Introduction
Lhermitte-Duclos disease (LDD), also referred to as
dysplastic cerebellar gangliocytoma, is a rare, but clinically
significant type of cerebellar disorder that corresponds
histologically to a hamartoma. Lhermitte and Duclos (1) were the first to describe this
cerebellar condition in 1920. LDD is a slowly enlarging,
non-cancerous entity, characterized by enlarged, abnormally
developed cerebellar folia, containing dysplastic rather than
neoplastic cells. Symptomatic LDD may occur at any age, but most
commonly affects adults (adult-onset LDD, aLDD), as an isolated
condition or associated with Cowden’s disease (CD). Accumulating
evidence indicates that aLDD is pathognomonic for CD (2–5),
which is a multiple hamartoma syndrome with an increased risk for
benign and malignant tumors of the thyroid gland, breast,
endometrium and other organs (5–7). To
the best of our knowledge, ∼250 LDD cases have been reported in the
medical literature to date, 53 of which were associated with CD
(3,8–10).
Magnetic resonance imaging (MRI) is considered to be
the imaging modality of choice for the diagnosis of LDD and an Aunt
Minnie diagnosis of LDD may be confirmed due to the characteristic
‘tiger-striped’ appearance of LDD on MRI (11–14).
An improved understanding of the underlying molecular pathogenesis
and affected signaling pathways of aLDD and its association with CD
may have direct implications for optimizing current treatments and
developing novel therapeutic approaches.
This study aimed to investigate the MRI
characteristics and underlying histopathological findings in 7
cases of aLDD, with emphasis on the association between aLDD and CD
and the characteristic MRI appearance as an imaging marker for
active cancer surveillance and preventive care of this rare
condition.
Materials and methods
Patients and data
The medical records of 7 patients diagnosed with LDD
between January, 1999 and December, 2011 were retrieved from our
hospital database in accordance with human subject research
protocols and were retrospectively reviewed. All the patients had
been treated with surgical resection and the diagnosis was
histopathologically confirmed. All the patients underwent complete
examination, including thyroid and breast ultrasonography, in order
to collect clinical evidence suggestive of associated CD. A
clinical diagnosis of CD was made when an individual met the
operational criteria established by the International Cowden
Consortium (6). The PubMed
database was searched for previous cases of LDD, particularly those
associated with CD, using the key terms ‘Lhermitte-Duclos disease’
and ‘Cowden’s disease’.
Our retrospective study was approved by the
Institutional Ethics Review Board of the Fourth Military Medical
University (Xi’an, China) and all the patients provided informed
consent to the use of their clinical and imaging data for research
purposes.
MRI protocol and analysis
All 7 patients underwent MRI in our hospital, which
was performed with different scanners: two with a Gyroscan Intera
Master 1.5T MR system (Philips Medical Systems, Best, The
Netherlands) and five 3-Tesla Siemens Trio MRI scanners (Siemens
Medical Solutions, Erlangen, Germany). Images were obtained in the
transverse plane with a T2-weighted turbo-spin-echo sequence of
4,774/110 (repetition time/echo time), a T1-weighted spin-echo
sequence of 476/15 and a fluid-attenuated inversion-recovery
(FLAIR) turbo-spin-echo sequence of 6,000/100/2,000 (repetition
time/echo time/inversion time). Images were also obtained in the
sagittal plane with a T1-weighted spin-echo sequence of 476/15
(repetition time/echo time), with 5- and 3-mm sections for the
transverse and sagittal plane, respectively. The corresponding
contrast-enhanced axial and coronal T1 spin-echo sequence was
obtained in 3 patients following intravenous injection of
gadopentetate dimeglu-mine (0.1 mmol/kg; Magnevist; Bayer Schering
Pharma AG, Berlin, Germany). In addition to conventional sequences,
diffusion-weighted imaging (DWI) was performed with the echo planar
imaging sequence with a repetition time/echo time of
5,000–6,000/90–100 ms, 5-mm contiguous sections, a field-of-view of
220×220 to 240×240 mm and a matrix size of 128×128 to 192×192.
Diffusion was measured in the 6 orthogonal directions with two
b-values (0 and 1,000 sec/mm2). The images were
retrospectively reviewed by 3 neuroradiologists who were blinded to
the histopathological findings.
Histopathological and immunohistochemical
analysis
In all 7 cases, tissue blocks were retrieved for
histopathological and immunohistochemical analysis. The resected
tissues were subjected to routine processing and paraffin embedding
following fixation in 10% buffered formalin. Hematoxylin and eosin
staining was routinely performed. In addition, immunohistochemical
assays were performed with the Dako EnVision system (including
peroxidase and 3,3’-diaminobenzidine; DAKO, Carpinteria, CA, USA).
All the primary antibodies used were mouse monoclonal antibodies,
unless otherwise stated. These included antibodies to glial
fibrillary acidic protein (1:400; 6F2, American Diagnostica GmbH,
Stamford, CT, USA), synaptophysin (1:50; SY38, DakoCytomation,
Glostrup Denmark), vimentin (1:100; V9, DakoCytomation),
chromogranin A (1:50; DAK-A3, DakoCytomation), neurofilament
protein (1:50; 2F11, DakoCytomation), S-100 (1:50; 4C4.9, rabbit
polyclonal, DakoCytomation), neuron-specific enolase (1:100;
BBS/NCA/I-H14, DakoCytomation), CD34 (1:100; QBEnd10; Immunotech,
Marseille, France) and Ki-67 (1:50; MIB-1, DakoCytomation). All the
histopathological slides were retrospectively reviewed and analyzed
by a specialized neuropathologist who was blinded to the
radiological findings.
Results
Clinical characteristics
The major clinical manifestations and neuroimaging
findings of the 7 patients are summarized in Table I. The patients included 2 men and 5
women with a mean age of 37.6 years (range, 23–48 years) at
diagnosis. The most frequent signs and symptoms included dizziness,
unsteadiness of gait, headache and loss of vision, which were
attributed to increased intracranial pressure or cerebellar
deficits. The duration of the symptoms ranged from 4 months to 4
years, with a median duration of 20 months.
 | Table I.Patient characteristics, MRI findings
and follow-up data. |
Table I.
Patient characteristics, MRI findings
and follow-up data.
| No. | Agea/gender | Presenting symptoms
and signs/duration | MRI findings | Associated lesions
suggestive of CD |
|---|
| 1 | 23/M | Headache, dizziness/4
months | Typicalb; hydrocephalus; L;
tonsillar herniation | - |
| 2 | 48/F | Intermittent
dizziness/3 years; unsteady gait/6 months | Typicalb; hydrocephalus; L;
tonsillar herniation | Tubular carcinoma of
the breast; multinodular goiter; leiomyoma uteri; papillomatous
lesions |
| 3 | 44/F | Unsteady gait/4
years | Typicalb + hydro/Chiari; R;
tonsillar herniation | Ductal carcinoma
in situ of the breast; facial papules |
| 4 | 46/F | Visual disturbance of
the left eye/1 year | Typicalb; R; tonsillar
herniation | Tubular adenoma of
the colon; mucous cell carcinoma of the stomach; acral
keratoses |
| 5 | 32/F | Intermittent
dizziness/2 years | Typicalb; R; tonsillar
herniation | Tubular carcinoma of
the breast; uterine fibroids |
| 6 | 36/M | Headache, nausea,
vomiting/6 months | Typicalb; L; hydrocephalus;
tonsillar herniation | - |
| 7 | 34/F | Headache, nauseA/1
year | Typicalb; L | Follicular thyroid
cancer; fibromas of the breast; uterine fibroids |
MRI analysis
All the lesions identified in the 7 patients
involved the unilateral cerebellar cortex. Among these, 3 lesions
were located in the right and 4 in the left cerebellar hemisphere,
with involvement of the vermis in 3 patients. Hydrocephalus was
observed in 4 cases and secondary tonsillar herniation was present
in 4 cases. The size of the lesions ranged from 29 to 68 mm in
greatest diameter, with a mean diameter of 50 mm. Gross total or
subtotal resections were performed in all the patients. There was
no reported recurrence during a follow-up period of 7 months to 11
years. Of the 7 patients, 5 met the operational criteria
established by the International Cowden Consortium. The patients
developed or presented with signs of additional abnormalities
associated with CD, the most common being breast tumor (n=4). All 5
patients with aLDD associated with CD were female (Table I).
On non-enhanced computed tomography (CT) of 3
patients, 1 lesion was not clearly seen and the other 2 lesions
exhibited non-specific hypoattenuation. No calcification was
observed. By contrast, MRI depicted the mass lesions more
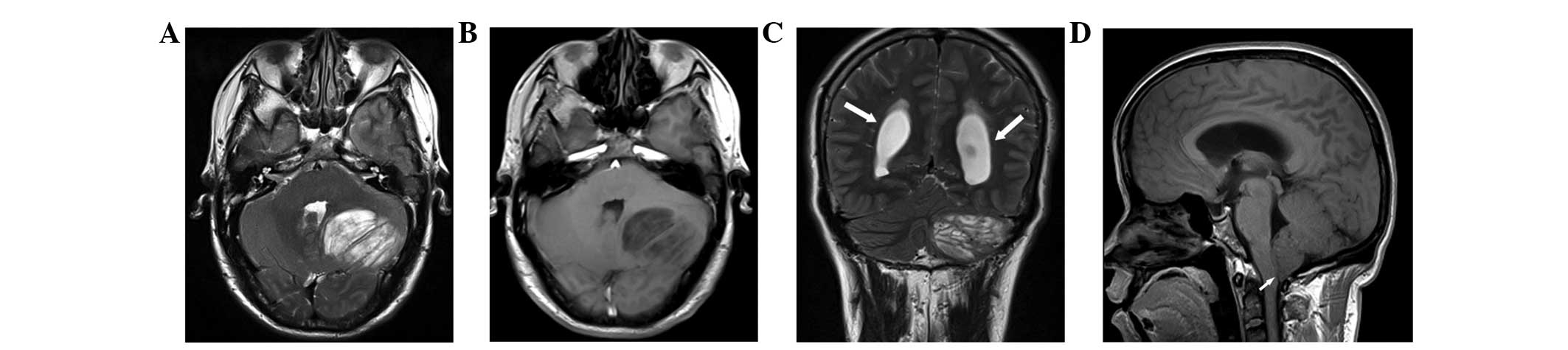
accurately compared to CT scanning. Multiplanar MRI images revealed
a ‘striated’ mass involving an enlarged cerebellar hemisphere, with
alternating bands of hyper- and isointensity compared to the gray
matter on T2-weighted imaging (Fig.
1). The striated appearance on T2-weighted imaging presented
with an alteration of iso- and hypointense stripes on T1-weighted
imaging. A total of 4 and 3 lesions were located in the left and
right cerebellar hemispheres, respectively. There were no
intratumoral necrotic regions or significant peritumoral edema in
any of the patients.
Our patients exhibited additional MRI abnormalities,
including involvement of the vermis (n=2) and variable
hydrocephalus (n=5), which was a frequent finding in these
patients. Furthermore, tonsillar herniation was associated with
variable hydrocephalus (n=5). Three lesions were evaluated by
postcontrast T1-weighted MR images and exhibited no enhancement
(Fig. 1D). The morphological
characteristics on MRI were uniform in the 7 patients, irrespective
of coexisting CD. An illustrative case of isolated aLDD (case 4) is
shown in Fig. 2. On FLAIR imaging,
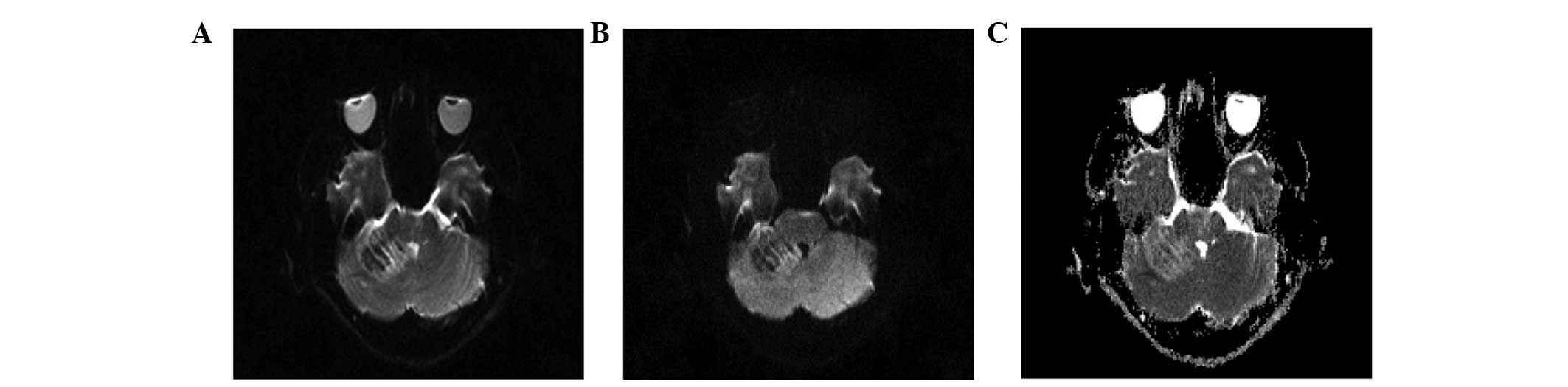
the mass was hyperintense in 1 of the 7 cases. In addition to
conventional MRI, DWI was performed in 2 patients. DWI of high
b-value (b=1,000 sec/mm2) revealed no restricted
diffusion in the lesion and the apparent diffusion coefficient
(ADC) maps demonstrated increased signal intensity compared to the
contralateral normal cerebellum (Fig.
3), reflecting an increased diffusion of water. Proton MR
spectroscopy was performed in 1 patient. Reduced NA/Cho and NA/Cr
ratios were observed compared to the controls, with peaks
attributable to lactate.
Histopathological and immunohistochemical
analysis
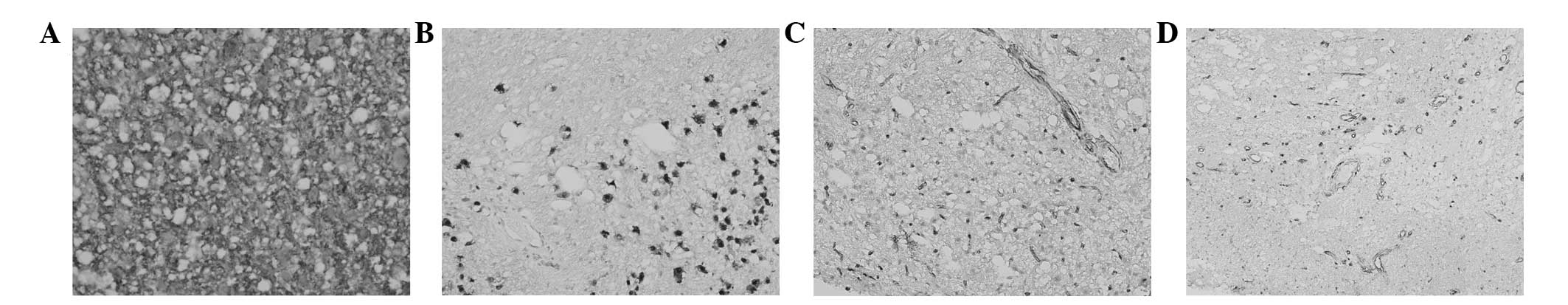
Macroscopically, a discrete region of hypertrophy
was observed within the affected cerebellum. The involved
cerebellar folia were distorted and enlarged on gross examination.
There was no sharp demarcation of abnormal cerebellar tissue from
the adjacent tissues. Histopathologically, the widened molecular
layer was occupied by abnormally hypertrophic ganglion cells.
Moreover, an attenuation in the number or absence of Purkinje
cells, abnormal molecular layer myelination and hypertrophy of the
granular cell layer were identified, with the cellular enlargement
being mainly restricted to the internal granular cell layer. These
findings, together with the relative preservation of the cerebellar
architecture, were consistent with the diagnosis of LDD (Fig. 4A and B). Ectopic cells
morphologically consistent with granular neurons were sporadically
identified in the molecular layer. Variable levels of vacuolization
of the white matter were observed in all 7 cases, which was mainly
located in the molecular layer (Fig.
4C). Other accompanying histopathological characteristics
included calcification and ectatic vessels (Fig. 5D). There were no mitoses or
pleomorphism in these cell populations.
The immunohistochemical analysis revealed no
proliferative activity in the lesions, as determined by staining
with monoclonal antibodies to Ki-67 (MIB-1). The ganglionic
component variably expressed synaptophysin (Fig. 5A) and Neu-N (Fig. 5B). Notably, relatively diffuse
immunoreactivity for CD34 was commonly observed within the lesions
as determined by CD34 immunohistochemical analysis (Fig. 5C).
Our systematic retrospective search revealed that
aLDD associated with CD exhibited no distinct pathological or
immunohistochemical characteristics compared to those of isolated
aLDD.
Discussion
LDD is a rare, slowly enlarging, benign entity that
may occur at any age, with a peak onset in later life. In this
study, 6 of our 7 patients who were diagnosed with LDD were aged
23–48 years. LDD exhibits a female preponderance; in the present
series, 5 of the 7 patients were women. LDD may be manifested by a
spectrum of clinical symptoms associated with the size and anatomic
location of the disorder. Notably, all 7 patients suffered from
long-standing symptoms, which is considered to be indicative of the
slowly progressive nature of LDD.
All the cases in our study shared similar MRI
characteristics, whether aLDD was sporadic or associated with CD.
The appearance of LDD on MRI is pathognomonic and enables a
definitive preoperative diagnosis (11–14).
The hyperintense signal observed on T2-weighted images corresponds
to the inner molecular and granular cell layers and the loss of
central white matter within the folia (12). In addition, vacuolization of the
white matter and the molecular layer may contribute to the striped
pattern of the lesions on MRI (15). Enhancement in postcontrast images
was not common; however, it is occasionally reported in the
literature, partly due to abnormal thin-walled blood vessels
(16). Our patients also exhibited
MRI abnormalities such as involvement of the vermis, hydrocephalus
and secondary tonsillar herniation.
Advanced MRI with novel imaging capabilities may
provide insight into the underlying pathophysiology and the nature
of LDD (13). Variable diffusion
properties of LDD have been reported. In one of our patients, the
lesion exhibited diffuse hyperintensity on the ADC maps, indicative
of a benign lesion. Proton MR spectroscopy demonstrated reduced
NA/Cho and NA/Cr ratios compared to the controls, with peaks
attributable to lactate (13,17),
which was consistent with one of our cases. The low NA/Cho and
NA/Cr ratios may be attributed to the apparent lack of neuronal
architecture (a hallmark of hamartoma) and the presence of
embryonic neural tissue, which indicates a benign hamartoma rather
than a malignant tumor (17).
LDD is pathologically characterized by regional and
obvious thickening of the cerebellar folia, resulting from
dysplastic replacement of cerebellar Purkinje and granular cells
with hamartomatous overgrowth of hypertrophic ganglion cells.
Mitotic activity and necrosis is uncommon and malignant
transformation was not observed, which suggested that LDD is a
hamartoma rather than a malignant tumor. Moreover, ectatic vessels
are sporadically seen. Vacuolation of varying size and variable
levels was also observed in the white matter, which was consistent
with the findings of Abel et al (15). Following careful retrospective
histopathological analysis, we observed similar histological
characteristics between sporadic cases of aLDD and those with
coexisting CD. Furthermore, pathological angiogenesis is commonly
associated with malignant neoplasms. However, angiogenesis was
frequently detected by CD34 immunohistochemistry in our series,
which may be attributed to the loss of phosphatase and tensin
homolog (PTEN) function on the phosphoinositol 3-kinase (PI3K)/Akt
pathway and PTEN-mediated control of angiogenesis (18).
CD is an autosomal dominant hereditary cancer
syndrome characterized by hamartomatous overgrowth of tissues of
all three embryonic origins and increased risk of thyroid, breast
and possibly other types of cancer (5–7,19,20).
In 1995, the International Cowden Consortium established a set of
strict operational diagnostic criteria for CD. Furthermore, the
diagnostic criteria were revised in 2000 and 2004 and are now
updated annually by the US National Comprehensive Cancer Network
(NCCN) (20,21).
LDD occurs either sporadically or in association
with CD. It was reported that aLDD is strongly associated with CD
and the presence of a PTEN mutation (2,5,15,20),
as a cancer-prone disease. Based on recent data, aLDD is currently
considered to be pathognomonic for CD (2–5), as
the majority of patients with LDD either eventually fulfill the
diagnostic criteria of CD or harbor at least one other
characteristic. Zhou et al (2) performed PTEN analysis on the
DNA of 18 patients with LDD; PTEN mutations were identified
in all the aLDD patients (15/15), but in none of the three control
children. Germline PTEN mutations may account for the majority of
the cases of LDD (20), with or
without signs indicative of CD. Abel et al (15) reviewed 31 cases of LDD and analyzed
the key members of the PTEN/Akt/mammalian target of rapamycin
(mTOR) pathway by immunohistochemistry. Their data indicated
activation of this pathway and suggested a central role for mTOR in
the pathogenesis of LDD. Due to the high prevalence of PTEN
mutations observed in patients with aLDD, the diagnosis of aLDD
alone is sufficient for the clinical diagnosis of CD (5). A previous study reviewed 14 patients
with childhood-onset LDD; however, only 3 patients were diagnosed
with CD (4).
The improved awareness regarding the
under-recognized association between LDD and CD, prompts a
systematic search of manifestations suggestive of CD when aLDD is
diagnosed. A recent literature review identified 208 LDD cases
reported after 1981, of which only 53 were associated with CD
(3,8,9,22–25).
Notably, Robinson and Cohen (3)
reported that 5 patients with LDD who were treated at their
institution over a period of 40 years exhibited manifestations of
CD, including 3 previously unrecognized cases. In our series, 5 of
the 7 cases of LDD presented evidence suggestive of CD.
Furthermore, all these 5 patients were women, suggesting a female
preponderance in the incidence of LDD coexisting with CD, which is
in concordance with previous findings (n=53; 39 women and 14 men)
(3,8–10).
It is of great significance and clinical relevance
to acknowledge the association between LDD and CD, although further
data collection is required at the molecular level to elucidate the
nature of this association. Individuals with CD are at an increased
risk of developing benign and malignant tumors of the breast,
thyroid gland and endometrium (5,6).
More recently, patients with PTEN mutations were also
reported to exhibit an increased lifetime risk of colorectal and
kidney cancer, melanoma and gastrointestinal polyps (7,26).
The coexistence of LDD and CD highlights several
issues. First, the high frequency of aLDD occurrence in the context
of CD warrants the exclusion of this latter disorder in all cases
of aLDD, or rather, the diagnosis of either disorder in a patient
should prompt a thorough search for the other. Moreover,
individuals diagnosed with aLDD coexisting with CD or with germline
PTEN mutations require long-term follow-up in order to
preclude malignancies associated with this condition. Second,
molecular evaluation, genetic counseling and gene-informed risk
assessment of patients with aLDD should become evidence-based and
warranted. Third, for patients with aLDD associated with CD,
particulary women, active cancer surveillance and preventive care
is required following the guidelines set by the NCCN (20,27).
Gross total resection is currently the treatment of
choice for aLDD, with a limited range of alternative treatment
options. Concomitant with a better understanding of the
disease-causing gene mutations, the biology of PTEN and the
PI3K/Akt/mTOR pathway, further optimization of personal management
of this entity with etiology-based molecular intervention may be
anticipated.
In conclusion, all the patients included in this
series shared similar neuroimaging characteristics that are highly
pathognomonic of LDD. aLDD is strongly associated with CD and the
MRI diagnosis of aLDD may serve as a predictor of increased risk of
malignancies and entail active cancer surveillance and preventive
care of associated neoplasms. Increasing knowledge regarding the
nature of the association between aLDD and CD, its underlying
molecular pathogenesis and the affected signaling pathways, may
enable a simplified classification and individualized management of
such patients in the future.
References
|
1.
|
Lhermitte J and Duclos P: Diffuse
cerebellar cortex ganglioneuromas. Bull Assoc Fr Etude Cancer.
9:99–107. 1920.(In French).
|
|
2.
|
Zhou XP, Marsh DJ, Morrison CD, et al:
Germline inactivation of PTEN and dysregulation of the
phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos
disease in adults. Am J Hum Genet. 73:1191–1198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Robinson S and Cohen AR: Cowden disease
and Lhermitte-Duclos disease: characterization of a new
phakomatosis. Neurosurgery. 46:371–383. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Robinson S and Cohen AR: Cowden disease
and Lhermitte-Duclos disease: an update. Case report and review of
the literature. Neurosurg Focus. 20:E62006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Hobert JA and Eng C: PTEN hamartoma tumor
syndrome: an overview. Genet Med. 11:687–694. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Eng C: Mendelian genetics of rare - and
not so rare - cancers. Ann NY Acad Sci. 1214:70–82. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Heald B, Mester J, Rybicki LA, et al:
Frequent gastrointestinal polyps and colorectal adenocarcinomas in
a prospective series of PTEN mutation carriers. Gastroenterology.
139:1927–1933. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Derrey S, Proust F, Debono B, et al:
Association between Cowden syndrome and Lhermitte-Duclos disease:
report of two cases and review of the literature. Surg Neurol.
61:447–454. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Nayil K, Wani M, Ramzan A, et al:
Lhermitte-Duclos disease with syrinx: case report and literature
review. Turk Neurosurg. 21:651–654. 2011.PubMed/NCBI
|
|
10.
|
Calabria F, Grillea G, Zinzi M, et al:
Lhermitte-Duclos disease presenting with positron emission
tomography-magnetic resonance fusion imaging: a case report. J Med
Case Rep. View Article : Google Scholar
|
|
11.
|
Meltzer CC, Smirniotopoulos JG and Jones
RV: The striated cerebellum: an MR imaging sign in Lhermitte-Duclos
disease (dysplastic gangliocytoma). Radiology. 194:699–703. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kulkantrakorn K, Awwad EE, Levy B, et al:
MRI in Lhermitte-Duclos disease. Neurology. 48:725–731. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Thomas B, Krishnamoorthy T, Radhakrishnan
VV and Kesavadas C: Advanced MR imaging in Lhermitte-Duclos
disease: moving closer to pathology and pathophysiology.
Neuroradiology. 49:733–738. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Shinagare AB, Patil NK and Sorte SZ: Case
144: Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos
disease). Radiology. 251:298–303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Abel TW, Baker SJ, Fraser MM, et al:
Lhermitte-Duclos disease: a report of 31 cases with
immunohistochemical analysis of the PTEN/AKT/mTOR pathway. J
Neuropathol Exp Neurol. 64:341–349. 2005.PubMed/NCBI
|
|
16.
|
Spaargaren L, Cras P, Bomhof MA, et al:
Contrast enhancement in Lhermitte-Duclos disease of the cerebellum:
correlation of imaging with neuropathology in two cases.
Neuroradiology. 45:381–385. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Nagaraja S, Powell T, Griffiths PD and
Wilkinson ID: MR imaging and spectroscopy in Lhermitte-Duclos
disease. Neuroradiology. 46:355–358. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Wen S, Stolarov J, Myers MP, et al: PTEN
controls tumor-induced angiogenesis. Proc Natl Acad Sci USA.
98:4622–4627. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Marsh DJ, Coulon V, Lunetta KL, et al:
Mutation spectrum and genotype-phenotype analyses in Cowden disease
and Bannayan-Zonana syndrome, two hamartoma syndromes with germline
PTEN mutation. Hum Mol Genet. 7:507–515. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Pilarski R and Eng C: Will the real Cowden
syndrome please stand up (again)? Expanding mutational and clinical
spectra of the PTEN hamartoma tumour syndrome. J Med Genet.
41:323–326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Blumenthal GM and Dennis PA: PTEN
hamartoma tumor syndromes. Eur J Hum Genet. 16:1289–1300. 2008.
View Article : Google Scholar
|
|
22.
|
Peltier J, Lok C, Fichten A, et al:
Lhermitte-Duclos disease and Cowden’s syndrome. Report of two
cases. Neurochirurgie. 52:407–414. 2006.
|
|
23.
|
Tan TC and Ho LC: Lhermitte-Duclos disease
associated with Cowden syndrome. J Clin Neurosci. 14:801–805. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zhou L, Luo L, Hui X, et al: Three
adolescents with Lhermitte-Duclos disease. J Clin Neurosci. Nov
14–2008.(Epub ahead of print). View Article : Google Scholar
|
|
25.
|
Govindan A, Premkumar S and Alapatt JP:
Lhermitte-Duclos disease (dysplastic gangliocytoma of the
cerebellum) as a component of Cowden syndrome. Indian J Pathol
Microbiol. View Article : Google Scholar
|
|
26.
|
Tan MH, Mester JL, Ngeow J, et al:
Lifetime cancer risks in individuals with germline PTEN mutations.
Clin Cancer Res. 18:400–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
National Comprehensive Cancer Network:
Genetic/Familial High-Risk Assessment: Breast and Ovarian.
http:/www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf.
Accessed July 29, 2011.
|