Introduction
Breast cancer is currently the most common type of
cancer in females. Approximately 5–10% of oncological cases are due
to inherited genetic defects in germ cells (1). The hereditary forms of breast cancer
are mainly caused by mutations in the BRCA1 and BRCA2
tumor-suppressor genes, resulting in the production of
non-functional proteins (2,3).
Sequencing of the BRCA1 and BRCA2
genes is currently considered the gold standard method for
determining the mutation status in breast cancer patients. Due to
the high prevalence of breast cancer, BRCA1 and BRCA2
are currently among the most sequenced genes worldwide (4). BRCA1 and BRCA2 are
responsible for accurate DNA repair of double-strand breaks
(5–7). In addition to the compromised DNA
repair function, mutations in the BRCA1 and BRCA2
genes are likely to affect cell cycle regulation and
transcriptional activity.
However, not all BRCA1 and BRCA2
mutations are pathological and their impact may vary depending on
the extent to which the normal protein function is compromised.
Furthermore, the frequency and type of mutations may vary among
different populations (http://www.breastcancerdatabase.org/).
In order to determine the frequency and type of
variants in the target exon sequences of the two genes, we
sequenced and analyzed BRCA1 and BRCA2 using
next-generation sequencing (NGS) technology in Bulgarian breast
cancer patients and healthy controls. To the best of our knowledge,
this study was the first to assess the genetic predisposition to
breast cancer in the Bulgarian population. Elucidating the effects
of BRCA1 and BRCA2 mutations is crucial for the
prevention of breast cancer.
Materials and methods
Subjects
A total of 24 Bulgarian patients (mean age, 35±10
years) diagnosed with breast cancer and with a positive family
history for this disease and 71 age-matched healthy controls
without a positive family history were recruited in this study. DNA
samples were collected from the subjects at the National
Oncological Hospital (Sofia, Bulgaria) and the BRCA1 and
BRCA2 genes were sequenced. Written informed consent was
obtained from all subjects. The study was approved by the Ethical
Comitee of Specialized Hospital for Active Treatment in Oncology,
Sofia, Bulgaria.
NGS analysis
The first step for NGS technology is to use the
TruSeq Custom Amplicon method to design oligo probes that are
specific for the target regions of BRCA1 and BRCA2,
using Illumina DesignStudio (Illumina, Inc., San Diego, CA, USA).
For each 150-bp sequence of the target region, a pair of oligo
probes were synthesized to hybridize with the 5’ and 3’ ends of the
sequence at one end (the other end was complementary to the
polymerase chain reaction primers). These oligo probes were used to
construct a library containing the necessary nucleotide sequences.
The target regions were determined by selecting all exons of the
BRCA1 and BRCA2 genes; however, in order to include
sections of the intron-exon regions, the regions also included 50
nucleotides upstream and downstream of the exon.
Sequencing was performed using the NGS MiSeq
Illumina sequencer (Illumina, Inc.). Obtained sequences were
aligned to the reference genome (GRCh37/hg19) using MiSeq Reporter
software (Illumina, Inc.), which detected discrepancies determining
their type, such as deletions, insertions and SNPs. The sequences
were analyzed using MiSeq software. As an acceptance threshold
value we selected a Q-score of 30, corresponding to a 1:1,000 error
rate.
Analysis of variants
In order to determine whether a given variant was
situated in a coding or non-coding region, we used the University
of California, Santa Cruz (UCSC) genomic browser (http:/genome.ucsc.edu/). The mutation positions were
identified by determining: i) whether the mutation was situated in
an intron or an exon; ii) if it was situated in an intron, whether
it affected the splice acceptor or donor, or the consensus splicing
sequence; and iii) if it was located in an exon, whether it
resulted in an alteration of the amino acid sequence.
The established variants were cross-checked with the
Breast Cancer Information Core (BIC) database (http://lgdfm3.ncifcrf.gov/bic/BIC.html),
which theoretically contains all identified BRCA1 and
BRCA2 mutations. The variants were also cross-checked in the
Database of Single-Nucleotide Polymorphisms (dbSNP) in order to
verify our results. In order to elucidate the effects of the
different variants with no clear clinical significance, we used the
PROVEAN (8), PolyPhen-2 (9) and SIFT (10) web-based platforms.
Results
NGS analysis
NGS analysis identified several types of variants,
which were classified according to their potential degree of
pathogenicity as follows:
Class 5 (pathological). Variants harbouring
mutations of verified clinical significance. These are usually
non-sense mutations (causing truncation of the protein, as a
portion of the amino acid sequence is lost), frame-shift, splice
(causing incorrect splicing) and pathological missense mutations,
experimentally verified to exert pathological effects.
Class 4 (presumably pathological). Variants
harbouring mutations that are likely to exert negative pathological
effects. For example, missense mutations have been identified in
breast cancer patients, although they have not been verified as
disease-causing mutations.
Class 3 [variants of unknown clinical
significance (VUS)]. Variants harbouring rare missense
mutations and triple nucleotide in-frame insertions and deletions.
This class also includes variants with mutations in the introns
that are often overlooked as possible causes for cancer development
(11). When deciding whether a
mutation belongs to this class or whether it is a polymorphism, its
conservation in among-species comparative analysis has to be
considered.
Class 2 (likely polymorphic variants).
Variants with no or marginal clinical significance. This class
includes missense mutations that are rare, but with an observable
frequency in the population.
Class 1 (common polymorphisms). Variants
without clinical significance. These can be synonymous mutations,
polymorphisms with high frequencies and missense variants, which
were established as not exerting any pathological effects.
Pathological mutations
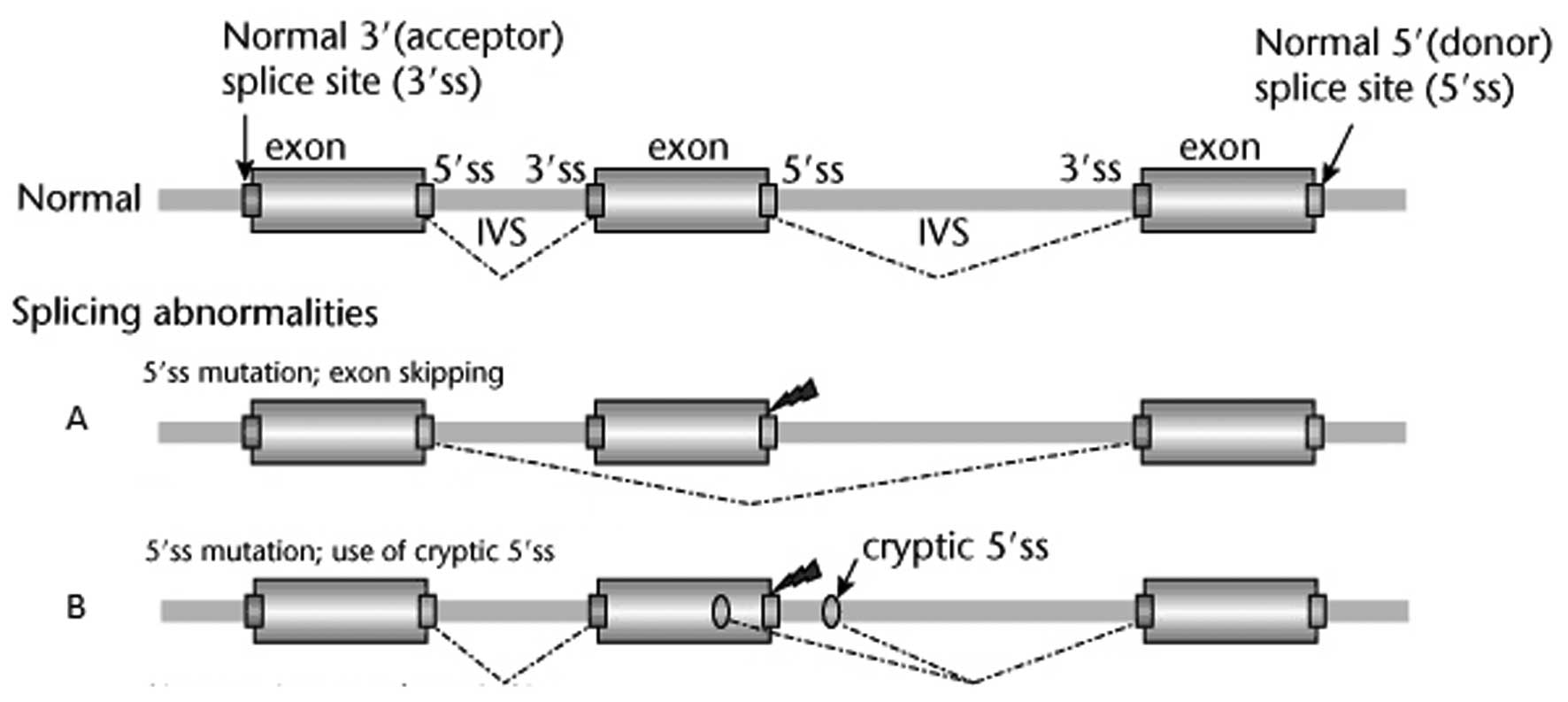
The only pathological mutation was identified a
patient with breast cancer and early-age diagnosis. This mutation
was identified in the BRCA2 gene at the chromosomal (chr)
position chr13:32890665 and affected the first position of the 5’
splice region following exon 2 (Fig.
1). The consensus 5’ GT sequence at the beginning of the intron
was replaced by a 5’ AT sequence. There are two possible outcomes
in such a case: skipping exon 2 (Fig.
1A) or using an alternative cryptic donor locus (Fig. 1B).
However, exon 2 contains the start codon that
initiates translation. Therefore, we cross-checked in the UniProt
database and established that the next start codon was at position
124 (Me124) and embedded in exon 5. This codon serves as an
initiator of the translation of the other transcript of
BRCA2 (ENST00000380152), although the latter is rarely
expressed. In case exon 2 is skipped, mRNA translation may commence
from this codon; however, the protein sequence will lack the first
123 amino acids, of which the first 40 are operational in the
interaction with the PALB2 protein (partner and localizer of
BRCA2). Single-nucleotide mutations in this region (G25R, W31C and
W31R) were shown to disrupt the interaction between BRCA2 and PALB2
(12), which is a key factor for
the effective repair of double-strand breaks through homologous
recombination. If the cryptic splice donor locus is used, the
effects may be less predictable, but will likely result in
frame-shift mutations in the majority of cases. Even if the reading
frame on the BRCA2 gene remains intact, the N-terminal amino
acids that are required for the interaction with PALB2 would be
lost. Bonatti et al (13)
demonstrated that this mutation indeed resulted in aberrant
transcripts and a consequent full loss of function. This particular
mutation, c.67+1G>A, was also described in 5 patients, two of
whom are from Western Europe (BIC database). The dbSNP
identification number of the mutation is rs81002796 and it is
described as ‘pathological’ in this database, meaning that this
mutation has been verified to cause breast cancer. This finding was
also confirmed by our study.
Possibly pathological mutations
This group includes mutations that are highly likely
to exert a detrimental effect and have been identified in
individuals with breast cancer, although without any direct
disease-causing evidence. Such a mutation was detected in 1 patient
in the BRCA1 gene at position 41219635; it is an in-frame
triple nucleotide deletion of valine 1688 in exon 17 and is
classified in the BIC database as a mutation of unknown effect
(14). The deleted amino acid is
part of the BRCT 1 functional domain (1642–1736) and mutations in
adjacent amino acids have been detected in patients with breast or
ovarian cancer (T1685A, T1685I, M1689R, K1690Q, D1692N, C1697R and
R1699W) (UniProt). Multivariate analyses predicted a pathological
effect of this mutation (LOVD database) and, based on the
multivariate analyses, we inferred that this mutation also exerted
a pathological effect in our study.
VUS.
Two VUS were detected in the control group. The
first variant was located in exon 12 of BRCA1 in position
chr17:41234509, wherein the guanine was replaced by cytosine. This
mutation resulted in a missense substitution at position 1423 of
the protein, wherein the serine was replaced by arginine
(Ser1423Arg). Serine 1423 is crucial, as this amino acid is
phosphorylated by the protein kinase ataxia-telangiectasia mutated
(ATM) (15). Patients with this
genotype have been identified worldwide and this mutation was
located in the BIC database. However, such mutation variants may be
generated by mutagenesis (16). It
was demonstrated that, by exposing the cells to ionizing radiation,
this mutation inhibited the insertion of BRCA1 during the G2
phase and disrupted the repair of accumulated radiation-induced DNA
damage. When serine 1423 is mutated, ATM cannot phosphorylate
BRCA1 and this modification is required for the activation
of the G2/M checkpoint signaling pathway. Thus, the function of
BRCA1 in regulating the cell cycle is disrupted and the cell
enters the M phase, despite the possible DNA damage. Furthermore,
the area covering amino acids 1397–1424 is responsible for the
interaction of BRCA1 with PALB2 (UniProt, 2013). The formation of
the BRCA1-PALB2-BRCA2 complex is a repair mechanism for
double-strand breaks by homologous recombination. PolyPhen-2 also
suggested that the Ser1423Arg mutation was ‘probably abnormal’,
while the SIFT algorithm considered the mutation as pathological
and PROVEAN - as a common polymorphism.
The second VUS was detected in the BRCA2 gene
at position chr13:32930634, G>A. This is an Arg2502His missense
mutation in exon 15 and the BIC database indicated 22 cases of
unknown effect. The missense mutation and consequent amino acid
replacement involves similar hydrophilic amino acids; however, the
mutation has also been detected in patients with breast and ovarian
cancer. PolyPhen-2, SIFT and PROVEAN predict a neutral effect.
Likely polymorphic variants
Likely polymorphic variants, as defined above, are
presented in Table I for patients
and controls.
 | Table I.Likely polymorphic variants in the
BRCA1 and BRCA2 genes in patients and controls. |
Table I.
Likely polymorphic variants in the
BRCA1 and BRCA2 genes in patients and controls.
| Gene | Position | Variant | Description |
|---|
| Patients | | | |
| BRCA1 (2
females) | 41277354 | G>A | 5’-UTR variant in
exon 1. The position is not conserved among mammals (PhyloP, GERP).
The risk of abnormal translation of the transcript is low. |
| BRCA2 | 32889548 | C>T | Variant upstream of
exon 1, few bases upstream of the highly conserved region of the
promoter. The base itself is not conserved among mammals (PhyloP,
GERP). The risk of pathological changes in the transcription and
expression of BRCA2 is low. |
| Controls | | | |
| BRCA1 | 41246812 | A>C | Leu246Val missense
variant in exon 11; BIC-unknown effect, it was detected 70 times,
mostly in individuals from Western Europe. |
| BRCA2 | 32973748 | A>G | Variant in the
3’-UTR. The position is not conserved among mammals (PhyloP, GERP)
and it is unlikely that the variant leads to a change in truncation
of mRNA. |
| BRCA2 | 32973660 | C>T | Variant in the
3’-UTR. The position has been moderately conserved among mammals
(PhyloP, GERP). The risk for truncation of mRNA and destabilization
of the transcript is low. |
| BRCA2 | 32889593 | G>A | Variant upstream of
exon 1, a CpG-rich region of the promoter. Optionally, binding site
for transcription factors. The position is not conserved among
mammals (PhyloP, GERP). The risk of abnormal expression of
BRCA2 is low. |
| BRCA2 | 32889548 | C>T | Variant upstream of
exon 1, few bases upstream of the highly conserved region of the
promoter. The base itself is not conserved among mammals (PhyloP,
GERP). The risk for pathological changes in the transcription and
expression of BRCA2 is low. |
Common polymorphic variants
The common polymorphic variants detected in our
study are presented in Table II
for BRCA1 and in Table III
for BRCA2.
| Table II.BRCA1 common variants in
Bulgarian females. |
Table II.
BRCA1 common variants in
Bulgarian females.
| Start | SNP
description | Total frequency
(%) | Patients (%) | Controls (%) |
|---|
| 41196408 | G>A; SNP within
3’-UTR of exon 27, no effects, worldwide polymorphism | 43.16 | 37.5 | 45.07 |
| 41197274 | C>A; SNP within
3’-UTR of exon 24, no effects, worldwide polymorphism | 43.16 | 37.5 | 45.07 |
| 41234470 | A>G; Ser>Ser,
synonymous SNP in exon 12, no effects, worldwide polymorphism | 43.16 | 37.5 | 45.07 |
| Table III.BRCA2 common variants in
Bulgarian females. |
Table III.
BRCA2 common variants in
Bulgarian females.
| Start | SNP
description | Total frequency
(%) | Patients (%) | Controls (%) |
|---|
| 32889792 | A>G; upstream of
gene sequence, within promoter sequence, binding site for
transcription factors, polymorphisms observed at this site, no
effects | 22.11 | 20.83 | 22.54 |
| 32890572 | G>A; SNP within
5’-UTR of exon 2, no effects, worldwide polymorphism | 41.05 | 50.00 | 38.07 |
| 32929232 | A>G; Ser>Ser,
synonymous SNP in exon 14, no effects, worldwide polymorphism | 34.74 | 33.33 | 35.21 |
| 32929387 | T>C; Val>Ala,
non-synonymous SNP in exon 14, similar (small, hydrophobic) amino
acids substituted, no effects, worldwide polymorphism | 97.89 | 91.67 | 100 |
| 32973276 | A>G; SNP within
3’-UTR of exon 27, no effects, worldwide polymorphism | 27.37 | 50.00 | 19.72 |
| 32973280 | A/-, mononucleotide
deletion within 5’-UTR of exon 27, polymorphism, no effects,
undescribed, Bulgarian polymorphism | 90.53 | 83.33 | 92.96 |
| 32973439 | A>G; SNP within
3’-UTR of exon 27, no effects, worldwide polymorphism | 33.68 | 33.33 | 33.80 |
| 32973737 | T/-; SNP within
3’-UTR of exon 27, no effects, undescribed, Bulgarian
polymorphism | 78.95 | 95.83 | 73.24 |
| 32973924 | T/-, mononucleotide
deletion, downstream of gene sequence, no effects, undescribed,
Bulgarian polymorphism | 21.05 | 16.67 | 22.54 |
| 32973924 | -/T, mononucleotide
insertion, downstream of gene sequence, no effects | 52.63 | 70.83 | 46.48 |
Discussion
To the best of our knowledge, this study was the
first to the perform BRCA1 and BRCA2 gene sequencing
using NGS methods in 24 Bulgarian breast cancer patients with a
family history of breast cancer and 71 healthy controls. A wide
range of variants were detected in the BRCA1 and
BRCA2 genes. In the patient group, we identified two
pathological/presumably pathological variants, including a mutation
in BRCA2 at position chr13:32890665 that affected the first
position of the 5’ splice region following exon 2 and a mutation in
BRCA1 at position chr17:41219635, which was an in-frame
triple nucleotide deletion of valine 1688 (8.3%).
According to a previous study, BRCA1 and
BRCA2 mutations are responsible for 16% of breast cancer
cases with a positive family history (17). We hypothesized that Bulgarian
patients with a family history of breast cancer, but without
verified pathological BRCA1 and BRCA2 mutations, may
harbour mutations in other genes, including CHEK2, PTEN, TP53,
ATM, STK11, CDH1, NBS1, RAD50, BRIP1 and PALB2 (18). Stratton and Rahman (19) classified the mutations responsible
for the hereditary form of breast cancer into three categories: i)
rare mutations in high-penetrance genes (BRCA1 and
BRCA2); ii) mutations in genes of moderate penetrance
(CHEK2, ATM, BRIP1 and PALB2); and iii) common
mutations in a large number of low-penetrance genes. Whole-genome
sequencing of patients with breast cancer and a positive family
history that was not a result of mutations in the BRCA1 or
BRCA2 genes may elucidate the genetic architecture that
predisposes to the development of breast cancer. However,
considering the various types of mutations, it is likely that
environmental factors also modify the penetrance of this type of
cancer.
In this study, we detected 2 VUS in the control
group. The first variant was located in exon 12 of BRCA1 at
position chr17:41234509, wherein the guanine was replaced by
cytosine, and the second was detected in BRCA2 at position
chr13:32930634, G>A, resulting in Arg2502His missense mutation
in exon 15. Rare VUS mean that it is not possible to make an
accurate clinical prediction. It is hypothesized that the genome of
each individual contains a large number of rare missense alleles
and it was estimated that 70% of these allelles may be of clinical
significance (20). In such cases,
co-segregation analyses are required to establish a correlation
between the variant and the disease in large families.
In the patient and control groups, 7 likely
polymorphic variants and 13 common variants were detected in the
BRCA1 and BRCA2 genes. This study was the first to
detect 3 common polymorphisms of BRCA2, characteristic
solely of the Bulgarian population: position chr13:32973737, T/-,
an SNP within the 3’-UTR of exon 27; position chr13:32973280, A/-,
a mononucleotide deletion within the 5’-UTR of exon 27; and
position chr13:32973924, T/-, a mononucleotide deletion downstream
of the gene sequence.
In conclusion, the creation of a database for the
type and frequency of BRCA1 and BRCA2 gene variants
in the Bulgarian population is crucial, in order to enable accurate
interpretation and genetic counseling regarding the genetic
predisposition to breast cancer.
References
|
1.
|
Anand P, Kunnumakkara AB, Sundaram C,
Harikumar KB, Tharakan ST, Lai OS, Sung B and Aggarwal BB: Cancer
is a preventable disease that requires major lifestyle changes.
Pharm Res. 25:2097–2116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Gage M, Wattendorf D and Henry LR:
Translational advances regarding hereditary breast cancer
syndromes. J Surg Oncol. 105:444–451. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Salmena L and Narod S: BRCA1
haploinsufficiency: consequences for breast cancer. Womens Health
(Lond Engl). 8:127–129. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Tavtigian SV, Byrnes GB, Goldgar DE and
Thomas A: Classification of rare missense substitutions, using risk
surfaces, with genetic- and molecular-epidemiology applications.
Hum Mutat. 29:1342–1354. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bunting SF, Callén E, Wong N, et al: 53BP1
inhibits homologous recombination in Brca1-deficient cells by
blocking resection of DNA breaks. Cell. 141:243–254. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lieber MR: The mechanism of human
nonhomologous DNA end joining. J Biol Chem. 283:1–5. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Tutt A, Bertwistle D, Valentine J, Gabriel
A, Swift S, Ross G, Griffin C, Thacker J and Ashworth A: Mutation
in Brca2 stimulates error-prone homology-directed repair of DNA
double-strand breaks occurring between repeated sequences. EMBO J.
20:4704–4716. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Choi Y, Sims GE, Murphy S, Miller JR and
Chan AP: Predicting the functional effect of amino acid
substitutions and indels. PLoS One. 7:e466882012. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome Res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Homolova K, Zavadakova P, Doktor TK,
Schroeder LD, Kozich V and Andresen BS: The deep intronic
c.903+469T>C mutation in the MTRR gene creates an SF2/ASF
binding exonic splicing enhancer, which leads to pseudoexon
activation and causes the cblE type of homocystinuria. Hum Mutat.
31:437–444. 2010.
|
|
12.
|
Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu
J, Christ N, Liu X, Jasin M, Couch FJ and Livingston DM: Control of
BRCA2 cellular and clinical functions by a nuclear partner, PALB2.
Mol Cell. 22:719–729. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Bonatti F, Pepe C, Tancredi M, Lombardi G,
Aretini P, Sensi E, Falaschi E, Cipollini G, Bevilacqua G and
Caligo MA: RNA-based analysis of BRCA1 and BRCA2 gene alterations.
Cancer Genet Cytogenet. 170:93–101. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Antonarakis SE and Cooper DN: Mutations in
human genetic disease. eLS. 2006. View Article : Google Scholar
|
|
15.
|
Tibbetts RS, Cortez D, Brumbaugh KM,
Scully R, Livingston D, Elledge SJ and Abraham RT: Functional
interactions between BRCA1 and the checkpoint kinase ATR during
genotoxic stress. Genes Dev. 14:2989–3002. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Xu B, Kim St and Kastan MB: Involvement of
Brca1 in S-phase and G2-phase checkpoints after ionizing
irradiation. Mol Cell Biol. 21:3445–3450. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
No authors listed. Prevalence and
penetrance of BRCA1 and BRCA2 mutations in a population-based
series of breast cancer cases. Anglian Breast Cancer Study Group.
Br J Cancer. 83:1301–1308. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
van der Groep P, van der Wall E and van
Diest PJ: Pathology of hereditary breast cancer. Cell Oncol
(Dordr). 34:71–88. 2011.
|
|
19.
|
Stratton MR and Rahman N: The emerging
landscape of breast cancer susceptibility. Nat Genet. 40:17–22.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kryukov GV, Pennacchio LA and Sunyaev SR:
Most rare missense alleles are deleterious in humans: implications
for complex disease and association studies. Am J Hum Genet.
80:727–739. 2007. View
Article : Google Scholar : PubMed/NCBI
|