells were cultured at 37°C in a humidified
atmosphere with 5% CO2. All HNSCC cell lines (except for
the UT-SCC-33), HaCaT, HeLa and MCF-7 were cultured in Dulbecco's
Modified Eagle Medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.)
containing 10% fetal calf serum (FCS; Bodinco BV). UT-SCC-33 and
SiHa cells were cultured in MEM (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FCS. CasKi was cultured in Roswell Park
Memorial Institute (RPMI) with 10% FCS. The NOK cell line was
cultured in keratinocyte serum-free medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with bovine pituitary extract (2.6
μg/ml) and recombinant EGF (0.16 ng/ml). The
clinicopathological cell line characteristics, including genetic
alterations, are presented in Table
SI. To confirm p16/HPV status and determine mutational status
of pathways that may be relevant for inhibitor efficacy,
immunocytochemical staining for p16 and mutation analysis were
performed as previously described (6,30).
All cell lines were regularly tested and found to be
mycoplasma-free. All cell lines were confirmed to have unique
genotypes, as determined using the ProfilerPlus assay (31). The presence of HPV DNA was
detected using PCR as previously described (32,33). Stocks of palbocliclib and
gedatolisib were provided by Pfizer, Inc. and stocks of alpelisib,
buparlisib, and ribociclib were provided by Novartis International
AG.
In vitro cell viability assay
The MTT assay is used to measure cellular metabolic
activity as an indicator of cell viability, proliferation and
cytotoxicity. This colorimetric assay is based on the reduction of
a yellow tetrazolium salt
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide or
MTT] to purple formazan crystals in metabolically active cells by
mitochondrial dehydrogenases, predominantly succinate dehydrogenase
(34,35). In this present study, cells were
seeded in 96-well flat-bottom plates at densities that allowed for
exponential growth throughout the experiment. The cells were placed
in the cell culture incubator overnight at 37°C allowing the cells
to attach, after which they were treated with different
concentrations of the different test compounds (Table SII). The compounds were resolved
in DMSO at a non-toxic (0.1%; 0.2% for palbociclib) concentration
of DMSO at the cellular level. At the indicated time points (PI3Ki:
day 3, CDKi: day 3 and 5), the MTT assay (Sigma-Aldrich; Merck
KGaA) was performed as previously described (34). Purple formazan crystals were
dissolved in ethanol/DMSO solution (1:1) and the absorbance was
measured at a wavelength of 595 nm with a spectrophotometer. The
experiments were performed in triplicate.
Western blot analysis
Cells treated with the compounds or control were
lysed with RIPA buffer (Cell Signaling Technology, Inc.) containing
Protease/Phosphatase Inhibitor Cocktail for 5 min on ice, followed
by brief sonication (30 sec, 47 kHz, 4°C). After centrifugation (10
min, 14,000 × g, 4°C), the pellet was discarded, and the protein
extracts were quantified using a Pierce BCA Protein Assay Kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Equal amounts of the extracts (10-30 μg) were
separated on 8-12% SDS-PAGE gels and transferred to nitrocellulose
membranes according to the manufacturers' instructions using a
Mini-Protean Tetra System (Bio-Rad Laboratories, Inc.). Membranes
were blocked with 5% bovine serum albumin (BSA; Sigma-Aldrich;
Merck KGaA) for 1 h at room temperature and incubated with primary
antibodies diluted in blocking buffer (5% BSA diluted in TBS/Tween
0.1%) overnight at 4°C. For detection, secondary antibodies labeled
with horseradish peroxidase were incubated with the membranes for 1
h at room temperature. The bands were visualized with enhanced
chemiluminescence (SuperSignal West Dura Extended Duration
Substrate; Thermo Fisher Scientific, Inc.) using an Image reader
LAS-3000 (FUJIFILM Wako Pure Chemical Corporation). The primary and
secondary antibodies, including the dilutions used, are listed in
Table SIII. The experiments were
performed in triplicate.
Cell cycle analysis
Cells were seeded in 6-well culture plates, placed
in the cell culture incubator at 37°C and allowed to attach
overnight. The culture medium containing the inhibitor or DMSO was
added to the cells. After 24 h, the cells were washed with PBS and
trypsinized to form a cell pellet. Ice-cold 70% ethanol was added
to the cell pellet while vortexing, ensuring cell fixation and
minimizing cell clumping. Cells in 70% ethanol were stored at −20°C
for a minimum of 30 min. The cells were then washed with PBS and
resuspended in 0.5 ml propidium iodide (PI)/RNAse staining solution
(100 μg/ml PI and 1 mg/ml RNAse in PBS). The cells were
incubated for 30 min at room temperature and analyzed by flow
cytometry using a FACSCanto II (BD Biosciences). Data analysis was
performed using FACSdiva software version 6.1.2 (BD Biosciences).
The different cell cycle regions were set to those defined by the
untreated control cells for each individual cell line.
Annexin-V apoptosis assay
For the Annexin V assay, cells were seeded in
96-well imaging microplates and allowed to attach overnight at
37°C. Cells were treated with 500 nM staurosporine and two
different concentrations of PI3Ki for 24 h. The cells were then
stained with Hoechst 33342 (200 μg/ml; Sigma-Aldrich; Merck
KGaA) in culture medium for 15 min at 37°C. Cells were washed with
Annexin-V binding buffer (10 mM HEPES pH 7.4, 140 mM NaCl, 5 mM
CaCl2 made up in PBS) and stained with Annexin-V-FITC
(2.5 μg/ml in Annexin-V binding buffer) for 15 min at 37°C.
Imaging was performed using a BDpathway 855 High-Content Bioimager
(BD Biosciences). Digitalization and segmentation of the acquired
data were performed using Attovision software Version 1.6 (BD
Biosciences). The processed data were evaluated using FACSDiva
software version 6.1.2 (BD Biosciences).
β-galactosidase staining
Cells were seeded in 24-well plates and allowed to
attach overnight. The cells were treated with palbociclib (2
μM), alpelisib (10 μM) or a combination of the two
for 6 days. Expression of β-galactosidase was determined using the
Senescence Detection Kit (Abcam; cat. no. ab65351), following the
manufacturer's instructions. Briefly, cells were washed with PBS
and fixed, followed by staining with the provided X-gal solution.
The plate was covered and incubated in a ziplock bag at 37°C
overnight. The stained cells were observed under a light microscope
(magnification, ×200).
Measurement of glucose uptake and lactate
release
Cells were seeded in 96-well plates and treated with
PI3Ki. After 24 h, the cell culture medium was collected and pooled
to a volume of 300 μl per condition. The glucose and lactate
concentrations were determined using a D-Glucose Enzymatic Assay
Kit and an L-Lactate Acid Enzymatic Assay Kit from BioSenTec, by
measuring absorbance at 340 nm. The reaction volumes were optimized
for use in 96-well plates, and a standard curve was used to
determine the glucose and lactate concentrations. To measure the
lactate content, the cell culture medium was deproteinized prior to
the assay using 10 kDa filter units (Merck KGaA; MRCPRT010). The
determined glucose and lactate concentrations were normalized to
μg of protein, measured using the Pierce BSA protein assay
kit (Thermo Fisher Scientific, Inc.).
Measurement of oxygen consumption rate
(OCR) and extracellular acidification rate (ECAR)
OCR and ECAR measurements were performed using an
XF96 Extracellular Flux analyzer and Mito Stress Test and Glyco
Stress test assays (all Seahorse Bioscience; Agilent Technologies,
Inc.), according to the manufacturer's instructions. Briefly, the
cells were seeded at optimized densities (7,300 for UM-SCC-47,
7,500 for UPCI-SCC-003 and 38,000 for UD-SCC-2) in 80 μl of
growth medium in an XF96 culture plate well. After attachment
overnight, the cells were treated with vehicle control or the
IC50 of the PI3Ki for 24 h, washed in XF assay medium
and kept in XF assay medium (with inhibitors) at 37°C in a
non-CO2 incubator for 1 h prior to the assay.
Mitochondrial respiration was determined using the Mito Stress Test
after the subsequent injection of oligomycin (1 μM), FCCP (1
μM) and a mixture of rotenone and antimycin A (both 1
μM) (all from Sigma-Aldrich; Merck KGaA) to determine the
basal and ATP-coupled respiration. Baseline respiration was
determined by subtracting non-mitochondrial respiration (OCR values
obtained after injection of antimycin A/rotenone) from the initial
OCR. ATP-linked respiration is the OCR decrease after the injection
of oligomycin. Injection of FCCP collapses the mitochondrial
membrane potential and results in maximal OCR. Finally, the
injection of rotenone/antimycin A (inhibitors of complex III and I,
respectively) block the mitochondrial respiratory chain and
strongly inhibit respiration. A Glycolysis Stress Test was
performed to measure changes in ECAR following the addition of
glucose (10 mM), oligomycin (1 μM) and 2-deoxyglucose (2-DG;
0.1 M) to determine glycolysis and glycolytic capacity. Glycolysis
was measured after adding saturating glucose. Oligomycin inhibits
mitochondrial ATP production, thereby pushing cells to use
glycolysis maximally. Glycolytic capacity was calculated using the
following equation: maximum rate measurement after oligomycin
injection-last rate measurement before glucose injection, expressed
as mpH/min. Finally, 2-DG, a glucose analog that inhibits
glycolysis through competitive binding to hexokinase, was added.
The decrease in ECAR after 2-DG injection confirmed that glycolysis
was the cause of the increase in ECAR during the experiment. Both
OCR and ECAR were corrected for total protein content using a
Pierce BCA protein assay kit (Thermo Fisher Scientific, Inc.).
Experiments were performed in triplicate with at least 7 technical
replicates per condition.
Analysis of inhibitor synergism
Cell viability after treatment with (combinations
of) inhibitors was determined using the MTT assay as described
previously. The interaction between alpelisib and ribociclib was
evaluated by comparing the observed response to the combination of
inhibitors to the expected response using the SynergyFinder V3
calculator (https://synergyfinder.org/). The expected response was
based on the highest single-agent (HSA) reference model, which
states that the expected combination effect is the maximum of the
single-drug responses at corresponding concentrations (36,37). The most synergistic area (MSA) was
determined by the most synergistic 3-by-3 dose window in the dose
response matrix. HSA synergy scores can be interpreted as the
average excess response due to drug interactions. A synergy score
>10 is considered a synergistic interaction.
Statistical analysis
GraphPad Prism software (version 8; Dotmatics) was
used to perform all statistical analyses. All results are presented
as the mean ± standard error of the mean. All experiments were
performed in triplicate, and statistical analysis was performed
using unpaired Student's t-tests and one-way ANOVA with a Dunnett's
post hoc tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
CDK4/6 inhibition suppresses the
viability of HPV-negative HNSCC cells
To identify differences in the response to CDK4/6
inhibition between cell lines, three HPV-negative and five
HPV-positive cell lines were treated with ribociclib and
palbociclib. In addition, immortalized human keratinocyte (HaCaT)
and NOK cell lines were included in the analysis. The MCF-7 breast
cancer cell line was used as a positive control as these cells are
dependent on the CDK pathway for proliferation (38). All cell lines were cultured for 3
days with increasing CDKi concentrations. Since no reduction of 50%
in cell viability was achieved, cell lines were treated for 5 days
with increasing concentrations of CDKi. The IC50 values
are listed in Table I. For
comparison, cell lines were also treated with increasing
concentrations of cisplatin, which is currently the standard
treatment along with radiotherapy for patients with HNSCC. The
positive control MCF-7 cell line showed an effective response to
both CDKi in a dose-dependent manner. All HPV-negative HNSCC cell
lines showed decreased cell viability following incubation with the
CDKi, which was not observed in the HPV-positive HNSCC cell lines.
The immortalized HaCaT and NOK cell lines were also sensitive to
both CDKi. In comparison with cisplatin, CDKi had comparable
IC50 values in HPV-negative HNSCC cells.
 | Table IIC50 values for CDKi (5
day-treatment), PI3Ki (3 day-treatment) and cisplatin 3
(day-treatment) in all cell lines. |
Table I
IC50 values for CDKi (5
day-treatment), PI3Ki (3 day-treatment) and cisplatin 3
(day-treatment) in all cell lines.
A, HPV-negative
HNSCC
|
|---|
| Cell line | IC50
CDKi, μM
| IC50
PI3Ki, μM
| IC50
cisplatin, μM |
|---|
| Ribociclib | Palbociclib | Alpelisib | Buparlisib | Gedatolisib |
|---|
| UPCI-SCC-72 | 6.5 | 2.1 | 4.7 | 0.6 | 0.005 | 22.9 |
| UPCI-SCC-003 | 4.7 | 0.5 | 4.1 | 0.6 | 0.011 | 5.9 |
| UT-SCC-33 | >10a | 2 | >10a | 1.8 | 0.032 | 5.1 |
|
| B, HPV-positive
HNSCC |
|
| Cell line | IC50
CDKi, μM
| IC50
PI3Ki, μM
| IC50
cisplatin, μM |
| Ribociclib | Palbociclib | Alpelisib | Buparlisib | Gedatolisib |
|
| 93-VU-147T | NA | NA | 3.3 | 0.4 | 0.009 | 3.7 |
| UM-SCC-104 | NA | NA | 5.8 | 0.3 | 0.029 | 17.3 |
| UM-SCC-47 | 7.1 | NA | 8.2 | 0.3 | 0.0057 | 2.8 |
| UD-SCC-2 | NA | NA | >10a | 1.1 | 0.021 | 11.8 |
| UPCI-SCC-090 | NA | NA | >10a | 1.0 | 0.014 | 5.3 |
|
| C, HPV-positive
UCC |
|
| Cell line | IC50
CDKi, μM
| IC50
PI3Ki, μM
| IC50
cisplatin, μM |
| Ribociclib | Palbociclib | Alpelisib | Buparlisib | Gedatolisib |
|
| CaSki | NA | NA | >10a | 0.5 | 0.01 | 6.2 |
| HeLa | NA | NA | 9.1 | 0.2 | 0.014 | 1.1 |
| SiHa | NA | NA | >10a | 2.0 | 0.02 | 5.5 |
|
| D, Controls |
|
| Cell line | IC50
CDKi, μM
| IC50
PI3Ki, μM
| IC50
cisplatin, μM |
| Ribociclib | Palbociclib | Alpelisib | Buparlisib | Gedatolisib |
|
| HaCaT | 4 | 0.3 | 4.7 | 0.3 | 0.0037 | 3.8 |
| NOK | 1.4 | 0.1 | 2.4 | 1.2 | 0.0061 | 0.8 |
| MCF-7 | 0.7 | 0.6 | - | - | - | - |
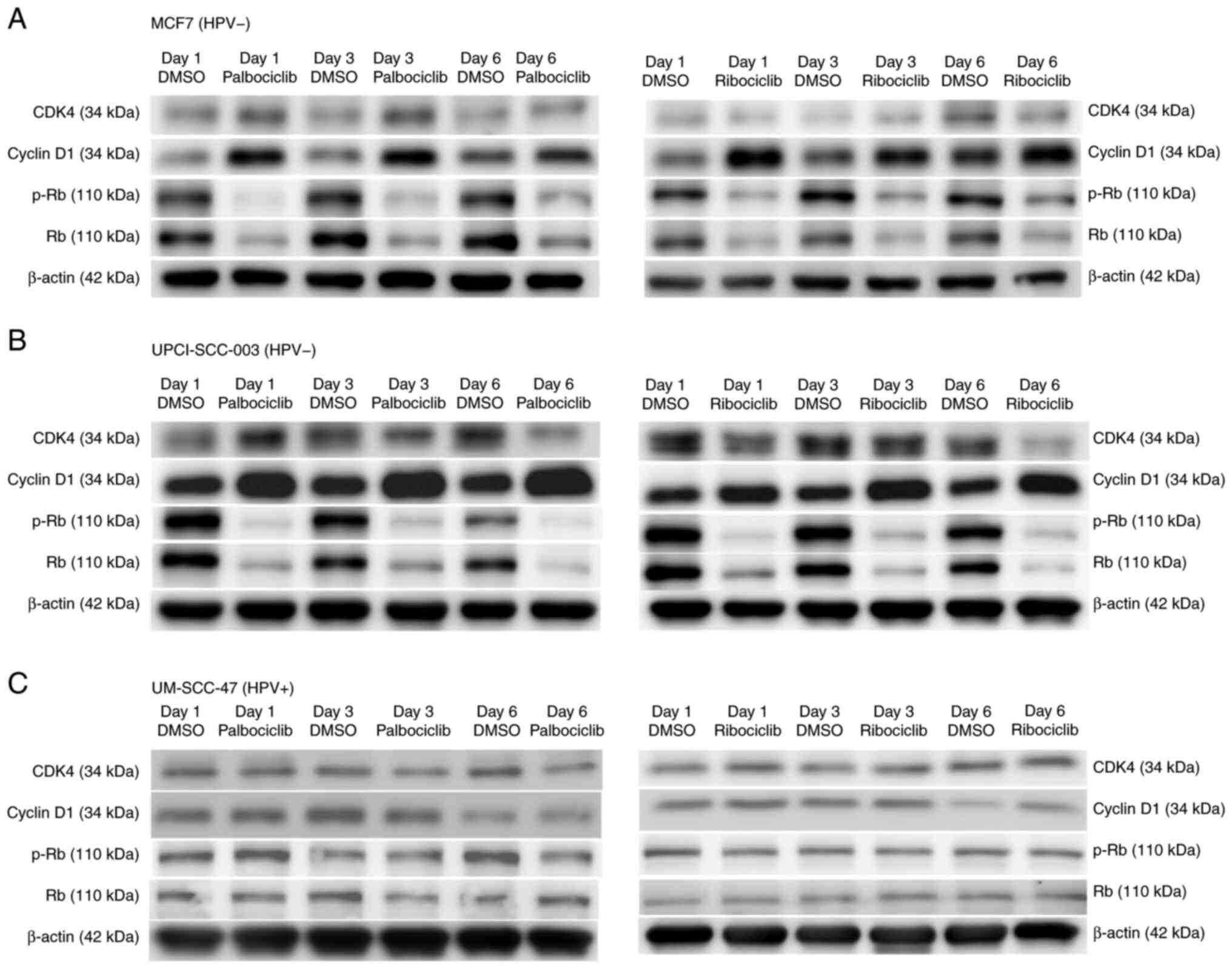
CDK4/6 inhibition downregulates the
levels of cell cycle-relevant proteins
The cell lines MCF-7, UPCI-SCC-003 (HPV negative)
and UM-SCC-47 (HPV positive) were used to investigate changes in
protein expression involved in the CDK pathway induced by CDKi
treatment. Upregulation of Cyclin D1 and downregulation of Rb1 and
phosphorylated (p-)Rb1 were observed in the MCF-7 and UPCI-SCC-003
cell lines upon CDKi administration, which was in agreement with
earlier studies (39) (Fig. 1). There was greater expression of
CDK4 in UPCI-SCC-003 cells than in MCF-7 cells, but under CDKi
treatment, no notable changes were observed. In the HPV-positive
UM-SCC-47 cell line, the baseline expression of CDK4, cyclin D1 and
p-Rb1 was lower than that in the other two cell lines, most likely
as a result of the activity of viral oncoprotein E7 (31), and no marked changes were observed
under CDKi treatment.
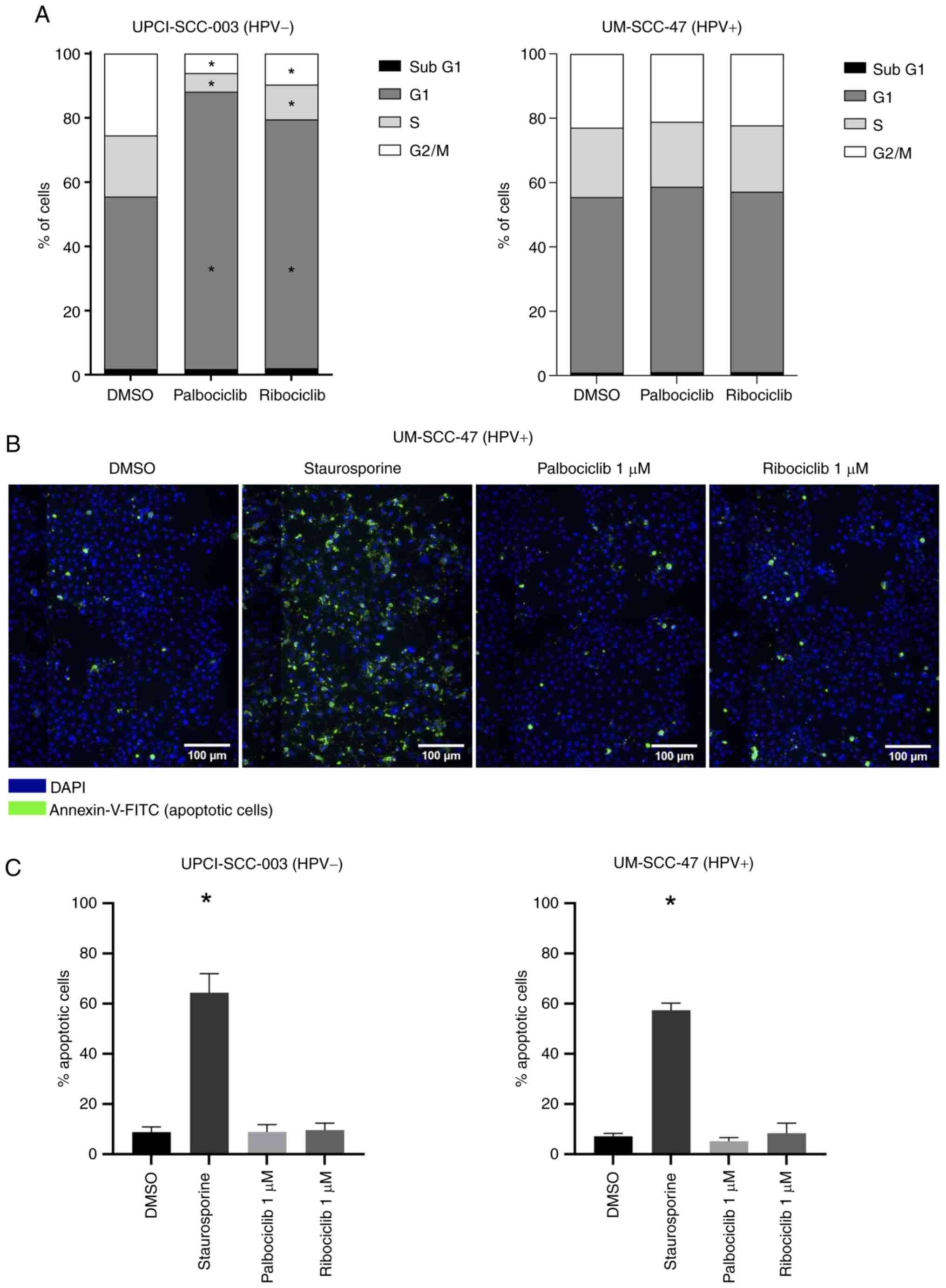
CDK4/6 inhibition results in G1-phase
arrest without apoptosis
A known consequence of CDK4/6 inhibition is cell
cycle phase arrest. For this purpose, the cell cycle distribution
was analyzed by flow cytometry after a 24-h treatment with
palbociclib and ribociclib (Figs.
2A and S1). In HPV-negative
UPCI-SCC-003 cells, treatment with CDKi resulted in an increase in
the proportion of cells in the G1 phase and a decrease in cells in
the S- and G2/M phases. In addition, increased levels of the
senescence marker β-galactosidase were observed in this cell line
after treatment with palbociclib (Fig. S2). In HPV-positive UM-SCC-47
cells, there was no difference in the distribution between
untreated and treated cells. These results underscore the western
blotting results. To assess whether cells undergo apoptosis after
treatment with CDKi, an Annexin-V assay was performed. In
UPCI-SCC-003 and UM-SCC-47 cells (Figs. 2B, C and S3), there was no significant increase
in the number of apoptotic cells after treatment.
PI3Ki suppress the viability of
HPV-positive and -negative HNSCC cell lines
To determine the efficacy of the PI3K pathway
inhibitors, all cell lines were cultured for 3 days at increasing
concentrations. The IC50 values of the cell lines are
presented in Table I. All three
inhibitors significantly decreased the viability of the
HPV-negative HNSCC, HPV-positive HNSCC and the immortalized HaCaT
and NOK cell lines. This effect was dose-dependent. Gedatolisib had
the lowest and alpelisib the highest IC50 values.
Notably, two HPV-positive cell lines (UD-SCC-2 and UPCI-SCC-090)
and the HPV-negative UT-SCC-33 cell line were less responsive to
all three PI3Ki. There was no statistically significant difference
in the activity of PI3Ki between the HPV-positive and -negative
cell lines (data not shown). In comparison with the IC50
values of cisplatin, PI3Ki appeared to be more potent in
HPV-positive and -negative HNSCC cells.
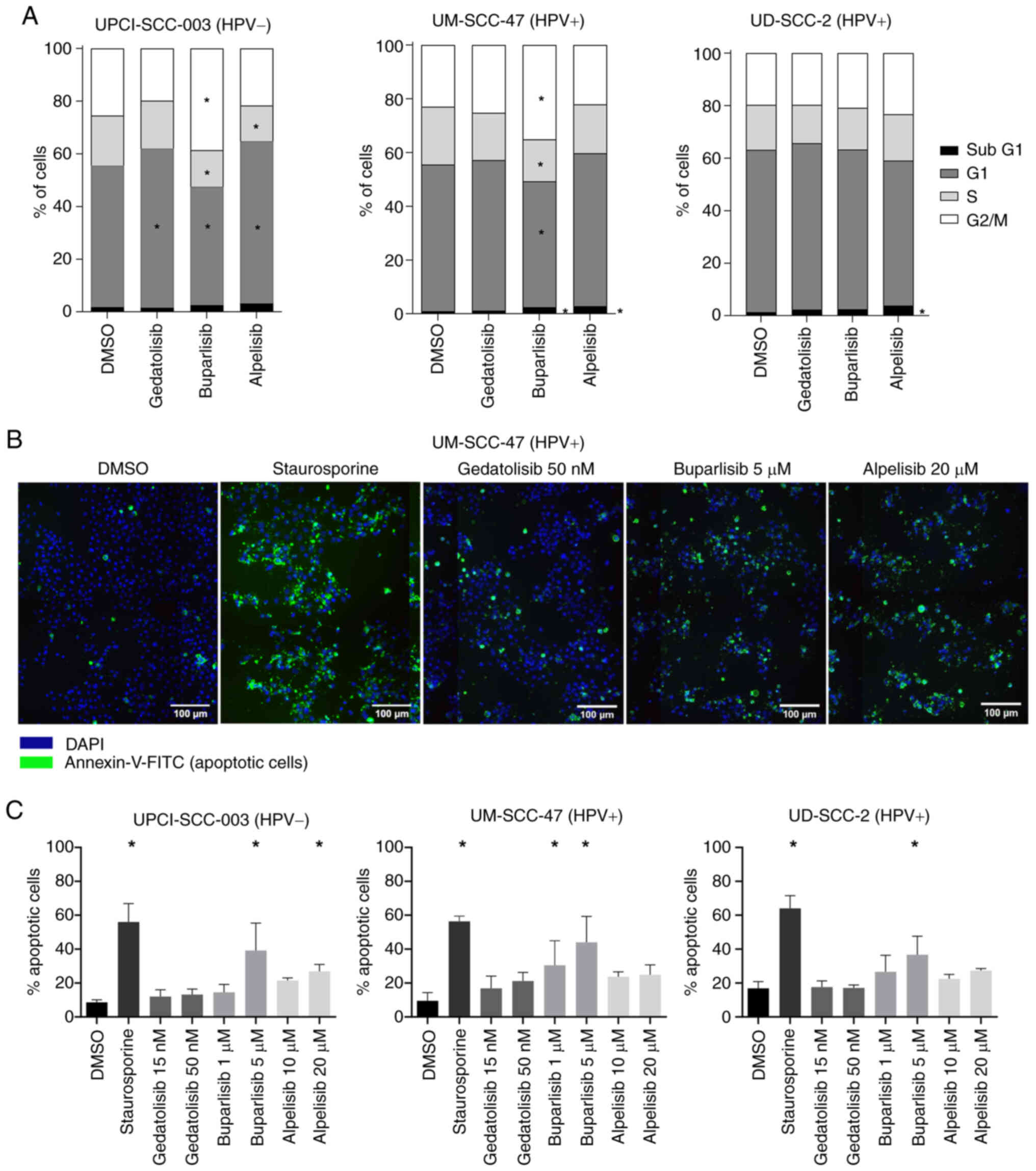
Effects of PI3Ki on cell cycle
distribution and apoptosis
To determine whether PI3Ki causes cell cycle arrest,
flow cytometry analysis was performed on the HPV-negative
UPCI-SCC-003 and HPV-positive UM-SCC-47 and UD-SCC-2 cell lines
(Figs. 3A and S1). In UPCI-SCC-003 and UM-SCC-47
cells, treatment with alpelisib resulted in an increase in G1 and
SubG1, whereas treatment with buparlisib resulted in an increase in
cells in G2/M phase. In UD-SCC-2 cells, none of the inhibitors
induced a notable change in the cell cycle (Figs. 3A and S1). In addition, all three PI3Ki
induced an increase in apoptosis in all cell lines, which was only
statistically significant for buparlisib (Figs. 3B, C and S3).
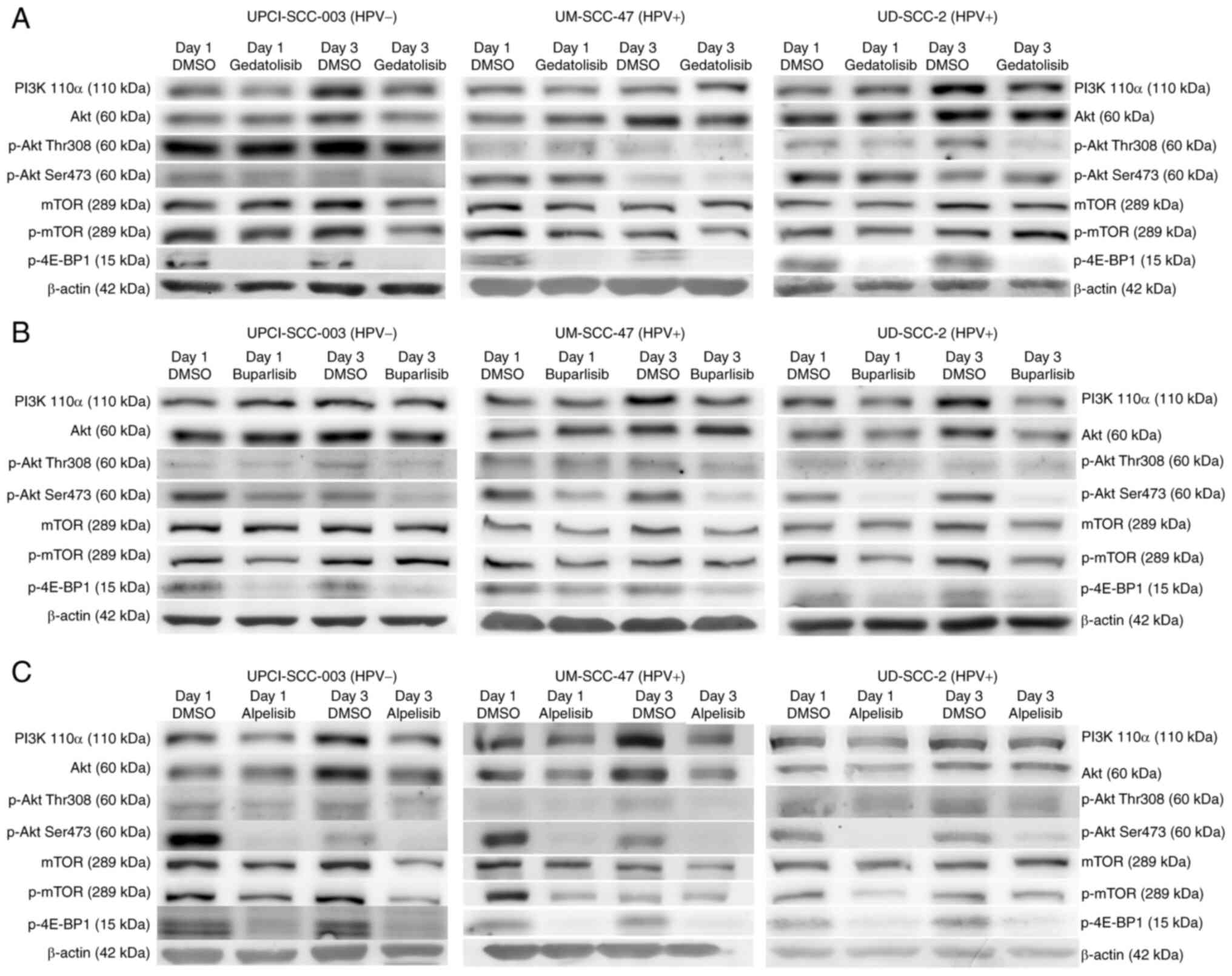
PI3K inhibition downregulates the levels
of PI3K-Akt-mTOR pathway proteins
To investigate the expression of proteins involved
in the PI3K pathway, western blotting of cell lysates after
treatment with PI3Ki was performed. Treatment with all three
inhibitors resulted in the expected downregulation p-4E-BP1 and a
slight downregulation of p-Akt (Thr308) (Fig. 4). In addition, treatment with
alpelisib and buparlisib resulted in the downregulation of p-Akt
(Ser473), and treatment with alpelisib and gedatolisib also
downregulated PI3K itself. Treatment with gedatolisib resulted in
less downregulation of the proteins compared with alpelisib and
buparlisib. Therefore, the cell lines were also treated with
concentrations of gedatolisib above the IC50 (5, 15 and
30 nM), producing similar results (Fig. S4).
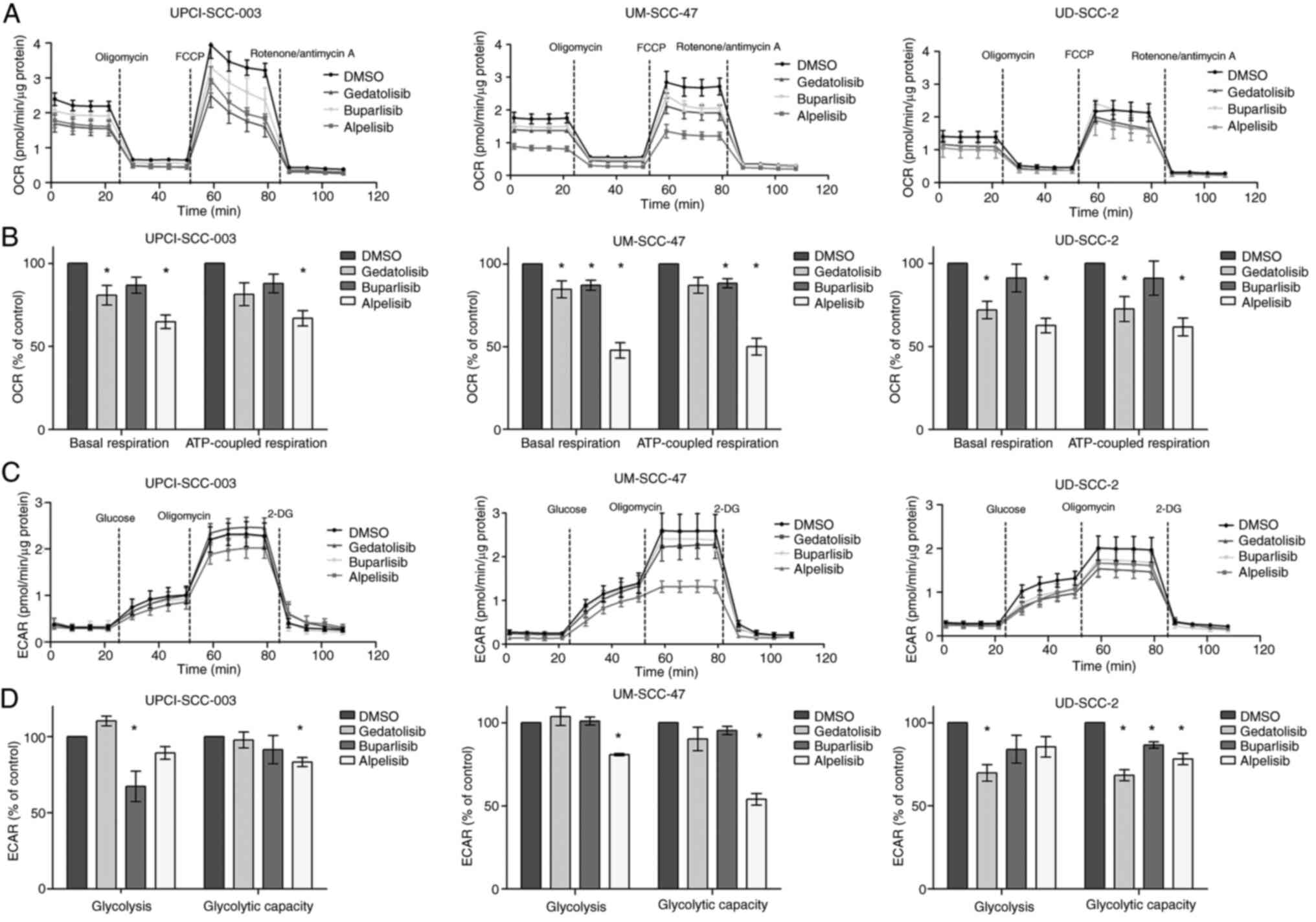
PI3K inhibition decreases cellular
metabolism
The PI3K/Akt/mTOR pathway is crucial in the
regulation of cell energy metabolism and is involved in both
glucose uptake and the coordination of glucose fate within the
cell. Therefore, it was evaluated whether the viability-inhibitory
effects of PI3Ki were associated with alterations in both oxidative
and glycolytic energy metabolism. To investigate the impact of
PI3Ki on mitochondrial respiration, a Seahorse Mito Stress Test was
performed. The baseline oxidative metabolism was the highest in
UPCI-SCC-003 and lowest in UD-SCC-2 cells (Fig. 5A). Treatment with alpelisib
significantly reduced basal respiration and the respiration
associated with ATP production in all cell lines. Buparlisib only
downregulated basal respiration and ATP production in UM-SCC-47
cells (Fig. 5A and B).
Gedatolisib downregulated basal respiration in all cell lines and
ATP production-coupled respiration in UD-SCC-2 cells. The impact of
PI3Ki treatment on cell glycolytic capacity was also examined by
performing the SeaHorse Glycolysis Stress Test. The glycolytic
capacity, measured as the maximum ECAR rate achieved upon the
inhibition of oxidative phosphorylation by oligomycin, was
significantly downregulated by alpelisib in the three cell lines.
Gedatolisib and buparlisib downregulated glycolytic capacity in
UD-SCC-2 cells (Fig. 5C and D).
These results were confirmed using glucose and lactate assays
(Fig. S5). Taken together, these
data show that alpelisib in particular has the capacity to inhibit
both oxidative metabolism and glycolysis.
Effect of the combination of PI3Ki
alpelisib and CDKi ribociclib on cell viability
Dual inhibition of PI3Ki and CDKi might
synergistically decrease cell viability, especially in HPV-negative
HNSCC cell lines. Therefore, the efficacy of the combination of
PI3Ki alpelisib and CDKi ribociclib in UPCI-SCC-003 and UM-SCC-47
cells were investigated. The choice for this inhibitor combination
was based on the subunit selectivity of alpelisib and the
pronounced effect of this inhibitor in the aforementioned
(functional) assays, as well as the enhanced solubility of
ribociclib compared with palbociclib, requiring lower
concentrations of DMSO. The cell lines were treated for 3 days with
increasing concentrations of ribociclib (0 nM, 10 nM, 100 nM, 1
μM and 10 μM), with the addition of increasing
concentrations of alpelisib (0 nM, 10 nM, 100 nM, 1 μM and 5
μM). In comparison with ribociclib alone, the combination
treatment resulted in lower IC50 values (Figs. 6A-E and S6). Interactions between ribociclib and
alpelisib were evaluated using the HSA reference model to determine
possible synergism. For the UPCI-SCC-003 cell line, the overall HSA
synergy score was 8.712, which corresponds to 8.712% of the
response beyond the expectation for all concentrations (Fig. 6C), indicating a moderate
synergistic effect. The MSA had a synergy score of 12.66 and the
highest score of 27.2 for the combination of 10 μM
ribociclib and 5 μM alpelisib. For the UM-SCC-47 cells, the
overall HSA synergy score was 5.6, indicating an additive effect of
the combination therapy (Fig.
6D-F). The MSA represented a synergy score of 9.15, with the
highest score of 28.1 obtained for the combination of 10 μM
ribociclib and 5 μM alpelisib. However, no increase in
apoptotic cells was observed with the combinational treatment
compared to alpelisib treatment alone (Fig. S7).
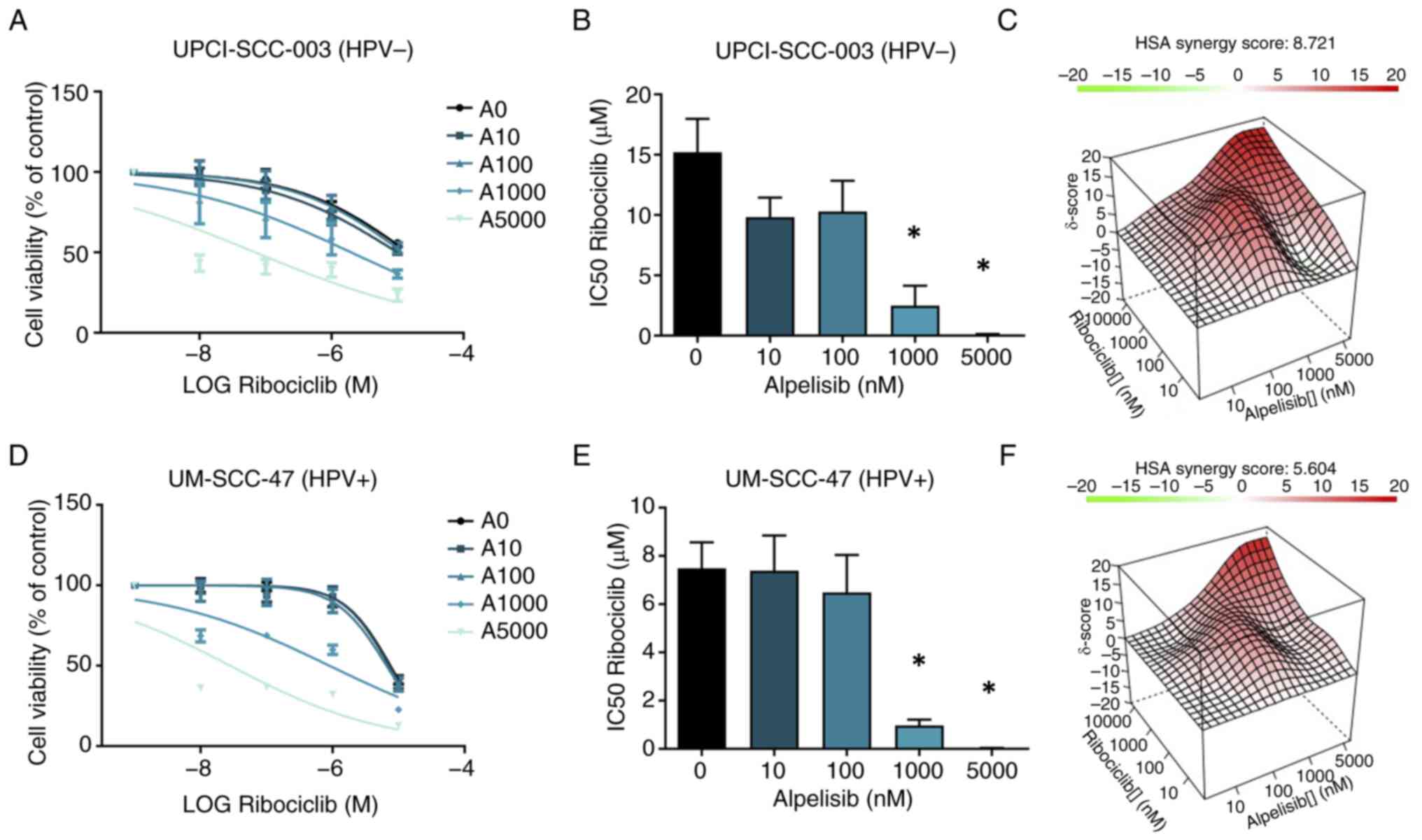 | Figure 6Combination therapy (3 days) of
ribociclib with alpelisib in the HPV-negative UPCI-SCC-003 and
HPV-positive UM-SCC-47 cell lines. (A and D) The dose-response
curves for ribociclib (0 nM, 10 nM, 100 nM, 1 μM and 10
μM) without alpelisib are illustrated by the A0 curves. The
increasing concentrations of alpelisib (0 nM, 10 nM, 100 nM, 1
μM and 5 μM) are indicated by the A10, A100, A1000
and A5000 curves. The calculated IC50 of ribociclib
alone and in combination with different alpelisib concentrations
(nM) are shown in (B) for UPCI-SCC-003 and in (E) for UM-SCC-47
cells. (C and F) The HSA synergy score graph shows the combined
concentration of ribociclib and alpelisib which are synergistic
(red), additive (white) and antagonistic (green). Each bar
represents the mean ± SEM. *P< 0.05. HSA, highest
single-agent; HPV, human papillomavirus. |
Discussion
A large body of preclinical data indicates that the
cell cycle and PI3K/Akt/mTOR pathways are deregulated and may
provide therapeutic targets for patients with HNSCC (11,12,13). Several studies are currently
evaluating CDKi and PI3Ki monotherapy or in combination with
chemotherapy, targeted therapy, immunotherapy or radiotherapy [such
as a phase III trial with buparlisib in combination with paclitaxel
(NCT04338399) and a phase III trial with palbociclib combined with
cetuximab (NCT04966481)]. The results thus far are diverse
regarding the advantages of the combination of CDKi and standard
cytotoxic chemotherapy (40). For
PI3Ki, most of these compounds have not advanced to late-stage
clinical trials, and the results of phase II studies are awaited in
the next couple of years. A possible explanation for these results
might involve the complexity of PI3K signaling and intrinsic
adaptive responses, leading to inadequate pathway inhibition
(41). The aim of the present
study was to investigate the in vitro effects of several
CDKi (palbociclib and ribociclib) and PI3Ki (alpelisib, buparlisib
and gedatolisib) on the viability of five HPV-positive and three
HPV-negative HNSCC cell lines, together with the consequences and
underlying mechanisms.
In the present study, CDKi showed particular
efficacy in HPV-negative HNSCC cell lines, of which two harbor a
CCND1 amplification, represented by reduced cell viability
and downregulation of the proteins, Rb1 and p-Rb1, leading to G1
cell cycle arrest without increased apoptosis. Cyclin-D1 was found
to be upregulated after CDKi treatment, which was possibly induced
by a positive feedback mechanism, as was also found in another
study (39). The observations of
the present study were also in accordance with an earlier study in
which CDKi treatment with palbociclib showed efficacy in oral
squamous cell carcinoma cell lines (42). Together, these data support CDKi
as cytostatic agents involving the prevention of cell cycle
progression by blocking hyperphosphorylation of Rb1 (43). Most likely, cells may enter
senescence, an irreversible arrest of cell proliferation, while
maintaining metabolic function (44,45). This was also supported by the
increased levels of the senescence maker β-galactosidase in the
HPV-negative UPCI-SCC-003 cell line after treatment with
palbociclib, observed in the present study. These findings suggest
that CDKi should be combined with other therapies to achieve
cytotoxic effects at clinically relevant concentrations. This is
underscored by a phase II study demonstrating a synergistic
treatment response of palbociclib combined with cetuximab in
patients with platinum- or cetuximab-resistant HNSCC (46). In accordance with our hypothesis,
CDKi treatment did not inhibit the viability of HPV-positive HNSCC
cell lines, and differences in cell cycle distribution were not
detected between untreated and treated cells in the present
study.
In the present study, PI3K inhibition resulted in a
strong reduction in cell viability in both HPV-positive and
-negative HNSCC cell lines and downregulation of PI3K/Akt/mTOR
pathway protein expression. Only moderate cell cycle arrest and
apoptosis were observed after treatment with PI3Ki. The differences
in therapeutic efficacy between PI3Ki may be explained by several
features. First, the PI3K subunits p110α and p110β are ubiquitously
expressed in mammalian cells, whereas p110γ and p110δ are mainly
expressed in leukocytes (47).
Thus, this may have consequences when using PI3Ki specific to one
or more PI3K subunits. In addition, the structure of the PI3K
inhibitors determines (subunit) selectivity, protein binding
affinity and flexibility, cellular uptake and thereby the activity
of the protein inhibitor (48,49).
As aforementioned, alpelisib selectively inhibits
p110α (23,24). In luminal HER2-amplified and
PIK3CA mutant breast cancer, the initial efficacy of p110α
inhibition appears to be mitigated by the rapid re-accumulation of
the PI3K product, PIP3, produced by the p110β isoform. The
combination of a p110β inhibitor with alpelisib therefore prevented
PIP3 rebound and induced greater antitumor efficacy in a luminal
breast cancer study (50).
Treatment with buparlisib significantly inhibited wild-type and
mutant PI3K catalytic subunit p110α, β, δ and γ (25), and showed lower IC50
values than treatment with alpelisib in the present study. However,
there are concerns regarding the safety profile of buparlisib. In
the BELLE-3 breast cancer trial, in which patients with advanced
breast cancer following progression on prior mTOR inhibition and
endocrine therapy were included and treated with fulvestrant with
or without the addition of buparlisib, treatment with buparlisib
resulted in frequent grade 3 and 4 adverse events (hyperglycemia,
elevated liver enzymes and major psychiatric symptoms) (51). A possible explanation for the
(severe) side effects of buparlisib observed in clinical trials
could be the off-target effects of this inhibitor. Buparlisib was
found to cause cell death in various cellular systems independent
of PI3K pathway dependence by influencing the expression of mitotic
genes. In addition, buparlisib inhibited microtubule dynamics
following direct binding to tubulin (52). This is supported by the G2/M phase
arrest and increased apoptosis observed in the present study and
other studies. Specifically, in both glioma and acute myeloid
leukemia cell lines, G2/M phase arrest and apoptosis were observed
after treatment with buparlisib (53,54).
Gedatolisib is a highly potent dual inhibitor of
PI3K (α, β, δ and γ) and mTOR (TORC1 and TORC2), showing low
IC50 values in the present study. Akt-mediated
stimulation of mTORC1 serves as a key point in the regulation of
anabolic metabolism, by stimulating, for example, pyrimidine and
de novo purine synthesis. mTORC1 activation also increases
the protein synthesis capacity of cells through multiple mechanisms
(55,56). Gedatolisib, on the one hand,
blocks DNA and protein synthesis. On the other hand, inhibiting
mTOR might repress a negative feedback loop that activates the PI3K
and MAPK pathways, which has also been reported in other breast
cancer cell lines (57,58). Together, these findings may
explain the lower IC50 value of gedatolisib observed
compared with the other two PI3K inhibitors.
Mutation analysis revealed several genetic
alterations in the PI3K/Akt/mTOR pathway in the studied cell lines,
in line with TCGA data (59).
Cell lines with amplification of PIK3CA (UPCI-SCC-72) or
amplification of AKT1 and AKT2 (UM-SCC-47) responded
well to PI3Ki in the present study, according to their
IC50 values. By contrast, the UPCI-SCC-090 cell line
harboring a double PTEN mutation was observed to be more
resistant to PI3Ki, which could be expected based on the literature
(59,60). Another relatively resistant cell
line (UT-SCC-33) harbors mutations in FGFR3 and HRAS,
among others, suggesting the activation of other compensatory
oncogenic signaling pathways. The UD-SCC-2 cell line was relatively
PI3Ki-resistant, and no pathogenic mutations were observed via the
sequencing panel. These results suggest that the presence of an
activating PIK3CA mutation is not a prerequisite for the
therapeutic effect of PI3Ki in these cell lines and that the
complexity of this pathway and its interaction with other
compensatory pathways, may play a role. Moreover, the oncogenic
activity of HPV E5, E6 and E7 oncogenes may directly and/or
indirectly interact with the PI3K pathway (61).
The PI3K pathway is central to most deregulated
metabolic pathways supporting the anabolic needs of cancer cells.
Therefore, the cellular metabolism of PI3Ki-treated cell lines were
investigated. Cells use two major pathways to produce ATP:
Glycolysis and mitochondrial respiration via oxidative
phosphorylation. Cancer cells heavily depend on glycolysis,
balancing ATP production with anabolic needs, which is also the
case for HNSCC, supported by the increased lactate levels observed
in HNSCC tumors (62). In the
present study, it was found that PI3Ki decreased both glycolysis
and mitochondrial oxidative metabolism in HNSCC cells.
The PI3K isoform, p110α, is suggested to be the key
mediator of glucose metabolism in multiple tissues (63,64), which is supported by the strongest
metabolic effect observed with p110α-specific inhibitor, alpelisib,
in the present study. Although the pan-PI3Ki, gedatolisib and
buparlisib, also inhibit this isoform, dual- and pan-PI3Ki in
general have been found to exhibit more off-target effects (such as
on mitotic genes or on members of the PI3K-related kinase family),
thereby reducing efficacy in the inhibition of the actual target
pathway (65,66). Isoform-selective inhibitors (such
as alpelisib) may achieve greater efficacy and have the potential
to block the relevant target more completely while limiting
toxicities (65,66). The findings of the present study
underscore this principle, as alpelisib treatment most prominently
affected Akt phosphorylation and cellular metabolism. There are few
studies investigating the effects of PI3K inhibition on metabolism
in HNSCC cell lines. In colorectal cancer, PI3Ki LY294002 reduced
glycolysis by significantly decreasing hexokinase-2 levels as a
result of PI3K-Akt inhibition (67). Treatment of glioblastoma cell
lines with NVP-BEZ235 resulted in a significant reduction in
lactate secretion and lower glucose uptake, indicating a strong
effect on glycolytic activity by this PI3Ki (68). Taken together, PI3K/Akt/mTOR
pathway inhibitors were effective in both HPV-negative and
HPV-positive cell lines, with varying efficacy between inhibitors,
inducing apoptosis, attenuating cellular metabolism and only
moderate cell cycle arrest. Although the induction of apoptosis is
the most frequently described mechanism of cell death in response
to PI3Ki, other cell death mechanisms, such as autophagy-mediated
cell death, may also play a role, which could be considered in
future research (69,70).
It has been reported that inhibition of the cell
cycle or MAPK pathway in cancer cells, including breast and
pancreatic cancer cells, leads to compensatory upregulation of
other oncogenic signaling pathways, including the PI3K/mTOR
pathway. Therefore, dual inhibition of CDKi and PI3Ki could
represent an interesting treatment option (39,71,72). The cell viability inhibitory
effects of combined treatment with CDKi ribociclib and the PI3Ki
alpelisib in HPV-negative UPCI-SCC-003 and HPV-positive UM-SCC-47
cell lines were analyzed in the present study. The results showed a
synergistic effect of the combination of ribociclib and alpelisib,
which was higher in the HPV-negative cell line, as expected, since
CDKi alone only affected HPV-negative HNSCC cells. Despite the
apparent synergistic effect of both inhibitors on cell viability,
no increase in apoptotic cells was observed with the combinational
treatment compared with alpelisib treatment alone, indicating that
other mechanisms may play a role in the observed synergistic
reduction in cell viability. Furthermore, treatment duration and
sequence (simultaneously or sequentially) are important
considerations that may influence the therapeutic effect of
combination therapy. Further studies to confirm compensatory
mechanisms, including PI3K/Akt/mTOR pathway upregulation, after
CDKi in HNSCC and the efficacy of combinational therapy at
clinically relevant concentrations are required. In this respect,
the effects of these targeted therapies may also be investigated in
combination with radiotherapy, which may offer opportunities for
treatment de-escalation, as a replacement for chemotherapy, or in
combination with radiotherapy dose reduction.
In addition to PI3Ki, mTOR inhibitors (mTORi) have
been investigated for the treatment of HNSCC, resulting in
decreased tumor cell proliferation and apoptosis in cell lines and
xenograft models (73,74). A phase II clinical trial showed
promising response rates to the mTORi, temsirolimus, combined with
cetuximab in patients with cetuximab-resistant HNSCC, but no
improvement in progression-free survival was observed (75). Furthermore, the combination of
CDKi ribociclib with mTORi everolimus showed acceptable safety
profiles and promising tumor responses in breast cancer and
multiple pediatric cancer types (76,77). Although it might be expected that
downstream inhibition of mTOR results in fewer off-target effects
than upstream PI3Ki, studies have shown that mTORi leads to
compensatory feedback loops. For example, mTORi can upregulate
tyrosine kinase receptors, leading to increased PI3K/Akt and MAPK
signaling (58,78). Dual PI3K and mTOR inhibitors,
including gedatolisib, may limit these compensatory feedback
mechanisms.
The present study may be limited by the absence of
PIK3CA mutations in the tested cell lines, which hampers the
evaluation of these specific genetic changes in response to
inhibitors. Nevertheless, the IC50 values of alpelisib
and gedatolisib to PI3Kα-wildtype and -mutant cell lines were
similar to that observed in other studies (26,79), thus extending the effect of PI3Ki.
It will be important to identify biomarkers that can predict the
response to PI3Ki in HNSCC and accelerate the integration of novel
targeted agents into the treatment of these cancer types.
In conclusion, the present study described multiple
mechanisms and consequences of CDK4/6 and PI3K/Akt/mTOR pathway
inhibition and provided the basis for further research into the
targeting of these oncogenic signaling pathways and possible
resistance mechanisms. Future preclinical studies should focus on
identifying optimal concentrations, treatment durations and
treatment sequences, as well as the mechanisms underlying the
synergistic effects of combination treatment approaches.
Furthermore, the use of ex vivo culture models, which are
directly derived from tumor tissue, will be an interesting next
step to facilitate the translation of in vitro findings to
patients (80). The combination
of CDKi with PI3Ki and combinational treatment with radiotherapy
could be a promising new treatment approach and may offer
opportunities for treatment de-escalation, as a replacement for
chemotherapy, or in combination with radiotherapy dose
reduction.
Supplementary Data
Availability of data and materials
The data generated in this study may be obtained
from the corresponding author.
Authors' contributions
Conceptualization and design were conducted by FV,
ID, BK, AH and EJS. Acquisition and analysis of data were conducted
by FV, ID, DL and RJ. FV, ID, EJS and BK confirm the authenticity
of all the raw data. Data interpretation was conducted by FV, ID,
DL, RJ and EJS. Writing of the original manuscript draft was
conducted by FV and ID. Reviewing and editing of the manuscript was
conducted by FV, ID, DL, RJ, AH, BK and EJS. Construction of the
figures was conducted by FV and ID. Study supervision was conducted
by AH, BK and EJS. All authors read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
Although Pfizer and Novartis funded this study and
the drugs, palbociclib, ribociclib, gedatolisib, buparlisib and
alpelisib, were provided by them, the funders were not involved in
the study design, collection, analysis and interpretation of data,
the writing of this article, or the decision to submit it for
publication.
Acknowledgements
The authors would like to thank Dr Ludwig Dubois
(Department of Precision Medicine, GROW Research Institute for
Oncology and Reproduction, Maastricht University, Maastricht) and
Dr Marike van Gisbergen (Department of Dermatology, Maastricht
University Medical Centre, GROW School for Oncology and
Developmental Biology, Maastricht University, Maastricht) for their
helpful discussions regarding the cellular metabolism
experiments.
The abstract was presented at the Annual Meeting of
the American Association for Cancer Research April 8-Apr 13, 2022,
in New Orleans, LA, and published as abstract no. 2573 in Cancer
Res 82 (Suppl 12): 2022.
Funding
This study was funded by Pfizer (grant no. WI194733) and
Novartis (grant no. CBYL719NCMUMC01).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bann DV, Deschler DG and Goyal N: Novel
immunotherapeutic approaches for head and neck squamous cell
carcinoma. Cancers (Basel). 8:872016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gillison ML, Chaturvedi AK, Anderson WF
and Fakhry C: Epidemiology of human papillomavirus-positive head
and neck squamous cell carcinoma. J Clin Oncol. 33:3235–3242. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stransky N, Egloff AM, Tward AD, Kostic
AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C,
McKenna A, et al: The mutational landscape of head and neck
squamous cell carcinoma. Science. 333:1157–1160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hafkamp HC, Manni JJ, Haesevoets A, Voogd
AC, Schepers M, Bot FJ, Hopman AHN, Ramaekers FC and Speel EJM:
Marked differences in survival rate between smokers and nonsmokers
with HPV 16-associated tonsillar carcinomas. Int J Cancer.
122:2656–2664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rosenquist K, Wennerberg J, Annertz K,
Schildt EB, Hansson BG, Bladström A and Andersson G: Recurrence in
patients with oral and oropharyngeal squamous cell carcinoma: Human
papillomavirus and other risk factors. Acta Otolaryngol.
127:980–987. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Furness S, Glenny AM, Worthington HV,
Pavitt S, Oliver R, Clarkson JE, Macluskey M, Chan KK and Conway
DI: Interventions for the treatment of oral cavity and
oropharyngeal cancer: Chemotherapy. Cochrane Database Syst Rev.
Cd0063862011.PubMed/NCBI
|
|
9
|
Machiels JP, René Leemans C, Golusinski W,
Grau C, Licitra L and Gregoire V; EHNS Executive Board; ESTRO
Executive Board: Electronic address: simplesecretariat@ehns.org;
ESMO Guidelines Committee. Electronic address: simpleclinicalguidelines@esmo.org.
Electronic address: simpleinfo@estro.org. Squamous
cell carcinoma of the oral cavitylarynx, oropharynx and
hypopharynx: EHNS-ESMO-ESTRO clinical practice guidelines for
diagnosis, treatment and follow-up. Ann Oncol. 31:1462–1475. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Agrawal N, Frederick MJ, Pickering CR,
Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et
al: Exome sequencing of head and neck squamous cell carcinoma
reveals inactivating mutations in NOTCH1. Science. 333:1154–1157.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cancer Genome Atlas Network: Comprehensive
genomic characterization of head and neck squamous cell carcinomas.
Nature. 517:576–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu L, Timmers C, Maiti B, Saavedra HI,
Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, et
al: The E2F1-3 transcription factors are essential for cellular
proliferation. Nature. 414:457–462. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lim AM, Do H, Young RJ, Wong SQ, Angel C,
Collins M, Takano EA, Corry J, Wiesenfeld D, Kleid S, et al:
Differential mechanisms of CDKN2A (p16) alteration in oral tongue
squamous cell carcinomas and correlation with patient outcome. Int
J Cancer. 135:887–895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smeets SJ, Braakhuis BJM, Abbas S,
Snijders PJF, Ylstra B, van de Wiel MA, Meijer GA, Leemans CR and
Brakenhoff RH: Genome-wide DNA copy number alterations in head and
neck squamous cell carcinomas with or without oncogene-expressing
human papillomavirus. Oncogene. 25:2558–2564. 2006. View Article : Google Scholar
|
|
17
|
White JS, Weissfeld JL, Ragin CCR, Rossie
KM, Martin CL, Shuster M, Ishwad CS, Law JC, Myers EN, Johnson JT
and Gollin SM: The influence of clinical and demographic risk
factors on the establishment of head and neck squamous cell
carcinoma cell lines. Oral Oncol. 43:701–712. 2007. View Article : Google Scholar
|
|
18
|
Huang WC and Hung MC: Induction of Akt
activity by chemotherapy confers acquired resistance. J Formos Med
Assoc. 108:180–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soulières D, Faivre S, Mesía R, Remenár É,
Li SH, Karpenko A, Dechaphunkul A, Ochsenreither S, Kiss LA, Lin
JC, et al: Buparlisib and paclitaxel in patients with
platinum-pretreated recurrent or metastatic squamous cell carcinoma
of the head and neck (BERIL-1): A randomised, double-blind,
placebo-controlled phase 2 trial. Lancet Oncol. 18:323–335. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
André F, Ciruelos EM, Juric D, Loibl S,
Campone M, Mayer IA, Rubovszky G, Yamashita T, Kaufman B, Lu YS, et
al: Alpelisib plus fulvestrant for PIK3CA-mutated, hormone
receptor-positive, human epidermal growth factor
receptor-2-negative advanced breast cancer: Final overall survival
results from SOLAR-1. Ann Oncol. 32:208–217. 2021. View Article : Google Scholar
|
|
21
|
Fry DW, Harvey PJ, Keller PR, Elliott WL,
Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK and
Toogood PL: Specific inhibition of cyclin-dependent kinase 4/6 by
PD 0332991 and associated antitumor activity in human tumor
xenografts. Mol Cancer Ther. 3:1427–1438. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim S, Tiedt R, Loo A, Horn T, Delach S,
Kovats S, Haas K, Engstler BS, Cao A, Pinzon-Ortiz M, et al: The
potent and selective cyclin-dependent kinases 4 and 6 inhibitor
ribociclib (LEE011) is a versatile combination partner in
preclinical cancer models. Oncotarget. 9:35226–35240. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Furet P, Guagnano V, Fairhurst RA,
Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J,
Aichholz R, et al: Discovery of NVP-BYL719 a potent and selective
phosphatidylinositol-3 kinase alpha inhibitor selected for clinical
evaluation. Bioorg Med Chem Lett. 23:3741–3748. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fritsch C, Huang A, Chatenay-Rivauday C,
Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De
Pover A, et al: Characterization of the novel and specific PI3Kα
inhibitor NVP-BYL719 and development of the patient stratification
strategy for clinical trials. Mol Cancer Ther. 13:1117–1129. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maira SM, Pecchi S, Huang A, Burger M,
Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, et al:
Identification and characterization of NVP-BKM120, an orally
available pan-class I PI3-kinase inhibitor. Mol Cancer Ther.
11:317–328. 2012. View Article : Google Scholar
|
|
26
|
Mallon R, Feldberg LR, Lucas J, Chaudhary
I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian
S, Venkatesan A and Hollander I: Antitumor efficacy of PKI-587, a
highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res.
17:3193–3203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Piboonniyom SO, Duensing S, Swilling NW,
Hasskarl J, Hinds PW and Münger K: Abrogation of the retinoblastoma
tumor suppressor checkpoint during keratinocyte immortalization is
not sufficient for induction of centrosome-mediated genomic
instability. Cancer Res. 63:476–483. 2003.PubMed/NCBI
|
|
28
|
Krisanaprakornkit S, Weinberg A, Perez CN
and Dale BA: Expression of the peptide antibiotic human
beta-defensin 1 in cultured gingival epithelial cells and gingival
tissue. Infect Immun. 66:4222–4228. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Piboonniyom SO, Timmermann S, Hinds P and
Münger K: Aberrations in the MTS1 tumor suppressor locus in oral
squamous cell carcinoma lines preferentially affect the INK4A gene
and result in increased cdk6 activity. Oral Oncol. 38:179–186.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eijkelenboom A, Kamping EJ, Kastner-van
Raaij AW, Hendriks-Cornelissen SJ, Neveling K, Kuiper RP, Hoischen
A, Nelen MR, Ligtenberg MJJ and Tops BBJ: Reliable next-generation
sequencing of formalin-fixed, paraffin-embedded tissue using single
molecule tags. J Mol Diagn. 18:851–863. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olthof NC, Huebbers CU, Kolligs J,
Henfling M, Ramaekers FCS, Cornet I, van Lent-Albrechts JA,
Stegmann APA, Silling S, Wieland U, et al: Viral load, gene
expression and mapping of viral integration sites in
HPV16-associated HNSCC cell lines. Int J Cancer. 136:E207–E218.
2015. View Article : Google Scholar
|
|
32
|
Hafkamp HC, Speel EJM, Haesevoets A, Bot
FJ, Dinjens WNM, Ramaekers FCS, Hopman AHN and Manni JJ: A subset
of head and neck squamous cell carcinomas exhibits integration of
HPV 16/18 DNA and overexpression of p16INK4A and p53 in the absence
of mutations in p53 exons 5-8. Int J Cancer. 107:394–400. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
de Roda Husman AM, Walboomers JM, van den
Brule AJ, Meijer CJ and Snijders PJ: The use of general primers GP5
and GP6 elongated at their 3′ ends with adjacent highly conserved
sequences improves human papillomavirus detection by PCR. J Gen
Virol. 76:1057–1062. 1995. View Article : Google Scholar
|
|
34
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maehara Y, Anai H, Tamada R and Sugimachi
K: The ATP assay is more sensitive than the succinate dehydrogenase
inhibition test for predicting cell viability. Eur J Cancer Clin
Oncol. 23:273–276. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Duarte D, Cardoso A and Vale N:
Synergistic growth inhibition of HT-29 colon and MCF-7 breast
cancer cells with simultaneous and sequential combinations of
antineoplastics and CNS drugs. Int J Mol Sci. 22:74082021.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ianevski A, Giri AK and Aittokallio T:
SynergyFinder 2.0: Visual analytics of multi-drug combination
synergies. Nucleic Acids Res. 48(W1): W488–W493. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grillo M, Bott MJ, Khandke N, McGinnis JP,
Miranda M, Meyyappan M, Rosfjord EC and Rabindran SK: Validation of
cyclin D1/CDK4 as an anticancer drug target in MCF-7 breast cancer
cells: Effect of regulated overexpression of cyclin D1 and
siRNA-mediated inhibition of endogenous cyclin D1 and CDK4
expression. Breast Cancer Res Treat. 95:185–194. 2006. View Article : Google Scholar
|
|
39
|
Herrera-Abreu MT, Palafox M, Asghar U,
Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M,
Rodriguez O, Grueso J, et al: Early adaptation and acquired
resistance to CDK4/6 inhibition in estrogen receptor-positive
breast cancer. Cancer Res. 76:2301–2313. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Billard-Sandu C, Tao YG, Sablin MP,
Dumitrescu G, Billard D and Deutsch E: CDK4/6 inhibitors in
P16/HPV16-negative squamous cell carcinoma of the head and neck.
Eur Arch Otorhinolaryngol. 277:1273–1280. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marquard FE and Jücker M: PI3K/AKT/mTOR
signaling as a molecular target in head and neck cancer. Biochem
Pharmacol. 172:1137292020. View Article : Google Scholar
|
|
42
|
Zainal NS, Lee BKB, Wong ZW, Chin IS, Yee
PS, Gan CP, Mun KS, Rahman ZAA, Gutkind JS, Patel V and Cheong SC:
Effects of palbociclib on oral squamous cell carcinoma and the role
of PIK3CA in conferring resistance. Cancer Biol Med. 16:264–275.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Finn RS, Dering J, Conklin D, Kalous O,
Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al: PD
0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially
inhibits proliferation of luminal estrogen receptor-positive human
breast cancer cell lines in vitro. Breast Cancer Res. 11:R772009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wagner V and Gil J: Senescence as a
therapeutically relevant response to CDK4/6 inhibitors. Oncogene.
39:5165–5176. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang TH, Chen CC, Leu YL, Lee YS, Lian JH,
Hsieh HL and Chen CY: Palbociclib induces DNA damage and inhibits
DNA repair to induce cellular senescence and apoptosis in oral
squamous cell carcinoma. J Formos Med Assoc. 120:1695–1705. 2021.
View Article : Google Scholar
|
|
46
|
Adkins D, Ley J, Neupane P, Worden F,
Sacco AG, Palka K, Grilley-Olson JE, Maggiore R, Salama NN,
Trinkaus K, et al: Palbociclib and cetuximab in platinum-resistant
and in cetuximab-resistant human papillomavirus-unrelated head and
neck cancer: A multicentre, multigroup, phase 2 trial. Lancet
Oncol. 20:1295–1305. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hawkins PT, Anderson KE, Davidson K and
Stephens LR: Signalling through class I PI3Ks in mammalian cells.
Biochem Soc Trans. 34:647–662. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Garces AE and Stocks MJ: Class 1 PI3K
clinical candidates and recent inhibitor design strategies: A
medicinal chemistry perspective. J Med Chem. 62:4815–4850. 2019.
View Article : Google Scholar
|
|
49
|
Miller MS, Thompson PE and Gabelli SB:
Structural determinants of isoform selectivity in PI3K inhibitors.
Biomolecules. 9:822019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Costa C, Ebi H, Martini M, Beausoleil SA,
Faber AC, Jakubik CT, Huang A, Wang Y, Nishtala M, Hall B, et al:
Measurement of PIP3 levels reveals an unexpected role for p110β in
early adaptive responses to p110α-specific inhibitors in luminal
breast cancer. Cancer Cell. 27:97–108. 2015. View Article : Google Scholar
|
|
51
|
Di Leo A, Johnston S, Lee KS, Ciruelos E,
Lønning PE, Janni W, O'Regan R, Mouret-Reynier MA, Kalev D, Egle D,
et al: Buparlisib plus fulvestrant in postmenopausal women with
hormone-receptor-positive, HER2-negative, advanced breast cancer
progressing on or after mTOR inhibition (BELLE-3): A randomised,
double-blind, placebo-controlled, phase 3 trial. Lancet Oncol.
19:87–100. 2018. View Article : Google Scholar
|
|
52
|
Brachmann SM, Kleylein-Sohn J, Gaulis S,
Kauffmann A, Blommers MJJ, Kazic-Legueux M, Laborde L, Hattenberger
M, Stauffer F, Vaxelaire J, et al: Characterization of the
mechanism of action of the pan class I PI3K inhibitor NVP-BKM120
across a broad range of concentrations. Mol Cancer Ther.
11:1747–1757. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Koul D, Shen R, LaFortune TA, Tiao N, Kim
YW, Liu JL, Maira SM, Garcia-Echeverria C and Yung WKA: Abstract
350: NVP-BKM120: A selective pan-PI3 kinase inhibitor induces G2/M
arrest in glioma cell lines via FOXO3a and GADD45a loop. Cancer
Res. 70(8 Suppl): S3502010. View Article : Google Scholar
|
|
54
|
Sadeghi S, Esmaeili S,
Pourbagheri-Sigaroodi A, Safaroghli-Azar A and Bashash D: PI3K
abrogation using pan-PI3K inhibitor BKM120 gives rise to a
significant anticancer effect on AML-Derived KG-1 cells by inducing
apoptosis and G2/M arrest. Turk J Haematol. 37:167–176.
2020.PubMed/NCBI
|
|
55
|
Hoxhaj G and Manning BD: The PI3K-AKT
network at the interface of oncogenic signalling and cancer
metabolism. Nat Rev Cancer. 20:74–88. 2020. View Article : Google Scholar :
|
|
56
|
Valvezan AJ and Manning BD: Molecular
logic of mTORC1 signalling as a metabolic rheostat. Nat Metab.
1:321–333. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hay N: The Akt-mTOR tango and its
relevance to cancer. Cancer Cell. 8:179–183. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
O'Reilly KE, Rojo F, She QB, Solit D,
Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al:
mTOR inhibition induces upstream receptor tyrosine kinase signaling
and activates Akt. Cancer Res. 66:1500–1508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cheng H, Yang X, Si H, Saleh AD, Xiao W,
Coupar J, Gollin SM, Ferris RL, Issaeva N, Yarbrough WG, et al:
Genomic and transcriptomic characterization links cell lines with
aggressive head and neck cancers. Cell Rep. 25:1332–1345.e5. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Huw LY, O'Brien C, Pandita A, Mohan S,
Spoerke JM, Lu S, Wang Y, Hampton GM, Wilson TR and Lackner MR:
Acquired PIK3CA amplification causes resistance to selective
phosphoinositide 3-kinase inhibitors in breast cancer. Oncogenesis.
2:e832013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang L, Wu J, Ling MT, Zhao L and Zhao
KN: The role of the PI3K/Akt/mTOR signalling pathway in human
cancers induced by infection with human papillomaviruses. Mol
Cancer. 14:872015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Curry JM, Tuluc M, Whitaker-Menezes D,
Ames JA, Anantharaman A, Butera A, Leiby B, Cognetti DM, Sotgia F,
Lisanti MP and Martinez-Outschoorn UE: Cancer metabolism, stemness
and tumor recurrence: MCT1 and MCT4 are functional biomarkers of
metabolic symbiosis in head and neck cancer. Cell Cycle.
12:1371–1384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Knight ZA, Gonzalez B, Feldman ME, Zunder
ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth
B, et al: A pharmacological map of the PI3-K family defines a role
for p110alpha in insulin signaling. Cell. 125:733–747. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sopasakis VR, Liu P, Suzuki R, Kondo T,
Winnay J, Tran TT, Asano T, Smyth G, Sajan MP, Farese RV, et al:
Specific roles of the p110alpha isoform of phosphatidylinsositol
3-kinase in hepatic insulin signaling and metabolic regulation.
Cell Metab. 11:220–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar :
|
|
67
|
Karim S, Burzangi AS, Ahmad A, Siddiqui
NA, Ibrahim IM, Sharma P, Abualsunun WA and Gabr GA: PI3K-AKT
pathway modulation by thymoquinone limits tumor growth and
glyco-lytic metabolism in colorectal cancer. Int J Mol Sci.
23:23052022. View Article : Google Scholar
|
|
68
|
Udawant S, Litif C, Lopez A, Gunn B,
Schuenzel E and Keniry M: PI3K Pathway inhibition with NVP-BEZ235
hinders glycolytic metabolism in glioblastoma multiforme cells.
Cells. 10:30652021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Alves LB, Moura AC, Amorim Dos Santos J,
Borges GA and Guerra ENS: Pharmacological PI3K inhibition in head
and neck squamous cell carcinoma: A systematic review. Toxicol In
Vitro. 88:1055582023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Xu J, Li Y, Kang M, Chang C, Wei H, Zhang
C and Chen Y: Multiple forms of cell death: A focus on the PI3K/AKT
pathway. J Cell Physiol. 238:2026–2038. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
O'Brien NA, McDermott MSJ, Conklin D, Luo
T, Ayala R, Salgar S, Chau K, DiTomaso E, Babbar N, Su F, et al:
Targeting activated PI3K/mTOR signaling overcomes acquired
resistance to CDK4/6-based therapies in preclinical models of
hormone receptor-positive breast cancer. Breast Cancer Res.
22:892020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Goodwin CM, Waters AM, Klomp JE, Javaid S,
Bryant KL, Stalnecker CA, Drizyte-Miller K, Papke B, Yang R, Amparo
AM, et al: Combination therapies with CDK4/6 inhibitors to treat
KRAS-mutant pancreatic cancer. Cancer Res. 83:141–157. 2023.
View Article : Google Scholar :
|
|
73
|
Coppock JD, Wieking BG, Molinolo AA,
Gutkind JS, Miskimins WK and Lee JH: Improved clearance during
treatment of HPV-positive head and neck cancer through mTOR
inhibition. Neoplasia. 15:620–630. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cassell A, Freilino ML, Lee J, Barr S,
Wang L, Panahandeh MC, Thomas SM and Grandis JR: Targeting TORC1/2
enhances sensitivity to EGFR inhibitors in head and neck cancer
preclinical models. Neoplasia. 14:1005–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Seiwert TY, Kochanny S, Wood K, Worden FP,
Adkins D, Wade JL, Sleckman BG, Anderson D, Brisson RJ, Karrison T,
et al: A randomized phase 2 study of temsirolimus and cetuximab
versus temsirolimus alone in recurrent/metastatic,
cetuximab-resistant head and neck cancer: The MAESTRO study.
Cancer. 126:3237–3243. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bautista F, Paoletti X, Rubino J, Brard C,
Rezai K, Nebchi S, Andre N, Aerts I, De Carli E, van Eijkelenburg
N, et al: Phase I or II study of ribociclib in combination with
topotecan-temozolomide or everolimus in children with advanced
malignancies: Arms A and B of the AcSé-ESMART trial. J Clin Oncol.
39:3546–3560. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bardia A, Modi S, Oliveira M, Cortes J,
Campone M, Ma B, Dirix L, Weise A, Hewes B, Diaz-Padilla I, et al:
Phase Ib dose-escalation/expansion trial of ribociclib in
combination with everolimus and exemestane in postmenopausal women
with HR+, HER2-advanced breast cancer. Clin Cancer Res.
26:6417–6428. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wright SCE, Vasilevski N, Serra V, Rodon J
and Eichhorn PJA: Mechanisms of resistance to PI3K inhibitors in
cancer: Adaptive responses, drug tolerance and cellular plasticity.
Cancers (Basel). 13:15382021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Song KW, Edgar KA, Hanan EJ, Hafner M, Oeh
J, Merchant M, Sampath D, Nannini MA, Hong R, Phu L, et al:
RTK-dependent inducible degradation of mutant PI3Kα drives GDC-0077
(Inavolisib) efficacy. Cancer Discov. 12:204–219. 2022. View Article : Google Scholar
|
|
80
|
Demers I, Donkers J, Kremer B and Speel
EJ: Ex vivo culture models to indicate therapy response in head and
neck squamous cell carcinoma. Cells. 9:25272020. View Article : Google Scholar : PubMed/NCBI
|