Introduction
A novel approach to the treatment of advanced renal
cancer is needed, as, despite the wide use of kinase inhibitors or
mammalian target of rapamycin inhibitors, there is currently no
curative treatment. Increasing histone acetylation is an attractive
epigenetic approach to cancer treatment and panobinostat is a novel
histone deacetylase (HDAC) inhibitor that has been shown to exert
beneficial antitumor effects in phase II trials in patients with
hematological malignancies (1,2).
Panobinostat was also clinically tested in patients with solid
tumors (3,4), but no complete or partial response
was observed in those studies. Ritonavir is a human
immunodeficiency virus protease inhibitor widely used for the
treatment of acquired immune deficiency syndrome, which is also a
potent CYP3A4 inhibitor (5). As
panobinostat is one of the substrates of CYP3A4 (6), we hypothesized that ritonavir may
enhance the activity of panobinostat by increasing its
intracellular accumulation through inhibiting its degradation.
In the present study, we aimed to evaluate the
combined effect of ritonavir and panobinostat on renal cancer cells
in vitro and in vivo and investigate their mechanism
of action.
Materials and methods
Cell lines
The Caki-1, ACHN, 769-P and 786-O renal cancer cell
lines were purchased from the American Type Culture Collection
(Rockville, MD, USA) and renal proximal tubule epithelial cells
(RPTECs) were purchased from Lonza (Basel, Switzerland). The cells
were cultured in either minimum essential medium, Dulbecco’s
modified Eagle’s medium, RPMI-1640 or renal epithelial cell basal
medium (depending on the cell line) supplemented with 10% fetal
bovine serum and 0.3% penicillin/streptomycin (Invitrogen,
Carlsbad, CA, USA) at 37°C under 5% CO2 in a humidified
incubator.
Reagents
Panobinostat, purchased from Cayman Chemical Company
(Ann Arbor, MI, USA) and ritonavir, purchased from Toronto Research
Chemicals (Toronto, ON, Canada), were dissolved in dimethyl
sulfoxide and stored at −20°C until use. The pan-caspase inhibitor
Z-VAD-FMK was purchased from Enzo Life Sciences (Farmingdale, NY,
USA).
Cell viability assay
Starting one day after 5×103 cells were
seeded into a 96-well culture plate, they were cultured for 48 h in
medium containing 10, 25 or 50 nM panobinostat and/or 25 or 50 μM
ritonavir. Cell viability was then evaluated with the MTS assay
(CellTiter 96 AQueous kit; Promega, Madison, WI, USA) according to
the manufacturer’s protocol.
Colony formation assay
For the colony formation assay, 100 individual cells
were seeded in 6-well plates 1 day prior to treatment and treated
with 25 nM panobinostat and/or 50 μM ritonavir for 48 h. The cells
were then given fresh media and cultured for 1–2 weeks. The
colonies were fixed with 100% methanol, stained with Giemsa’s
solution and counted.
Murine xenograft model
The efficacy of the combination of ritonavir and
panobinostat in vivo was assessed using the murine
subcutaneous xenograft model. The procedures were performed
according to a protocol approved by the Institutional Animal Care
and Use Committee. Ten million Caki-1 cells were implanted
subcutaneously in nude mice purchased from CLEA Japan, Inc. (Tokyo,
Japan) and treatment was initiated 7 days later (day 1), when all
the mice exhibited measurable tumors. The mice were divided into
control and treatment groups (n=5 per group). The treated mice
received intraperitoneal injection of either panobinostat (2
mg/kg), or ritonavir (50 mg/kg), or both, while the control mice
received vehicle only. The injections were given once a day, 5 days
a week, for 2 weeks. Tumor growth was measured using a digital
caliper and tumor volume was calculated as volume = 0.5 × length ×
width2.
Annexin V assay
Induction of apoptosis was assayed using the Annexin
V assay. Briefly, 1.5×105 cells were seeded in a 6-well
culture plate 1 day prior to treatment. The cells were then
cultured in medium containing 25 nM panobinostat and/or 50 μM
ritonavir for 48 h, stained with Annexin V according to the
manufacturer’s protocol (Beckman Coulter, Marseille, France) and
analyzed by flow cytometry using the CellQuest Pro software (BD
Biosciences, San Jose, CA, USA). To evaluate whether the apoptosis
induced by the combination of ritonavir and panobinostat was
caspase-dependent, the cells were treated with 25 nM panobinostat
combined with 50 μM ritonavir, with or without 40 μM Z-VAD-FMK.
After 48 h, induction of apoptosis was evaluated with the Annexin V
assay using flow cytometry.
Western blotting
The changes in protein expression induced by the
combination were evaluated using western blotting. The cells were
treated under the indicated conditions for 48 h and whole-cell
lysates were obtained using radioimmunoprecipitation assay buffer.
The proteins were resolved on 12.5% SDS-polyacrylamide gels and
electrophoretically transferred to nitrocellulose membranes. After
the membranes were blocked by 5% (w/v) skimmed milk according to
standard procedures, they were first incubated with either
anti-acetylated histone (Abcam, Cambridge, UK), anti-HDAC1,
anti-HDAC3, anti-HDAC6 (Santa Cruz Biotechnology, Santa Cruz, CA,
USA) or anti-actin (Millipore, Billerica, MA, USA) primary
antibodies and subsequently with horseradish-tagged secondary
antibodies (Bio-Rad, Hercules, CA, USA). The bands were visualized
by chemiluminescence with the ECL Plus system (GE Healthcare,
Wauwatosa, WI, USA) according to the manufacturer’s
instructions.
Statistical analysis
CalcuSyn software (Biosoft, Cambridge, UK) was used
for calculating the combination indices. The statistical
significance of observed differences between samples was determined
using the Mann-Whitney U test (StatView software; SAS Institute,
Cary, NC, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Ritonavir and panobinostat
synergistically inhibited renal cancer cell growth
According to the MTS assay, the combination of
panobinostat and ritonavir effectively inhibited the growth of
renal cancer cells, while only slightly inhibiting the growth of
RPTECs (Fig. 1A). The
morphological changes in the renal cancer cells were notable (the
majority of the cells treated by the combination were floating),
while there was no apparent change in the RPTECs (Fig. 1B). Using the Chou-Talalay method,
an isobologram analysis was performed and combination indices were
calculated, demonstrating that the combined effect on cell growth
was synergistic (combination indices <1) under most treatment
conditions (Table I). We then
investigated whether the combination of ritonavir and panobinostat
affects the clonogenicity of renal cancer cells. The colony
formation assay revealed that this combination significantly
inhibited the clonogenic survival of renal cancer cells (Fig. 1C). Thus, the combination of
ritonavir with panobinostat was found to be effective in inhibiting
renal cancer cell growth in vitro.
 | Figure 1The combination of ritonavir and
panobinostat synergistically inhibited renal cancer cell growth,
while only slightly inhibiting renal proximal tubule epithelial
cells (RPTECs). (A) MTS assay after a 48-h treatment (n=6, mean ±
SD). The combination of ritonavir and panobinostat synergistically
inhibited renal cancer cell growth, while only slightly inhibiting
renal proximal tubule epithelial cell (RPTEC) growth. (B)
Photomicrographs of ACHN cells and RPTECs after a 48-h treatment
(original magnification, ×100). The majority of the ACHN cells
treated with the combination are floating. (C) Colony formation
assay. The cells were treated for 48 h under the indicated
conditions and allowed to grow for 1–2 weeks. C, control; P, 25 nM
panobinostat; and R, 50 μM ritonavir. *P=0.0495,
**P=0.0463, ***P=0.0431. (D) The combination
significantly suppressed tumor growth in vivo. A murine
xenograft model was established using Caki-1 cells. The control
group received intraperitoneal injections of dimethyl sulfoxide and
the treatment groups received 2 mg/kg panobinostat, or 50 mg/kg
ritonavir, or both. The injections were given once a day, 5 days a
week for 2 weeks. P=0.0472 at day 12. |
 | Table ICombination indices for ritonavir and
panobinostat in renal cancer cells. |
Table I
Combination indices for ritonavir and
panobinostat in renal cancer cells.
| Panobinostat
(nM) |
|---|
|
|
|---|
| Ritonavir (μM) | 10 | 25 | 50 |
|---|
| Caki-1 |
| 25 | 0.963 | 0.735 | 1.074 |
| 50 | 0.652 | 0.155 | 0.309 |
| ACHN |
| 25 | 0.858 | 0.874 | 1.108 |
| 50 | 1.034 | 0.852 | 0.177 |
| 769-P |
| 25 | 0.874 | 1.001 | 0.550 |
| 50 | 0.758 | 0.458 | 0.224 |
| 786-O |
| 25 | 1.456 | 1.245 | 0.921 |
| 50 | 1.204 | 0.775 | 0.081 |
Ritonavir combined with panobinostat
significantly suppressed renal cancer cell growth in a murine
xenograft model
Following the in vitro experiments, we
evaluated the efficacy of the combination of ritonavir and
panobinostat in vivo. In murine xenograft tumor models, a
10-day treatment with ritonavir and panobinostat was well tolerated
and significantly suppressed tumor growth (P=0.0472 at day 12)
(Fig. 1D). The average tumor size
at day 15 was 652±184 mm3 (mean ± standard error) in the
vehicle-treated mice and 220±67 mm3 in the
combination-treated mice.
Ritonavir combined with panobinostat
induced apoptosis
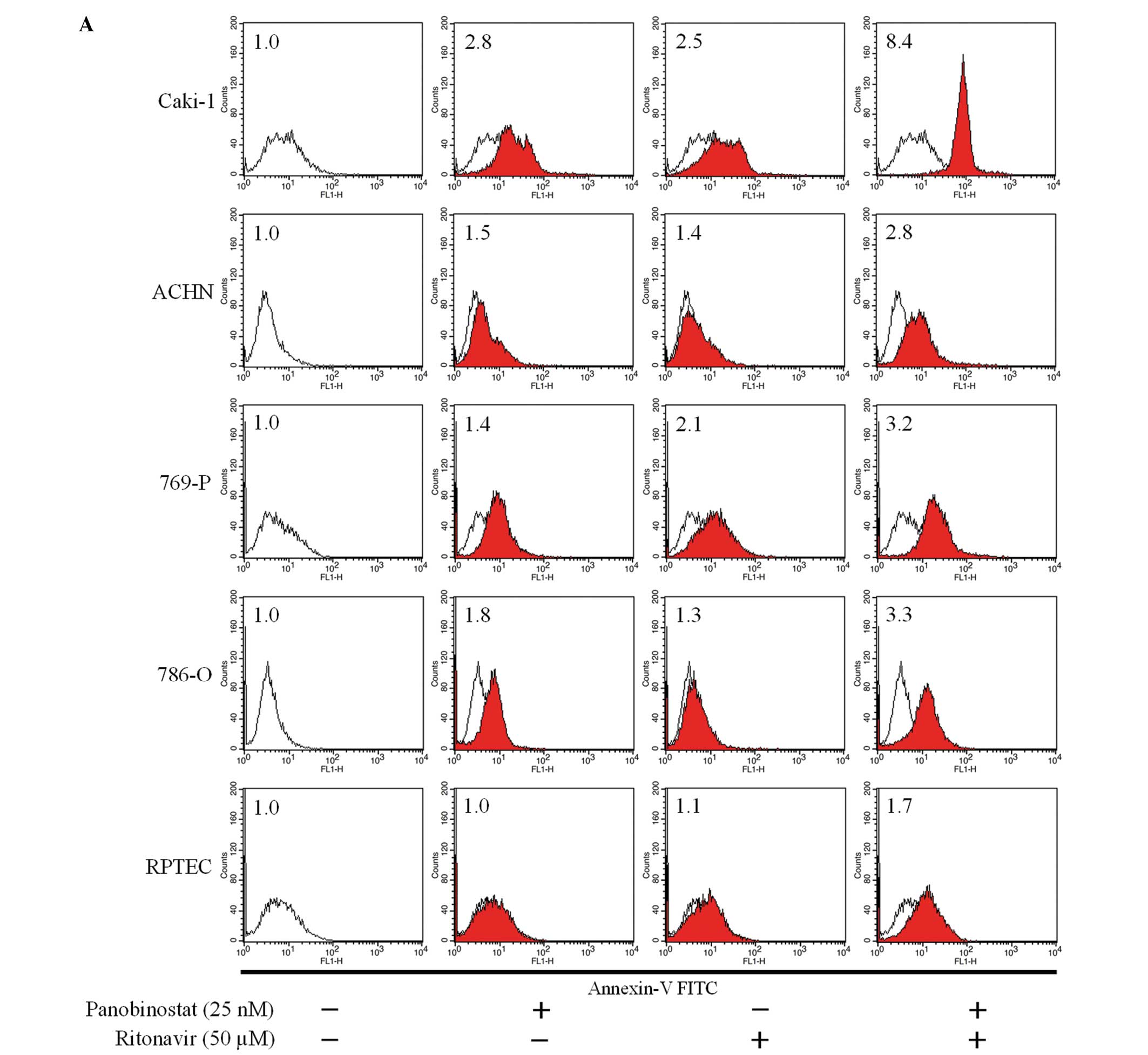
The combination of ritonavir and panobinostat
significantly increased Annexin V-fluorescein isothiocyanate (FITC)
fluorescence intensity in renal cancer cells and was thus shown to
induce apoptosis. In accordance with the results of the MTS assay,
this combination induced apoptosis only slightly in RPTECs
(Fig. 2A). We then investigated
whether the combination-induced apoptosis was caspase-dependent. In
Caki-1 and 769-P cells, co-incubation with the pan-caspase
inhibitor Z-VAD-FMK markedly reduced the degree to which the
combination increased Annexin V-FITC fluorescence intensity
(Fig. 2B), indicating that the
combination-induced apoptosis was indeed caspase-dependent.
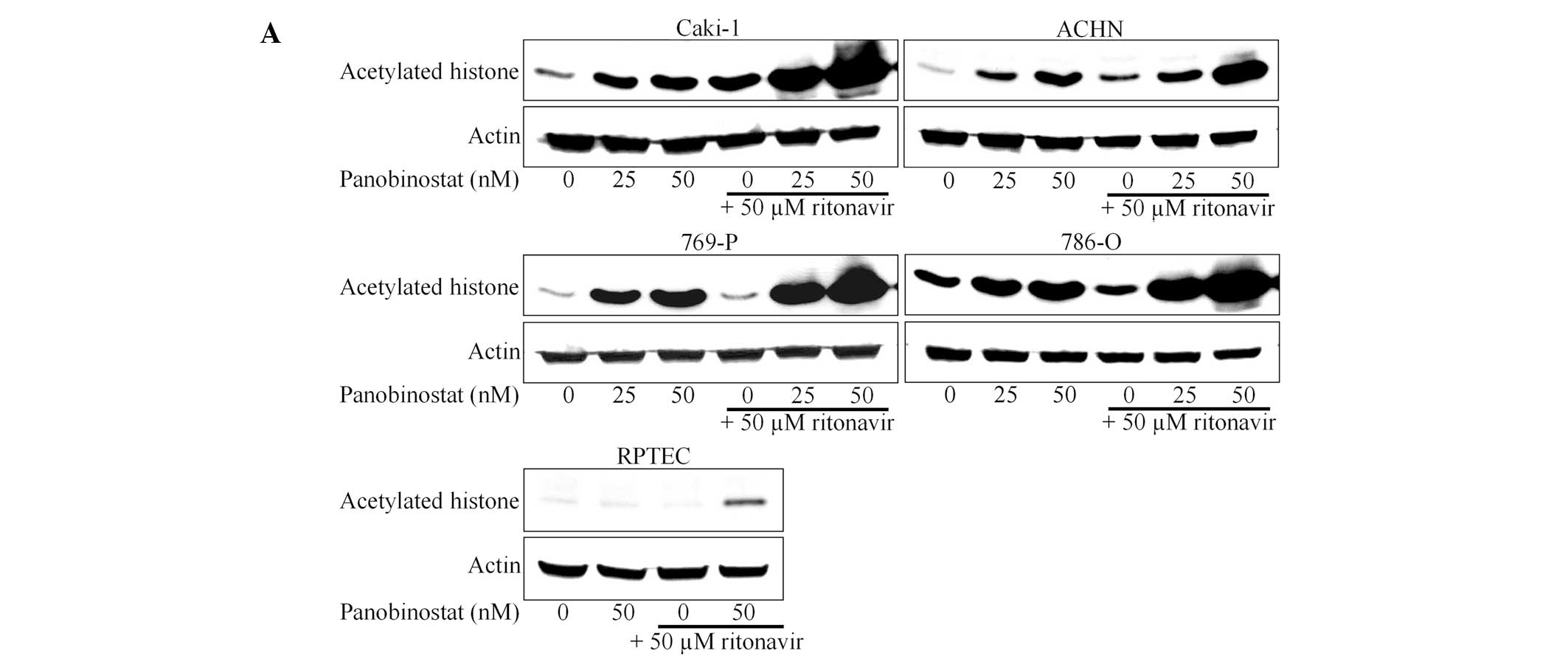
Ritonavir enhanced histone acetylation
induced by panobinostat
We next evaluated whether ritonavir enhanced the
activity of panobinostat. Since panobinostat is an HDAC inhibitor,
we hypothesized that the degree of induction of histone acetylation
reflects its activity. Panobinostat alone increased histone
acetylation in a dose-dependent manner and ritonavir
synergistically enhanced this acetylation (Fig. 3A). Thus, ritonavir was shown to
enhance the activity of panobinostat in renal cancer cells. Of
note, in RPTECs, even 50 nM panobinostat failed to induce histone
acetylation and the combined effect of ritonavir and panobinostat
was weaker compared to that in renal cancer cells. This is
consistent with the results of the MTS and the Annexin V assays.
Interestingly, this combination decreased the expression of HDACs
(Fig. 3B), which may also play a
role in enhancing histone acetylation.
Discussion
Targeted therapy using kinase inhibitors and
mammalian target of rapamycin inhibitors is a mainstay in the
treatment of metastatic renal cancer; however, a new treatment
approach is needed, as, although these inhibitors increase
progression-free survival to some extent, they are not
curative.
The acetylation and deacetylation of histones is
crucial in the modulation of chromatin structure (7). The levels of histone acetylation are
determined by the balance between the activities of histone
acetyltransferases and HDACs (8)
and deacetylation of histones tightens their interaction with DNA,
leading to a closed chromatin structure that inhibits gene
transcription (9). HDACs are
associated with a number of cellular oncogenes and tumor suppressor
genes (10); thus, compounds
targeting HDACs have attracted significant attention as anticancer
drugs (11). Panobinostat is one
such compound that has been clinically tested in patients with
refractory metastatic renal cell carcinoma (4). In that study, however, panobinostat
was not found to be effective. Since panobinostat is deactivated by
CYP3A4 (6), we hypothesized that
inhibiting this drug-eliminating machinery may enhance the activity
of panobinostat.
As expected, the combination of ritonavir and
panobinostat significantly induced apoptosis and synergistically
inhibited renal cancer cell growth, as shown by combination indices
of <1 under most treatment conditions. Of note, this combination
induced minimal apoptosis in RPTECs and only slightly inhibited
their growth, suggesting that it is advantageous in terms of side
effects, despite its drastic anticancer cell effects.
Panobinostat caused histone acetylation and this
acetylation was enhanced by ritonavir, which is consistent with the
hypothesis that ritonavir enhances the activity of panobinostat.
Furthermore, in RPTECs, even treatment with 50 nM panobinostat
failed to cause histone acetylation and the acetylation-enhancing
effect of the combination appeared to be weaker compared to that in
cancer cells. This result suggests that normal epithelial cells
tolerate panobinostat well and the present combination therapy acts
more specifically against renal cancer cells. Interestingly, we
also observed that the combination decreased the expression of
HDACs, which may be another important mechanism underlying its
enhancement of histone acetylation. This decreased HDAC expression
may also be a consequence of the enhanced histone acetylation, as
HDAC inhibitors themselves may decrease the expression of HDACs
(12,13).
Although the enhancement of panobinostat activity is
an important mechanism of action of ritonavir, the combination of
the two is considered to inhibit cancer growth by diverse
mechanisms. In the present study, ritonavir itself exhibited
antiproliferative activity against renal cancer cells, suggesting
that it may not only act as a CYP3A4 inhibitor. Ritonavir was
recently shown to exert antitumor effects through the inhibition of
proteins such as nuclear factor-κB (14) and heat shock protein (HSP) 90
(15) and it was also reported to
inhibit renal cancer growth by inhibiting heat shock factor 1, a
transcription factor of HSP 90, when used in combination with
17-allylamino-17-demethoxygeldanamycin (16). Furthermore, the inhibition of
HDACs, particularly HDAC6, acetylates HSP 90 and suppresses its
function as a molecular chaperone (17). It is considered that the
combination of ritonavir and panobinostat may cooperatively
suppress HSP 90, causing unfolded protein accumulation and,
thereby, endoplasmic reticulum stress. However, further study is
required to prove this mechanism.
The combination of ritonavir and panobinostat may be
one of the candidates for a clinical trial in patients with
advanced renal cancer. However, as CYP3A4 is also a major liver
enzyme catalyzing drug metabolism, there is a major concern that
ritonavir may increase the serum concentration of panobinostat
excessively and cause severe adverse events. However, a clinical
study using panobinostat with ketoconazole as a CYP3A4 inhibitor,
demonstrated that the combination increased the maximum
concentration (Cmax) of panobinostat 1.6-fold and the area under
the curve 1.8-fold, without significantly altering the time
required to reach Cmax or the half-life (6). The authors of that study concluded
that co-administration of panobinostat with CYP3A inhibitors is
feasible, as the increases in the parameters of panobinostat
pharmacokinetics were not clinically relevant. In addition,
considering our results that the combination of ritonavir and
panobinostat was not associated with lethal side effects in
vivo and affected the growth of RPTECs only slightly, the side
effects of this combination are expected to be minimal. Optimal
concentrations, however, must be carefully determined in phase I
trials with strict monitoring of the drugs’ serum
concentrations.
In conclusion, ritonavir enhanced the activity of
panobinostat and the combination of the two synergistically
inhibited renal cancer growth. The inhibition of the expression of
HDACs by this combination may further enhance histone acetylation.
To the best of our knowledge, this is the first study demonstrating
the beneficial combined effect of ritonavir and panobinostat on
renal cancer cells and it may provide a basis for clinical studies
with this combination in patients with advanced renal cancer.
References
|
1
|
Younes A, Sureda A, Ben-Yehuda D, et al:
Panobinostat in patients with relapsed/refractory Hodgkin’s
lymphoma after autologous stem-cell transplantation: results of a
phase II study. J Clin Oncol. 30:2197–2203. 2012.
|
|
2
|
Ghobrial IM, Campigotto F, Murphy TJ, et
al: Results of a phase 2 trial of the single-agent histone
deacetylase inhibitor panobinostat in patients with
relapsed/refractory Waldenström macroglobulinemia. Blood.
121:1296–1303. 2013.PubMed/NCBI
|
|
3
|
Morita S, Oizumi S, Minami H, et al: Phase
I dose-escalating study of panobinostat (LBH589) administered
intravenously to Japanese patients with advanced solid tumors.
Invest New Drugs. 30:1950–1957. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hainsworth JD, Infante JR, Spigel DR, et
al: A phase II trial of panobinostat, a histone deacetylase
inhibitor, in the treatment of patients with refractory metastatic
renal cell carcinoma. Cancer Invest. 29:451–455. 2011.PubMed/NCBI
|
|
5
|
Eagling VA, Back DJ and Barry MG:
Differential inhibition of cytochrome P450 isoforms by the protease
inhibitors, ritonavir, saquinavir and indinavir. Br J Clin
Pharmacol. 190–194. 1997.PubMed/NCBI
|
|
6
|
Hamberg P, Woo MM, Chen LC, Verweij J,
Porro MG, Zhao L, Li W, van der Biessen D, Sharma S, Hengelage T
and de Jonge M: Effect of ketoconazole-mediated CYP3A4 inhibition
on clinical pharmacokinetics of panobinostat (LBH589), an orally
active histone deacetylase inhibitor. Cancer Chemother Pharmacol.
68:805–813. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marks P, Rifkind RA, Richon VM, Breslow R,
Miller T and Kelly WK: Histone deacetylases and cancer: causes and
therapies. Nat Rev Cancer. 1:194–202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wade PA: Transcriptional control at
regulatory checkpoints by histone deacetylases: molecular
connections between cancer and chromatin. Hum Mol Genet.
10:693–698. 2001. View Article : Google Scholar
|
|
9
|
Grunstein M: Histone acetylation in
chromatin structure and transcription. Nature. 389:349–352. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cress WD and Seto E: Histone deacetylases,
transcriptional control, and cancer. J Cell Physiol. 184:1–16.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoo CB and Jones PA: Epigenetic therapy of
cancer: past, present and future. Nat Rev Drug Discov. 5:37–50.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hrzenjak A, Moinfar F, Kremser ML, et al:
Valproate inhibition of histone deacetylase 2 affects
differentiation and decreases proliferation of endometrial stromal
sarcoma cells. Mol Cancer Ther. 5:2203–2210. 2006. View Article : Google Scholar
|
|
13
|
Gui CY, Ngo L, Xu WS, Richon VM and Marks
PA: Histone deacetylase (HDAC) inhibitor activation of p21WAF1
involves changes in promoter-associated proteins, including HDAC1.
Proc Natl Acad Sci USA. 101:1241–1246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dewan MZ, Tomita M, Katano H, et al: An
HIV protease inhibitor, ritonavir targets the nuclear factor-kappaB
and inhibits the tumor growth and infiltration of EBV-positive
lymphoblastoid B cells. Int J Cancer. 124:622–629. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Srirangam A, Mitra R, Wang M, et al:
Effects of HIV protease inhibitor ritonavir on Akt-regulated cell
proliferation in breast cancer. Clin Cancer Res. 12:1883–1896.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sato A and Asano T, Ito K and Asano T:
17-allylamino-17-demethoxygeldanamycin and ritonavir inhibit renal
cancer growth by inhibiting the expression of heat shock factor-1.
Int J Oncol. 41:46–52. 2012.PubMed/NCBI
|
|
17
|
Bali P, Pranpat M, Bradner J, et al:
Inhibition of histone deacetylase 6 acetylates and disrupts the
chaperone function of heat shock protein 90: a novel basis for
antileukemia activity of histone deacetylase inhibitors. J Biol
Chem. 280:26729–26734. 2005. View Article : Google Scholar : PubMed/NCBI
|