Introduction
Melanoma arises from the transformation of the
pigment-producing melanocytes of the skin and is often initiated by
deregulation of the MAPK pathway, which is known to modulate
melanoma cell survival (1,2). According to the National Cancer
Institute (NCI), melanoma remains the most fatal type of skin
cancer and is the fifth most commonly occurring cancer in men and
the seventh in women. In 2011, it was estimated that over 70,000
individuals would be diagnosed with melanoma and approximately
9,000 individuals would succumb to the disease in the USA (3). Surgery remains the standard treatment
for localized melanoma skin lesions, whereas the standard of care
for metastatic melanoma remains a single chemotherapeutic agent,
known as dacarbazine, which has a response rate of 10–15% in
patients and is not associated with long-lasting responses, since
the median survival rate is approximately nine months (4,5).
This lack of effective chemotherapeutic compounds has led
researchers to focus on the development of immunotherapeutic
methods to treat melanoma.
Immunotherapeutic melanoma-treatment strategies have
been investigated over the past 30 years and resulted in various
clinical trials (6). Immunotherapy
has focused on four major areas: immuno-activating cytokines,
immuno-modulating antibodies, adoptive T-cell therapy and vaccines.
The immuno-activating cytokine, interleukin-2 (IL-2), is one of
three FDA-approved compounds used to treat melanoma; it is,
however, associated with high toxicity (7). The FDA has recently approved an
immunotherapeutic compound that targets melanoma, known as
ipilimumab (anti-CTLA-4 antibody) (8), which has shown promising results in
clinical trials. In addition, although it is not an
immunotherapeutic compound, it should be noted that the FDA has
approved a small molecule inhibitor, known as vemurafenib (BRAF
inhibitor) (9), which has also
shown promising results in clinical trials. Although there are
ongoing clinical trials aimed at assessing the effectiveness of
various immunotherapeutic compounds, further research on melanoma
vaccines is required, since melanoma is one of the most immunogenic
types of cancer and several clinically relevant melanoma-associated
antigens (MAAs) have already been identified (10–13).
The possibilities for developing antigen-based
vaccines against melanoma are numerous, as multiple MAAs have been
identified since the 1970s, cloned (14,15)
and well-characterized, in vivo and in vitro, with
respect to their antigenicity. Despite their characterization, not
all antigens elicit an optimal immune response, which may be
attributed to the fact that the selected antigens were not
HLA-typed to match the vaccinated individuals or that an adjuvant
is required to help initiate an antigen-specific immune response.
Examples of antigens used in vaccines against melanoma are MAAs
such as gp100, tyrosinase, MAGE-3 and MART-1 (16–18);
however, phase I and II studies have not shown promising results
when administered as a single antigen/protein-containing vaccine
(19,20). Other clinical trials have focused
on the generation of vaccines that incorporate one or more
antigens, combined with the administration of an immuno-activating
cytokine, such as IL-2 or granulocyte-macrophage colony-stimulating
factor (GM-CSF) (19,21,22).
One such clinical trial recently showed a high response rate and
progression-free survival in patients with advanced melanoma who
were vaccinated with gp100 and IL-2 (23). Melanoma vaccines using a
multivalent antigen approach appear promising, although further
studies should be performed to determine the conditions that are
likely to foster a productive immune response, such as selecting an
adjuvant with optimal immuno-stimulatory properties.
Previously, we developed a melanoma multivalent
vaccine using live vaccinia virus-augmented melanoma cell lysates,
in order to generate the vaccinia melanoma oncolysate (VMO), or
first generation melanoma vaccine. The VMO is composed of four
established allogeneic melanoma cell lines, providing a variety of
MAAs that help generate a robust immune response, using vaccinia
virus as an adjuvant (24,25). Vaccinia virus was incorporated as
an adjuvant in VMO since i) it modifies and re-expresses host
membrane-associated melanoma antigens, thus making these antigens
more recognizable to the immune system of the host (26) and ii) it non-specifically induces
tumor-specific cytotoxic T lymphocytes (CTLs), thus initiating the
host immune response (27).
The efficacy of VMO in the treatment of melanoma was
studied in several preliminary trials (24,28),
followed by the Southeastern Cancer Study Group-sponsored phase I
and II trials (29,30). Given the favorable results of these
trials, we proceeded with the first randomized, prospective,
double-blind, multi-institutional, pharma-produced, FDA-approved,
NCI-funded melanoma vaccine phase III trial (1988–1991), in order
to evaluate the effects of VMO on disease-free interval (DFI) and
overall survival (OS) in patients with stage II melanoma (based on
the International Union Against Cancer criteria). Although the
results of the first interim analysis (performed in May 1994)
showed no statistical difference in OS between patients receiving
VMO and those receiving the control vaccinia virus vaccine (V),
there was a 10% increased survival advantage in favor of VMO.
Moreover, although the number of patients that were enrolled in
this trial (n=250) was not sufficient to generate statistically
significant results in specific subset analyses, the final analysis
demonstrated increased OS in VMO-treated patients, specifically in
i) VMO-treated males, aged 44–57 years, with one to five positive
nodes and ii) VMO-treated males and females who had undergone
prophylactic lymph node dissection which demonstrated positive
nodes (clinical stage I and pathological stage II disease)
(31). A second interim analysis
(performed in May 1995) demonstrated no difference in survival
between VMO-and V-treated groups; however, further analysis
revealed i) a subset of males aged 44–57 years, with one to five
positive nodes, treated with VMO, that had a statistically
significant OS compared to those treated with the placebo V
(p=0.037) and ii) patients with clinical stage I disease, treated
with VMO, that had a statistically significant OS compared to those
treated with the placebo V (p=0.05) (32). The third analysis of this phase III
trial (performed in May 1996) showed no significant difference in
either DFI or OS in patients treated with VMO, compared to those
treated with V; however, the same subset of males, aged 44–57
years, with one to five positive nodes, treated with VMO, showed a
statistically significant improvement in survival at two-, three-
and five-year intervals (p=0.046).
Based on the results and trends observed in our
clinical trial, we report on a 10-year follow-up analysis on the
patients originally involved in the phase III trial. We have now
compiled follow-up data on the surviving patients originally
enrolled in the phase III trial and present data that reflect upon
the validity and efficacy of the trial design originally
envisioned.
Materials and methods
Patients
As previously described (31–33),
250 patients were accrued from 11 participating institutions, from
June 1988 to January 1991. These institutions are listed as
follows: MD Anderson Cancer Center, Houston, TX; University of
Alabama, Birmingham, AL; Emory University, Atlanta, GA; University
of Pennsylvania, Philadelphia, PA; Wayne State University, Detroit,
MI; University of Florida, Gainesville, FL; University of Chicago,
Chicago, IL; University of Colorado, Denver, CO; Duke University,
Durham, NC; Brown University, Providence, RI; and Mount Sinai
Medical Center, Miami Beach, FL, USA. Patients were selected
according to strict eligibility criteria, which included
histologically-positive nodes, age range 15–70 years, Karnofsky
performance status <70% and no concomitant malignancy, apart
from basal and squamous cell carcinoma of the skin. Originally,
patients with any number of positive nodes were eligible for the
trial during the first year (June 1988–June 1989). Subsequently,
only patients with one to five positive nodes were accepted, since
the protocol advisory panel (including Dr Charles Balch,
statistician; Dr Al Bartolucci, medical advisor; and Dr Marc
Wallack, principal investigator) considered more than five positive
nodes to represent excessive tumor burden. Further criteria for
ineligibility included patients with: lentigo maligna melanoma;
satellite lesions at a distance >2 cm from the primary lesion;
previous chemotherapy, radiotherapy, or immunotherapy; matted nodes
(except during the first year of the trial); more than one primary
lesion; and pregnancy. Patients were enrolled in the study eight
weeks after surgery.
Informed consent was obtained from all patients
enrolled in our study and the procedures followed were in
accordance with the ethical standards of the responsible committee
on human experimentation and with the Helsinki Declaration of 1975,
as revised in 1983.
VMO, V and attenuated smallpox
preparation
As previously described (31–33),
the VMO vaccine for the treatment arm, the V vaccine for the
control arm and the smallpox vaccine for both arms (used as an
immune booster), were prepared by the Mérieux Institute, Lyon,
France. The preparation of the VMO has been extensively described
in our previous publication (31).
Essentially, four allogeneic cell lines (Mel-2, Mel-3, Mel-4 and
Mel-B) were established from four patients treated in our clinic.
These cell lines were assessed using standard assays to confirm the
absence of bacteria, fungi, mycoplasma and all human viruses
(including human immunodeficiency virus).
The V vaccine was prepared by infecting the MRC5
human diploid cell line with a seed vaccinia virus that was
developed in this laboratory, using smallpox vaccine ‘Dryvax’
(Wyeth Laboratories, Inc., Marietta, PA, USA), containing the New
York City Board of Health strain of vaccinia virus. Melanoma cells
were infected with the vaccinia virus at a ratio of 1 cell to 10
TCID50 (50% tissue culture infectious dose) of vaccinia
virus, then incubated overnight at 37°C. Virus-infected cells were
lysed by sonication and a nucleus-free cell lysate was obtained by
centrifugation to create the final product, VMO.
Study enrollment
Surgical review of the wide excision and regional
lymphadenectomy was performed for each patient prior to
registration. Pathological review of the biopsy specimens was
performed by the study pathologist to establish the diagnosis of
stage III melanoma (based on 1988 AJCC criteria). All the patients
had complete physicals and laboratory work-up, including complete
blood count, liver function tests and urinalysis, as well as
baseline chest X-ray. Each patient signed an informed consent form,
that was approved by the respective institution. Patients were then
randomized between the VMO and V treatment groups by the
Statistical Center at the University of Alabama.
VMO, V and attenuated smallpox vaccine
administration
The administration of the respective vaccines has
been described in detail in previous publications (31–33).
Briefly, each patient was given a smallpox booster injection
subcutaneously ≥48 h prior to the initiation of treatment. The VMO
or V was injected intradermally into the sites in proximity to the
regional lymph node groups, excluding the sites of node
dissections. One dose of VMO or V was divided into four to six
aliquots, with two to four aliquots injected above the waist in the
infraclavicular region bilaterally and two aliquots injected below
the waist in the infrainguinal region bilaterally. These injections
were given once a week for 13 weeks, then once every two weeks for
one year or until recurrence. These patients were closely monitored
throughout the trial for signs and symptoms of toxicity, as well as
recurrence.
Statistical analysis
For the follow-up analysis, the Social Security
Death Index was accessed to obtain additional information on the
patients. Additionally, each institution involved in the original
trial was contacted regarding the status of these patients, as well
as any additional therapies these patients may have received after
VMO. Statistical analysis was performed by the Statistical Center
at the Department of Biostatistics, University of Alabama, using
log-rank statistics. Patient data were analyzed using 2002 AJCC
staging criteria.
Results
Patient characteristics and survival
analysis
Records were available for 109 out of the 111
patients evaluated. Patient characteristics of the VMO group and
the V group are presented in Table
I. In April 2008, 78 patients were still alive in both VMO and
V groups, with 35 surviving patients (45%) in the VMO group and 43
(55%) in the V group. Thirty-one patients (19 patients in the VMO
group and 12 in the V group) had succumbed in the time period
between this analysis and the third analysis (performed in May
1996).
 | Table IPatient physical data comparing VMO
and V. |
Table I
Patient physical data comparing VMO
and V.
| VMO | V |
|---|
| Number of
patients | 54 | 55 |
| Number of patients
alive (April 2008) | 35 | 43 |
| Gender | 35 | 19 |
| Male | 35 | 31 |
| Female | 19 | 24 |
| Primary tumor
site | | |
| Head/neck | 4 | 3 |
| Upper
extremity | 13 | 11 |
| Trunk | 21 | 28 |
| Lower
extremity | 8 | 8 |
| Unknown | 8 | 3 |
| Type of
melanoma | | |
| Nodular | 14 | 13 |
| Radial | 23 | 25 |
| Other (incl.
lentigo maligna) | 7 | 4 |
| Unknown | 10 | 13 |
| Ulceration | | |
| Absent | 34 | 33 |
| Present | 12 | 18 |
| Unknown | 8 | 4 |
| Location of lymph
nodes | | |
| Axillary | 37 | 38 |
| Cervical | 7 | 3 |
| Inguinal | 10 | 14 |
| Nodal status | | |
| Macroscopic | 40 | 38 |
| Microscopic | 14 | 17 |
| Clinical stage
(based on 2002 AJCC criteria) | | |
| IIIA | 10 | 7 |
| IIIB | 22 | 33 |
| IIIC | 22 | 15 |
| Disease status | | |
| NED | 41 | 44 |
| REC | 13 | 11 |
The VMO and V groups were further subdivided into
alive and deceased patient subsets (VMOalive,
VMOdeceased, Valive and Vdeceased)
and the demographics of these respective subsets are presented in
Table II. There was no significant
difference in distribution of age (p=0.3286), number of nodes
(p=0.6855), time (p=0.5525), or size of primary lesion (p=0.4008)
among VMOalive, VMOdeceased,
Valive and Vdeceased groups. Subsequent
subset analysis revealed a statistically significant survival
advantage in females, regardless of whether they were administered
VMO or V (p=0.047). However, there was no statistically significant
association between type of melanoma (p=0.9311), ulceration
(p=0.3798), node location (p=0.8303), disease status (p=0.0521),
stage (p=0.1198), location of primary lesion (p=0.9295) or age
(specifically, males aged 44–57 years) with survivability among the
VMOalive, VMOdeceased, Valive and
Vdeceased groups.
| Table IIPatient physical data for VMO and V
alive, compared to VMO and V deceased patient subsets. |
Table II
Patient physical data for VMO and V
alive, compared to VMO and V deceased patient subsets.
|
VMOalive |
VMOdeceased |
Valive |
Vdeceased |
|---|
| Number of
patients | 35 | 19 | 43 | 12 |
| Gender | | | | |
| Male | 20 | 15 | 21 | 10 |
| Female | 15 | 4 | 22 | 2 |
| Primary tumor
site | | | | |
| Head/neck | 2 | 2 | 2 | 1 |
| Upper
extremity | 6 | 2 | 6 | 2 |
| Trunk | 13 | 8 | 13 | 7 |
| Lower
extremity | 5 | 3 | 5 | 0 |
| Unknown | 9 | 4 | 11 | 2 |
| Ulceration | | | | |
| Absent | 23 | 11 | 23 | 10 |
| Present | 7 | 5 | 16 | 2 |
| Unknown | 5 | 3 | 4 | 0 |
| Location of lymph
nodes | | | | |
| Axillary | 23 | 14 | 30 | 8 |
| Cervical | 5 | 2 | 2 | 1 |
| Inguinal | 7 | 3 | 11 | 3 |
| Nodal status | | | | |
| Macroscopic | 28 | 12 | 33 | 5 |
| Microscopic | 7 | 7 | 10 | 7 |
| Clinical stage
(based on 2002 AJCC criteria) | | | | |
| IIIA | 6 | 4 | 3 | 4 |
| IIIB | 14 | 8 | 26 | 7 |
| IIIC | 15 | 7 | 14 | 1 |
| Disease status | | | | |
| NED | 31 | 10 | 37 | 7 |
| REC | 4 | 9 | 6 | 5 |
| Age | | | | |
| Males 44–57
years | 7 | 5 | 2 | 3 |
| Other | 28 | 14 | 41 | 9 |
Additional subset analysis, evaluating melanoma
stage and disease spread, demonstrated a statistically significant
association between melanoma stage and nodal status (i.e.,
microscopic vs. macroscopic disease) (p=0.0001), with a higher
incidence of microscopic disease in stage IIIA patients and a
higher incidence of macroscopic disease in stage IIIC patients.
However, there was no statistically significant association between
melanoma stage and node location (p=0.8641), thus demonstrating
that the location of the melanoma-positive lymph node basins had no
impact on the OS of melanoma.
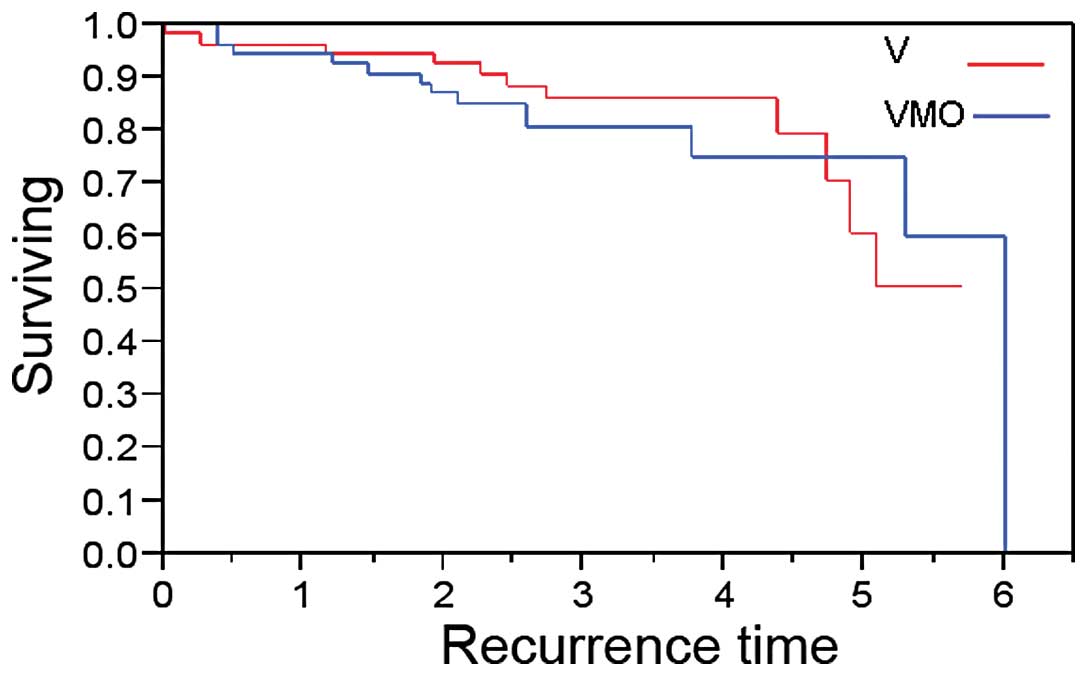
As shown in Fig. 1,
there was no statistically significant difference in DFI between
patients receiving VMO and those receiving V (p=0.76). The median
DFI for the VMO group was six years, while the median DFI for the V
group has not yet been reached.
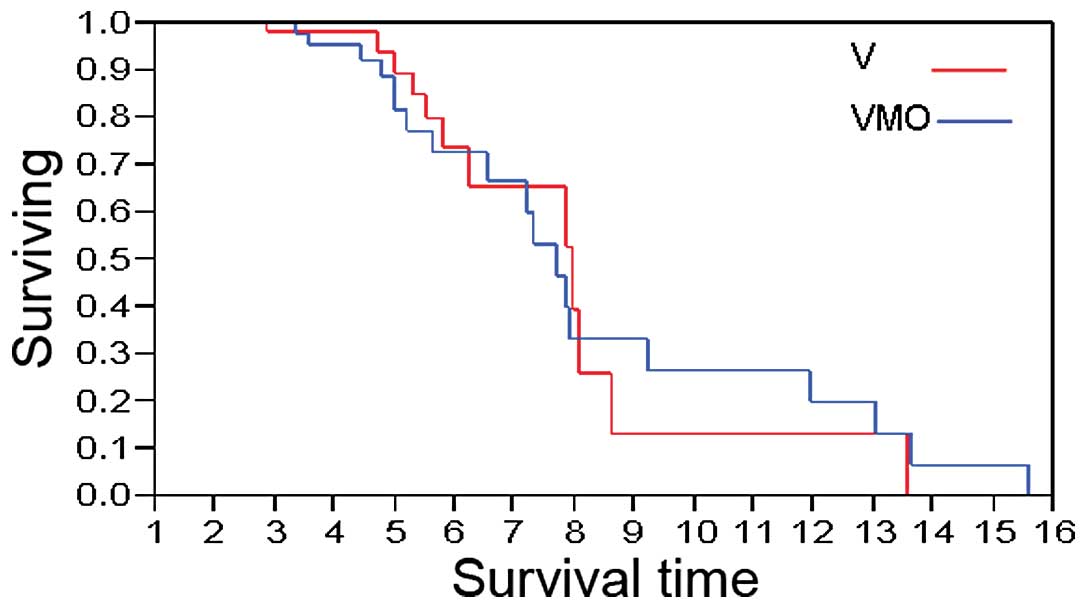
The OS of patients treated with VMO or V is shown in
Fig. 2. In patients administered
VMO, the median OS was 7.71 years, compared to 7.95 years in those
administered V (p=0.70).
Discussion
Identification of new immunotherapeutic strategies
to treat melanoma remains an area of intense research. In addition
to the FDA-approved immuno-activating IL-2 (7) and the immuno-modulatory agent
ipilimumab (9) as treatments for
melanoma patients, research on developing melanoma vaccines remains
a field of interest, since melanoma is the only type of cancer
which is highly immunogenic, by virtue of its expression of a
plethora of MAAs (16–18). Previous studies on identification
of melanoma vaccines have focused on either the use of a single
MAA, such as gp100 or MART-1, or multiple MAAs, although the
optimal conditions required to initiate and sustain an
anti-melanoma immune response were not met. Our laboratory has
created and characterized a multivalent MAA vaccine using four
primary melanoma cell lines and vaccinia virus as a potent adjuvant
(24,25), which was evaluated in an
FDA-approved, NCI-funded clinical trial.
To the best of our knowledge, our first generation
melanoma vaccine (VMO) phase III trial was the first FDA-approved
and NCI-funded, randomized, prospective, multi-institutional and
double-blind cancer vaccine trial to employ a polyvalent vaccinia
virus-augmented melanoma cell lysate against stage III melanoma.
This trial was the first cancer vaccine trial designed to assess
the efficacy of VMO, without any bias in the selection of patients,
distribution of VMO and V and collection or analysis of patient
data. Thus, this series is unique, not only in terms of the
treatment provided, but also in its design, which was a novel
strategy to improve the only standard of care that was previously
available, which was surgery alone.
This follow-up analysis demonstrated no differences
in DFI or OS in patients who received VMO compared to those who
received V, although it should be noted that a significant number
of patients from the original trial (78) were still alive at the
time of this follow-up analysis. The fact that such a large number
of VMO patients (35), as well as
V patients, survived, demonstrates the possible benefits of either
method of immune stimulation and, more importantly, highlights the
longevity of the immune response that was initially generated (as
detected by delayed-type hypersensitivity reactions) in both the
VMO and V treatment arms. Furthermore, it should be stated that, at
the time of the vetting of this phase III trial, through the FDA
and NCI, there was intense discussion on the use of V as a
treatment arm, since standard of care dictated surgery alone as the
second arm. More importantly, V by itself has been shown to produce
‘therapeutic efficacy’ in melanoma patients (34). In fact, there should not have been
any other placebo used in this trial except a true no-treatment
arm, as it is our opinion that such a placebo for this trial would
have helped elucidate the efficacy of the VMO. Thus, it is
plausible that the VMO arm may have demonstrated statistically
significant results if a ‘true’ no-treatment arm was included
(35).
Of note, in this follow-up analysis, the phase III
trial demonstrated a statistically significant increase in OS in
VMO- and V-treated females compared to males in both arms, even
though in the first three analyses, males had exhibited an
increased OS, particularly the VMO-treated, aged 44–57 years
subset. In general, female melanoma patients exhibit significantly
increased survival rates compared to males, which is usually
attributed to earlier detection among women (36) and it can also be attributed to the
fact that this trial did not include a sufficient number of
patients, in order to perform significant survival analyses in
subsets with prognostic significance. Therefore, we strongly
recommend that future ASI trials enroll a sufficient number of
patients in each of the melanoma patient subsets, so that valid
conclusions may be drawn from the comparison of factors such as
gender (male vs. female), age, number of microscopic vs.
macroscopic nodes and location of primary tumor, among others.
Moreover, although our phase III trial demonstrated
no difference in DFI or OS between patients receiving VMO and those
receiving V, our long-term results are in clear opposition to the
vaccine trial conducted by Morton et al(37). These authors investigated Canvaxin,
an antigen-rich, allogeneic whole-cell vaccine developed from three
melanoma cell lines, which had appeared promising in phase I and II
trials. However, in October 2005, CancerVax announced the
discontinuation of its phase III clinical trial of Canvaxin in
patients with stage III melanoma, since it was determined that
Canvaxin-treated patients [those that received bacillus
Calmette-Guérin (BCG) plus allogeneic melanoma cell vaccine],
exhibited no improved survival benefit compared to patients that
received BCG plus placebo. This was not the case in our trial,
since the FDA did not detect any marked differences in OS or DFS
among the VMO and V treatment groups. In fact, at the three-year
mark, VMO showed a 10% survival advantage compared to V, thus
allowing for the continuation of the trial (31). However, at the five-year mark, the
two arms were similar.
Another melanoma vaccine trial, which used a
somewhat similar vaccine to ours, was one that Hersey and
associates developed and further conducted as a prospective,
randomized, multicenter trial, with the treatment arm consisting of
vaccinia melanoma cell lysates (VMCL) and the control arm being a
‘true’ placebo (i.e., no-treatment arm) (38). The results of this trial showed
that patients who were administered the vaccine did not exhibit a
statistically significant improvement in OS or relapse-free
survival, compared to the no-treatment arm. Although no
statistically significant observations were made in the clinical
trial conducted by Hersey et al, it is difficult to directly
compare the results of our trial with theirs, since our vaccine
consisted of four allogeneic melanoma cell lines with significant
immunological activity (24,25),
as opposed to their single allogeneic melanoma cell line
vaccine.
This phase III trial demonstrated an unexpected
long-term survival in both arms; however, there are components of
the vaccine that require improvement, which may allow for the
optimal presentation of MAAs to the immune system, i.e., the
inclusion of melanoma cell lines expressing HLA antigens. Over the
past few years, we have designed a new vaccine incorporating the
latest advances in cell and molecular biology. We have previously
demonstrated the ability of dendritic cells (DCs) pulsed with an
IL-2 gene-encoded vaccinia virus (VV) melanoma oncolysate
(DC-IL-2VMO or DC-MelVac) to generate a cellular immune response
in vitro(39) and in murine
tumor models (40). This second
generation melanoma vaccine possesses key immunogenic properties
that may render it a potent therapeutic agent in the treatment of
patients with stage III and IV melanoma and was approved by the FDA
for a phase I trial in February 2005. We are currently in the
development phase of this second generation melanoma vaccine, which
consists of components that increase its adjuvanticity, compared to
the first generation melanoma vaccine. Furthermore, we are striving
for the complete characterization of the antigens within the VMO
preparation, which is pivotal for developing an effective
pan-antigen multivalent vaccine, using vaccinia virus as an
adjuvant.
Acknowledgements
This clinical trial was funded by NCI
(RO1CA4538-01A1).
References
|
1.
|
Haass NK, Smalley KS and Herlyn M: The
role of altered cell-cell communication in melanoma progression. J
Mol Histol. 35:309–318. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Inamdar GS, Madhunapantula SV and
Robertson GP: Targeting the MAPK pathway in melanoma: why some
approaches succeed and other fail. Biochem Pharmacol. 80:624–637.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Atkins MB, Hsu J, Lee S, Cohen GI,
Flaherty LE, Sosman JA, Sondak VK and Kirkwood JM; Eastern
Cooperative Oncology Group: Phase III trial comparing concurrent
biochemotherapy with cisplatin, vinblastine, dacarbazine,
interleukin-2, and inter-feron alfa-2b with cisplatin, vinblastine,
and dacarbazine alone in patients with metastatic malignant
melanoma (E3695): a trial coordinated by the Eastern Cooperative
Oncology Group. J Clin Oncol. 26:5748–5754. 2008.
|
|
5.
|
Bedikian AY, Millward M, Pehamberger H,
Conry R, Gore M, Trefzer U, et al: Bcl-2 antisense (oblimersen
sodium) plus dacarbazine in patients with advanced melanoma: the
Oblimersen Melanoma Study Group. J Clin Oncol. 24:4738–4745. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Weber J: Immunotherapy for melanoma. Curr
Opin Oncol. 23:163–169. 2011. View Article : Google Scholar
|
|
7.
|
Block MS, Suman VJ, Nevala WK, Kottschade
LA, Creagan ET, Kaur JS, et al: Pilot study of
granulocyte-macrophage colony-stimulating factor and interleukin-2
as immune adjuvants for a melanoma peptide vaccine. Melanoma Res.
21:438–445. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Hodi FS, O’Day SJ, McDermott DF, Weber RW,
Sosman JA, Haanen JB, et al: Improved survival with ipilimumab in
patients with metastatic melanoma. N Engl J Med. 363:711–723. 2010.
View Article : Google Scholar
|
|
9.
|
Flaherty KT, Yasothan U and Kirkpatrick P:
Vemurafenib. Nat Rev Drug Discov. 10:811–812. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
McCardle TW, Messina JL and Sondak VK:
Completely regressed cutaneous melanocytic lesion revisited. Semin
Oncol. 36:498–503. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Overwijk WW and Restifo NP: Autoimmunity
and the immunotherapy of cancer: targeting the ‘self’ to destroy
the ‘other’. Crit Rev Immunol. 20:433–450. 2000.
|
|
12.
|
Pittet MJ, Zippelius A, Valmori D, Speiser
DE, Cerottini JC and Romero P: Melan-A/MART-1-specific CD8 T cells:
from thymus to tumor. Trends Immunol. 23:325–328. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Gnjatic S, Nishikawa H, Jungbluth AA, Güre
AO, Ritter G, Jäger E, et al: NY-ESO-1: review of an immunogenic
tumor antigen. Adv Cancer Res. 95:1–30. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Boon T: Tumor antigens recognized by
cytolytic T lymphocytes: present perspectives for specific
immunotherapy. Int J Cancer. 54:177–180. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Rosenberg SA: A new era of cancer
immunotherapy: converting theory to performance. CA Cancer J Clin.
49:70–73. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Reynolds SR, Celis E, Sette A, Oratz R,
Shapiro RL, Johnston D, Fotino M and Bystryn JC: Identification of
HLA-A*03, A*11 and B*07-restricted
melanoma-associated peptides that are immunogenic in vivo by
vaccine-induced immune response (VIIR) analysis. J Immunol Methods.
244:59–67. 2000.
|
|
17.
|
Reynolds SR, Celis E, Sette A, Oratz R,
Shapiro RL, Johnston D, Fotino M and Bystryn JC: HLA-independent
heterogeneity of CD8+ T cell responses to MAGE-3, Melan-A/MART-1,
gp100, tyrosinase, MC1R, and TRP-2 in vaccine-treated melanoma
patients. J Immunol. 161:6970–6976. 1998.PubMed/NCBI
|
|
18.
|
Reynolds SR, Oratz R, Shapiro RL, Hao P,
Yun Z, Fotino M, Vukmanović S and Bystryn JC: Stimulation of CD8+ T
cell responses to MAGE-3 and Melan A/MART-1 by immunization to a
polyvalent melanoma vaccine. Int J Cancer. 72:972–976. 1997.
|
|
19.
|
Baurain J, Stas M, Hammouch F, Gillain A,
Feyens A, Van Baren N, et al: Association of primary melanoma
ulceration and clinical benefit of adjuvant vaccination with
tumor-specific antigenic peptides. J Clin Oncol. 27:abs. 3022.
2009.
|
|
20.
|
Kruit WH, van Ojik HH, Brichard VG,
Escudier B, Dorval T, Dréno B, et al: Phase 1/2 study of
subcutaneous and intradermal immunization with a recombinant MAGE-3
protein in patients with detectable metastatic melanoma. Int J
Cancer. 117:596–604. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Slingluff CL Jr, Petroni GR, Olson WC,
Smolkin ME, Ross MI, Haas NB, et al: Effect of
granulocyte/macrophage colony-stimulating factor on circulating
CD8+ and CD4+ T-cell responses to a multipeptide melanoma vaccine:
outcome of a multicenter randomized trial. Clin Cancer Res.
15:7036–7044. 2009.PubMed/NCBI
|
|
22.
|
Filipazzi P, Pilla L, Patuzzo R, Castelli
C, Maurichi A, Tragni G, et al: Adjuvant multipeptide vaccination
in high-risk early melanoma patients. J Clin Oncol. 26:abs. 3014.
2008.
|
|
23.
|
Schwartzentruber DJ, Lawson DH, Richards
JM, Conry RM, Miller DM, Treisman J, et al: gp100 peptide vaccine
and interleukin-2 in patients with advanced melanoma. N Engl J Med.
364:2119–2127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Wallack MK, Steplewski Z, Koprowski H,
Rosato E, George J, Hulihan B and Johnson J: A new approach in
specific, active immunotherapy. Cancer. 39:560–564. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wallack MK: Specific active immunotherapy
with vaccinia oncolysates. Tumor Progression. Crispen R:
Elsevier/North Holland Inc.; New York: pp. 277–287. 1980
|
|
26.
|
Berthier-Vergnes O, Portoukalian J,
Lefthériotis E and Doré JF: Induction of IgG antibodies directed to
a M(r) 31,000 melanoma antigen in patients immunized with vaccinia
virus melanoma oncolysates. Cancer Res. 54:2433–2439.
1994.PubMed/NCBI
|
|
27.
|
Shimizu Y, Hasumi K, Masubuchi K and
Okudaira Y: Immunotherapy of tumor-bearing mice utilizing virus
help. Cancer Immunol Immunother. 27:223–227. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Wallack MK, Meyer M, Bourgoin A, Doré JF,
Leftheriotis E, Carcagne J and Koprowski H: A preliminary trial of
vaccinia oncolysates in the treatment of recurrent melanoma with
serologic responses to the treatment. J Biol Response Mod.
2:586–596. 1983.PubMed/NCBI
|
|
29.
|
Wallack MK, McNally KR, Leftheriotis E,
Seigler H, Balch C, Wanebo H, Bartolucci AA and Bash JA: A
Southeastern Cancer Study Group phase I/Il trial with vaccinia
melanoma oncolysates. Cancer. 57:649–655. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Wallack MK, Bash JA, Leftheriotis E,
Seigler H, Bland K, Wanebo H, Balch C and Bartolucci AA: Positive
relationship of clinical and serological responses to vaccinia
melanoma oncolysate. Arch Surg. 122:1460–1463. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wallack MK, Sivanandham M, Balch CM, Urist
MM, Bland KI, Murray D, et al: A phase III randomized, double-blind
multiinstitutional trial of vaccinia melanoma oncolysate-active
specific immunotherapy for patients with stage II melanoma. Cancer.
75:34–42. 1995. View Article : Google Scholar
|
|
32.
|
Wallack MK, Sivanandham M, Ditaranto K,
Shaw P, Balch CM, Urist MM, et al: Increased survival of patients
treated with a vaccinia melanoma oncolysate vaccine. Second interim
analysis of data from a phase III, multi-institutional trial. Ann
Surg. 226:198–206. 1997. View Article : Google Scholar
|
|
33.
|
Wallack MK, Sivanandham M, Balch CM, Urist
MM, Bland KI, Murray D, et al: Surgical adjuvant active specific
immunotherapy for patients with stage III melanoma: the final
analysis of data from a phase III, randomized, double-blind,
multicenter vaccinia melanoma oncolysate trial. J Am Coll Surg.
187:69–79. 1998. View Article : Google Scholar
|
|
34.
|
Roenigk HH Jr, Deodhar S, Jacques R and
Burdick K: Immunotherapy of malignant melanoma with vaccinia virus.
Arch Dermatol. 109:668–673. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Linge C, Gewert D, Rossmann C, Bishop JA
and Crowe JS: Interferon system defects in human malignant
melanoma. Cancer Res. 55:4099–4104. 1995.PubMed/NCBI
|
|
36.
|
Joosse A, de Vries E, Eckel R, Nijsten T,
Eggermont AM, Hölzel D, Coebergh JW and Engel J; Munich Melanoma
Group: Gender differences in melanoma survival: female patients
have a decreased risk of metastasis. J Invest Dermatol.
131:719–726. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Morton DL, Hsueh EC, Essner R, Foshag LJ,
O’Day SJ, Bilchik A, et al: Prolonged survival of patients
receiving active immunotherapy with Canvaxin therapeutic polyvalent
vaccine after complete resection of melanoma metastatic to regional
lymph nodes. Ann Surg. 236:438–449. 2002. View Article : Google Scholar
|
|
38.
|
Hersey P, Coates AS, McCarthy WH, Thompson
JF, Sillar RW, McLeod R, et al: Adjuvant immunotherapy of patients
with high-risk melanoma using vaccinia viral lysates of melanoma:
results of a randomized trial. J Clin Oncol. 20:4181–4190. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Aydin N, Jack A, Montenegro G, Boyes C,
Alam K and Wallack MW: Expression of melanoma-associated antigens
in human dendritic cells pulsed with an interleukin-2 gene encoded
vaccinia melanoma oncolysate (rIL-2VMO). Cancer Biol Ther.
5:1654–1657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Jack AM, Aydin N, Montenegro G, Alam K and
Wallack MK: A novel dendritic cell-based cancer vaccine produces
promising results in a syngenic CC-36 murine colon adenocarcinoma
model. J Surg Res. 139:164–169. 2007. View Article : Google Scholar : PubMed/NCBI
|