Introduction
Renal cell carcinoma (RCC) accounts for ~2% of all
cancer cases and is characterized by diverse clinical
manifestations, few early warning signs and a resistance to
radiotherapy and chemotherapy (1).
Clear cell RCC accounts for the majority of RCC cases (2) and one-third of the patients present
with metastases at initial diagnosis. Due to the resistance of RCC
to radiotherapy and chemotherapy, the 5-year survival rate for
patients with metastatic RCC is <10% (3). The responsiveness of RCC to treatment
with conventional anticancer agents, such as 5-fluorouracil (5-FU)
and cisplatin (CDDP), was reported to be lower compared to other
types of cancer (4,5). Despite the development of various
chemotherapeutic strategies, RCC remains a challenging tumor
entity. A few patients were reported to exhibit complete or partial
response to frequently used chemotherapeutic agents, such as
gemcitabine, 5-FU, capecitabine and vinblastine (6). As RCC is known to be immunogenic,
several clinical trials investigated the potency of cytokines,
mainly interleukin 2 and/or interferon-α (7,8).
Targeted therapies, including monoclonal antibodies and
small-molecule inhibitors, have significantly modified the
treatment of cancer over the last 10 years through inhibiting
tyrosine kinase activity or vascular endothelial growth factor
receptors (9). However, despite
these novel therapies, the clinical outcome of patients with
metastatic RCC remains poor (6).
Thus, there is a pressing need to establish alternative therapeutic
modalities against RCC.
In our previous study, we reported that the
topoisomerase-I inhibitor camptothecin exhibited toxicity
synergistically with a peroxisome proliferator-activated receptor-γ
(PPARγ) agonist (10).
15-Deoxy-Δ12,14-prostaglandin J2
(15d-PGJ2) is an endogenous carcinostatic agent, whose
nuclear receptor is a PPARγ. PPARγ activation was shown to induce
growth inhibition in human RCC cells (11). Furthermore, 15d-PGJ2 was
also implicated in antiproliferation independently of PPARγ
(12). The antitumor activity of
15d-PGJ2 was also found to be associated with the
inhibition of topoisomerase-II (13). We previously identified novel
binding sites for 15d-PGJ2 on the cell surface (14). With regard to targets for
15d-PGJ2 in the plasma membrane, molecular chaperones,
glycolytic enzymes and cytoskeletal components, such as β-actin,
were also identified (15). PPARγ
agonists were shown to enhance 5-FU-, CDDP- or topoisomerase-II
inhibitor-induced apoptosis in cancer cell types other than RCC
(16–19). The aim of the present study was to
evaluate the therapeutic efficacy of the combination treatment with
15d-PGJ2 and the topoisomerase-II inhibitor etoposide
(VP-16) in RCC.
Materials and methods
Cell lines and cell culture
The Caki-2 human RCC cell line was obtained from
Summit Pharmaceuticals International (Tokyo, Japan). The Caki-2
cells were routinely cultured in RPMI-1640 medium supplemented with
10% fetal bovine serum, 50 mg/l penicillin G and 50 mg/l
streptomycin (Invitrogen, Tokyo, Japan), at 37°C in a 5%
CO2 atmosphere.
Reagents
15d-PGJ2 was obtained from Cayman
Chemicals (Ann Arbor, MI; Cabru, Milan, Italy). Etoposide (VP-16)
was purchased from Wako Pure Chemical Industries, Ltd. (Osaka,
Japan). GW9662 was obtained from Sigma-Aldrich (St. Louis, MO,
USA); and MTT was purchased from Dojindo Laboratories (Kumamoto,
Japan).
Cell viability analysis
To evaluate the effects of 15d-PGJ2 and
VP-16, alone or in combination, on the growth of Caki-2 cells, cell
viability was determined by the MTT assay. The cells were seeded on
a 96-well tissue culture plate at 10,000 cells/cm2 and
incubated for 24 h prior to drug exposure. The cells were incubated
with 15d-PGJ2 and VP-16 at increasing concentrations (0,
10, 20, 30, 40 and 50 μM of 15d-PGJ2; and 0, 10, 20, 30,
40, 50, 60 and 70 μM of VP-16) for 24 h. After 24 h, the cells were
incubated with MTT solution (0.1 mg/ml in phosphate-buffered
saline) for an additional 3 h at 37°C. The MTT solution was then
aspirated off. To dissolve the formazan crystals formed in viable
cells, 100 μl dimethyl sulfoxide was added to each well. Absorbance
was measured at 570 nm using a spectrophotometer (iMark Microplate
Reader, Bio-Rad Laboratories, Hercules, CA, USA).
Detection of chromatin condensation
(fluorescence microscopy)
For nuclear staining, the cells were treated with
15d-PGJ2 and VP-16 for 24 h. Immediately following
treatment, the nuclear chromatin of trypsinized cells was stained
with 80 μg/ml Hoechst 33342 (Nacalai Tesque, Kyoto, Japan) for 15
min at room temperature. The cells were then observed under a
brightfield fluorescent microscope (Vanox; Olympus, Tokyo, Japan)
under UV excitation. The percentage of chromatin-condensed cells
was determined by counting >100 cells in each experiment.
Fluorimetric assay of caspase-3
activity
Caspase-3 activity was assessed using a Caspase-3
Fluorimetric Assay kit, (Sigma-Aldrich), according to the
manufacturer’s instructions. Briefly, the cells were seeded into
96-well plates at a density of 10,000 cells/cm2 and
incubated with 15d-PGJ2 and VP-16 for 24 h. After
exposure to the drugs for 24 h, the supernatants were aspirated and
the cells were harvested with lysis buffer [50 mM HEPES (pH 7.4), 5
mM CHAPS and 5 mM DTT]. The reaction buffer, including
acetyl-Asp-Glu-Val-Asp-7-amido4-methylcoumarin (Ac-DEVD-AMC), a
caspase-3 specific substrate, was added to the wells and the
production of AMC was sequentially detected with a
CytoFluor® Plate reader (MTX Lab Systems, Vienna, VA,
USA) at an excitation wavelength of 360 nm and at an emission
wavelength of 460 nm. The enzyme activities were determined as
initial velocities expressed as nmol AMC/min/ml and were then
corrected by the quantity of protein in each well detected by
bicinchoninic acid protein assays (Thermo Fisher Scientific,
Waltham, MA, USA).
Statistical analysis
Data were statistically analyzed with the Student’s
t-test for comparison with the control group and are expressed as
means ± SEM. Data on various drugs were statistically analyzed by
one-way analysis of variance, followed by Scheffe’s F test for
comparison between the groups.
Results
VP-16 enhanced the antiproliferative
effects of 15d-PGJ2 in Caki-2 cells
RCCs are chemoresistant to conventional anticancer
agents (3), but are sensitive to
the endogenous anticancer agent 15d-PGJ2 (20). It was previously confirmed that
15d-PGJ2 induced apoptosis in RCCs (10,11,20,21).
Therefore, we investigated the cytotoxic effects of
15d-PGJ2 in the Caki-2 human RCC cell line with the MTT
assay. Following incubation with 15d-PGJ2 for 24 h, we
observed that the viability of Caki-2 cells was significantly
reduced in a dose-dependent manner (from 30 to 50 μM; P<0.01;
Fig. 1A). By contrast, incubation
with VP-16 alone (10–70 μM) for 24 h did not affect the viability
of Caki-2 cells (Fig. 1B).
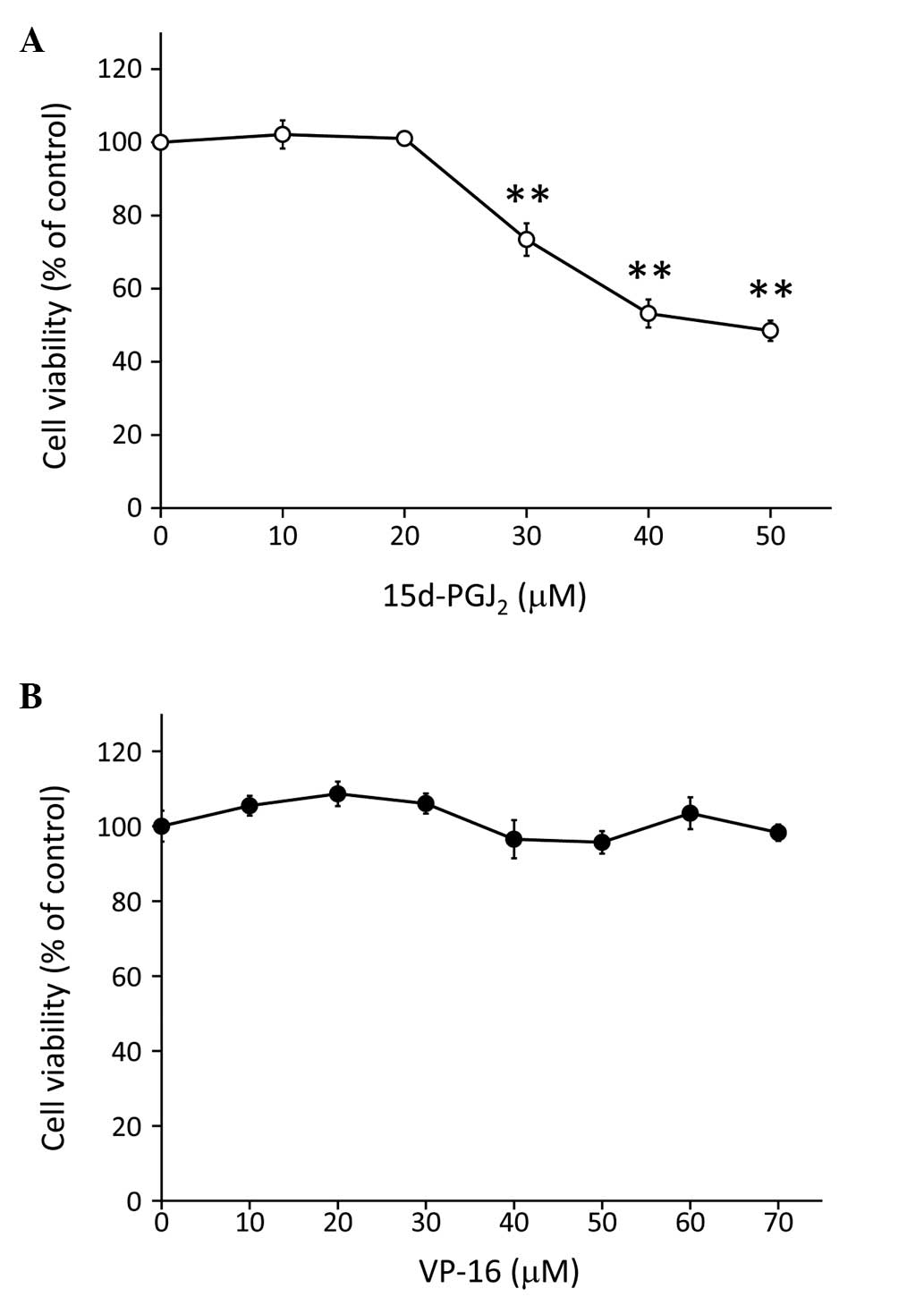 | Figure 1Treatment with 15d-PGJ2
inhibited the proliferation of Caki-2 cells, but treatment with
VP-16 alone did not affect cell viability. The cells were assayed
for viability using the MTT assay after treatment for 24 h with
increasing doses of (A) 15d-PGJ2 (0, 10, 20, 30, 40 and
50 μM) and (B) VP-16 (0, 10, 20, 30, 40, 50, 60 and 70 μM).
**P<0.01 vs. control cells. Data are expressed as
means ± standard error of the mean of three independent
experiments. 15d-PGJ2,
15-deoxy-Δ12,14-prostaglandin J2; VP-16,
etoposide. |
To investigate the synergistic cytotoxicity of VP-16
and 15d-PGJ2, we exposed Caki-2 cells to VP-16 and
15d-PGJ2 combination treatment (Fig. 2). The Caki-2 cells were co-treated
with VP-16 (10–70 μM) and 15d-PGJ2 (20 μM)
simultaneously and we observed that cell viability was
significantly lower compared to that of cells treated with
15d-PGJ2 alone (Fig.
2A). Furthermore, the phase contrast microscopy analysis
indicated that the combination of VP-16 and 15d-PGJ2
induced more prominent morphological changes compared to
15d-PGJ2 alone (Fig.
2B). Caki-2 cell cultures treated with either VP-16 or
15d-PGJ2 exhibited marginally broadened cells, whereas
cells treated with VP-16 and 15d-PGJ2 exhibited
significant atrophy. As previously reported, topoisomerase-II
activity is inhibited by 15d-PGJ2 (13). Thus, the synergistic antitumor
activity of 15d-PGJ2 and VP-16 may be mediated via the
topoisomerase-II inhibition pathway.
 | Figure 2Treatment with VP-16 enhanced the
antiproliferative effect of 15d-PGJ2 in Caki-2 cells.
(A) The cells were assayed for viability using MTT following
treatment with VP-16 (0, 10, 20, 30, 40, 50, 60 and 70 μM) (open
circles) and combination treatment with VP-16 and
15d-PGJ2 (20 μM) (closed triangles) for 24 h. The
results are expressed as the means ± SEM of three independent
experiments. **P<0.01, vs. control cells. (B) The
combination of 15d-PGJ2 and VP-16 induced morphological
changes in Caki-2 cells. The cells were treated with
15d-PGJ2 alone (20 μM), VP-16 alone (70 μM) and the
combination of the two. Caki-2 cells were then examined by phase
contrast microscopy following 24 h of incubation. Scale bar, 100
μm. 15d-PGJ2, 15-deoxy-Δ12,14-prostaglandin
J2; VP-16, etoposide. |
15d-PGJ2 enhanced
VP-16-induced apoptosis via the activation of caspase-3
To elucidate whether the inhibition of cell
proliferation induced by the combined treatment of VP-16 and
15d-PGJ2 is associated with apoptosis, we assessed
nuclear chromatin condensation in Caki-2 cells treated with VP-16
(70 μM) and/or 15d-PGJ2 (20 μM) (Fig. 3A). Treatment with either VP-16 or
15d-PGJ2 exhibited a tendency to increase chromatin
condensation, whereas a combination of the two was found to
strongly induce chromatin condensation (P<0.05). We then
assessed caspase-3 activity in Caki-2 cells treated with VP-16 (70
μM) and/or 15d-PGJ2 (20 μM) (Fig. 3B). Cells treated with either VP-16
or 15d-PGJ2 exhibited a tendency to activate caspase-3,
whereas the combination of the two significantly induced caspase-3
activation (P<0.05). These results suggested that
15d-PGJ2 enhanced VP-16-induced apoptosis via the
activation of caspase-3. Topoisomerase-II inhibitor-induced
apoptosis was shown to be mediated by caspase-3 (22). Thus, the synergistic inhibition of
topoisomerase-II by the combination of 15d-PGJ2 and
VP-16 may induce caspase-3 activation.
15d-PGJ2 enhanced the
antitumor activity of VP-16 independently of PPARγ
It was previously reported that 15d-PGJ2
treatment inhibits cell proliferation in several types of cancer
cells via PPARγ (23–26). In addition, topoisomerase-II
inhibitors enhance the cytotoxicity of 15d-PGJ2 in RCC.
However, whether the inhibition of topoisomerase-II and the
activation of PPARγ result in synergistic toxicity, has not been
fully elucidated. To determine whether 15d-PGJ2 enhanced
the antitumor activity of VP-16 via PPARγ activation, Caki-2 cells
were co-treated with VP-16 (70 μM), 15d-PGJ2 (20 μM) and
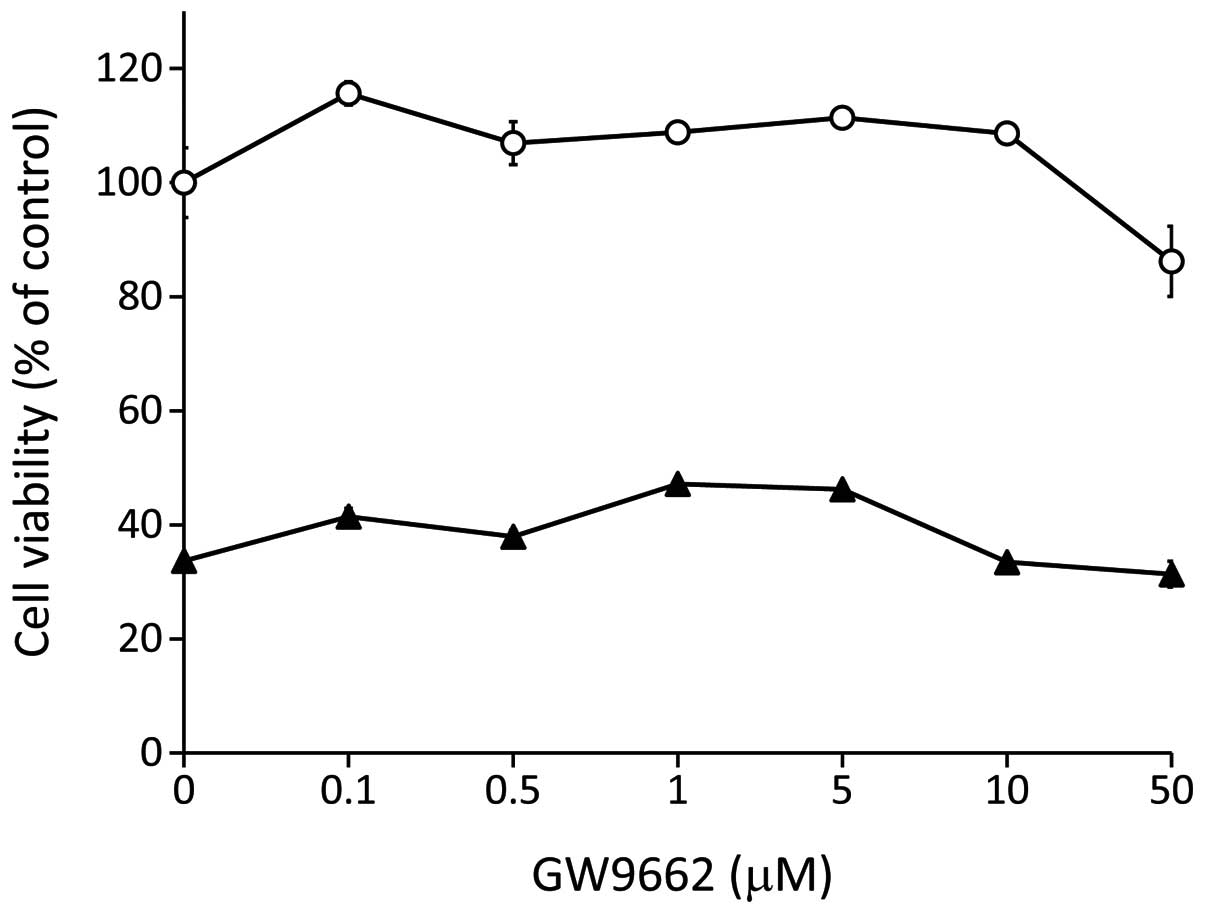
the PPARγ inhibitor, GW9662 (0.1–50 μM) (Fig. 4). Our results demonstrated that the
synergistic cytotoxic effects of VP-16 and 15d-PGJ2
combination treatment were not decreased by PPARγ inhibition,
suggesting that 15d-PGJ2 enhanced the antitumor activity
of VP-16 independently of PPARγ. 15d-PGJ2 was also
reported to induce apoptosis via the activation of caspase-3
independently of PPARγ (26).
Discussion
In the present study, we demonstrated that the
topoisomerase-II inhibitor, VP-16, enhanced the cytotoxicity of
15d-PGJ2 in RCC. Moreover, the PPARγ antagonist, GW9662,
did not protect Caki-2 cells against 15d-PGJ2-induced
cytotoxicity. These findings suggested that 15d-PGJ2
exhibited synergistic antitumor activity with VP-16 independently
of the PPARγ pathway.
RCCs are chemoresistant to conventional anticancer
agents (3), but are sensitive to
the endogenous anticancer agent 15d-PGJ2 (20). Several previous studies confirmed
that 15d-PGJ2 induced apoptosis in RCCs (11,20,21).
Additionally, the responsiveness of RCC cells to treatment with
5-FU and CDDP was found to be lower compared to that of other types
of cancer cells (4,5), whereas cancer cells other than RCC
cells were found to be sensitive to conventional anticancer agents
when co-treated with 15d-PGJ2 (27).
Topoisomerase-II inhibitors enhance the cytotoxicity
of 15d-PGJ2 in RCC. However, whether the inhibition of
topoisomerase-II and the activation of PPARγ synergistically
produce toxicity, has not been fully elucidated. The PPARγ
antagonist, GW9662, did not affect the responsiveness of RCC to the
combined treatment with 15d-PGJ2 and VP-16, suggesting
that 15d-PGJ2 exhibited synergistic antitumor activity
with VP-16 independently of PPARγ. Topoisomerase-II was shown to be
inhibited by 15d-PGJ2 (13). Thus, 15d-PGJ2 may
exhibit synergistic antitumor activity with VP-16 via the
inhibition of topoisomerase-II. Topoisomerase-II introduces
double-strand breaks in DNA, which may subsequently be converted
into chromosomal damage following chromatin condensation (28). In this study, increased chromatin
condensation was observed following 15d-PGJ2 and VP-16
combination treatment for RCC. However, chromatin condensation was
not significantly increased following treatment with
15d-PGJ2 alone. Furthermore, 15d-PGJ2
treatment induced marked morphological changes in Caki-2 cells,
whereas treatment with VP-16 alone did not affect cell morphology.
Cytoskeletal proteins are responsible for maintaining cell
morphology. The effects of 15d-PGJ2 on the organization
of the actin cytoskeleton were shown to be mediated by a direct
covalent modification of proteins through electrophilic
cyclopentenone binding (15,29).
It has been hypothesized that VP-16 and 15d-PGJ2 induce
chromatin condensation and morphological changes via the inhibition
of topoisomerase-II and the disruption of the actin cytoskeleton,
respectively.
It was reported that 15d-PGJ2 may induce
apoptosis via the activation of caspase-3 independently of PPARγ
(26). Topoisomerase-II
inhibitor-induced apoptosis is also mediated by caspase-3 (30). In the present study,
15d-PGJ2 and VP-16 increased caspase-3 activity, both
individually and synergistically.
In conclusion, we demonstrated that
15d-PGJ2 and VP-16 synergistically inhibited the
proliferation of RCC independently of the PPARγ pathway.
Furthermore, 15d-PGJ2 enhanced VP-16-induced apoptosis.
We hypothesized that 15d-PGJ2 induced changes in cell
morphology independent of the PPARγ pathway and that VP-16 induced
chromatin condensation via topoisomerase-II inhibition; thus, the
combination of 15d-PGJ2 and VP-16 exerted synergistic
anticancer effects involving caspase-3 activation. Our results
suggested that the 15d-PGJ2 and VP-16 combination
treatment may be a novel chemotherapeutic option for the treatment
of RCC.
Acknowledgements
This study was supported by a Grants-in-Aid for
Scientific Research from the Japan Society for the Promotion of
Science (grant no. 25860072). The authors would like to thank
Tsutomu Minami, Daichi Yamaki, Yoshiya Kobayasi, Akihiro Yamada and
Tomonori Nakagawa from the Hyogo Prefectural Kobe High School for
supporting this study.
References
|
1
|
Motzer RJ, Bander NH and Nanus DM:
Renal-cell carcinoma. N Engl J Med. 335:865–875. 1996. View Article : Google Scholar
|
|
2
|
Costa LJ and Drabkin HA: Renal cell
carcinoma: new developments in molecular biology and potential for
targeted therapies. Oncologist. 12:1404–1415. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mickisch GH: Gene therapy on renal-cell
carcinoma: magic bullet or tragic insanity? World J Urol.
13:178–185. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen XL, Cao LQ, She MR, Wang Q, Huang XH
and Fu XH: Gli-1 siRNA induced apoptosis in Huh7 cells. World J
Gastroenterol. 14:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jin KL, Park JY, Noh EJ, et al: The effect
of combined treatment with cisplatin and histone deacetylase
inhibitors on HeLa cells. J Gynecol Oncol. 21:262–268. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Motzer RJ and Russo P: Systemic therapy
for renal cell carcinoma. J Urol. 163:408–417. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aso Y, Tazaki H, Umeda T and Marumo K:
Treatment of renal cell carcinoma with systemic administration of
intermediate doses of recombinant human interleukin-2 alone.
Recombinant Human Interleukin-2 (S-6820) Research Group on Renal
Cell Carcinoma. Prog Clin Biol Res. 303:681–688. 1989.
|
|
8
|
Hayashi T, Miyagawa Y, Tsujimura A,
Nonomura N, Minami M and Okuyama A: A case of renal cell carcinoma
with multiple lung metastases refractory to interferon-α showing
complete remission by interleukin-2 monotherapy. Int J Urol.
13:805–808. 2006.PubMed/NCBI
|
|
9
|
Logan JE, Rampersaud EN, Sonn GA, et al:
Systemic therapy for metastatic renal cell carcinoma: a review and
update. Rev Urol. 14:65–78. 2012.PubMed/NCBI
|
|
10
|
Yamamoto Y, Fujita M, Koma H, et al:
15-Deoxy-Δ12,14-prostaglandin J2enhanced the
anti-tumor activity of camptothecin against renal cell carcinoma
independently of topoisomerase-II and PPARγ pathways. Biochem
Biophys Res Commun. 410:563–567. 2011.
|
|
11
|
Inoue K, Kawahito Y, Tsubouchi Y, et al:
Expression of peroxisome proliferator-activated receptor gamma in
renal cell carcinoma and growth inhibition by its agonists. Biochem
Biophys Res Commun. 287:727–732. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ward JE, Gould H, Harris T, Bonacci JV and
Stewart AG: PPARγ ligands, 15-deoxy-Δ12,14-prostaglandin
J2and rosiglitazone regulate human cultured airway
smooth muscle proliferation through different mechanisms. Br J
Pharmacol. 141:517–525. 2004.
|
|
13
|
Suzuki K and Uyeda M: Inhibitory
properties of antitumor prostaglandins against topoisomerases.
Biosci Biotechnol Biochem. 66:1706–1712. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yagami T, Ueda K, Asakura K, et al: Novel
binding sites of 15-deoxy-Δ12,14-prostaglandin
J2in plasma membranes from primary rat cortical neurons.
Exp Cell Res. 291:212–227. 2003.
|
|
15
|
Yamamoto Y, Takase K, Kishino J, et al:
Proteomic identification of protein targets for
15-deoxy-Δ12,14-prostaglandin J2in neuronal
plasma membrane. PLoS One. 6:e175522011.
|
|
16
|
Zhang YQ, Tang XQ, Sun L, et al:
Rosiglitazone enhances fluorouracil-induced apoptosis of HT-29
cells by activating peroxisome proliferator-activated receptor γ.
World J Gastroenterol. 13:1534–1540. 2007.PubMed/NCBI
|
|
17
|
Hamaguchi N, Hamada H, Miyoshi S, et al:
In vitro and in vivo therapeutic efficacy of the PPARγ agonist
troglitazone in combination with cisplatin against malignant
pleural mesothelioma cell growth. Cancer Sci. 101:1955–1964.
2010.
|
|
18
|
Tikoo K, Kumar P and Gupta J:
Rosiglitazone synergizes anticancer activity of cisplatin and
reduces its nephrotoxicity in 7,12-dimethyl benz{a}anthracene
(DMBA) induced breast cancer rats. BMC Cancer. 9:1072009.PubMed/NCBI
|
|
19
|
Kanbe E, Abe A, Towatari M, Kawabe T,
Saito H and Emi N: DR1-like element in human topoisomerase IIα gene
involved in enhancement of etoposide-induced apoptosis by PPARγ
ligand. Exp Hematol. 31:300–308. 2003.PubMed/NCBI
|
|
20
|
Yuan J, Takahashi A, Masumori N, et al:
Ligands for peroxisome proliferator-activated receptor gamma have
potent antitumor effect against human renal cell carcinoma.
Urology. 65:594–599. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoshimura R, Matsuyama M, Hase T, et al:
The effect of peroxisome proliferator-activated receptor-γ ligand
on urological cancer cells. Int J Mol Med. 12:861–865. 2003.
|
|
22
|
Khelifa T and Beck WT: Merbarone, a
catalytic inhibitor of DNA topoisomerase II, induces apoptosis in
CEM cells through activation of ICE/CED-3-like protease. Mol
Pharmacol. 55:548–556. 1999.PubMed/NCBI
|
|
23
|
Brockman JA, Gupta RA and Dubois RN:
Activation of PPARγ leads to inhibition of anchorage-independent
growth of human colorectal cancer cells. Gastroenterology.
115:1049–1055. 1998.
|
|
24
|
Chen YX, Zhong XY, Qin YF, Bing W and He
LZ: 15d-PGJ2inhibits cell growth and induces apoptosis
of MCG-803 human gastric cancer cell line. World J Gastroenterol.
9:2149–2153. 2003.
|
|
25
|
Clay CE, Atsumi GI, High KP and Chilton
FH: Early de novo gene expression is required for
15-deoxy-Δ12,14-prostaglandin J2-induced
apoptosis in breast cancer cells. J Biol Chem. 276:47131–47135.
2001.
|
|
26
|
Li L, Tao J, Davaille J, et al:
15-deoxy-Δ12,14-prostaglandin J2induces
apoptosis of human hepatic myofibroblasts. A pathway involving
oxidative stress independently of peroxisome-proliferator-activated
receptors. J Biol Chem. 276:38152–38158. 2001.
|
|
27
|
McClay EF, Winski PJ, Jones JA, Jennerette
J III and Gattoni-Celli S:
Δ12-Prostaglandin-J2is cytotoxic in human
malignancies and synergizes with both cisplatin and radiation.
Cancer Res. 56:3866–3869. 1996.
|
|
28
|
Huang X, Okafuji M, Traganos F, Luther E,
Holden E and Darzynkiewicz Z: Assessment of histone H2AX
phosphorylation induced by DNA topoisomerase I and II inhibitors
topotecan and mitoxantrone and by the DNA cross-linking agent
cisplatin. Cytometry A. 58:99–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aldini G, Carini M, Vistoli G, et al:
Identification of actin as a
15-deoxy-Δ12,14-prostaglandin J2target in
neuroblastoma cells: mass spectrometric, computational, and
functional approaches to investigate the effect on cytoskeletal
derangement. Biochemistry. 46:2707–2718. 2007.
|
|
30
|
Mizutani H, Tada-Oikawa S, Hiraku Y,
Oikawa S, Kojima M and Kawanishi S: Mechanism of apoptosis induced
by a new topoisomerase inhibitor through the generation of hydrogen
peroxide. J Biol Chem. 277:30684–30689. 2002. View Article : Google Scholar
|