Introduction
Pancreatic cancer (PC) is one of the most common
malignant tumors of the digestive tract. The 5-year survival rate
for pancreatic cancer is estimated to be 10% after diagnosis,
making it one of the malignancies with the poorest prognosis
(1). In the clinical setting, the
common treatment strategy for PC is local surgical resection and
adjuvant chemotherapy. However, toxicity and side effects of
chemotherapeutic drugs often decrease quality of life (2). Therefore, the discovery,
identification and development of novel antitumor drugs from safe,
natural products are urgently needed to improve treatment and
prognosis of patients with PC.
Fucoxanthin (FX) is a xanthophyll carotenoid
extracted from marine aquatic brown seaweed such as Hizikia
fusifarme and Laminaria japonica. It has antioxidant,
anti-inflammatory, anti-obesity and antidiabetic properties
(3). Furthermore, FX is
incorporated as a functional ingredient in certain food, cosmetic
and pharmaceutical products, suggesting it is a safe therapeutic
natural extract with no adverse effects on the human body (3). FX has antitumor activity against
various types of cancer (4). It
has also been found to inhibit tumor progression by inducing cell
cycle arrest and apoptosis in vitro and suppress
carcinogenesis in vivo in various types of cancer, such as
colorectal and breast cancer, lymphoma and leukemia (5). Moreover, FX triggers LC3-dependent
autophagy, which may be associated with apoptosis in cervical,
gastric and nasopharyngeal cancers. However, the precise role of
autophagy in different types of cancer remains controversial
(6-8). FX has been shown to prevent
tumorigenesis of murine PC by suppressing the chemokine (C-C motif)
ligand 21/chemokine receptor 7 axis, T-lymphocyte attenuator, tumor
microenvironment, epithelial-mesenchymal transition and adhesion
(9). The metabolite of FX,
fucoxanthinol (FxOH), has been shown to promote human PC PANC-1
cell proliferation while proliferation of MIA PaCa-2 PC cells
(10). This suggests that cell
heterogeneity may contribute to the differential anti-tumor effects
of FXOH, potentially associated with genetic background of the
cells. However, the effects of FX on human PC and its mode of
action remain unclear. Therefore, the present study aims to
investigate the potential antitumor effects of FX on human PC.
One potential mechanism by which FX may promote
anticancer effects is altering cancer cell metabolism. Metabolic
alterations are a key hallmark of cancer cells, enabling them to
meet biosynthetic and energetic requirements (11). Therefore, targeting tumor
metabolism may represent a promising strategy for cancer treatment.
Tumor cells preferentially rely on anaerobic glucose metabolism
(aerobic glycolysis) over mitochondrial oxidative phosphorylation
(OXPHOS) to meet energy needs, even in the presence of oxygen
(12). Most cancer cells rely on
glycolysis for energy and macromolecule synthesis to sustain
uncontrolled proliferation, contributing to decreased or
dysfunction of mitochondrial OXPHOS (13,14). However, studies show that numerous
cancer cell subpopulations rely on OXPHOS for bioenergetic and
biosynthetic processes (15,16). For example, certain types of
cancers (such as leukemia, lymphoma and PC) have increased
mitochondrial function, even during active glycolysis (14-17). Furthermore, cancer cells retaining
mitochondrial OXPHOS metabolism use glucose-derived pyruvate and
also derive energy from alternative substrates, such as fatty acids
and glutamine (Gln), in mitochondria (18). These cancer cells can upregulate
the uptake of Gln and its conversion into a-ketoglutarate. Gln
metabolism sustains the tricarboxylic acid (TCA) cycle, thereby
supporting mitochondrial generation of the nucleotides and amino
acids necessary for tumor progression (19-24). Overall, these observations suggest
that aerobic glycolysis and mitochondria-associated metabolism
(including OXPHOS and Gln metabolism) are key for development of
certain types of tumor.
Aerobic glycolysis and mitochondria-associated
metabolism are crucial for cancer cell proliferation, with Gln
serving as an alternative metabolite during glucose limitation,
suggesting that inhibiting either process may be a potential
treatment. However, although this strategy may work in in
vitro cultured cells, its clinical efficiency for in
vivo cancer therapy is unsatisfactory (25). Therefore, it was hypothesized that
the in vivo therapeutic effect of PC treatment could be
enhanced by a targeted metabolic strategy combining dietary glucose
restriction and drug-induced inhibition of mitochondrial
metabolism. FX can attenuate the reprogramming of energy metabolism
during activation of hepatic stellate cells, suggesting a potential
role of FX in metabolism regulation (26). To the best of our knowledge,
however, FX regulation of the bioenergetic metabolism of cancer
cells, especially PC cells, has not been investigated. The present
study aimed to investigate the effects of FX on aerobic glycolysis,
mitochondrial OXPHOS and Gln metabolism to develop a safe, natural
and efficient therapeutic strategy for PC.
Materials and methods
Cell lines and culture
PATU-8988T (cat. no. CL0254) and PATU-8988S (cat.
no. CL0908) cell lines were purchased from Hunan Fenghui
Biotechnology, whereas PANC-1 (cat. no. CL-0184) and 293T (cat. no.
CL-0005) cell lines were obtained from Procell Life Science &
Technology and Cell Bank of the Chinese Academy of Science,
respectively. All cell lines were cultured in a humidified
incubator with 5% CO2 at 37°C in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) containing 10% fetal bovine serum
(BIOVISTECH; cat. no. SE100-B) and penicillin-streptomycin.
The genetic background of PATU-8988T and PATU-8988S
is similar in k-ras; for example, the mutation site of K-RAS is the
same (k-ras-p.G12V) between the two cell lines. PANC-1 is a
k-ras-p.G12D mutated cell line. Therefore, PATU-8988T and
PATU-8988S were used for experiments.
Primary antibodies and reagents
The primary antibodies and reagents are all listed
in Tables SI and SII,
respectively. FX was obtained from Shanghai Macklin Biochemical
Co., Ltd. (cat. no. F861277; purity, 98%).
Assessment of cell viability
The half-maximal inhibitory concentration of FX (0,
5, 10, 20, 40, 80, 160 and 320 µM) on PC cells treated for
72 h at 37°C was determined by MTT Cell Proliferation and
Cytotoxicity Assay Kit according to the manufacturer's instructions
(Beyotime Biotechnology, C0009S). Furthermore, changes in cells
treated for 24 h with FX (25 µM) in the presence or absence
of 7.5 mM glutathione (GSH) under glucose-limited (medium
containing 1 mM glucose) conditions at 37°C were also evaluated by
MTT assay. In addition, cells treated for 24 h with FX (50
µM) in the presence or absence of 30 µM DDP at 37°C
were also analyzed by MTT assay. Then the purple formazan was
dissolved by using Formazan solvent in this Kit and then detected
at wavelength 570 nm.
To select concentrations of ZVAD, Z-VAD-FMK
(27); Fer-1, Ferrostatin-1
(27); NEC-1, Necrostatin-1
(28); NSA, Necrosulfonamide
(28); DFO, Deferoxamine mesylate
(29); TTM, Ammonium
Tetrathiomolybdate (30,31); 3-MA, 3-Methyladenine (32-34); BafA1, Bafilomycin A1 (35) and GSH, (36), cells were treated with these
compounds (ZVAD: 0, 10, 20, 40, and 80 µM; Fer-1: 0, 0.5, 1,
2, and 4 µM; DFO: 0, 25, 50, 100, and 200 µM; NEC-1:
0, 1.25, 2.5, 5, and 10 µM; NSA: 0, 1.25, 2.5, 5, and 10
µM; TTM: 0, 2.5, 5, 10, and 20 µM; 3-MA: 0, 2.5, 5,
10, and 20 mM; BafA1: 0, 5, 10, 20, and 40 nM) in a 12-well plate
at 37°C for 48 h, observed using optical microscope (Nikon
Corporation; ECLIPSE TS 100, light) and non-lethal maximum
concentrations were selected for subsequent experiments.
Measurement of cell proliferation by
real-time cell analysis (RTCA) assay
The suppressive effect of FX on proliferation of PC
cell lines was determined using RTCA assay according to the
manufacturer's instructions.
Clonogenic cell survival assay
PC cells (1,000 cells/well) were seeded in 6-well
plates 1,000 cells/well and treated with or without FX (0, 25, 50,
100 µM) for 2 weeks at 37°C. The colonies (>50 cells)
were washed with PBS, fixed with methanol 4% for 30 min at Room
temperature, stained with crystal violet for 30 min at Room
temperature, and quantified by Image J (National Institutes of
Health (NIH), V1.8.0.112).
FACS (Fluorescence-activated cell
Sorting) nalysis of cell death rate
Following 48 h FX (0, 25, 50, 100 µM)
monotreatment or 24 h FX (25 µM) treatment in the presence
or absence of 7.5 mM GSH under GL conditions at 37°C, cells were
stained with Annexin V-FITC/PI in the dark for 15 min and then
analyzed by BD FACSCanto™ Clinical Flow Cytometry System (BD
Biosciences, BDCanto II) according to the manufacturer's
instruction using Annexin V-FITC/PI apoptosis detection kit (KeyGEN
Bio TECH, KGA108). Results were analyzed by Flowjo (BD Biosciences,
FlowJo_v10.8.1_CL). Cell death rate was quantified based on the
formula as (Q2+Q3+Q4) ×100%.
Western blot analysis
PC cells were harvested, lysed with lysis buffer
(Beyotime Institute of Biotechnology; cat. no. P0013) for 20 min
and centrifuged at 13,400 g for 20 min at 4°C to collect protein in
the supernatant. The proteins were quantified using the Pierce™ BCA
Protein Assay kit, according to the manufacturer's instructions,
mixed with 5X DualColor Protein Loading Buffer to adjust the
protein concentration to 1 µg/µl and heated at 95°C
for 5 min. A total of 20 µg protein/lane were loaded onto
10% SDS-PAGE gel, electrophoresed in running buffer and transferred
to a 0.22 µM PVDF membrane in transfer buffer. The membranes
were treated with 5% non-fat dry milk to block the non-specific
binding at room temperature for 90 min, incubated with primary
antibody (GSDME, GSDMD, GSDMC, OPA1, SLC31A1 and GLS; all 1:1,000)
overnight at 4°C and washed three times with 1X TBST at room
temperature, followed by incubation with HRP-labeled Goat
Anti-Rabbit or Anti-Mouse IgG(H+L) (all 1:1,000) for 90 min at room
temperature. After washing the membrane three times with 1X TBST at
room temperature, it was visualized using the SuperSignal™ West
Pico PLUS kit and UVP ChemStudio PLUS (Jena GmbH, H116457),and
quantified using ImageJ software (National Institutes of Health
(NIH), V1.8.0.112).
Determination of oxygen consumption rate
(OCR)
The effect of FX on mitochondrial respiration in PC
cells was determined using an XFe96 extracellular flux analyzer
according to the manufacturer's instructions. Briefly, cells were
seeded into culture plates (20,000 cells/well) and incubated
overnight at 37°C. Adherent cells were treated with FX (0, 25, 50
and 100 µM) for 8 h at 37°C. The medium was replaced with a
Seahorse XF Base Medium (Seahorse Bioscience, 102353-100)
containing 25 mM glucose and 2 mM pyruvic acid sodium and cultured
for 1-2 h at 37°C. Working solutions containing oligomycin, FCCP
(Carbonyl cyanide 4-(trifluoromethoxy) or rotenone/antimycin A were
prepared and added according to the manufacturer's protocol. OCR
was detected using the XFe96 extracellular flux analyzer (Seahorse
Bioscience, XFe96, S7800B).
Measurement of extracellular
acidification rate (ECAR)
The effect of FX on aerobic glycolysis in PC cells
was measured using the XFe96 extracellular flux analyzer according
to the manufacturer's guidelines. Briefly, cells were seeded in a
culture plate (20,000 cells/well) overnight and treated with FX (0,
25, 50, 100 µM) for 8 h at 37°C. The medium was then
replaced with Seahorse XF Base Medium containing Gln (2.24 mM), and
cells were incubated for 1-2 h at 37°C. Working solutions
containing glucose, oligomycin and 2-DG (2-Deoxy-D-glucose) were
added according to the manufacturer's protocol. ECAR was detected
using the XFe96 extracellular flux analyzer according to the
manufacturer's instructions (Seahorse Bioscience; cat. no.
S7800B).
FACS analysis for whole-cell and
mitochondrial ROS
Following 48 h FX (0, 25, 50, 100 µM)
treatment at 37°C, cells were stained for 15 min in the dark with
DCFH-DA (for whole-cell) or Mitosox (for mitochondrial ROS) at
37°C. The stained cells were analyzed by BD FACSCanto™ Clinical
Flow Cytometry System (BD Biosciences, BDCanto II) according to the
manufacturer's instructions using Reactive Oxygen Species Assay
kit, Beyotime Biotechnology, S0033; Mitosox Red, Invitrogen
M36008). Results were analyzed by Flowjo (BD Biosciences,
FlowJo_v10.8.1).
Whole-cell and mitochondrial ROS observed
by fluorescence microscopy
Following 48 h FX (0, 25, 50, 100 µM)
treatment at 37°C, cells were stained for 15 min in the dark with
DCFH-DA (for whole-cell) or Mitosox (for mitochondrial ROS) at 37°C
and then analyzed by fluorescence microscopy according to the
manufacturer's instructions using Reactive Oxygen Species Assay kit
(Beyotime Biotechnology, S0033; Mitosox Red, Invitrogen
M36008).
FACS analysis of cell death
Following 48 h FX (0, 25, 50 and 100 µM)
monotreatment or 24 h FX (25 µM) treatment in the presence
or absence of 7.5 mM GSH under GL conditions at 37°C, cells were
stained with Annexin V-FITC/PI for 20 min in the dark. The stained
cells were then analyzed by BD FACSCanto™ Clinical Flow Cytometry
System (BD Biosciences, BDCanto II) according to the manufacturer's
instructions (Annexin V-FITC/PI apoptosis detection kit, KeyGEN Bio
TECH, KGA108). Results were further analyzed by Flowjo (BD
Biosciences, FlowJo_v10.8.1).
RNA interference
The lentiviral plasmid of pPLK/GFP + Puro-SLC31A1
short hairpin (sh)RNA was obtained from PPL (Public Protein/Plasmid
Library) and 293T cells were used to package lentivirus. The
generation system (2nd) was used. A total of 2 target plasmid + 2
µg psPAX2+1 µg pMD2G was transfectd to 293T cells for
6 h at 37°C to package lentivirus. Collection was performed after
48 h, then centrifuged at 500 g for 5 min at 4°C, and filtered by
using 0.45 µM filter membrane. PATU-8988T cells were
infected with packaged lentivirus (the multiplicity of infection
(~3 used in PATU-8988T cells) for 48 h and screened by puromycin (2
µg/ml) for 7 days. The efficiency of transfection was
assessed by western blotting. And the 0.5 µg/ml of puromycin
was used to maintain the efficiency of transfection. The western
blot and MTT assay were performed in transfected PATU-8988T cells
to confirm the whether the SLC31A1 knockdown can attenuate
cytotoxicity caused by the combination of FX and DDP. Sequences of
sh-SLC31A1 clone were as follows: NC, 5′-GTT CTC CGA ACG TGT CAC
GTT-3′; shSLC31A1#1, 5′-CGG TAC AGG ATA CTT CCT CTT-3′ and
shSLC31A1#2, 5′-GAT GCC TAT GAC CTT CTA CTT-3′.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 5 (Dotmatics) and data are expressed as the mean ±
SD. One-way ANOVA followed by Tukey post hoc test was performed to
analyze differences between >2 groups. Each experiment was
repeated ≥3 times. P<0.05 was considered to indicate a
statistically significant difference.
Results
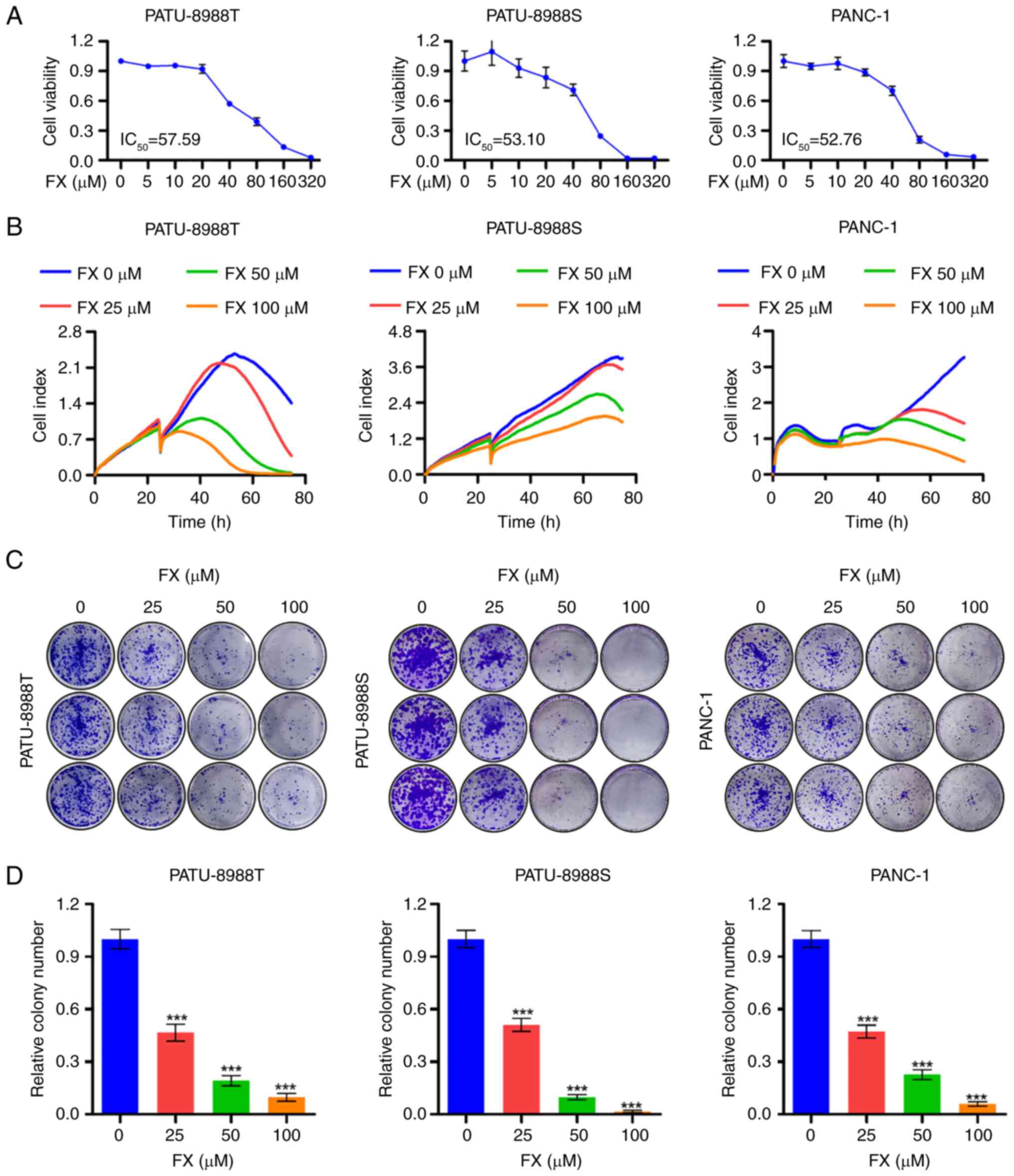
FX impedes PC cell proliferation
The structural formula of FX is shown in Fig. S1. The present study demonstrated
a dose-dependent reduction in PC cell viability, confirming the
tumor-suppressive effects of FX on PC cells (Fig. 1A). RTCA assay further indicated
that FX notably inhibited PC cell proliferation (Fig. 1B). Furthermore, FX inhibited the
colony-forming ability of PC cells (Fig. 1C and D). Overall, these results
suggested that FX significantly inhibited the in vitro
proliferation of PC cells.
FX induces a distinct form of regulated
cell death in PC cells
The present study further investigated the antitumor
activity of FX by flow cytometry analysis of PC cells labeled with
Annexin V-FITC and PI. There was significant dose-dependent
induction of PC cell death by FX (Fig. 2A). However, FX-induced cell death
was not prevented by inhibitors of cell death mechanisms, including
inhibitors of apoptosis (ZVAD), ferroptosis (Fer-1), necroptosis
(NEC-1 and NSA), cuproptosis (TTM) and autophagy (3-MA and BafA1;
Fig. 2B). Moreover, western blot
analysis revealed that FX did not induce the cleavage of GSDME,
GSDMD or GSDMC, which are key factors involved in pyroptosis
(37). These results suggested
that FX stimulated a novel type of cell death in PC cells, distinct
from apoptosis, pyroptosis, necroptosis, ferroptosis, cuproptosis
or autophagic cell death.
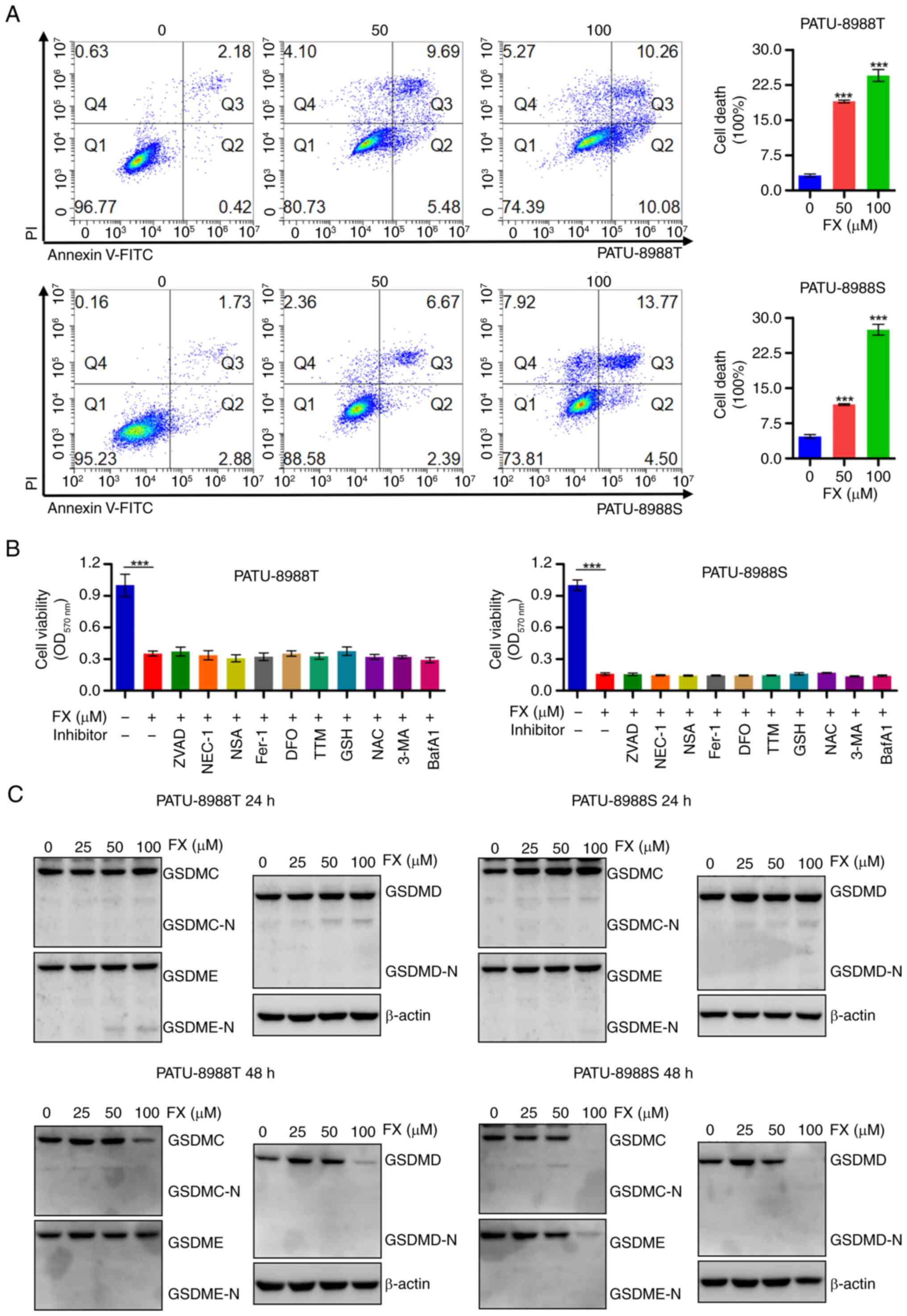 | Figure 2FX induces a distinct form of
regulated cell death in PC cells. (A) PC cells were stained with
FITC/PI and cell death was analyzed using flow cytometry. (B)
Viability was assessed in FX-treated cells in the presence or
absence of programmed cell death inhibitors, including inhibitors
of apoptosis (ZVAD, 20 µM), ferroptosis (Fer-1, 1 µM;
DFO, 50 µM), necroptosis (NEC-1, 5 µM; NSA, 5
µM), cuproptosis (TTM, 5 µM) and autophagic cell
death (3-MA, 5 mM; BafA1, 10 nM). (C) Changes in GSDMC, GSDMD and
GSDME expression in response to FX treatment were examined using
western blot analysis. ***P<0.001 vs. 0 µM).
FX, Fucoxanthin; PC, Pancreatic cancer; ZVAD, Z-VAD-FMK; Fer-1,
Ferrostatin-1; DFO, Deferoxamine mesylate; NEC-1, Necrostatin-1;
NSA, Necrosulfonamide; TTM, Ammonium Tetrathiomolybdate; 3-MA,
3-Methyladenine; BafA1, Bafilomycin A1; GSDMC, Gasdermin C; OD,
Optical density; -N, N-terminal. |
FX disrupts mitochondrial homeostasis and
causes oxidative stress in PC cells
The present study also evaluated mitochondria,
responsible for producing cellular ATP and key intermediate
metabolites (23), as potential
targets of FX-induced proliferation inhibition of PC cells.
Seahorse XF Cell Mito Stress test showed that FX treatment
significantly inhibited overall mitochondrial OCR (Figs. 3A and S2A) in PC cells and reduced basal
respiration, ATP production and maximal respiration (Figs. 3B and S2B). FX triggered accumulation of ROS
in whole cells and the mitochondria (Figs. 3C and D and S2C-F). FX significantly decreased the
whole-cell ATP production in PC cells (Fig. 3E). An increase in OPA1 cleavage in
response to FX supported the observed mitochondrial fragmentation
(Figs. 3F and S2G). Mitotracker staining results also
confirmed that FX increased morphological fragmentation of PC cells
(Fig. 3G). These observations
suggested that FX treatment disrupted mitochondrial homeostasis and
promoted oxidative stress in PC cells.
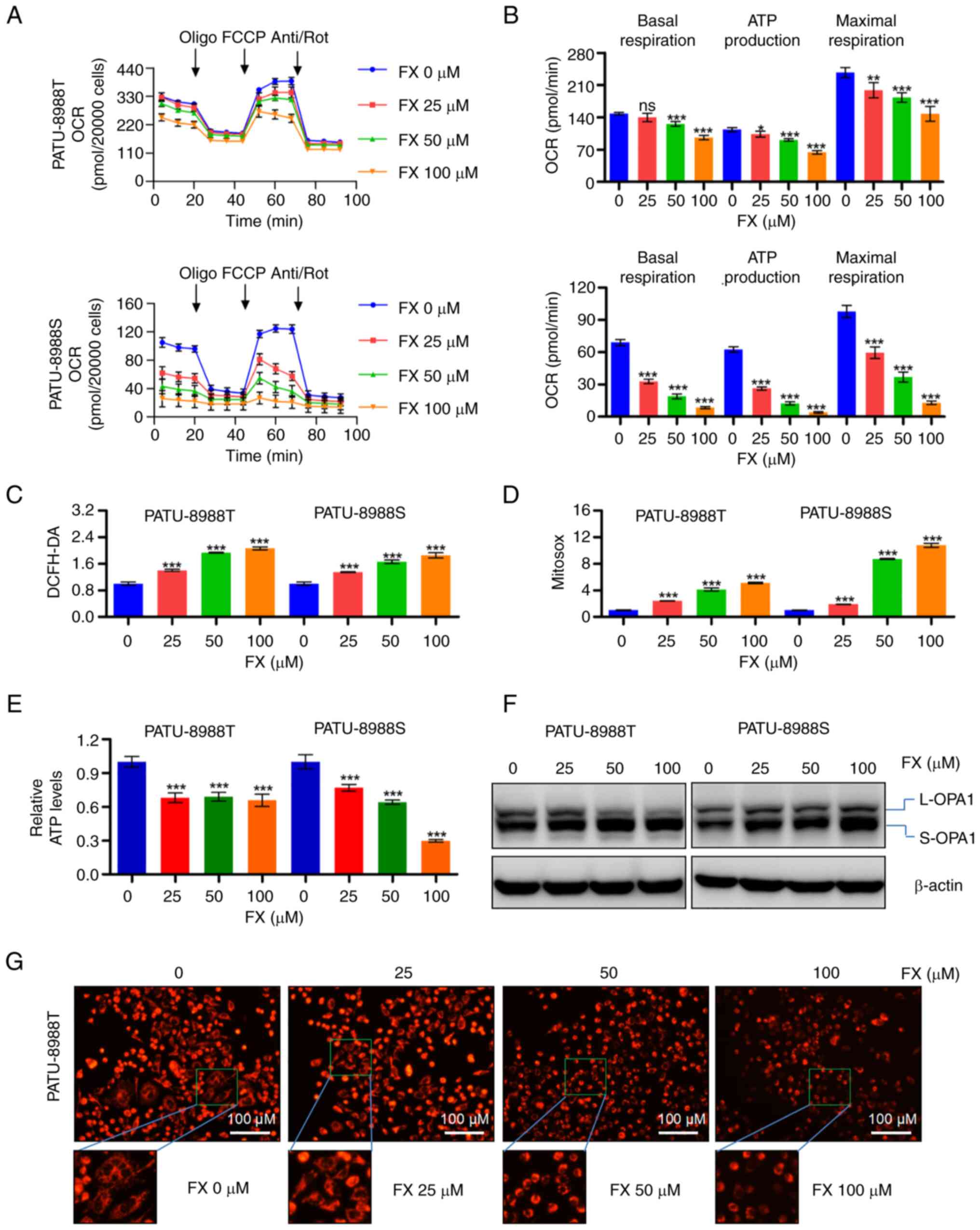 | Figure 3FX impairs mitochondrial homeostasis
and causes oxidative stress in PC cells. (A) OCR in FX-treated PC
cells was measured using the XFe96 extracellular flux analyzer to
evaluate changes in (B) basal respiration, ATP production and
maximal respiration in FX-treated PC cells. (C) ROS levels were
detected in PC cells stained with DCFH-DA using flow cytometry. (D)
Mitochondrial ROS production was measured in PC cells stained with
MitoSOX via flow cytometry. (E) Cellular ATP levels. (F) Western
blot analysis was performed to evaluate changes in OPA1 expression
in response to FX treatment in PC cells. (G) Changes in
mitochondrial morphology in PC cells following FX exposure were
observed using MitoTracker staining. *P<0.05,
**P<0.01, ***P<0.001 vs. 0 µM).
FX, Fucoxanthin; PC, Pancreatic cancer; OCR, Determination of
oxygen consumption rate; ROS, Reactive oxygen species; OPA1, Optic
atrophy 1; ns, no significant; FCCP, Carbonyl cyanide
4-(trifluoromethoxy)phenylhydrazone; Anti/Rot, Antimycin
a1/Rotenone; L-, Long-OPA1; S-OPA1, Short-OPA1. |
FX initiates energy metabolism
reprogramming and a switch from mitochondrial respiration to
aerobic glycolysis in PC cells
The present study analyzed the overall metabolic
profile based on the aforementioned bioenergetics analysis data. FX
decreased mitochondrial respiration in PC cells (as determined by
OCR), accompanied by an increase in capability for aerobic
glycolysis (as determined by ECAR; Fig. 4A). The overall increase in
ECAR/OCR ratio favored glycolysis in PC cells (Fig. 4B). Seahorse XF Glycolysis Stress
Test revealed that FX altered aerobic glycolysis and elevated
overall ECAR levels in PC cells (Figs. 4C and S3A), which were accompanied by an
increase in rate of glycolysis (Figs.
4D and S3B). The
aforementioned decrease in whole-cell ATP levels indicated that
this glycolysis promotion could not compensate for FX-induced
suppression of mitochondrial ATP production. Overall, these results
suggested that FX reprograms PC cell metabolism toward glycolysis
for ATP production; however, increased glycolysis does not
compensate for the ATP reduction caused by FX-mediated inhibition
of mitochondrial respiration.
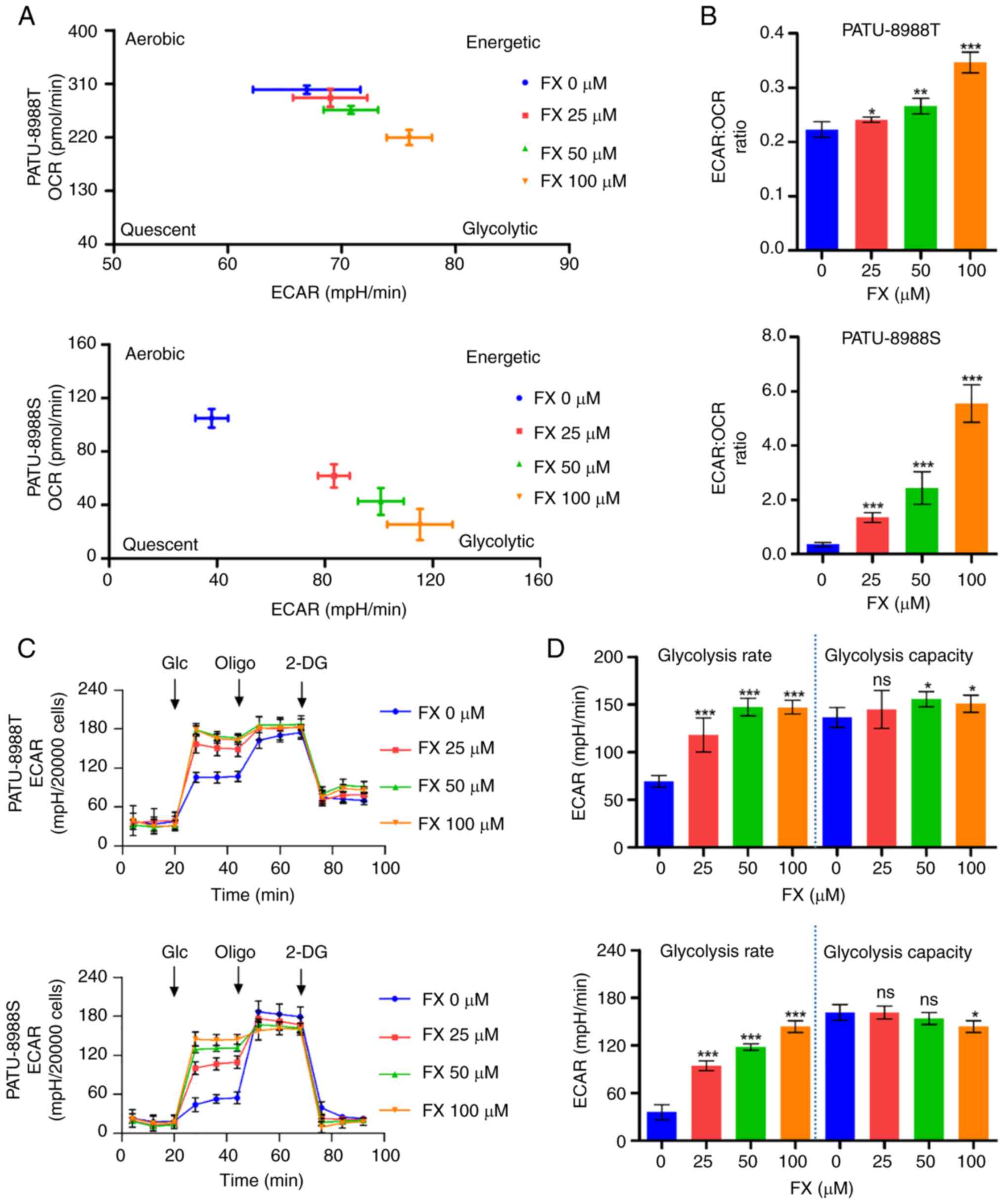 | Figure 4FX initiates reprogramming of energy
metabolism to convert mitochondrial respiration to aerobic
glycolysis in PC cells. (A) Metabolic phenotype profiles of PC
cells representing changes in response to FX treatment. (B)
ECAR/OCR ratio was analyzed to confirm changes in metabolic
profiles in FX-treated cells. (C) Changes in ECAR in FX-exposed PC
cells were measured using XFe96 extracellular flux analyzer. (D)
Changes in glycolysis rate and capacity in FX-treated PC cells.
*P<0.05, **P<0.01,
***P<0.001 vs. 0 µM. FX, Fucoxanthin; PC,
Pancreatic cancer; ECAR, Measurement of extracellular acidification
rate; OCR, Determination of oxygen consumption rate; Glc, Glucose;
Oligo, Oligomycin; 2-DG, 2-Deoxy-D-glucose; ns, no significant. |
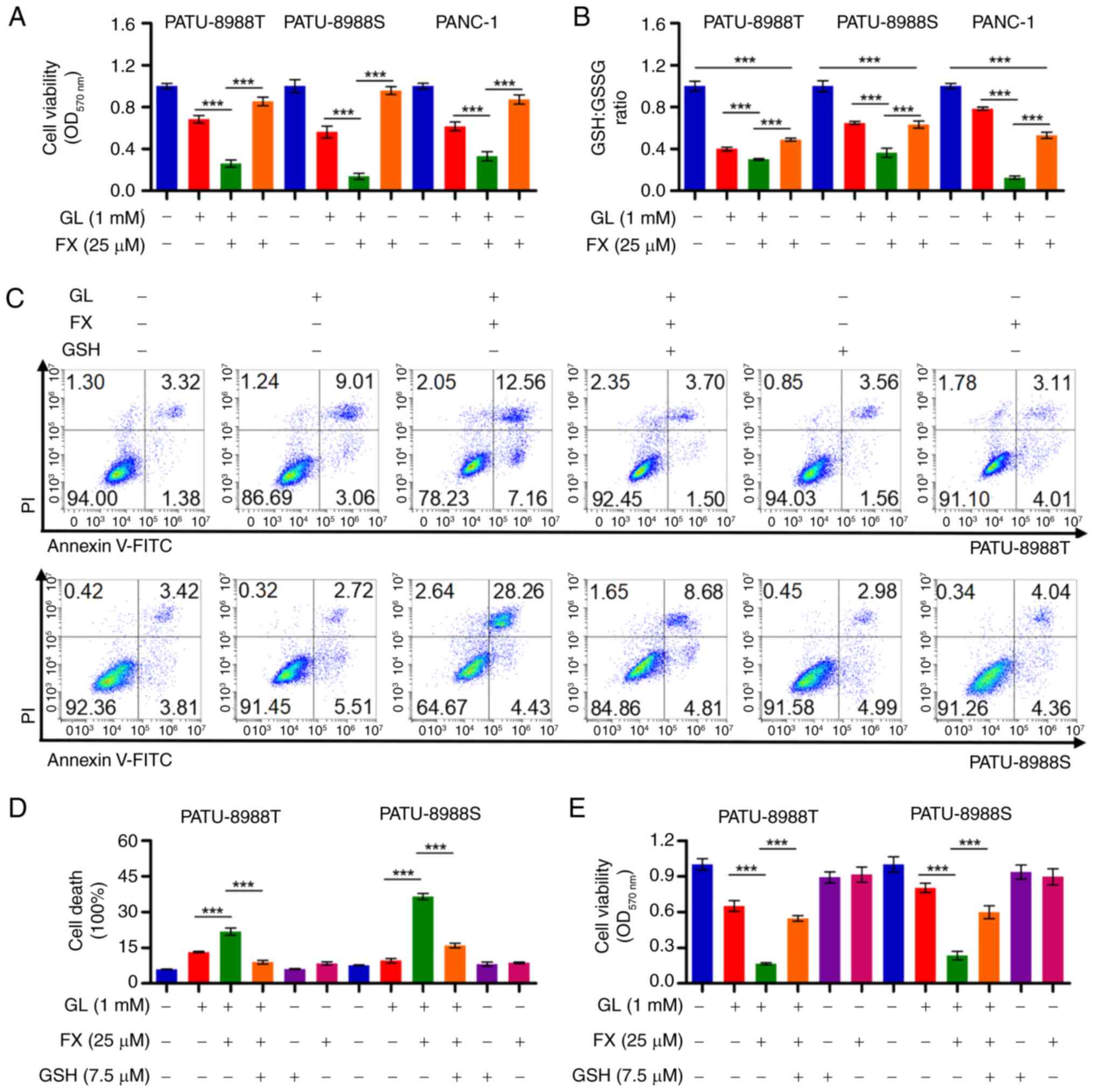
FX sensitizes PC cells to GL condition by
enhancing the decrease in GSH/GSSG ratio
FX-induced increased reliance on aerobic glycolysis
in PC cells suggests that this shift may decrease metabolic
flexibility in response to environmental stress, particularly
during glucose deficiency. Under GL conditions, FX significantly
decreased cell viability compared with PC cells treated with FX or
GL alone (Fig. 5A). There was a
significant decrease in GSH/GSSG ratio after the co-treatment
(Fig. 5B). GSH prevented the
decrease in cell viability and cell death in PC cells co-treated
with FX and GL (Figs. 5C-E and
S4A). These findings indicated
that FX treatment may suppress conversion of GSSG to GSH, thereby
increasing oxidative stress under GL conditions and promoting PC
cell death.
FX enhances PC cell sensitivity to DDP by
promoting expression of SLC31A1
FX has been shown to enhance DDP-induced
cytotoxicity in several cancer types (38-40), though the specific mechanism
remains unclear. Cell viability assay showed that FX increased the
cytotoxicity of DDP in PC cells (Fig.
6A). FX significantly promoted the expression of SLC31A1, a key
protein responsible for DDP uptake, in PC cells (Figs. 6B and S5A). Western blotting confirmed SLC31A1
knockdown (Figs. 6C and S5B), which attenuated cytotoxicity
caused by the combination of FX and DDP (Fig. 6D). These findings suggested that
FX enhanced the sensitivity of PC cells to DDP by increasing
SLC31A1-mediated cellular uptake of DDP.
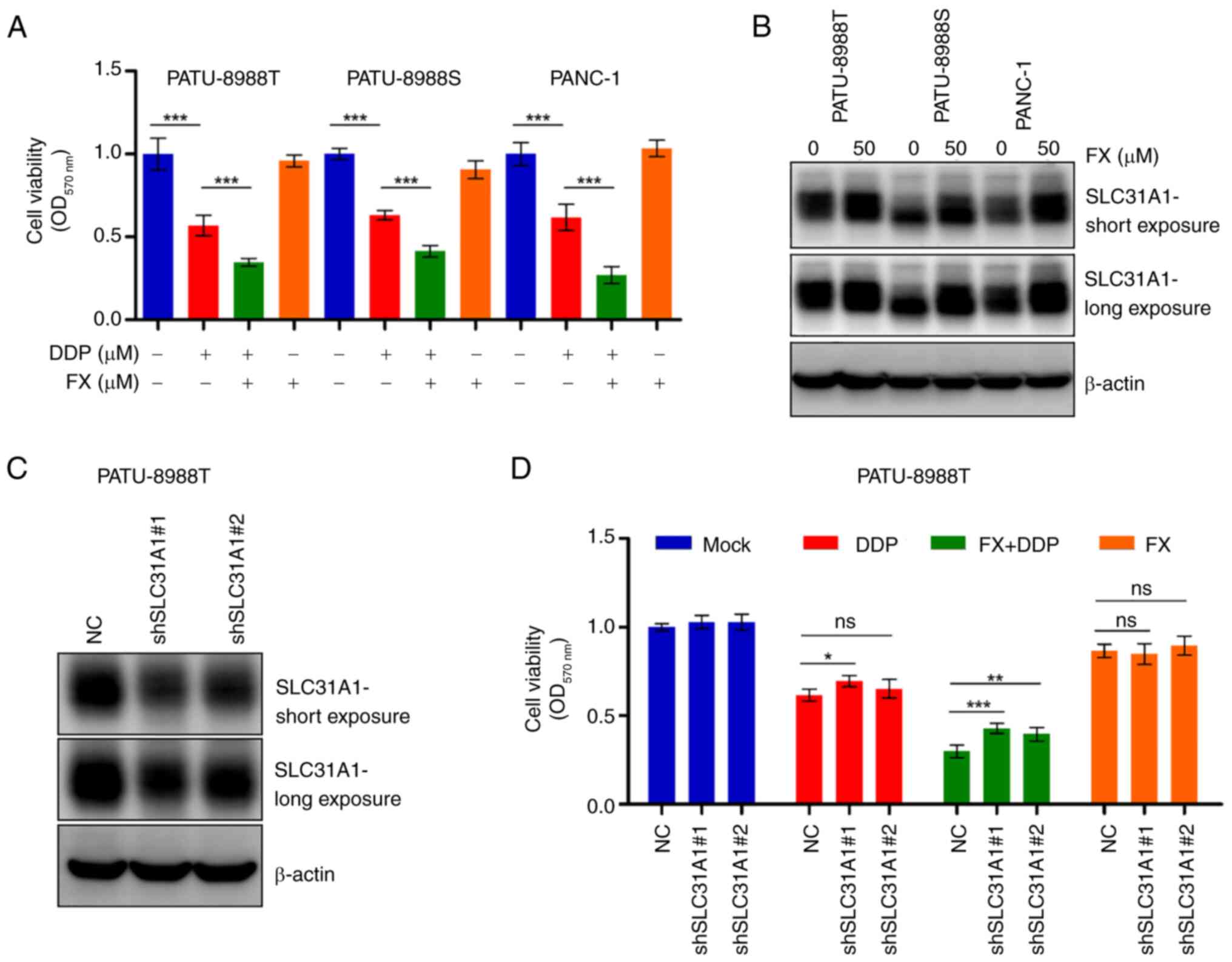 | Figure 6FX enhances cell sensitivity to DDP
by upregulating SLC31A1 expression. (A) Cell viability was measured
via MTT assay following 24 h treatment with FX (50 µM)
and/or DDP (30 µM). (B) Changes in SLC31A1 expression in PC
cells after 24 h FX treatment were analyzed by western blotting.
(C) PATU-8988T SLC31A1 knockdown cell lines were constructed, and
western blot assay was performed to confirm transfection
efficiency. (D) Cell viability was assessed using MTT assay
following treatment with FX in the presence or absence of DDP for
24 h. *P<0.05, **P<0.01,
***P<0.001 vs. NC. FX, Fucoxanthin; SLC31A1, Solute
carrier family 31 member 1; PC, pancreatic cancer; OD, Optical
density; sh, short hairpin; NC, Negative control; ns, no
significant; DDP, Cisplatin. |
Discussion
Although previous studies have shown that FX has
anti-tumor activity in various other cancer types (4-10),
its effects on human PC cells have not been investigated. The
present study showed that FX exerted anti-PC activity by
suppressing cell proliferation and inducing non-classical
programmed cell death (PCD). FX treatment of PC cells triggered
metabolic reprogramming switch from mitochondrial oxidative
phosphorylation to aerobic glycolysis, decreasing cellular ATP
levels. This FX-mediated metabolic alteration also decreased
metabolic flexibility in PC cells, increasing sensitivity to GL
conditions. Further investigation revealed that FX treatment
decreased GSH/GSSG ratio, resulting in cytotoxicity under
glucose-depleted conditions; however, these effects were alleviated
by GSH. FX treatment also significantly induced expression of
SLC31A1, a crucial execution factor required for DDP uptake
(41), indicating FX may improve
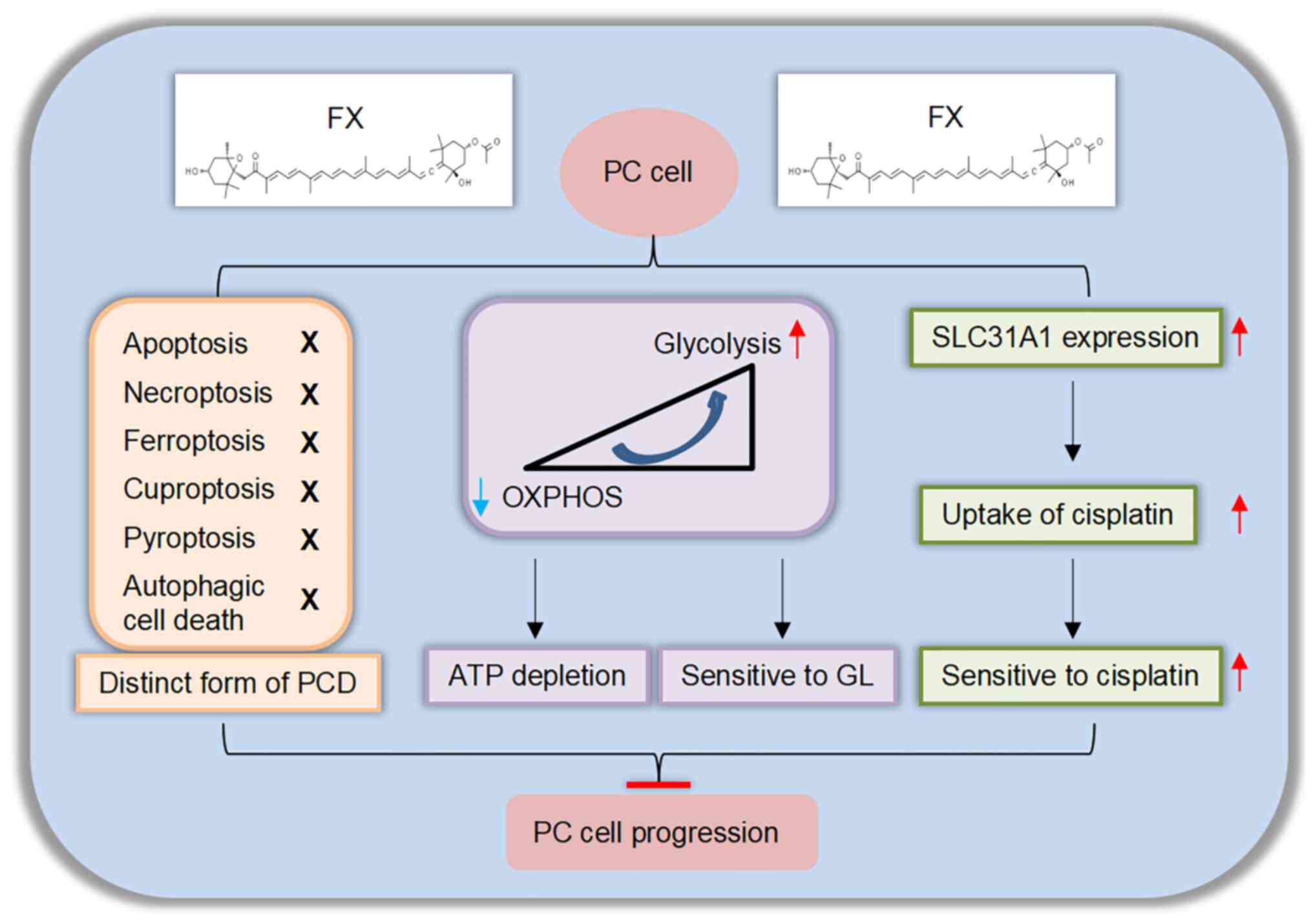
cell sensitivity to DDP. Taken together, these findings indicated
FX exerted its antitumor effects by initiating cellular energy
reprogramming and inducing a distinct form of regulated cell death.
Therefore, combining FX and either GL or DDP treatment may serve as
a potential therapeutic strategy for PC treatment (Fig. 7).
Previous studies have indicated that FX, similar to
other antineoplastic drugs, eliminates cancer cells by inducing
apoptosis (5,7,42,43). To the best of our knowledge,
however, the association between FX and other types of PCD, such as
necroptosis (44), ferroptosis
(45), pyroptosis (46,47), cuproptosis (48) or autophagic cell death (49), has not been evaluated. Here, FX
significantly induced death in PC cells. Furthermore, in PC cells
co-cultured with PCD inhibitors, FX induced cell death. Subsequent
cell viability assay showed that inhibitors of apoptosis (ZVAD),
ferroptosis (Fer-1 and DFO), necroptosis (NEC-1 and NSA),
cuproptosis (TTM) or autophagic cell death (3-MA and BafA1) did not
attenuate FX-induced cell death. Due to the unavailability of a
specific pyroptosis inhibitor, western blotting was used to detect
activation of GSDME, GSDMD and GSDMC, the key indicators of
pyroptosis (37), in response to
FX. FX did not induce cleavage of GSDME, GSDMD or GSDMC in PC
cells. These co-culture experiments indicated that FX induced a
novel type of cell death that differs from known PCD mechanisms.
Mitochondrial dysfunction, oxidative stress and ATP depletion
caused by FX occur in parthanatos (50-52), suggesting that cell death caused
by FX may be associated with parthanatos. Future studies should
assess DNA damage, PARP1 overactivation, oxidative NAD
(NAD+) depletion and apoptosis inducing factor transfer
from mitochondria to the nucleus, which are indicative of
parthanatos (50-52). Knockdown of PARP1 or related
inhibitors is also needed to perform rescue assays to confirm
whether PARP1 mediated parthanatos induced by FX. Overall, further
studies are warranted to explore the mechanism of FX-induced cell
death.
Mitochondria serve roles in ATP production and
macromolecule biosynthesis (28).
The role of mitochondria in bioenergetics, dynamics and signal
transduction in tumor development has been studied (17). Therefore, it was hypothesized that
mitochondria may be a potential FX target in PC cells. FX treatment
of PC cells caused mitochondrial-associated changes, including a
decrease in mitochondrial respiration, dysfunction of OPA1-mediated
mitochondrial dynamics and accumulation of mitochondrial ROS.
Moreover, PATU-8988S was more sensitive to inhibition of
mitochondrial function by FX compared with PATU-8988T and PANC-1.
Daemen et al (24) showed
that lipogenic subtype PC cells (PATU-8988S) have, on average,
higher O2 consumption and mitochondrial content than
glycolytic subtype cells (PATU-8988T and PANC-1) (24). These results indicated that FX may
be more effective for PC cells, which highly rely on mitochondria
function. Therefore, identifying the key regulatory factors
(histopathological markers) that lead to metabolic difference is
key for selecting specific treatment for PC, which is consistent
with the conclusion of Fraile-Martine et al (53) and Ortega et al (53,54). FX treatment also stimulated
aerobic glycolysis, a key pathway for the generation of energy and
intermediate metabolites needed for PC progression (55-57). These alterations in metabolic
signatures revealed that FX induced metabolic reprogramming, which
switched mitochondrial metabolism to aerobic glycolysis. However,
the increased glycolysis was insufficient to compensate for the ATP
loss caused by mitochondrial dysfunction. Thus, it was hypothesized
that promotion of glycolysis might allow glucose to serve as an
alternative anaplerotic metabolite when mitochondria are damaged
and increased aerobic glycolysis levels counteract the cytotoxicity
of FX (58). The present study
confirmed this by culturing PC cells treated with low FX doses in
GL conditions. The lower cell viability and a higher cell death
rate revealed that GL conditions significantly sensitized PC cells
to FX. FX promoted the reduction of GSH, ultimately decreasing the
GSH/GSSG ratio in GL conditions. Similarly, the addition of GSH
substantially alleviated the cytotoxicity caused by FX, suggesting
that FX may sensitize PC cells to GL conditions by promoting
consumption of reduced GSH under GL conditions.
The association between FX cytotoxicity and GL
conditions smay involve k-ras. K-ras is an oncogene frequently
mutated in PC that promotes aerobic glycolysis and Gln metabolism
(59,60). Most cancer cells convert Gln into
α-ketoglutarate to fuel the TCA cycle, thereby maintaining
mitochondria respiration. However, k-ras-mutated PC cells convert
Gln-derived aspartate to oxaloacetate and convert malate and
pyruvate to fuel the TCA cycle and upregulate the generation of
NADPH (12,61). Therefore, whether FX treatment of
PC cells causes mitochondrial respiratory depression and oxidative
stress by regulating Gln metabolism was evaluated. Western blot
analysis of key factors involved in Gln metabolism and Gln synthase
(GLS) activity assay revealed significant inhibition of GLS
expression in response to FX treatment, indicating decreased Gln
metabolism. Based on these results and FX-induced mitochondrial
inhibition and oxidative stress, it was hypothesized that
FX-induced mitochondrial damage is partly caused by GLS inhibition.
However, the specific mechanism requires further exploration.
Drug resistance is an inevitable problem of PC
(62). FX promotes cytotoxicity
of gemcitabine on PC cells at a safe concentration (63). FX has also been found to increase
sensitivity of liver and lung cancer to DDP; however, the
underlying mechanism remains undetermined (38-40) and has not been studied in PC. PC
cells have also shown a consistent increase in sensitivity to DDP
when co-cultured with FX (38-40). FX significantly induces SLC31A1,
an important transporter responsible for DDP uptake (41,64), in PC cells. Here, depletion of
SLC31A1 can mitigate the inhibition of cell viability caused by
combined treatment with FX and DDP. These data indicated that FX
promoted chemosensitivity of PC cells to DDP by inducing SLC31A1
expression.
The present study proposed a mechanistic model in
which FX significantly inhibited PC by impeding cell proliferation
and inducing cell death through a non-classical pathway. FX was
also found to switch mitochondrial respiration to glycolysis and
inhibit GSH metabolism, decreasing metabolic flexibility under GL
conditions. Therefore, FX may be more effective for PC cells that
rely on mitochondria, providing novel theoretical support for FX
clinical targeted therapy. Moreover, FX treatment induced SLC31A1
expression, promoting DDP uptake and enhancing PC cell
chemosensitivity to DDP. These results suggested that combining FX
with glucose restriction or DDP may be a potential therapeutic
strategy for PC.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
FS and HY conceived the study and wrote the
manuscript. NM and YC performed experiments and analyzed data. YZ
and TM performed experiments. JA and JL analyzed data and edited
the manuscript. All authors have read and approved the final
manuscript. FS and HY confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by Project of Zhejiang
Provincial Naturel Science Foundation of China (grant nos.
LQ23H310006 and LQ20H260003) and Science and Technology Bureau of
Wenzhou (grant no. Y20220171).
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jain T and Dudeja V: The war against
pancreatic cancer in 2020-advances on all fronts. Nat Rev
Gastroenterol Hepatol. 18:99–100. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Din NAS, Mohd Alayudin S, Sofian-Seng NS,
Rahman HA, Mohd Razali NS, Lim SJ and Wan Mustapha WA: Brown algae
as functional food source of fucoxanthin: A Review. Foods.
11:22352022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Méresse S, Fodil M, Fleury F and Chénais
B: Fucoxanthin, a Marine-derived carotenoid from brown seaweeds and
microalgae: A promising bioactive compound for cancer therapy. Int
J Mol Sci. 21:92732020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rengarajan T, Rajendran P, Nandakumar N,
Balasubramanian MP and Nishigaki I: Cancer preventive efficacy of
marine carotenoid fucoxanthin: Cell cycle arrest and apoptosis.
Nutrients. 5:4978–4989. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hou LL, Gao C, Chen L, Hu GQ and Xie SQ:
Essential role of autophagy in fucoxanthin-induced cytotoxicity to
human epithelial cervical cancer HeLa cells. Acta Pharmacol Sin.
34:1403–1410. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu Y, Cheng J, Min Z, Yin T, Zhang R,
Zhang W, Hu L, Cui Z, Gao C, Xu S, et al: Effects of fucoxanthin on
autophagy and apoptosis in SGC-7901 cells and the mechanism. J Cell
Biochem. 119:7274–7284. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Long Y, Cao X, Zhao R, Gong S, Jin L and
Feng C: Fucoxanthin treatment inhibits nasopharyngeal carcinoma
cell proliferation through induction of autophagy mechanism.
Environ Toxicol. 35:1082–1090. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Murase W, Kamakura Y, Kawakami S, Yasuda
A, Wagatsuma M, Kubota A, Kojima H, Ohta T, Takahashi M, Mutoh M,
et al: Fucoxanthin prevents pancreatic tumorigenesis in C57BL/6J
mice that received allogenic and orthotopic transplants of cancer
cells. Int J Mol Sci. 22:136202021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Terasaki M, Takahashi S, Nishimura R,
Kubota A, Kojima H, Ohta T, Hamada J, Kuramitsu Y, Maeda H, Mutoh
M, et al: A marine carotenoid of fucoxanthinol accelerates the
growth of human pancreatic cancer PANC-1 cells. Nut Cancer.
74:357–371. 2020. View Article : Google Scholar
|
|
11
|
Wang YP, Zhou W, Wang J, Huang X, Zuo Y,
Wang TS, Gao X, Xu YY, Zou SW, Liu YB, et al: Arginine methylation
of MDH1 by CARM1 inhibits glutamine metabolism and suppresses
pancreatic cancer. Mol Cell. 64:673–687. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liberti MV and Locasale JW: The Warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ganapathy-Kanniappan S and Geschwind JF:
Tumor glycolysis as a target for cancer therapy: Progress and
prospects. Mol Cancer. 12:1522013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ashton TM, McKenna WG, Kunz-Schughart LA
and Higgins GS: Oxidative phosphorylation as an emerging target in
cancer therapy. Clin Cancer Res. 24:2482–2490. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Molina JR, Sun Y, Protopopova M, Gera S,
Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et
al: An inhibitor of oxidative phosphorylation exploits cancer
vulnerability. Nat Med. 24:1036–1046. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Passaniti A, Kim MS, Polster BM and
Shapiro P: Targeting mitochondrial metabolism for metastatic cancer
therapy. Mol Carcinog. 61:827–838. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fu Y, Ricciardiello F, Yang G, Qiu J,
Huang H, Xiao J, Cao Z, Zhao F, Liu Y, Luo W, et al: The role of
mitochondria in the chemoresistance of pancreatic cancer cells.
Cells. 10:4972021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gentric G, Mieulet V and Mechta-Grigoriou
F: Heterogeneity in cancer metabolism: New concepts in an old
field. Antioxid Redox Signal. 26:462–485. 2017. View Article : Google Scholar :
|
|
19
|
Tong Y, Guo D, Lin SH, Liang J, Yang D, Ma
C, Shao F, Li M, Yu Q, Jiang Y, et al: SUCLA2-coupled regulation of
GLS succinylation and activity counteracts oxidative stress in
tumor cells. Mol Cell. 81:2303–2316.e8. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jacque N, Ronchetti AM, Larrue C, Meunier
G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M,
et al: Targeting glutaminolysis has antileukemic activity in acute
myeloid leukemia and synergizes with BCL-2 inhibition. Blood.
126:1346–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jeong SM, Xiao C, Finley LW, Lahusen T,
Souza AL, Pierce K, Li YH, Wang X, Laurent G, German NJ, et al:
SIRT4 has tumor-suppressive activity and regulates the cellular
metabolic response to DNA damage by inhibiting mitochondrial
glutamine metabolism. Cancer Cell. 23:450–463. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tardito S, Oudin A, Ahmed SU, Fack F,
Keunen O, Zheng L, Miletic H, Sakariassen PØ, Weinstock A, Wagner
A, et al: Glutamine synthetase activity fuels nucleotide
biosynthesis and supports growth of glutamine-restricted
glioblastoma. Nat Cell Biol. 17:1556–1568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu W, Zhang C, Wu R, Sun Y, Levine A and
Feng Z: Glutaminase 2, a novel p53 target gene regulating energy
metabolism and antioxidant function. Proc Natl Acad Sci USA.
107:7455–7460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Daemen A, Peterson D, Sahu N, McCord R, Du
X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, et al: Metabolite
profiling stratifies pancreatic ductal adenocarcinomas into
subtypes with distinct sensitivities to metabolic inhibitors. Proc
Natl Acad Sci USA. 112:E4410–E4417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu Y, Yu Z, Fu H, Guo Y, Hu P and Shi J:
Dual inhibitions on Glucose/glutamine metabolisms for nontoxic
pancreatic cancer therapy. ACS Appl Mater Interfaces.
14:21836–21847. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bae M, Kim MB and Lee JY: Fucoxanthin
attenuates the reprogramming of energy metabolism during the
activation of hepatic stellate cells. Nutrients. 14:19022022.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ye Z, Zhuo Q, Hu Q, Xu X, Mengqi Liu,
Zhang Z, Xu W, Liu W, Fan G, Qin Y, et al: FBW7-NRA41-SCD1 axis
synchronously regulates apoptosis and ferroptosis in pancreatic
cancer cells. Redox Biol. 38:1018072021. View Article : Google Scholar
|
|
28
|
Ma N, Shangguan F, Zhou H, Huang H, Lei J,
An J, Jin G, Zhuang W, Zhou S, Wu S, et al:
6-methoxydihydroavicine, the alkaloid extracted from Macleaya
cordata (Willd.) R. Br. (Papaveraceae), triggers
RIPK1/Caspase-dependent cell death in pancreatic cancer cells
through the disruption of oxaloacetic acid metabolism and
accumulation of reactive oxygen species. Phytomedicine.
102:1541642022. View Article : Google Scholar
|
|
29
|
Carvalho TMA, Audero MM, Greco MR, Ardone
M, Maggi T, Mallamaci R, Rolando B, Arpicco S, Ruffinatti FA, Pla
AF, et al: Tumor microenvironment modulates invadopodia activity of
Non-Selected and Acid-Selected pancreatic cancer cells and its
sensitivity to gemcitabine and C18-Gemcitabine. Cells. 13:7302024.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu Z, Zhou R, Zhao Y, Pan Y, Liang H,
Zhang JS, Tai S, Jin L and Teng CB: Blockage of SLC31A1-dependent
copper absorption increases pancreatic cancer cell autophagy to
resist cell death. Cell Prolif. 52:e125682019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Geng R, Ke N, Wang Z, Mou Y, Xiang B,
Zhang Z, Ji X, Zou J, Wang D, Yin Z, et al: Copper deprivation
enhances the chemosensitivity of pancreatic cancer to rapamycin by
mTORC1/2 inhibition. Chem Biol Interact. 382:1105462023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chiu HW, Lin SW, Lin LC, Hsu YH, Lin YF,
Ho SY, Wu YH and Wang YJ: Synergistic antitumor effects of
radiation and proteasome inhibitor treatment in pancreatic cancer
through the induction of autophagy and the downregulation of TRAF6.
Cancer Lett. 365:229–239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dasgupta A, Arneson-Wissink PC, Schmitt
RE, Cho DS, Ducharme AM, Hogenson TL, Krueger EW, Bamlet WR, Zhang
L, Razidlo GL, et al: Anticachectic regulator analysis reveals
Perp-dependent antitumorigenic properties of 3-methyladenine in
pancreatic cancer. JCI Insight. 7:e1538422022. View Article : Google Scholar :
|
|
34
|
Qi J, Xing Y, Liu Y, Wang MM, Wei X, Sui
Z, Ding L, Zhang Y, Lu C, Fei YH, et al: MCOLN1/TRPML1 finely
controls oncogenic autophagy in cancer by mediating zinc influx.
Autophagy. 17:4401–4422. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Song CF, Hu YH, Mang ZG, Ye Z, Chen HD,
Jing DS, Fan GX, Ji SR, Yu XJ, Xu XW, et al: Hernandezine induces
autophagic cell death in human pancreatic cancer cells via
activation of the ROS/AMPK signaling pathway. Acta Pharmacol Sin.
44:865–876. 2023. View Article : Google Scholar :
|
|
36
|
Liu J, Tang H, Chen F, Li C, Xie Y, Kang R
and Tang D: NFE2L2 and SLC25A39 drive cuproptosis resistance
through GSH metabolism. Sci Rep. 14:295792024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zou J, Zheng Y, Huang Y, Tang D, Kang R
and Chen R: The versatile gasdermin family: Their function and
roles in diseases. Front Immunol. 12:7515332021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nurcahyanti ADR, Kusmita L and Wink M:
Bixin and fucoxanthin sensitize human lung cancer and cervical
cancer cell to cisplatin in vitro. BMC Res Notes. 14:4542021.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu CL, Lim YP and Hu ML: Fucoxanthin
enhances cisplatin-induced cytotoxicity via NFκB-mediated pathway
and downregulates DNA repair gene expression in human hepatoma
HepG2 cells. Mar Drugs. 11:50–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chandra F, Tania TF and Nurcahyanti ADR:
Bixin and Fuxoxanthin alone and in combination with cisplatin
regulate ABCC1 and ABCC2 transcription in A549 lung cancer cells. J
Pharm Bioallied Sci. 15:15–20. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu G, Peng H, Tang M, Yang M, Wang J, Hu
Y, Li Z, Li J, Li Z and Song L: ZNF711 down-regulation promotes
CISPLATIN resistance in epithelial ovarian cancer via interacting
with JHDM2A and suppressing SLC31A1 expression. EBioMedicine.
71:1035582021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Terasaki M, Inoue T, Murase W, Kubota A,
Kojima H, Kojoma M, Ohta T, Maeda H, Miyashita K, Mutoh M, et al: A
fucoxanthinol induces apoptosis in a pancreatic intraepithelial
neoplasia cell. Cancer Genomics Proteomics. 18:133–146. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tang JY, Ou-Yang F, Hou MF, Huang HW, Wang
HR, Li KT, Fayyaz S, Shu CW and Chang HW: Oxidative
stress-modulating drugs have preferential anticancer
effects-involving the regulation of apoptosis, DNA damage,
endoplasmic reticulum stress, autophagy, metabolism, and migration.
Semin Cancer Biol. 58:109–117. 2019. View Article : Google Scholar
|
|
44
|
Su Z, Yang Z, Xie L, DeWitt JP and Chen Y:
Cancer therapy in the necroptosis era. Cell Death Differ.
23:748–756. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liang C, Zhang X, Yang M and Dong X:
Recent progress in ferroptosis inducers for cancer therapy. Adv
Mater. 31:e19041972019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu D, Wang S, Yu G and Chen X: Cell death
mediated by the pyroptosis pathway with the aid of nanotechnology:
Prospects for cancer therapy. Angew Chem Int Ed Engl. 60:8018–8034.
2021. View Article : Google Scholar
|
|
47
|
Wang Y, Gao W, Shi X, Ding J, Liu W, He H,
Wang K and Shao F: Chemotherapy drugs induce pyroptosis through
caspase-3 cleavage of a gasdermin. Nature. 547:99–103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tsvetkov P, Coy S, Petrova B, Dreishpoon
M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R,
Spangler RD, et al: Copper induces cell death by targeting
lipoylated TCA cycle proteins. Science. 375:1254–1261. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen L, Chen D, Li J, He L, Chen T, Song
D, Shan S, Wang J, Lu X and Lu B: Ciclopirox drives growth arrest
and autophagic cell death through STAT3 in gastric cancer cells.
Cell Death Dis. 13:10072022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Huang P, Chen G, Jin W, Mao K, Wan H and
He Y: Molecular mechanisms of parthanatos and its role in diverse
diseases. Int J Mol Sci. 23:72922022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou Y, Liu L, Tao S, Yao Y, Wang Y, Wei
Q, Shao A and Deng Y: Parthanatos and its associated components:
Promising therapeutic targets for cancer. Pharmacol Res.
163:1052992021. View Article : Google Scholar
|
|
52
|
Peng F, Liao M, Qin R, Zhu S, Peng C, Fu
L, Chen Y and Han B: Regulated cell death (RCD) in cancer: Key
pathways and targeted therapies. Signal Transduct Target Ther.
7:2862022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fraile-Martinez O, García-Montero C,
Pekarek L, Saz JV, Álvarez-Mon MÁ, Barrena-Blázquez S,
García-Honduvilla N, Buján J, Asúnsolo Á, Coca S, et al: Decreased
survival in patients with pancreatic cancer may be associated with
an increase in histopathological expression of inflammasome marker
NLRP3. Histol Histopathol. 39:35–40. 2024.
|
|
54
|
Ortega MA, Jiménez-Álvarez L,
Fraile-Martinez O, Garcia-Montero C, León-Oliva DD, Toledo-Lobo
MDV, Palacios E, Granado P, Esteban A, Guijarro LG, et al: Elevated
tissue expression of RANKL and RANK is associated with poorer
survival rates in pancreatic cancer patients. Histol Histopathol.
39:1133–1140. 2024.PubMed/NCBI
|
|
55
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic Kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Humpton TJ, Alagesan B, DeNicola GM, Lu D,
Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y,
et al: Oncogenic KRAS induces NIX-mediated mitophagy to promote
pancreatic cancer. Cancer Discov. 9:1268–1287. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dey P, Li J, Zhang J, Chaurasiya S, Strom
A, Wang H, Liao WT, Cavallaro F, Denz P, Bernard V, et al:
Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer
cells utilizes cytokines from the tumor microenvironment. Cancer
Discov. 10:608–625. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Teng R, Liu Z, Tang H, Zhang W, Chen Y, Xu
R, Chen L, Song J, Liu X and Deng H: HSP60 silencing promotes
Warburg-like phenotypes and switches the mitochondrial function
from ATP production to biosynthesis in ccRCC cells. Redox Biol.
24:1012182019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Qin S, Li J, Bai Y, Wang Z, Chen Z, Xu R,
Xu J, Zhang H, Chen J, Yuan Y, et al: Nimotuzumab plus gemcitabine
for K-Ras Wild-type locally advanced or metastatic pancreatic
cancer. J Clin Oncol. 41:5163–5173. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gaglio D, Metallo CM, Gameiro PA, Hiller
K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G and
Chiaradonna F: Oncogenic K-Ras decouples glucose and glutamine
metabolism to support cancer cell growth. Mol Syst Biol. 7:5232011.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Son J, Lyssiotis CA, Ying H, Wang X, Hua
S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et
al: Glutamine supports pancreatic cancer growth through a
KRAS-regulated metabolic pathway. Nature. 496:101–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jaaks P, Coker EA, Vis DJ, Edwards O,
Carpenter EF, Leto SM, Dwane L, Sassi F, Lightfoot H, Barthorpe S,
et al: Effective drug combinations in breast, colon and pancreatic
cancer cells. Nature. 603:166–173. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lu J, Wu XJ, Hassouna A, Wang KS, Li Y,
Feng T, Zhao Y, Jin MF, Zhang BH, Ying TL, et al:
Gemcitabine-fucoxanthin combination in human pancreatic cancer
cells. Biomed Rep. 19:462023. View Article : Google Scholar
|
|
64
|
Cheng C, Ding Q, Zhang Z, Wang S, Zhong B,
Huang X and Shao Z: PTBP1 modulates osteosarcoma chemoresistance to
cisplatin by regulating the expression of the copper transporter
SLC31A1. J Cell Mol Med. 24:5274–5289. 2020. View Article : Google Scholar : PubMed/NCBI
|