Introduction
As one of the most common malignant neoplasms in the
digestive tract, esophageal cancer (EC) remains an integral cause
of cancer-related death (1).
Although the incidence of EC varies considerably with location, the
number of global new cases in 2020 was ~604,100, and the mortality
caused by EC ranked tenth in terms of malignant tumors (2). EC can be separated by histological
type into squamous cell carcinoma, adenocarcinoma, small cell
carcinoma and carcinosarcoma. Esophageal squamous cell carcinoma
(ESCC) is the predominant histological type worldwide, and accounts
for ~90% of all EC cases (3). The
management of EC is complex and highly variable between countries
and medical centers. Clinically, chemotherapy has been one of the
major therapeutic approaches in the trimodal therapy of EC
(4). Multimodal therapy refers to
neoadjuvant chemoradiotherapy or perioperative chemotherapy
combined with esophagectomy, which has been increasingly used
worldwide due to its greater survival advantage compared with
surgery alone (5). Additionally,
despite multimodal treatment, most patients with EC and locally
advanced disease will eventually develop metastatic disease, and
most patients will receive palliative chemotherapy at some point
(6). However, although some
progress has been made in multidisciplinary management in recent
years, the treatment of EC is still a serious challenge, partly
because numerous patients are insensitive or adaptively resistant
to chemotherapy drugs (7).
Therefore, it is imperative to further elucidate the mechanisms of
drug resistance in EC and develop chemosensitizers and combined
therapies to decrease EC mortality.
Cisplatin is one of the principal chemotherapeutic
agents used for the first-line treatment of a number of
malignancies, including EC (8).
In addition, combination therapies with cisplatin and other
therapeutics such as 5-fluorouracil remain the most frequently used
chemotherapy for ESCC (9). The
main anticancer mechanism of cisplatin is that it interacts with
purine bases in DNA to form DNA-protein and DNA-DNA inter-strand
and intra-strand crosslinks, thereby inhibiting the proliferation
and apoptosis of tumor cells (10). It has been proposed that one
aspect of the molecular mechanism of cisplatin is the induction of
intracellular calcium efflux and subsequent triggering of apoptosis
(11). Intracellular calcium ions
(Ca2+), as a second messenger, regulate gene
transcription, and cell migration, proliferation and death.
Evidence has shown that the Ca2+ homeostasis in cancer
cells is altered, which is related to tumor initiation,
angiogenesis, progression and metastasis (11). It has been proposed that reduced
calcium influx in cancer cells prevents calcium overload in
response to proapoptotic stimuli, thereby impairing the
effectiveness of mitochondrial and cytoplasmic apoptotic pathways
(12). Moreover,
cisplatin-induced cell death is reported to be dependent on
calcium/calpain, store-operated calcium entry and calcium efflux in
ovarian (13), non-small cell
lung (14) and cervical (15) cancer. Thus, it appears that
approaches which enhance calcium efflux may contribute to the
apoptotic death of cancer cells. However, to the best of our
knowledge, how the intracellular calcium stock contributes to the
cisplatin-mediated death of EC cells, and whether excessive
intracellular calcium can sensitize EC cells to cisplatin remains
unexplored so far.
Angiotensin II receptor type 1, also termed the AT1
receptor (AGTR1), is the most characterized angiotensin receptor
(16) and is an important
effector controlling blood pressure and volume in the
cardiovascular system (16). An
entire class of AGTR1 receptor blockers are already clinically
available (17). AGTR1 has been
previously linked to cancer and is considered a therapeutic target
in estrogen receptor (ER)-positive and Erb-B2 receptor tyrosine
kinase 2-negative breast cancer, as it is upregulated in this
cancer type and promotes lymph node metastasis (18-20). In addition, AGTR1 blockade has
shown therapeutic effects in ovarian cancer (21). However, repression of AGTR1
contributes to the multi-chemoresistance of osteosarcoma (22), and high AGTR1 expression was
reported to induce apoptosis in β-cell and islet (23,24), intestinal epithelial (25) and cardiac (26) cells. These results suggest that
AGTR1 may function as an oncogenic factor in some cancer types
while its upregulation can induce apoptosis in multiple cell types.
However, to the best of our knowledge, utilization of the
pro-apoptotic property of AGTR1 to eliminate malignancies and the
potential mechanisms in EC have not yet been explored.
In the present study, the gene expression profiles
of EC and normal esophageal tissues were compared. To analyze the
association between AGTR1 expression and chemosensitivity of EC
tumors to cisplatin, KYSE-150 cells with a low AGTR1 expression
level and EC109 cells with a high AGTR1 expression level were used.
In addition, the effect of combined treatment with cisplatin and
the nonselective calcium channel blocker fendiline was evaluated
using an EC xenograft model. In clinical practice, the results of
the present study may provide an alternative treatment option with
an improved prognosis for patients with EC.
Materials and methods
Patients and clinical sample
preparation
The tumor tissue specimens were collected from
patients with EC who underwent surgical resection between December
2019 and June 2021 in the Department of Thoracic Surgery of
Nanchong Central Hospital (Nanchong, China). The age of patients
ranged between 48 and 75 years (mean age, 63.67 years; median age,
65 years), with 50% male and 50% female patients. The inclusion
criteria were: Age ≥18 years; the medical records were complete;
and the patient was diagnosed with esophageal cancer. The exclusion
criteria were: Combined with other malignant tumors; the telephone
number and other contact information were missing; and those with
incomplete follow-up information. These patients received a
standardized treatment protocol consisting of neoadjuvant
chemotherapy with cisplatin followed by appropriate surgical
management. The pathological staging was reassessed with the new
international tumor-node-metastasis staging system for EC approved
by the American Joint Committee on Cancer (Version 9) (27). The specimens from 30 cases of EC
were divided into the following groups based on the follow-up
visits from 2019: i) Subjects with a good response and a survival
of >3 years without recurrence (n=13), and subjects with a poor
response, who died within 3 years due to recurrence (n=17); and ii)
subjects without metastasis within 1 year after surgery (n=18), and
subjects with metastasis within 1 year after surgery (n=12). All
tissue specimens were paraffin-embedded for immunohistochemistry
staining or stored in liquid nitrogen for immunoblotting and
reverse transcription-quantitative (RT-qPCR) analysis. Ethical
approval was obtained from the Medical Ethics Committee of Nanchong
Central Hospital (approval no. 2019.095). The study protocol was
carefully explained to the participants and participation was fully
voluntary. Written informed consent was obtained from all
participants and they agreed to the publication of their individual
data.
Cell lines and cell culture
The human endometrial endothelial cell line, HEEC,
and the human ESCC cell lines, EC109, KYSE-150, KYSE-510 and TE-3,
were purchased from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences. The TE-3 cell line has been shown to
be identical to the following cell lines: TE-2, TE-7, TE-12 and
TE-13 (https://www.cellosaurus.org/CVCL_9971) (28,29). The EC109 cells used in the present
study were authenticated using short tandem repeat analysis
(Table SI). Cells were cultured
in Dulbecco's Modified Eagle's Medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (AccuRef
Scientific), 100 units/ml penicillin and 100 µg/ml
streptomycin (Gibco; Thermo Fisher Scientific, Inc.). The cells
were cultured under an atmosphere of 5% CO2 at 37°C. The
concentration of nucleic acid was 20 nM and transfection was
performed at room temperature, while subsequent experiments were
conducted 48 h after transfection.
The KYSE-150 cells were transfected with an
AGTR1-expressing pcDNA3.1 plasmid or empty vector (Thermo Fisher
Scientific, Inc.) or with AGTR1-targeting small interfering (si)RNA
oligonucleotides (antisense strand, 5′-CUG UAG AAU UGC AGA UAU UdT
dT-3′ and sense strand, 3′-dTd TGA CAU CUU AAC GUC UAU AA-5′) or
negative control (NC) oligonucleotides (antisense strand, 5′-UUC
UCC GAA CGU GUC ACG UdT dT-3′ and sense strand, 3′-dTd TAA GAG GCU
UGC ACA GUG CA-5′) (Shanghai GenePharma Co., Ltd.). Cell
transfection was performed using Lipofectamine 3000 reagent (Thermo
Fisher Scientific, Inc.) following the manufacturer's instructions.
After 48 h of transfection, cells were harvested for further
analysis.
The Tet-on cell line with inducible expression of
AGTR1 was constructed by transfection of KYSE-150 cells with the
AGTR1-expressing pCDH plasmid (Vazyme Biotech Co., Ltd.) using
Lipofectamine 3000 reagent as aforementioned and subsequent
screening by limiting dilution cloning. AGTR1-expression was
induced by treating the cells with 100 ng/ml doxycycline
(MilliporeSigma). In some experiments, cells were treated with
cisplatin (MilliporeSigma) at the indicated concentration and/or 5
µM fendiline (Thermo Scientific, Inc.) as specified.
Cell viability and colony formation
assay
Cells were seeded in 96-well plates at a density of
5×103 cells/well and incubated overnight. After the
indicated treatments, Cell Counting Kit-8 reagent (AccuRef
Scientific) was added to cells at the fixed time (24, 48 and 72 h)
and the cells were incubated for another 2 h at 37°C. The
absorbance of each well was measured at 450 nm by a plate reader
(SpectraMax iD3 Multi-Mode Microplate Reader; Molecular Devices
LLC). The half maximal inhibitory concentration (IC50)
of cisplatin was calculated using GraphPad Prism (version 8.0;
Dotmatics).
The colony forming ability of the cells was
determined using a clonogenic assay. Briefly, cells were diluted
and seeded in 12-well culture plates at a density of
1.5×104 cells/well, and the cells were incubated at 37°C
under 5% CO2 for 10 days. The cells were fixed with 4%
paraformaldehyde (Biosharp Life Sciences) at room temperature for
30 min, then stained with Diff-Quik stain set (Siemens
Healthineers) at room temperature for 10 min, washed with PBS
(Gibco; Thermo Fisher Scientific, Inc.) to remove the stain that
did not bind to the cells, imaged using a light microscope
(Eclipse-TS100; Nikon Corporation) and counted using ImageJ
software (version 1.53; National Institutes of Health). A cell
cluster with >10 cells was considered to be a single colony.
Transwell migration and invasion
assays
Transwell Boyden chambers (BD Biosciences) with
8-µm pore polycarbonate filters were used for in
vitro cell migration assays, and chambers pre-coated with
Matrigel (BD Biosciences) at 37°C for 2 h were used to evaluate the
in vitro invasive potential of EC cells. Briefly, cells were
seeded at a density of 1×105 per well in DMEM serum-free
culture medium (Gibco; Thermo Fisher Scientific, Inc.) into the
upper chamber that was coated with Matrigel (BD Biosciences) and
600 µl complete medium was added to the lower chamber.
Chambers were then incubated for 24 h at 37°C. At the time of
harvesting, cells remaining inside the upper chambers were removed,
while migrated and invaded cells on the lower surface of the
membrane were fixed in 4% paraformaldehyde for 20 min and stained
with crystal violet (MilliporeSigma) for 5 min (both at room
temperature), followed by visualization and counting under an
inverted microscope. Cells were imaged, and five visual fields per
well were randomly selected for cell counting using ImageJ software
(version 1.53; National Institutes of Health).
Wound healing assay
Cells were seeded in a 6-well plate at a
concentration of 1×106 cells per well, then cultured
overnight to allow adequate adherence to form a confluent
monolayer. The cell layers were wounded using a sterile 200
µl pipette tip and washed with serum-free medium to
eliminate dislodged cells. Cells were imaged immediately (time 0 h)
using an inverted microscope and then cultured with serum-free
medium for 24 h. The wounds were imaged at 24 h post wounding to
monitor the wound closure progression. The wound healing rate was
calculated as follows: Wound healing rate (%)=[1-(24 h scratch
width/0 h scratch width)] ×100%. The wound closure distance of
three independent wounds in each group was measured using ImageJ
software (version 1.53; National Institutes of Health).
RT-qPCR
Total RNA was extracted from cells or tumor tissues
using a High Pure RNA isolation kit (Roche Diagnostics) following
the manufacturer's instructions. The RNA concentration was measured
using a Nano Drop ONE spectrophotometer (Thermo Fisher Scientific,
Inc.). RT was carried out on 1 µg of RNA using the
SuperScript III RT kit with random hexamer primer (Thermo Fisher
Scientific, Inc.) per the manufacturer's instructions. qPCR was
then performed using SYBR Green reaction mix (AccuRef Scientific)
and a 7500 Real-Time PCR System instrument (Thermo Fisher
Scientific, Inc.) with the following conditions: 95°C for 10 min
and 40 cycles of 95°C for 10 sec, 58°C for 20 sec and 72°C for 15
sec. The specific primer sequences were as follows: ATGR1, forward:
5′-CGG GGC GCG GGT TTG-3′ and revers: 5′-TCA AAT ACA CCT GGT GCC
GA-3′; and β-actin, forward: 5′-TTC CTG GGC ATG GAG TCC-3′ and
revers: 5′-CTC GTT ACT AGA ACT AGA AGT-3′. The comparative
threshold cycles (Cq) of tested mRNA and GAPDH were measured and
their difference (ΔCq) was calculated. The relative expression was
then calculated by the 2−ΔΔCq method (30) with normalization to the control
sample.
Western blotting
Cells or tissue specimens were lysed using RIPA
buffer (AccuRef Scientific) supplemented with protease inhibitor
cocktail (AccuRef Scientific). The protein content was determined
by BCA Protein Assay Kit (AccuRef Scientific). A total of 40
µg protein was separated by 8% SDS-PAGE and then transferred
to polyvinylidene difluoride membranes (MilliporeSigma). The
membranes were blocked with 5% milk/PBS with Tween-20 (PBST;
PBS:Tween, 1,000:1) for 1 h at room temperature and probed with
antibodies against the targets overnight at 4°C. After washing with
PBST, the membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG (H+L) (1:10,000; cat.
no. SA00001-2; Proteintech Group, Inc.) or anti-mouse IgG
(1:10,000; cat. no. SA00001-1; Proteintech Group, Inc.) as the
secondary antibodies for 1 h at room temperature and then
visualized by ECL detection kit (AccuRef Scientific). The band
intensities of the proteins were analyzed using ImageJ (version
1.53) densitometry software (National Institutes of Health). The
primary antibodies used for the western blot assays are listed in
Table SII.
Apoptosis assays
Apoptosis of EC cells was determined by Annexin
V-FITC/propidium iodide (PI) double staining assay (AccuRef
Scientific) according to the manufacturer's instructions. Apoptotic
cells were counted with a FACSCalibur flow cytometer (BD
Biosciences) and data were analyzed by FlowJo (version 10.8.1)
software (BD Biosciences).
Paraffin-embedded cancer tissue was cut into slices
of 4 µm thickness and then affixed to glass slides. The
terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling
(TUNEL) assay was performed using a commercial TUNEL apoptosis
assay kit (Beyotime Institute of Biotechnology). Briefly, cell or
tissue slides were incubated with 2 µg/ml proteinase K
(MilliporeSigma) in 10 mM Tris-HCl (pH 7.4) buffer containing 5 mM
EDTA. Then, endogenous peroxidase was inactivated using 0.3%
hydrogen peroxide for 20 min at room temperature. After washing
with PBS three times, the slides were incubated with TUNEL solution
(2 µl TdT + 48 µl fluorescent labelling solution) at
37°C for 1 h in the dark. The slides were washed with PBS three
times and then mounted with mounting medium containing DAPI (Vector
laboratories, Inc.). Finally, the staining results were examined
using a fluorescence microscope (DM1,000; Leica Microsystems GmbH),
then five fields were randomly selected and the TUNEL-positive
cells were calculated using ImageJ (version 1.53) software
(National Institutes of Health).
Flow cytometry and ATP measurement
Cells were loaded with 1 µM Fluo-3, AM
(Thermo Fisher Scientific, Inc.) in phenol red-free culture medium
at 37°C for 30 min following the manufacturer's protocol. After
washing with PBS, KYSE-150 cells were kept in culture medium
without phenol red. The intensity of fluorescent signal was
measured with a FACSCalibur flow cytometer (BD Biosciences). The
mitochondrial membrane potential of EC cells was measured by
staining with
5,50,6,60-tetrachloro-1,10,3,30-tetraethyl-imidacarbocyanine iodide
(JC-1; MillipreSigma) at a final concentration of 2.5 µM.
After shaking in the dark at 37°C for 15 min, the cells were
analyzed using a FACSCalibur flow cytometer (BD Biosciences). Flow
cytometry data were analyzed with the FlowJo (v10.8.1) software (BD
Biosciences). The ATP levels were measured with the Molecular
Probes® ATP Determination Kit (Thermo Fisher Scientific,
Inc.) following the manufacturer's protocol.
Immunohistochemistry staining
After dissection, the tumor tissues were mounted in
paraffin embedding compound and frozen at -80°C. The cryostat
sections were prepared by cutting the tissues at a thickness of 5
µm using a cryostat microtome (Leica Microsystems GmbH). The
tumor tissues were soaked in xylene three times for 3-5 min each,
with new xylene added each time. Tissues were dehydrated in
anhydrous ethanol twice for 3-5 min each. Tissues were incubated
with 95% ethanol for 3-5 min, 90% ethanol for 3-5 min and 70%
ethanol for 3-5 min. The tissues were rinsed twice with distilled
water for 3-5 min each. Subsequently, the tissues were soaked in
antigen retrieval solution (1X) and heated at 96°C for ~20 min in a
water bath. The slides were fixed in 4% formalin solution for 10
min at 37°C and washed with PBS three times. An appropriate amount
of endogenous peroxidase blocking solution (undiluted; cat. no.
P0100A; Beyotime Institute of Biotechnology) was added dropwise to
completely cover the sample, and the samples were incubated at room
temperature for 5-10 min or 37°C for 5 min. The slides were then
incubated with PBS containing 0.1% Triton X-100 for 10 min and
washed in PBS three times. Next, the slides were incubated with 10%
goat serum (cat. no. ZLI-9022; Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd.) in PBST (0.1% Tween 20) for 30 min at room
temperature to block the unspecific binding of the antibodies.
After incubation with the primary antibodies against AGTR1 (2.5
µg/ml dilution; cat. no. 124505; Abcam) or Ki-67 (2.5
µg/ml; cat. no. ab15580; Abcam) overnight at 4°C, the slides
were washed three times in PBST and then incubated with
biotinylated secondary antibody (1:200; cat. no. PV-9005; Beijing
Zhongshan Jinqiao Biotechnology Co., Ltd.) at room temperature for
30 min. The signal was then visualized using the DAB method
(AccuRef Scientific) according to the manufacturer's protocol.
Next, the slides were counterstained with hematoxylin (Nanjing
Jiancheng Bioengineering Institute) for 20 sec at room temperature
to visualize the nuclei. After mounting with mounting medium, the
slides were imaged using a light microscope with ×20 and ×40
objective (NA1.3; Leica Microsystems GmbH) and analyzed using
GraphPad Prism (version 8.0; Dotmatics).
Animal experiments
Female athymic mice (BALB/c, nu/nu; 15-20 g; 6-8
weeks old) were purchased from Charles River Laboratories, Inc.,
and housed at the specific pathogen-free facility at the Animal
Center of North Sichuan Medical College (Nanchong, China). The mice
were maintained at room temperature (22±1°C) with a 12/12 h
light/dark cycle and ad libitum access to food and water.
The indicated KYSE-150 cell lines or EC109 cell lines were injected
subcutaneously into the right flank of each mouse (100 µl;
1×106 cells/mouse) to establish the xenograft model (20
mice per experiment for 4 groups for 2 experiments; 5 mice in each
group). Mice were divided into the following eight groups: pEmpty,
pAGTR1, pEMPty + cisplatin, pAGTR1 + cisplatin; or cisplatin,
cisplatin + Dox, cisplatin + fendiline and cisplatin + fendiline +
Dox, as indicated. In some experiments, mice received treatments,
alone or in combination as indicated, including intraperitoneal
injection of the vehicle PBS or cisplatin at a dose of 5 mg/kg body
weight twice every week, intraperitoneal injection of Dox at a dose
of 20 mg/kg body weight every other day and gastric gavage
administration of fendiline at a dose of 3 mg/kg body weight every
other day. All treatments began when the tumor mass reached a
diameter of 5 mm. Tumor size was measured once a week with calipers
and tumor volumes were calculated by the following formula:
length2 × width × π/6. Animals were checked daily, and
any animal found unexpectedly to be moribund, cachectic or unable
to obtain food or water was euthanized. In addition, mice with a
tumor burden exceeding 2 cm diameter in any direction or a tumor
volume >4.2 cm3 were euthanized. None of the mice
reached the humane endpoints in the present study.
In total, 5 weeks after the initiation of
treatments, mice were euthanized (no animals were found dead in
this study) and tumor tissues were subjected to further analyses.
Animals were euthanized using a 30-70% per min displacement of
chamber air with compressed CO2. After the completion of
euthanasia with CO2 inhalation, cervical dislocation
with the confirmation of a gap between the skull and spinal column
was used to verify the death of mice. All tumors were resected from
mice and the total tumor weight in each mouse was measured. Some
tumors were fixed in 10% formalin for 24 h at room temperature for
subsequent immunohistochemistry. Animal experiments were conducted
in accordance with the procedures and protocols of the Animal
Center of North Sichuan Medical College and approved by the Ethics
Committee of North Sichuan Medical College (approval no.
NSMC202144).
RNA sequencing (RNA-seq) and
bioinformatics
RNA was isolated from EC tumor and normal esophageal
tissues using the RNeasy Kit (cat. no. 74104; Qiagen GmbH)
according to the manufacturer's instructions. The integrity of
total RNA in samples was detected by 2% agarose gel
electrophoresis, and a NanoDrop (ND-1,000; Thermo Fisher
Scientific, Inc.) was used for quantification and further quality
inspection. For each sample, 1-2 µg total RNA was selected
for sequencing library construction. The total RNA was enriched
with the NEBNext Poly (a) mRNA Magnetic Isolation Module (cat. no.
E7490S; New England BioLabs, Inc.). After treatment, the RNA
library was constructed with a KAPA Stranded RNA-seq Library Prep
Kit (cat. no. KK8401; Illumina, Inc.). The KAPA kit used the
construction method of a chain-specific library based on dUTP. The
constructed library was inspected using an Agilent 2100 Bioanalyzer
(library concentration, fragment size 400-600 bp and whether there
were connectors), and the library was finally quantified by qPCR.
For qPCR, DNA was extracted using Qiagen DNeasy Blood & Tissue
kit (Qiagen GmbH) and the reaction was performed using Phusion
High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Inc.). The
random PCR primers, which were used for cDNA synthesis followed by
PCR amplification, were as follows: Forward, 5′-AGT ACC GTA CGA TGA
CT-3′ and reverse, 5′-CAG TGC ATA GTA CAG TC-3′. The thermocycling
conditions were as follows: Initial denaturation at 95°C for 3 min,
followed by denaturation at 95°C for 30 sec, annealing at 55°C for
30 sec and extension at 72°C for 1 min (35 cycles); final extension
at 72°C for 5-10 min; hold at 4°C indefinitely. PCR products were
analyzed using a 1.5% agarose gel and visualized using SYBR Green.
According to the quantitative results and the amount of final
sequencing data, the sequencing libraries of different samples were
required to be mixed in the sequencing process. The mixed
sequencing libraries of different samples were denatured with 0.1 M
NaOH to generate single-stranded DNA, diluted to 8 pM, and then
amplified in situ using a Truseq SR Cluster Kit (cat. no.
GD-401-3001; Illumina, Inc.). The ends of the generated fragments
were sequenced using an Illumina HiSeq2000™ Genome Analyzer
(Illumina, Inc.). Reference-based RNA-seq was performed with a
nucleotide length of 200-400 bp. The direction of sequencing was
forward and reverse strands. The loading concentration of the final
library was 10 nM, which was determined using a Qubit fluorometer
(Thermo Fisher Scientific, Inc).
Solexa Pipelime (Off Line Base Caller software;
version 1.8; Illumina, Inc) was used for image processing and base
recognition. FastQc software (version 0.11.6; Babraham
Bioinformatics) was used to evaluate the sequencing quality of
reads. Sequences were aligned to the reference genome using Hisat
software (version 2.2.1; https://daehwankimlab.github.io/hisat2/). StringTie
software (version 2.2.0; https://ccb.jhu.edu/software/stringtie/) was employed
to estimate transcriptional abundance by referencing official
database annotation information. R software (version 4.1.2;
https://cloud.r-project.org/bin/windows/) was used to
perform fragments per kilobase of transcript per million mapped
reads calculations at the gene and transcript levels, and to
calculate the expression differences at both the gene and
transcript levels. Differentially expressed genes were selected
between samples or groups. RNA-sequencing data were uploaded to the
publicly available Gene Expression Omnibus (GEO) database with the
accession number GSE263647 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE263647).
The differentially expressed genes were determined
using a false discovery rate of <5% and a fold change >1.5.
The ENCODE project database (www.encodeproject.org) was utilized to search for the
most enriched Kyoto Encyclopedia of Genes and Genomes pathways in
the selected gene lists. In addition, the Database for Annotation,
Visualization, and Integrated Discovery (https://davidbioinformatics.nih.gov/) was employed to
search for the most enriched biological processes, molecular
functions and cellular component terms of the Gene Ontology.
Enrichment scores were used to indicate the enrichment level of
genes assigned to the indicated Gene Ontology terms in each
subgroup. Related pathways were evaluated by Gene Set Enrichment
Analysis. To perform expression analysis, pan-cancer was selected
on the home page (https://starbase.sysu.edu.cn/; version 3.0) to enter
the gene differential expression and survival analysis platform,
and AGTR1 was compared between cancer and normal samples. The
conditions set for the Kmplot online website (https://kmplot.com/analysis/) were: Matching The
Cancer Genome Atlas dataset (RNA-seq pan-cancer); stage, all; sex,
all; ethnicity, all; grade, all; follow-up threshold, all; and
cutoff value used in the analysis, 10. Overall survival and
metastasis-free survival in patients with EC were computed with the
Kaplan-Meier method, and the log rank test was used to assess the
statistical significance of the results.
Statistical analysis
Data are presented as the mean ± standard deviation.
The differences between two groups were analyzed by unpaired
Student's t-test. ANOVA with Bonferroni correction was applied to
compare the differences between more than two groups. All
statistical analyses were performed with GraphPad Prism (version
8.0; Dotmatics) software. P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression of AGTR1 is downregulated in
recurrent and metastasized EC tissues
To explore the genes that are potentially associated
with the progression of EC, RNA-seq was performed using EC and
normal esophageal tissues. As shown in the volcano plot of Fig. 1A, RNA-seq analysis demonstrated
that 969 genes were differentially downregulated, while 1,408 genes
were significantly upregulated in EC tissues compared with the
normal tissues. Related pathways analysis showed that the 'calcium
signaling pathway' was among the top significantly enriched
pathways (Fig. 1B), and the
heatmap data indicated that most of the genes in the calcium
signaling pathway had downregulated expression in EC tissues
compared with normal tissues (Fig.
1C). AGTR1, a significantly downregulated gene, became the
subject focus, and the qPCR results in a validation cohort of
samples confirmed that the AGTR1 mRNA level in EC tissues was
significantly lower than that in adjacent normal esophageal tissues
(Fig. 1D). The expression of
AGTR1 and the disease progression of EC was further investigated
and it was found that patients at the later development stages
(Fig. 1E), with cancer recurrence
after cisplatin treatments (Fig.
1F) or with cancer metastasis (Fig. 1G) had significantly reduced
expression levels of AGTR1. Moreover, the data mining of public
databases (starBase) also revealed a lower AGTR1 expression level
in tumor tissues compared with normal tissues (Fig. 1H), and that patients with a high
AGTR1 expression level showed improved overall survival (Fig. 1I). Taken together, these results
suggest that AGTR1 may be a prognostic marker, the expression of
which is downregulated in recurrent and metastasized EC.
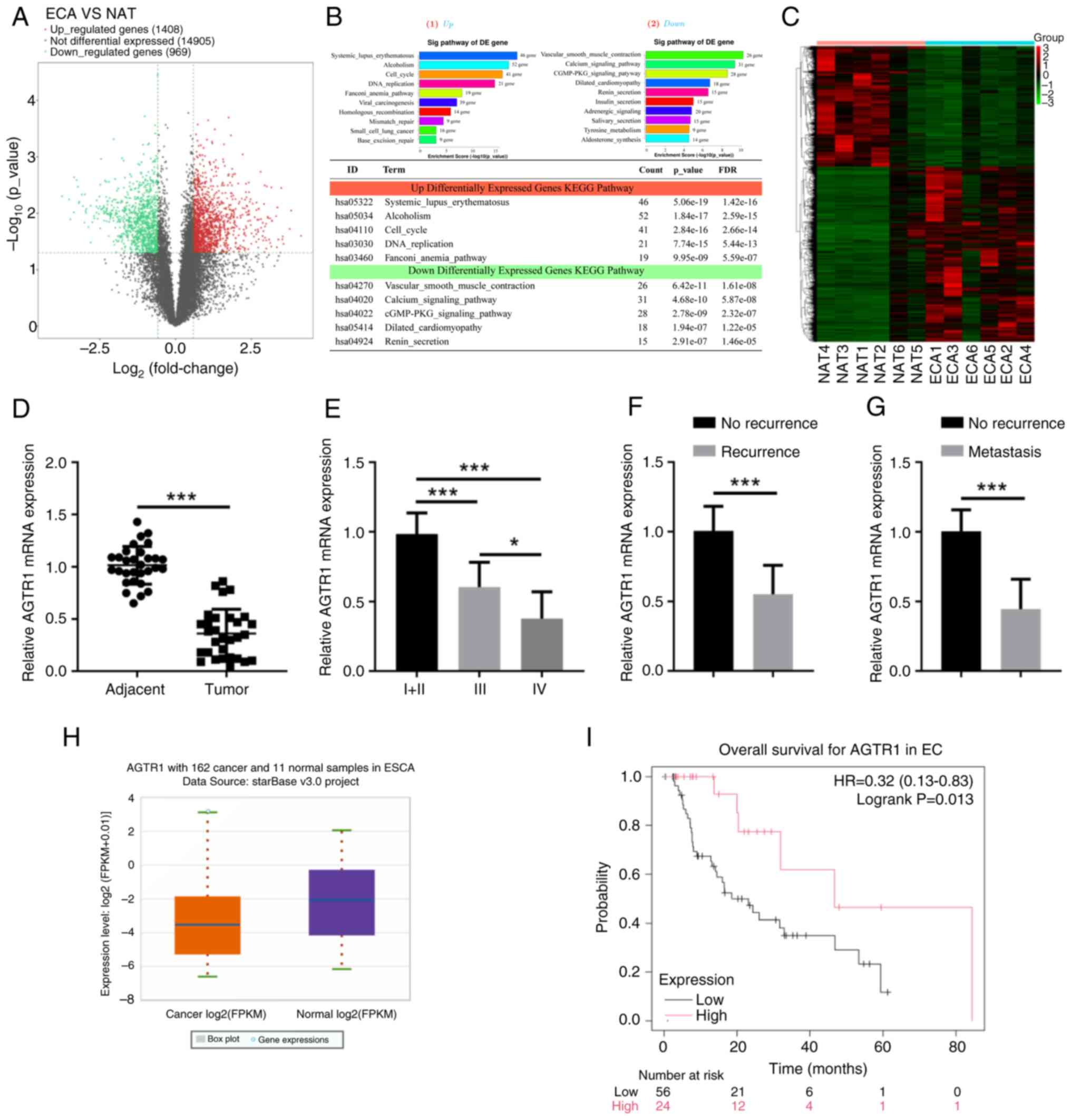 | Figure 1EC tissues have lower AGTR1
expression, which is a prognostic marker in recurrent and
metastasized EC. (A) ECA (n=5) and NAT (n=5) were subjected to
RNA-sequencing analyses, and the DE genes were plotted in a Volcano
Plot. (B) The top 10 significantly changed signaling pathways
involving the DE genes are shown. (C) The heatmap shows the
expression levels of 31 genes in the calcium signaling pathway. (D)
The mRNA levels of AGTR1 in adjacent normal tissues (n=30) and EC
tumor tissues (n=30) were determined by RT-qPCR. Relative mRNA
levels of AGTR1 in tumor samples from patients at (E) stages I/II
(n=14), III (n=9) and IV (n=7), (F) without (n=13) or with
recurrence (n=17) or (G) without (n=18) or with metastasis (n=12),
were determined by RT-qPCR. Bioinformatics analyses showing (H)
AGTR1 expression and (I) overall survival of patients with EC. Data
were retrieved from starbase (https://starbase.sysu.edu.cn/; https://kmplot.com/analysis/). *P<0.05,
***P<0.001. ESCA, esophageal carcinoma; EC,
esophageal cancer; ECA, EC tumor tissues; NAT, normal esophageal
tissues; DE, differentially expressed; RT-qPCR, reverse
transcription-quantitative PCR; AGTR1, angiotensin II receptor type
1. |
AGTR1 expression increases
chemosensitivity of EC cells to cisplatin in vitro and in vivo
To investigate the biological effect of AGTR1 on the
development of EC, the expression of AGTR1 in EC cell lines was
first evaluated. Compared with the control HEECs, the EC cell lines
(including KYSE-150, KYSE-510 and TE-3) showed significantly
downregulated expression of AGTR1 mRNA (Fig. 2A) and protein (Fig. 2B). Following this, the cisplatin
IC50 values of the tested EC cell lines were determined.
Notably, KYSE-150 cells with the lowest AGTR1 expression had the
largest IC50 value, while EC109 cells with a higher
AGTR1 expression displayed a smaller IC50 value
(Fig. 2C). To further confirm the
roles of AGTR1 expression in sensitizing EC cells to cisplatin, the
impacts on cell viability and colony formation in response to
cisplatin treatments in KYSE-150 cells and EC109 cells after AGTR1
ectopic expression and knockdown, respectively, were examined.
Overexpression of AGTR1 in KYSE-150 cells (Fig. 2D and E) significantly reduced the
cell viability upon treatment with cisplatin at various
concentrations (Fig. 2F), which
was accompanied by reduced colony-formation (Fig. 2G). On the contrary, knockdown of
AGTR1 in EC109 cells (Fig. 2H and
2I) significantly increased cell
viability (Fig. 2J) and the
colony formation ability (Fig.
2K) in response to cisplatin treatment.
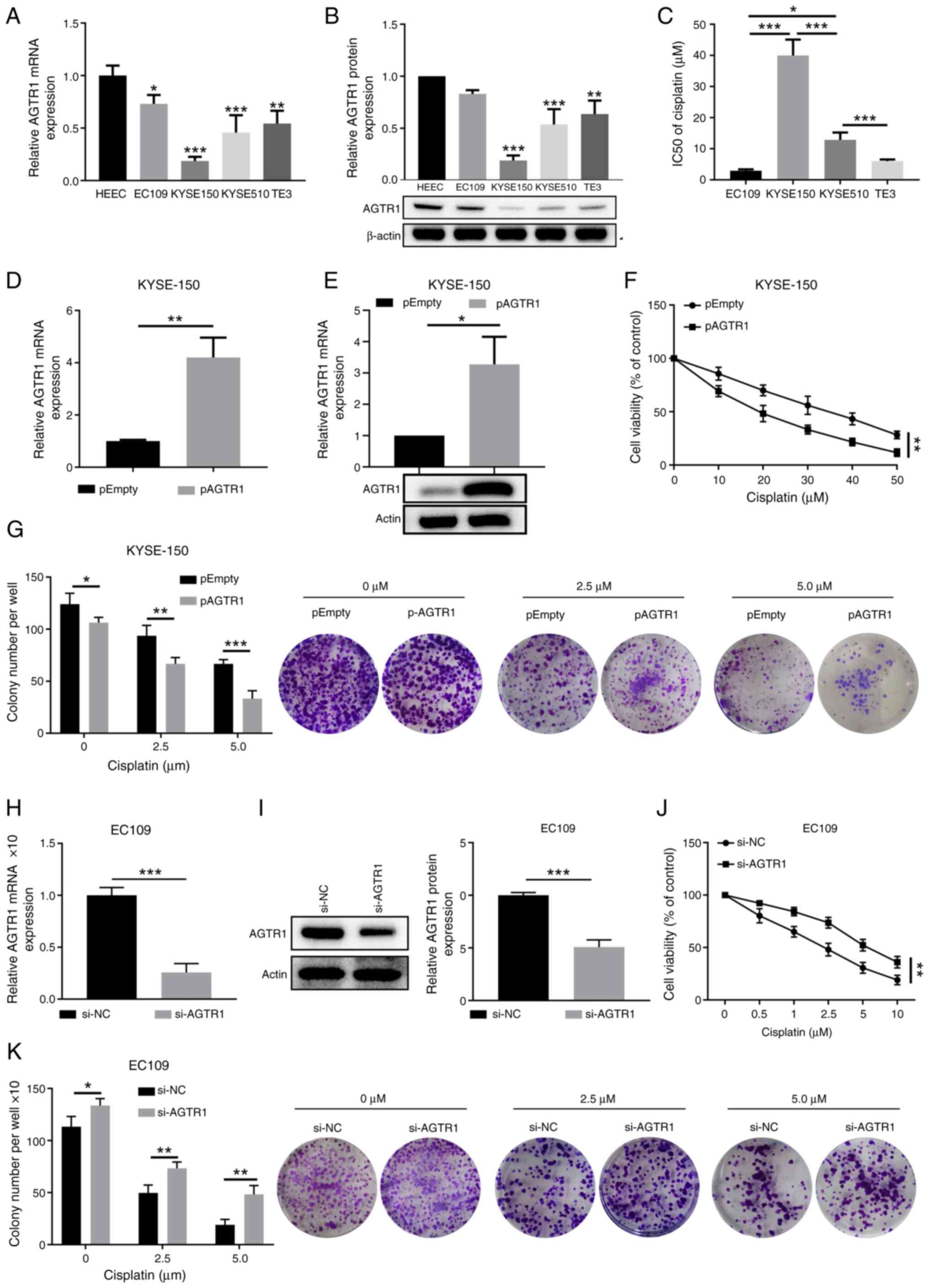 | Figure 2AGTR1 expression in EC tumors
promotes the chemosensitivity to cisplatin in vitro. (A) The
mRNA and (B) protein levels of AGTR1 in control HEECs and selected
EC cell lines (EC109, KYSE-150, KYSE-510 and TE-3) were quantitated
by RT-qPCR and western blotting, respectively. Representative
images of western blot bands are shown. (C) IC50 values
for cisplatin in selected EC cell lines were compared. Cells were
treated with cisplatin at a range of concentrations (0-50
µM) for 72 h and IC50 was calculated. KYSE-150
cells were transfected with empty vector plasmid or
AGTR1-overexpressing plasmid. After 48 h of transfection, the
expression of AGTR1 (D) mRNA was determined by RT-qPCR and (E)
protein was determined by western blotting. (F) Viability of
control and AGTR1-overexpressing KYSE-150 cells treated with
cisplatin at the indicated doses was measured by CCK-8 assays.
Control group, pEmpty (0 µM). (G) Colony formation ability
of control and AGTR1-overexpressing KYSE-150 cells upon treatment
with cisplatin at the indicated doses was determined.
Representative images of colony formation are shown, and colony
numbers per well were summarized. EC109 cells were transfected with
NC or AGTR1-specific siRNA oligos. After 48 h of transfection, the
expression of AGTR1 (H) mRNA was determined by RT-qPCR and (I)
protein was determined by western blotting. The (J) viability and
(K) colony formation ability of EC109 cells upon treatment with
cisplatin at the indicated concentrations were measured by CCK-8
and colony-formation assays, respectively. n=3 for each group.
Control group, si-NC (0 µM). (G and K) Magnification, ×10.
*P<0.05, **P<0.01,
***P<0.001. EC, esophageal cancer; AGTR1, angiotensin
II receptor type 1; HEEC, human esophageal epithelial cell;
RT-qPCR, reverse transcription-quantitative PCR; CCK-8, Cell
Counting Kit-8; NC, negative control; siRNA, small interfering
RNA. |
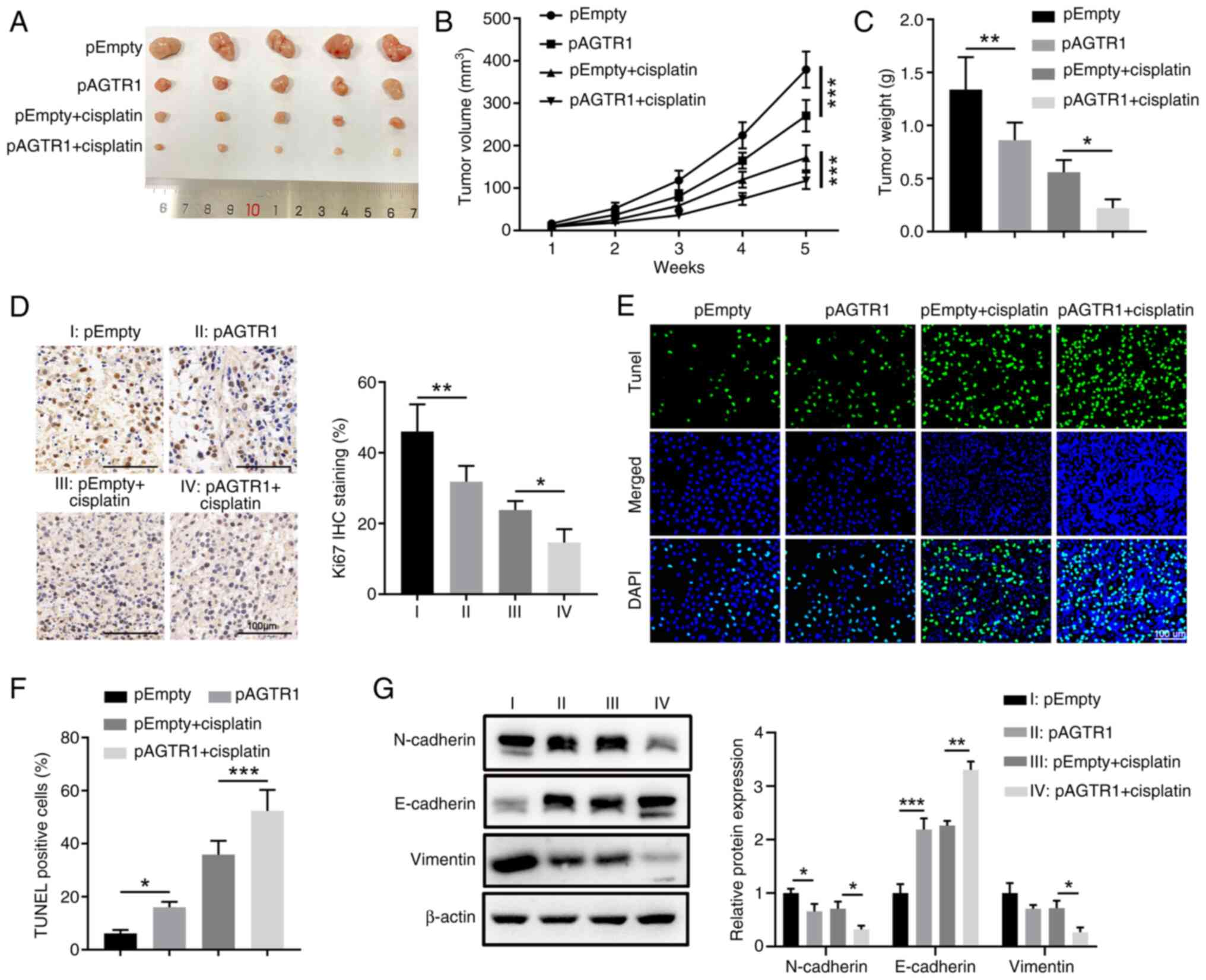
Animal experiments were further conducted with
xenografted EC cells to consolidate the chemosensitivity-promoting
role of AGTR1 expression in vivo. The growth of engrafted
control KYSE-150 cells and AGTR1-overexpressing KYSE-150 cells were
compared upon in vivo cisplatin therapy in nude mice
(Fig. 3A). Compared with the
control group without treatments, cisplatin administration reduced
the tumor volume and weight and a higher reduction was observed in
the AGTR1-overexpressing group (Fig.
3B and C). This trend was in-line with the lowest cell
proliferation as revealed by Ki-67 IHC (Fig. 3D) and highest level of apoptosis
shown by TUNEL staining (Fig. 3E and
F) in the cisplatin-treated AGTR1-overexpressing group.
Furthermore, the protein level of N-cadherin and Vimentin in the
AGTR1-overexpressing group, the cisplatin group and the
cisplatin-treated AGTR1-overexpressing group were decreased, while
the protein level of E-cadherin was increased (Fig. 3G).
Additionally, how AGTR1 knockdown impacted the
growth of engrafted EC109 cells in nude mice upon cisplatin
administration was also tested (Fig.
S1A). Consistently, EC109 cells with AGTR1 knockdown grew
larger than the control cells in mice with or without cisplatin
treatments (Fig. S1B and C),
which was accompanied by increased Ki-67 staining (Fig. S1D) and reduced apoptosis in tumor
tissues (Fig. S1E and F). In
addition, the protein levels of N-cadherin and Vimentin in the
AGTR1 knockdown group were increased and the protein level of
E-cadherin was decreased. However, cisplatin decreased the protein
level of N-cadherin and Vimentin, and increased the protein level
of E-cadherin in both control and AGTR1-konckdown cells (Fig. S1G). Therefore, these in
vitro and in vivo studies demonstrated that AGTR1
expression increased the chemosensitivity of EC cells to
cisplatin.
AGTR1 expression and cisplatin
administration affects the metastasis-relevant traits of EC
cells
Since a higher expression level of AGTR1 was found
in patients with EC without metastasis (Fig. 1G), the potential role of AGTR1 in
suppressing metastasis of EC cells was next evaluated. Transwell
experiments were performed using control KYSE-150 cells,
AGTR1-overexpressing KYSE-150 cells, cisplatin-treated
AGTR1-overexpressing KYSE-150 cells, control EC109 cells,
AGTR1-knockdown EC109 cells and cisplatin-treated AGTR1-knock down
EC109 cells. As shown in Fig. 4A,
ectopic expression of AGTR1 or cisplatin administration caused the
significantly reduced migration and invasion of KYSE-150 cells. In
line with this observation, EC109 cells with AGTR1 knockdown had
significantly enhanced migration and invasion (Fig. 4B), compared with the control EC109
cells. As expected, the wound healing assay demonstrated similar
trends. Compared with the control KYSE-150 cells, AGTR1
overexpression or cisplatin administration significantly diminished
the wound healing ability (Fig.
4C, upper). Consistently, AGTR1 knockdown in EC109 cells
resulted in enhanced wound healing ability (Fig. 4C, lower).
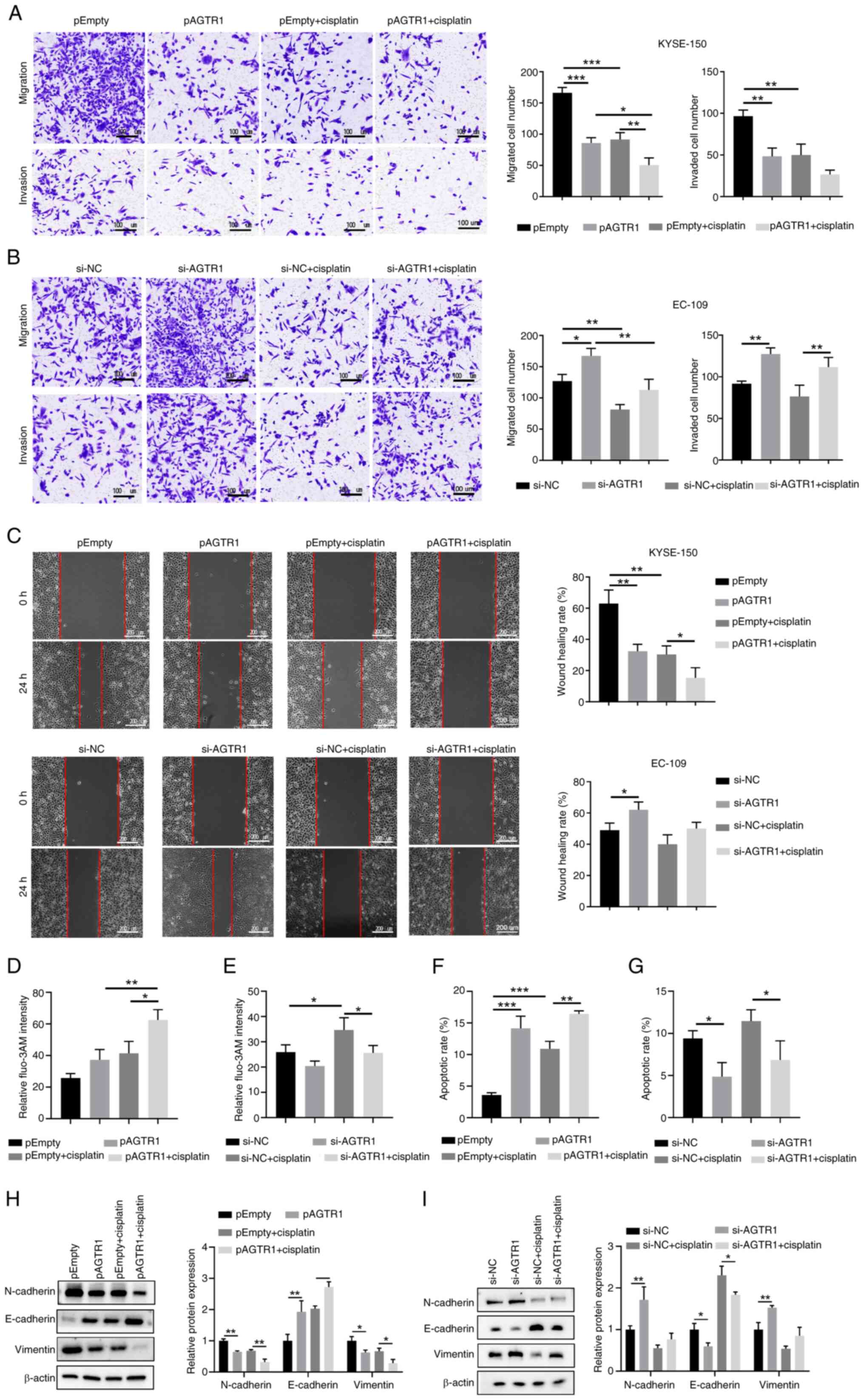 | Figure 4AGTR1 expression suppresses the
metastasis-relevant traits of EC cells and upregulates
intracellular Ca2+ levels as well as enhances
mitochondria-dependent apoptosis in vitro. (A) Control,
KYSE-150 cells with AGTR1 overexpression, cisplatin and
cisplatin-treated AGTR1-overexpressing cells, in addition to (B)
control, EC109 cells with AGTR1-knockdown, cisplatin and
cisplatin-treated AGTR1 knockdown cells were subjected to Transwell
assays. Representative images are shown and the migrated/invaded
cells numbers were quantified. (C) Wound healing assays of the
indicated EC cells. Wounds were imaged at 0 and 24 h following
wound formation. Representative images are shown and the extent of
wound closure was quantified. (D, E) The intracellular
Ca2+ levels of the indicated eight groups were
determined by flow cytometry with the calcium indicator, Fluo-3 AM.
(F, G) The apoptosis of the indicated eight groups were determined
by flow cytometry with Annexin V/PI staining. (H, I) The protein
level of N-cadherin, E-cadherin and Vimentin of the indicated eight
groups were detected by western blotting. n=5 for each group.
*P<0.05, **P<0.01,
***P<0.001. EC, esophageal cancer; AGTR1, angiotensin
II receptor type 1; NC, negative control; siRNA, small interfering
RNA. |
To unravel the molecular mechanisms underlying the
chemo-sensitizing roles of AGTR1 expression, the relationship
between AGTR1 expression and intracellular calcium level in EC
cells were further evaluated. AGTR1 overexpression in KYSE-150
cells enhanced the intracellular Ca2+ levels (Figs. 4D and S2A), while the AGTR1 knockdown in EC109
cells reduced the intracellular Ca2+ levels (Figs. 4E and S2A). Notably, AGTR1 overexpression in
KYSE-150 cells enhanced the apoptosis rate (Figs. 4F and S2B), while the AGTR1 knockdown in EC109
cells reduced the apoptosis rate (Figs. 4G and S2B). Furthermore, the protein level of
N-cadherin and Vimentin in the AGTR1-overexpressing group, the
cisplatin group and the cisplatin-treated AGTR1-overexpressing
group were decreased, while the protein level of E-cadherin was
increased (Fig. 4H). However, the
protein level of N-cadherin and Vimentin in the AGTR1 knockdown
group were increased and the protein level of E-cadherin was
decreased. However, cisplatin reduced the protein levels of
N-cadherin and Vimentin, while increasing the protein content of
E-cadherin in both control and AGTR1-konckdown cells (Fig. 4I). Collectively, these data
indicate that AGTR1 expression affects metastasis-relevant traits
in EC cells.
AGTR1 expression upregulates
intracellular Ca2+ levels and enhances
mitochondria-dependent apoptosis in EC cells
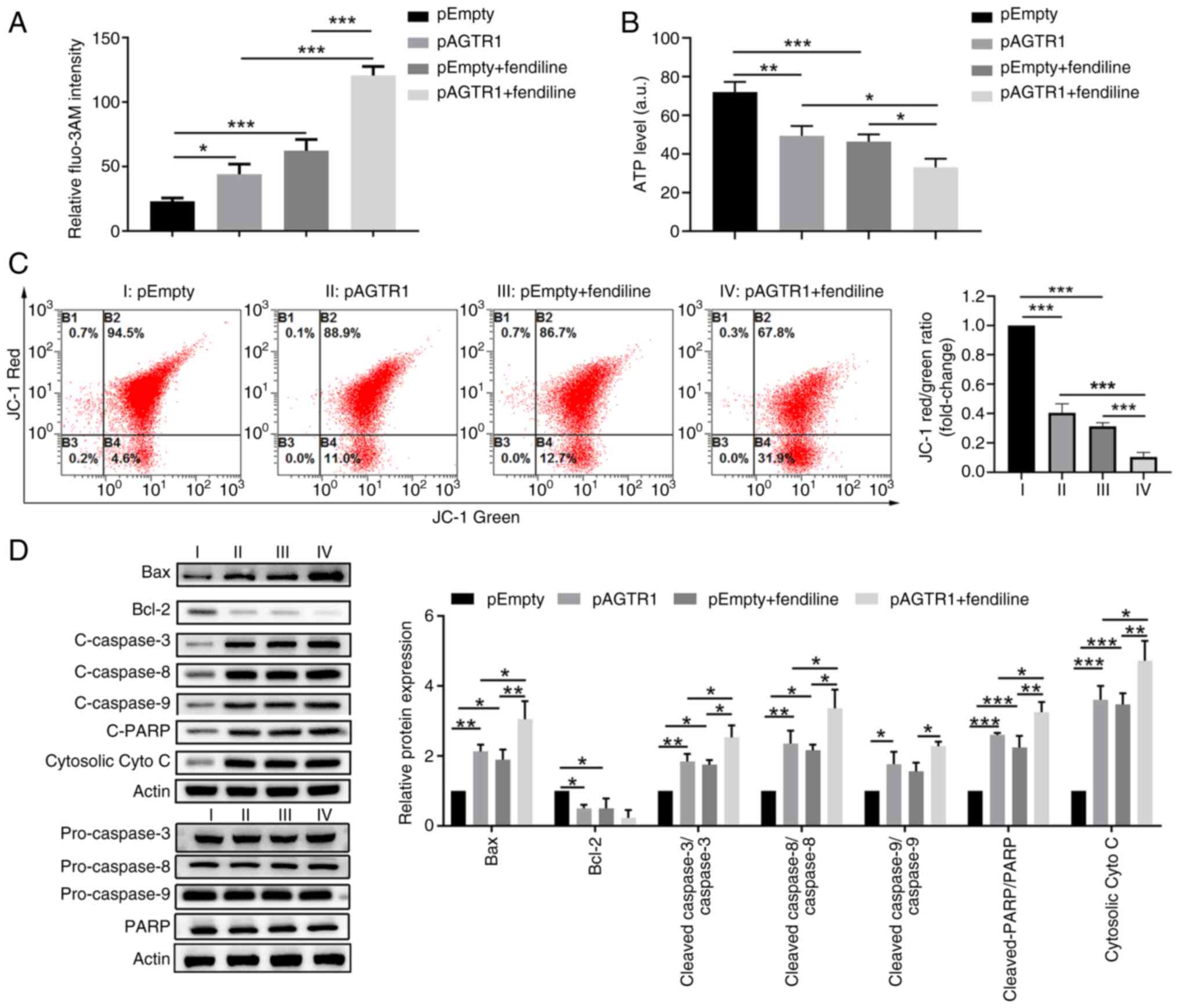
To verify the pro-apoptosis role of high
intracellular Ca2+ levels in EC cells, the cells were
treated with fendiline, which is a non-selective calcium channel
blocker and causes accumulation of Ca2+ in cells. First,
it was confirmed that the intracellular Ca2+ levels in
fendiline-treated cells were much higher than the corresponding
control cells (Figs. 5A and
S2C), which were accompanied by
a reversed trend in ATP levels (Fig.
5B) and a similar trend in mitochondrial membrane potential
loss, as indicated by JC-1 staining (Fig. 5C). Moreover, additional fendiline
treatments caused higher mitochondria-dependent apoptosis, as
indicated by the increased protein expression levels of Bax,
cleaved caspase 3/8/9, cleaved PARP and cytosolic cytochrome c, as
well as decreased protein expression of Bcl-2 in KYSE-150 cells
(Fig. 5D). Moreover, the impacts
of fendiline treatment on control EC109 cells and AGTR1-knockdown
EC109 cells were also evaluated. In agreement with that observed in
KYSE-150 cells, fendiline treatments led to increased intracellular
Ca2+ levels (Figs. S3A
and S2D), decreased ATP levels (Fig. S3B), increased mitochondrial
membrane potential loss (Fig.
S3C) and increased mitochondria pathway-mediated apoptosis
(Fig. S3D) in EC109 cells. Thus,
these results indicate that upregulation of intracellular
Ca2+ levels and enhanced mitochondria-dependent
apoptosis are associated with the beneficial roles of AGTR1
expression in chemo-sensitizing EC cells.
AGTR1 expression and fendiline
treatment-mediated increase in intracellular Ca2+ level
contributes to chemosensitivity to cisplatin in EC cells
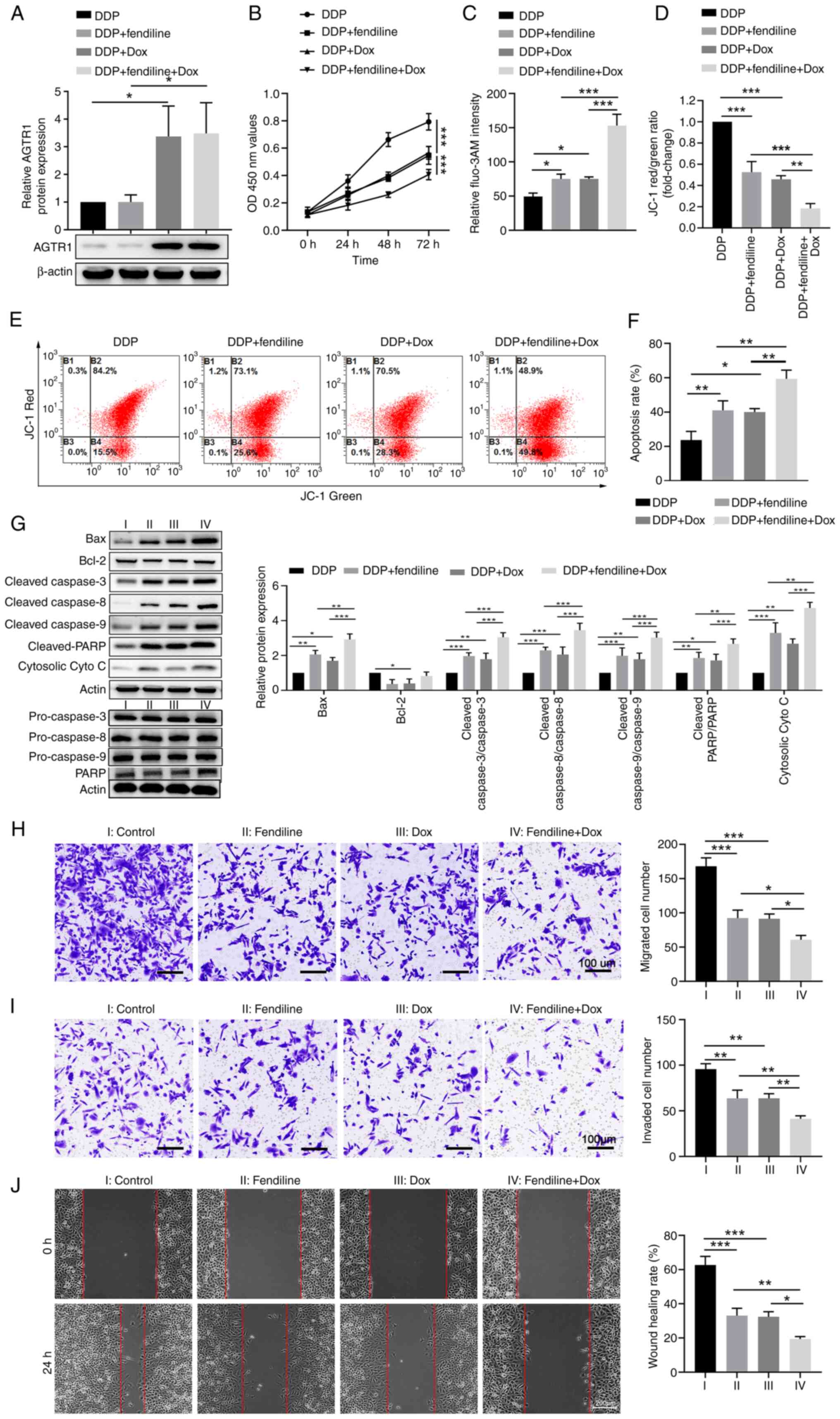
After the identification of increased intracellular
Ca2+ levels as an underlying mechanism in mediating the
AGTR1 expression-induced tumor suppressive effects in EC cells, it
was further evaluated whether this approach could be utilized to
enhance the chemo-sensitivity of EC cells to cisplatin. First, the
Tet-on KYSE-150 cell line was generated and it was confirmed that
Dox successfully induced expression of AGTR1 (Figs. 6A and S4A). This KYSE-150 cell line was
treated with cisplatin, alone or in the presence of fendiline
or/and Dox. Compared with the group with cisplatin stimulation
alone, fendiline treatment or AGTR1 overexpression induced by Dox
significantly reduced cell proliferation. Notably, the group with
the combinational treatments of both fendiline and Dox were
significantly more reduced (Fig.
6B). Compared with cisplatin stimulation alone, combinational
treatments of fendiline or Dox increased the intracellular
Ca2+ level, as indicated by fluo-3AM staining (Figs. 6C and S4B), and the level of mitochondrial
membrane potential loss, as measured by JC-1 staining (Figs. 6D and 6E). Moreover, the apoptosis rates
indicated by Annexin V/PI staining (Figs. 6F and S4C) and the changes in mitochondrial
apoptosis-related protein levels showed similar trends among the
four groups (Fig. 6G), that is,
compared with the cisplatin alone group, fendiline or Dox treatment
increased apoptosis, while the group with concurrent fendiline and
Dox administration displayed higher levels of apoptosis. It was
also evaluated how fendiline impacted the metastasis-relevant
traits in EC cells. Compared with the control Tet-on KYSE-150 cells
without treatment, single fendiline or single Dox stimulation
significantly reduced cell migration and invasion (Fig. 6H and I), as well as the wound
healing ability of cells (Fig.
6J), while simultaneous administration of fendiline and Dox
more notably reduced the metastasis-relevant traits of KYSE-150
cells. Taken together, these results suggest that the AGTR1
overexpression and fendiline treatment-mediated increase in
intracellular Ca2+ level contributes to chemosensitivity
to cisplatin in EC cells.
Xenograft animal model verifies the
feasibility of using fendiline to potentiate the therapeutic
efficacy of cisplatin in EC
To further clarify the beneficial effects of
synchronous administration of fendiline and cisplatin in treating
EC, a xenograft mice model was established by inoculating the
Tet-on KYSE-150 cells into nude mice and treating them with
cisplatin alone or together with fendiline or Dox or with both
fendiline and Dox (Fig. 7A).
Compared with the cisplatin treatment only group, either Dox
administration-induced AGTR1 expression or fendiline exposure,
significantly reduced the growth of engrafted EC cells, while
simultaneous administration of fendiline and Dox inhibited tumor
growth (Fig. 7B). This was also
evidenced by the lowest tumor weight among the four groups at the
end time-point (Fig. 7C). In
agreement with these findings, tumor cells that received both
cisplatin and fendiline or both cisplatin and Dox displayed
significantly lower cell proliferation and more apoptosis than the
cells that received only cisplatin (Figs. 6B and S4B). As expected, the mice that
received all the drugs (cisplatin, Dox and fendiline) exhibited the
slowest tumor cell proliferation according to Ki67 IHC staining of
tumor tissue (Fig. 7D) and the
highest level of tumor cell apoptosis (Fig. 7E).
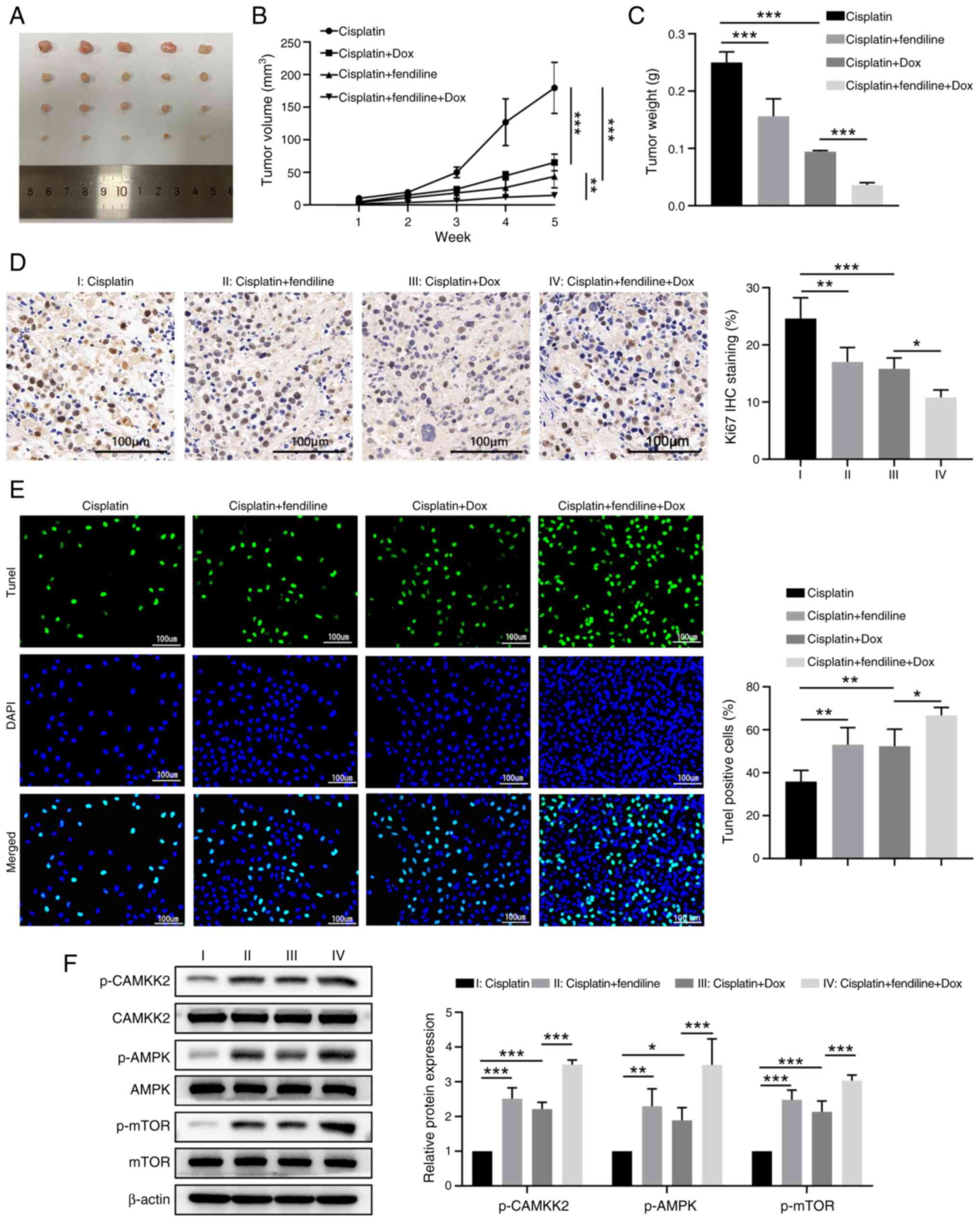 | Figure 7Fendiline administration and induced
AGTR1 overexpression sensitizes grafted esophageal cancer cells to
cisplatin chemotherapy in a mice model. Nude mice were injected
with the Tet-on KYSE-150 cells. At 7 days after tumor growth, nude
mice were treated with cisplatin alone, cisplatin + fendiline,
cisplatin + Dox or cisplatin + fendiline + Dox. Tumor volume was
monitored once per week and tumor tissues were collected at 36 days
after tumor inoculation. (A) Tumor image of the indicated four
groups. (B) Tumor growth curves of the indicated four groups. (C)
Tumor weight at the end time-point. (D) Cell proliferation in tumor
tissues was determined by Ki-67 staining. (E) Apoptosis of tumor
tissues was determined by TUNEL assay. (F) The levels of calcium
signaling-associated proteins in tumor tissues were determined by
western blotting. Representative images of the western blot bands
are shown and the relative protein levels were semi-quantified. n=5
for each group. *P<0.05, **P<0.01,
***P<0.001. Dox, doxycycline; AGTR1, angiotensin II
receptor type 1; IHC, immunohistochemistry; TUNEL, terminal
deoxynucleotidyl transferase dUTP nick-end labeling; p-,
phosphorylated. |
To verify that the enhanced chemo-sensitivity to
cisplatin by fendiline and AGTR1 overexpression was associated with
increased intracellular Ca2+ levels, the expression
levels of calcium signaling-associated proteins, including
phosphorylated (p-)CAMKK2 (Ser511), p-AMPK (Thr172) and p-mTOR
(Ser2448) were examined. As shown in Fig. 7F, a similar trend to that of the
apoptosis rate in EC tissues was found. That is, the tumor tissues
from mice that received fendiline or Dox had higher levels of the
aforementioned calcium signaling-associated proteins than that from
mice that received only cisplatin, while tissues from mice that
received all the drugs had the highest levels of the calcium
signaling-associated proteins. Collectively, these observations in
the animal model indicate that it is feasible to use fendiline to
increase the sensitivity of EC cells to cisplatin chemotherapy
in vivo.
Discussion
The incidence of EC shows an alarming increasing
trend in Asia and globally, and the mortality rate is still high
despite the available systemic treatments (31-33). Since cisplatin-based chemotherapy
is one part of the popular treatment regimens for EC (6,34),
it is urgent to find new therapeutic targets and develop new
treatment approaches to fully potentiate the antitumor efficacy of
cisplatin. In the present study, AGTR1 was identified as a
potential prognostic marker and therapeutic target. AGTR1
expression was found to enhance the chemosensitivity of KYSE-150
and EC109 cells, and markedly decrease their metastasis-relevant
traits. Notably, it was found that the tumor-suppressive function
of upregulated AGTR1 was associated with upregulated intracellular
Ca2+ levels and increased mitochondria-dependent
apoptosis. These results provide insights into the functional role
of the AGTR1 and Ca2+ influx in EC treatments and
suggest that they are potential targets for amplifying the efficacy
of cisplatin-mediated chemotherapy in EC.
AGTR1 is a component of the renin angiotensin
system. AGTR1 classically regulates cardiovascular homeostasis and
has shown the potential to stimulate cell proliferation, migration
or invasion, and promote angiogenesis, inflammation and immunity
(19). Increasing evidence shows
that the inhibitory effect of losartan and candesartan on AGTR1 can
inhibit angiogenesis, which helps to inhibit tumor growth and blood
metastasis (35,36). For instance, AGTR1-mediated
signaling through CXC chemokine receptor 4/stromal cell derived
factor 1α was reported to regulate breast cancer migration and
lymph node metastasis (19). It
was also found that AGTR1 activation in ovarian cancer
significantly enhanced multicellular spheroids formation and
increased peritoneal metastasis via upregulation of lipid
desaturation and suppression of endoplasmic reticulum stress
(37). It is of note that two
recent reports also revealed a positive correlation between AGTR1
expression and the proliferation of human EC cells (38,39). Li et al (38) found that AGTR1 upregulation in
ESCC was univariately associated with inferior overall survival and
remained multivariately independent, and angiotensin II stimulated
the growth of ESCC cells in vitro. Fujihara et al
(39) reported that the AGTR1
antagonist, telmisartan, suppressed the proliferation and tumor
growth of human esophageal adenocarcinoma cells by inducing cell
cycle arrest via the AMPK/mTOR pathway. Contrary to the report from
Li et al (38), an adverse
association between ATGR1 expression and the outcome of patients
with EC after treatment was identified in the present study. This
discrepancy may be due to the differences in geographical location,
disease stages and treatment regimens in the selected patient
cohorts. In addition, in their study models, either angiotensin II
or the AGTR1 antagonist, telmisartan, was employed, while the
models in the present study involved merely the manipulation of the
AGTR1 expression levels in EC cells, which might more faithfully
reflect the relationship between AGTR1 expression and EC disease
progression.
Multiple aspects of the molecular mechanisms of the
angiotensin II/AGTR1 axis and the use of an AGTR1 antagonist in
controlling cancer disease progression were proposed (37), such as upregulation of lipid
desaturation and suppression of endoplasmic reticulum stress. The
present study focused on whether AGTR1 regulates the intracellular
Ca2+ level and induces apoptosis in EC cells. After
activation by the vasoconstricting peptide, angiotensin II, AGTR1
is coupled with Gq/11 to activate phospholipase C and increase the
concentration of cytoplasmic Ca2+, which triggers
cellular responses, such as stimulation of protein kinase C,
inhibition of adenylate cyclase and activation of various tyrosine
kinases (40). The stored
Ca2+ is released from the endoplasmic reticulum and
taken up by mitochondria, triggering cell apoptosis (41,42). The accumulation of Ca2+
in mitochondria leads to an increase in mitochondrial membrane
permeability by stimulating the opening of the mitochondrial
permeability transition pore (mPTP). The opening of the mPTP leads
to the release of pro-apoptotic factors, particularly cytochrome C
(41,42). The results of the present study
showed that the expression levels of AGTR1 in both KYSE-150 cells
and EC109 cells were positively associated with their apoptotic
rates, intracellular Ca2+ levels, mitochondrial membrane
potential loss and level of released cytochrome C. These results
indicated that AGTR1 overexpression can induce Ca2+
influx in EC cells and the mitochondrial calcium overload can
trigger apoptotic cell death. Moreover, the reduced migration and
invasion abilities of EC cell lines upon AGTR1 overexpression might
at least be partially due to AGTR1-mediated apoptotic cell death
through the mitochondria-dependent pathway. Consistently,
repression of the AGTR1 gene was found to promote the
multi-chemoresistance of osteosarcoma, indicating that AGTR1 is a
potential target for the effective chemotherapy of osteosarcoma
(22). Additionally, in
accordance with the previous results showing AGTR1-induced
apoptosis in other cell types including intestinal epithelial
(25) and cardiac (26) cells, the results of the present
study suggest that AGTR1 is a new therapeutic target in EC, and
drug screening for activating the function of AGTR1 in triggering
Ca2+ influx-mediated apoptosis may be useful to develop
new drugs to treat EC.
Initially, as an anti-anginal agent for the
treatment of coronary heart disease (43,44), fendiline was shown to bind to the
Ca2+ channel and calmodulin, thus inhibiting the
transient outward current in rat ventricular cardiomyocytes
(45) and L-type Ca2+
channels in ventricular myocytes from rats and guinea pigs
(46,47). In contrast to its inhibition of
Ca2+ channels, fendiline was found to be able to induce
a rise in intracellular Ca2+ level in different cells by
causing extracellular Ca2+ influx and intracellular
Ca2+ release from the endoplasmic reticulum (48,49). The possibility of utilizing
fendiline to evoke Ca2+ influx and subsequent cell death
in multiple cancer types has been explored. The efficacy of
fendiline as anti-malignancy drug, including inhibition of
proliferation and invasion as well as the induction of cell death
were demonstrated in human oral cancer (50), pancreatic cancer (51), bladder carcinoma (52), hepatoma (53) and human osteosarcoma (54) cells. It is worth noting that
fendiline is reported to enhance the cytotoxic effects of other
therapeutic agents. For instance, the combinational use of
fendiline with inhibitors such as gemcitabine, visudyne, a
Yes-associated protein 1 inhibitor or tivantinib (ARQ197, a c-Met
inhibitor) can overcome the growth and oncogenic characteristics of
pancreatic cancer cells (55).
Additionally, co-administration of fendiline hydrochloride was
reported to enhance the chemotherapeutic efficacy of cisplatin in
neuroblastoma treatment, as evidenced by the enhanced ability of
cisplatin to induce the apoptosis of neuroblastoma cells and a
reduction in tumor growth and prolonged animal survival rate in a
xenograft mice model (56). In
line with these findings, the present study found that fendiline
treatment increased the cytotoxic potency of cisplatin in KYSE-150
cells both in vitro and in vivo, suggesting that
administration of fendilin may be a potential novel therapeutic
method to improve the efficacy of cisplatin in invasive and poorly
responsive EC cases.
Of note, in the xenograft mice model established in
the present study, increased expression levels of Ca2+
signaling-related molecules (including p-AMPK and p-mTOR) in tumor
tissues from the cisplatin + fendiline group and the Dox +
fendiline group were observed, compared with the cisplatin alone
group. This is similar to another study, in which telmisartan
inhibited human esophageal adenocarcinoma cell proliferation and
growth via the AMPK/mTOR pathway (39). Moreover, as this drug combination
showed enhanced efficacy, the dose of cisplatin could be decreased
to minimize the adverse effects of cisplatin treatments.
Furthermore, it has been reported that fendiline inhibited the
proliferation of lung, pancreatic, endometrial and colon tumor
cells expressing oncogenic mutant K-Ras more effectively than those
of tumor cells expressing wild-type K-Ras, which indicates that
fendiline is a selective inhibitor of carcinogenic K-Ras function,
regardless of tumor origin (56).
Therefore, the implication of the findings of the present study
could be extended to treating EC and other types of malignancies
with oncogenic mutant K-Ras expression in a more efficient way by
combinational administration of fendiline and other
therapeutics.
The present study has several limitations. First,
the sample size of patients with EC for the quantitation of AGTR1
expression was limited. The patients included in the present study
were also drawn from a single institution and thus were subject to
referral bias. A study with data from multiple centers with
different geographical locations, disease stages and treatment
regimens may be necessary to further validate the study findings.
Second, the present study did explore the signaling pathway
mechanisms involved in AGTR1 and the detailed molecular mechanisms
underlying the regulation of EC apoptosis. In the future,
predictions through bioinformatics analysis could be made, and the
combined in silico study and experimental investigation
could facilitate the identification of more interacting genes to
locate the detailed downstream pathways of AGTR1 in EC. Third, the
experiments merely showed the add-on effects of cisplatin,
fendiline and induced AGTR1 overexpression in EC cells. More
insensitive investigations are needed to clarify whether the
co-administration regimens with reduced doses still work
efficiently and how a synergistic effect could be achieved to
further minimize the therapeutic doses.
In summary, the results of the present study
indicated that AGTR1 expression-mediated Ca2+ influx
suppressed the oncogenic traits of EC cells and increased apoptosis
via the mitochondrial pathway, which can be utilized to enhance the
chemosensitivity of cisplatin in EC treatment. Notably, increasing
the intracellular Ca2+ levels by co-administration of
fendiline reinforced the cytotoxic efficacy of cisplatin both in
vitro and in vivo. These findings shed new light on the
functional role of Ca2+ influx-induced cell death in EC
and provide a mechanistic insight into using fendiline to
potentiate the chemotherapy efficacy of cisplatin in patients with
EC.
Supplementary Data
Availability of data and materials
The RNA sequencing data generated in the present
study may be found in the Gene Expression Omnibus database under
accession number GSE263647 or at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE263647.
All other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
KL, JB and RZ confirm the authenticity of all the
raw data. Conception and design were conducted by KL, JB, RZ, RX
and LP; acquisition of data (acquired and managed patients and
provided facilities) was conducted by YL, SY and GF; analysis and
interpretation of data (such as statistical, biostatistics and
computational analyses) was conducted by KL, GS and JB; writing
and/or revising the manuscript was conducted by RZ and RX;
administrative, technical or material support (such as reporting or
organizing data and constructing databases) was conducted by LP and
YL; study supervision was conducted by KL. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
patients and the study protocol was approved by the Medical Ethics
Committee of Nanchong Central Hospital (approval no. 2019.095;
Nanchong, China). Animal care and study were approved by the Ethics
Committee of North Sichuan Medical College (approval no.
NSMC202144; Nanchong, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This study was supported by the National Natural Science
Foundation of China (grant no. 82203851), Nanchong Science and
Technology Program (grant nos. 22SXQT0340, 22SXQT0336, 20SXQT0328,
20SXQT0325, 20SXPTJS0003, 20SXQT0074 and 20SXQT0181) and Doctoral
Fund of North Sichuan Medical College (grant no. CBY22-QDA01).
References
|
1
|
Huang FL and Yu SJ: Esophageal cancer:
Risk factors, genetic association, and treatment. Asian J Surg.
41:210–215. 2018. View Article : Google Scholar
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arnal MJ, Arenas AF and Arbeloa AL:
Esophageal cancer: Risk factors, screening and endoscopic treatment
in Western and Eastern countries. World J Gastroenterol.
21:7933–7943. 2015. View Article : Google Scholar
|
|
4
|
Yang YM, Hong P, Xu WW, He QY and Li B:
Advances in targeted therapy for esophageal cancer. Signal
Transduct Target Ther. 5:2292020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Borggreve AS, Kingma BF, Domrachev SA,
Koshkin MA, Ruurda JP, van Hillegersberg R, Takeda FR and Goense L:
Surgical treatment of esophageal cancer in the era of multimodality
management. Ann N Y Acad Sci. 1434:192–209. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ku GY: Systemic therapy for esophageal
cancer: Chemotherapy. Chin Clin Oncol. 6:492017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luan S, Zeng X, Zhang C, Qiu J, Yang Y,
Mao C, Xiao X, Zhou J, Zhang Y and Yuan Y: Advances in drug
resistance of esophageal cancer: From the perspective of tumor
microenvironment. Front Cell Dev Biol. 9:6648162021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ilson DH, Forastiere A, Arquette M, Costa
F, Heelan R, Huang Y and Kelsen DP: A phase II trial of paclitaxel
and cisplatin in patients with advanced carcinoma of the esophagus.
Cancer J. 6:316–323. 2000.PubMed/NCBI
|
|
9
|
Li Z, Zhang P, Ma Q, Wang D and Zhou T:
Cisplatin-based chemoradiotherapy with 5-fluorouracil or pemetrexed
in patients with locally advanced, unresectable esophageal squamous
cell carcinoma: A retrospective analysis. Mol Clin Oncol.
6:743–747. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cui C, Merritt R, Fu L and Pan Z:
Targeting calcium signaling in cancer therapy. Acta Pharm Sin B.
7:3–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dubois C, Abeele FV and Prevarskaya N:
Targeting apoptosis by the remodelling of calcium-transporting
proteins in cancerogenesis. FEBS J. 280:5500–5510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Al-Bahlani S, Fraser M, Wong AY, Sayan BS,
Bergeron R, Melino G and Tsang BK: P73 regulates cisplatin-induced
apoptosis in ovarian cancer cells via a calcium/calpain-dependent
mechanism. Oncogene. 30:4219–4230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gualdani R, de Clippele M, Ratbi I, Gailly
P and Tajeddine N: Store-Operated calcium entry contributes to
cisplatin-induced cell death in non-small cell lung carcinoma.
Cancers (Basel). 11:4302019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen L, Wen N, Xia M, Zhang YU, Liu W, Xu
YE and Sun L: Calcium efflux from the endoplasmic reticulum
regulates cisplatin-induced apoptosis in human cervical cancer HeLa
cells. Oncol Lett. 11:2411–2419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singh KD and Karnik SS: Angiotensin
receptors: Structure, function, signaling and clinical
applications. J Cell Signal (Los Angel). 1:1112016.PubMed/NCBI
|
|
17
|
Barreras A and Gurk-Turner C: Angiotensin
II receptor blockers. Proc (Bayl Univ Med Cent). 16:123–126.
2003.
|
|
18
|
Rhodes DR, Ateeq B, Cao Q, Tomlins SA,
Mehra R, Laxman B, Kalyana-Sundaram S, Lonigro RJ, Helgeson BE,
Bhojani MS, et al: AGTR1 overexpression defines a subset of breast
cancer and confers sensitivity to losartan, an AGTR1 antagonist.
Proc Natl Acad Sci USA. 106:10284–10289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma Y, Xia Z, Ye C, Lu C, Zhou S, Pan J,
Liu C, Zhang J, Liu T, Hu T, et al: AGTR1 promotes lymph node
metastasis in breast cancer by upregulating CXCR4/SDF-1alpha and
inducing cell migration and invasion. Aging (Albany NY).
11:3969–3992. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ateeq B, Tomlins SA and Chinnaiyan AM:
AGTR1 as a therapeutic target in ER-positive and ERBB2-negative
breast cancer cases. Cell Cycle. 8:3794–3795. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suganuma T, Ino K, Shibata K, Kajiyama H,
Nagasaka T, Mizutani S and Kikkawa F: Functional expression of the
angiotensin II type 1 receptor in human ovarian carcinoma cells and
its blockade therapy resulting in suppression of tumor invasion,
angiogenesis, and peritoneal dissemination. Clin Cancer Res.
11:2686–2694. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pu Y, Zhao F, Li Y, Cui M, Wang H, Meng X
and Cai S: The miR-34a-5p promotes the multi-chemoresistance of
osteosarcoma via repression of the AGTR1 gene. BMC Cancer.
17:452017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kooptiwut S, Hanchang W, Semprasert N,
Junking M, Limjindaporn T and Yenchitsomanus PT: Testosterone
reduces AGTR1 expression to prevent β-cell and islet apoptosis from
glucotoxicity. J Endocrinol. 224:215–224. 2015. View Article : Google Scholar
|
|
24
|
Kooptiwut S, Wanchai K, Semprasert N,
Srisawat C and Yenchitsomanus PT: Estrogen attenuates AGTR1
expression to reduce pancreatic β-cell death from high glucose. Sci
Rep. 7:166392017. View Article : Google Scholar
|
|
25
|
Wang W, Sun L, Xiao W and Yang H:
Essential role of angiotensin receptors in the modulation of
intestinal epithelial cell apoptosis. J Pediatr Gastroenterol Nutr.
57:562–569. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang Y, Zhou Y, Cao Z, Tong XZ, Xie HQ,
Luo T, Hua XP and Wang HQ: miR-155 functions downstream of
angiotensin II receptor subtype 1 and calcineurin to regulate
cardiac hypertrophy. Exp Ther Med. 12:1556–1562. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Berry MF: Esophageal cancer: Staging
system and guidelines for staging and treatment. J Thorac Dis.
6(Suppl 3): S289–S297. 2014.PubMed/NCBI
|
|
28
|
Boonstra JJ, van der Velden AW, Beerens
EC, van Marion R, Morita-Fujimura Y, Matsui Y, Nishihira T,
Tselepis C, Hainaut P, Lowe AW, et al: Mistaken identity of widely
used esophageal adenocarcinoma cell line TE-7. Cancer Res.
67:7996–8001. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Capes-Davis A, Theodosopoulos G, Atkin I,
Drexler HG, Kohara A, MacLeod RA, Masters JR, Nakamura Y, Reid YA,
Reddel RR and Freshney RI: Check your cultures! A list of
cross-contaminated or misidentified cell lines. Int J Cancer.
127:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
31
|
Liu CQ, Ma YL, Qin Q, Wang PH, Luo Y, Xu
PF and Cui Y: Epidemiology of esophageal cancer in 2020 and
projections to 2030 and 2040. Thorac Cancer. 14:3–11. 2023.
View Article : Google Scholar :
|
|
32
|
Zhu H, Wang Z, Deng B, Mo M, Wang H, Chen
K, Wu H, Ye T, Wang B, Ai D, et al: Epidemiological landscape of
esophageal cancer in Asia: Results from GLOBOCAN 2020. Thorac
Cancer. 14:992–1003. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Uhlenhopp DJ, Then EO, Sunkara T and
Gaduputi V: Epidemiology of esophageal cancer: Update in global
trends, etiology and risk factors. Clin J Gastroenterol.
13:1010–1021. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brown A, Kumar S and Tchounwou PB:
Cisplatin-Based chemotherapy of human cancers. J Cancer Sci Ther.
11:972019.PubMed/NCBI
|
|
35
|
Chauhan VP, Martin JD, Liu H, Lacorre DA,
Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X,
Adstamongkonkul P, et al: Angiotensin inhibition enhances drug
delivery and potentiates chemotherapy by decompressing tumour blood
vessels. Nat Commun. 4:25162013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Diop-Frimpong B, Chauhan VP, Krane S,
Boucher Y and Jain RK: Losartan inhibits collagen I synthesis and
improves the distribution and efficacy of nanotherapeutics in
tumors. Proc Natl Acad Sci USA. 108:2909–2914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang Q, Yu S, Lam MMT, Poon TCW, Sun L,
Jiao Y, Wong AST and Lee LT: Angiotensin II promotes ovarian cancer
spheroid formation and metastasis by upregulation of lipid
desaturation and suppression of endoplasmic reticulum stress. J Exp
Clin Cancer Res. 38:1162019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li SH, Lu HI, Chang AY, Huang WT, Lin WC,
Lee CC, Tien WY, Lan YC, Tsai HT and Chen CH: Angiotensin II type I
receptor (AT1R) is an independent prognosticator of esophageal
squamous cell carcinoma and promotes cells proliferation via mTOR
activation. Oncotarget. 7:67150–67165. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fujihara S, Morishita A, Ogawa K, Tadokoro
T, Chiyo T, Kato K, Kobara H, Mori H, Iwama H and Masaki T: The
angiotensin II type 1 receptor antagonist telmisartan inhibits cell
proliferation and tumor growth of esophageal adenocarcinoma via the
AMPKalpha/mTOR pathway in vitro and in vivo. Oncotarget.
8:8536–8549. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo DF, Sun YL, Hamet P and Inagami T: The
angiotensin II type 1 receptor and receptor-associated proteins.
Cell Res. 11:165–180. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rizzuto R, Pinton P, Ferrari D, Chami M,
Szabadkai G, Magalhães PJ, Di Virgilio F and Pozzan T: Calcium and
apoptosis: Facts and hypotheses. Oncogene. 22:8619–8627. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Giorgi C, Baldassari F, Bononi A, Bonora
M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A,
Suski JM, et al: Mitochondrial Ca(2+) and apoptosis. Cell Calcium.
52:36–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Schulz J, Lubnau E, Grossmann M and Rück
W: Double-blind, randomized study of the anti-anginal and
anti-ischaemic efficacy of fendiline and diltiazem in patients with
coronary heart disease. Curr Med Res Opin. 12:521–539. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bayer R and Mannhold R: Fendiline: A
review of its basic pharmacological and clinical properties.
Pharmatherapeutica. 5:103–136. 1987.PubMed/NCBI
|
|
45
|
Fassbender V, Wegener JW, Shainberg A and
Nawrath H: Inhibition by fendiline of the transient outward current
in rat ventricular cardiomyocytes. J Cardiovasc Pharmacol.
33:905–911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nawrath H, Klein G, Rupp J, Wegener JW and
Shainberg A: Open state block by fendiline of L-type Ca++ channels
in ventricular myocytes from rat heart. J Pharmacol Exp Ther.
285:546–552. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tripathi O, Schreibmayer W and Tritthart
HA: Fendiline inhibits L-type calcium channels in guinea-pig
ventricular myocytes: A whole-cell patch-clamp study. Br J
Pharmacol. 108:865–869. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lin MC and Jan CR: The anti-anginal drug
fendiline elevates cytosolic Ca(2+) in rabbit corneal epithelial
cells. Life Sci. 71:1071–1079. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jan CR, Lee KC, Chou KJ, Cheng JS, Wang
JL, Lo YK, Chang HT, Tang KY, Yu CC and Huang JK: Fendiline, an
anti-anginal drug, increases intracellular Ca2+ in PC3 human
prostate cancer cells. Cancer Chemother Pharmacol. 48:37–41. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Huang C, Huang C, Cheng J, Liu S, Chen I,
Tsai J, Chou C, Tseng P and Jan C: Fendiline-evoked [Ca2+]i rises
and non-Ca2+-triggered cell death in human oral cancer cells. Hum
Exp Toxicol. 28:41–48. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Woods N, Trevino J, Coppola D, Chellappan
S, Yang S and Padmanabhan J: Fendiline inhibits proliferation and
invasion of pancreatic cancer cells by interfering with ADAM10
activation and β-catenin signaling. Oncotarget. 6:35931–35948.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cheng JS, Wang JL, Lo YK, Chou KJ, Lee KC,
Liu CP, Chang HT and Jan CR: Effects of the antianginal drug
fendiline on Ca2+ movement in hepatoma cells. Hum Exp Toxicol.
20:359–364. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang J, Cheng J, Chan R, Tseng L, Chou K,
Tang K, Lee KC, Lo Y, Wang J and Jan C: The anti-anginal drug
fendiline increases intracellular Ca(2+) levels in MG63 human
osteosarcoma cells. Toxicol Lett. 119:227–233. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Alhothali M, Mathew M, Iyer G, Lawrence
HR, Yang S, Chellappan S and Padmanabhan J: Fendiline enhances the
cytotoxic effects of therapeutic agents on PDAC cells by inhibiting
tumor-promoting signaling events: A potential strategy to combat
PDAC. Int J Mol Sci. 20:24232019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Brizzolara A, Garbati P, Vella S,
Calderoni M, Quattrone A, Tonini GP, Capasso M, Longo L, Barbieri
R, Florio T and Pagano A: Co-Administration of fendiline
hydrochloride enhances chemotherapeutic efficacy of cisplatin in
neuroblastoma treatment. Molecules. 25:52342020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
van der Hoeven D, Cho KJ, Ma X,
Chigurupati S, Parton RG and Hancock JF: Fendiline inhibits K-Ras
plasma membrane localization and blocks K-Ras signal transmission.
Mol Cell Biol. 33:237–251. 2013. View Article : Google Scholar :
|