Introduction
A congenital anomaly syndrome including Wilms'
tumor, aniridia, genitourinary anomalies and mental retardation
(WAGR syndrome) is a rare, sporadic genetic disorder characterized
by a de novo deletion in the distal band of chromosome 11p13
(1). WAGR syndrome is often
diagnosed when aniridia is clinically evident and a chromosomal
analysis is performed. The precise incidence of WAGR syndrome
remains unclear. The incidence of Wilms' tumor in Caucasians is
1/10,000, and a previous study reported that 0.75% of patients with
Wilms' tumor have WAGR syndrome (2);
thus, WAGR syndrome is an extremely rare condition. We encountered
a case where a pelvic tumor in a female patient with WAGR syndrome
was diagnosed as dysgerminoma. The tumor is considered to have
developed from an ectopic ovary, which is defined as ovarian tissue
found outside a normal ovary. An ectopic ovary is extremely rare,
with an incidence of 1/93,000 (3).
In addition, reports of an ovarian tumor forming from an ectopic
ovary are also rare (4–6).
Case report
The present case involved a 24-year-old nulliparous
woman with a height of 159 cm, weight of 89 kg and BMI of 35.2
kg/m2. Aniridia at birth led to the diagnosis of WAGR
syndrome. At the age of 7 months, left nephrectomy and
postoperative chemotherapy with vincristine and actinomycin D were
performed to treat Wilms' tumor. At the age of 7 years, glaucoma
surgery was performed. Since the age of 20 years, the patient has
been receiving oral medication at the Department of Pediatrics of
our hospital to treat diabetes mellitus, hypertension and
hyperuricemia. The family history was unremarkable. The patient was
investigated at the Department of Pediatrics for oliguria, and her
blood pressure was 220/130 mmHg. Blood collected at the time was
tested, revealing elevated levels of blood urea nitrogen (BUN; 45
mg/dl) and creatinine (Cr; 5.4 mg/dl). The patient was admitted to
the hospital with renal hypertension and acute renal failure.
Blood test results upon admission revealed a white
blood cell count of 6,100/µl, hemoglobin (Hb) level 10.7 g/dl,
platelet count 279,000/µl, aspartate aminotransferase level 21 U/l,
alanine aminotransferase level 34 U/l, lactate dehydrogenase 162
U/l, total cholesterol 214 mg/dl, triglycerides 298 mg/dl, BUN 45
mg/dl, Cr 5.4 mg/dl, uric acid 7.3 mg/dl, Na 144 mEq/l, K 4.1
mEq/l, Cl 104 mEq/l, Ca 9.1 mEq/l, blood glucose 101 mg/dl, HbA1c
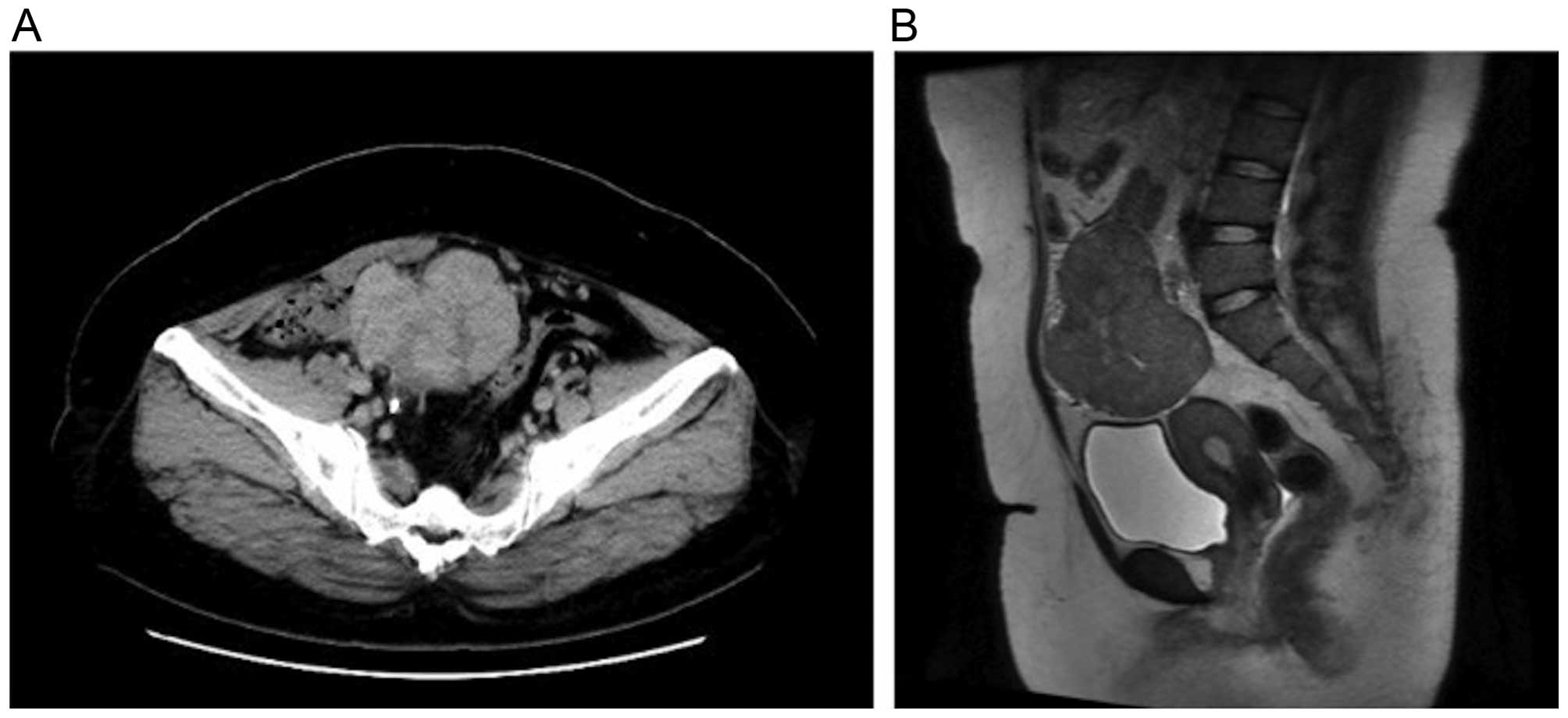
5.6% and C-reactive protein 2.52 mg/dl. A CT scan revealed a solid
mass with a long axis of ~80 mm in the pelvis. There were signs of
hydronephrosis in the right kidney due to ureteral compression by
the mass, which was hypothesized to have caused postrenal failure.
Placement of a ureteral stent in the right ureter alleviated
hydronephrosis; the urinary output and blood pressure were normal.
BUN and Cr returned to their previous levels and contrast-enhanced
CT was performed. The scan revealed a solid mass with mild contrast
enhancement that was heterogeneous (Fig.
1A). A tumor was hypothesized to have originated from the
adnexa, so the patient was referred to the Department of Obstetrics
and Gynecology. A magnetic resonance imaging scan revealed
hypointensity on T1-weighted images and a mixture of mildly
hypointense areas with some hyperintensity on T2-weighted images. A
solid tumor without a cystic component was identified; thus,
recurrence of Wilms' tumor, sex cord-stromal tumor and
leiomyosarcoma were considered in the differential diagnosis
(Fig. 1B). The levels of tumor
markers were as follows: Carbohydrate antigen (CA) 125 10 IU/ml,
CA19-9 4 U/ml, carcinoembryonic antigen 0.6 mg/ml, squamous cell
carcinoma antigen 1.2 ng/ml, neuron-specific enolase 31.8 ng/ml,
human chorionic gonadotropin <1.0 mIU/ml and α-fetoprotein 2.0
ng/ml.
The surgical plan was to treat a pelvic tumor
originating from the right adnexa. The left and right ovaries were
normal (Fig. 2A); the tumor was
located in the retroperitoneal space behind the right ovary
(Fig. 2B), and was not connected to
the ovary. The tumor was in contact with the right ureter, but
ureteral involvement was not observed. The greater omentum and
retroperitoneal space were dissected, the right adnexa was spared
and only the tumor was removed. The resected specimen was a solid
mass, which appeared whitish on cross sections (Fig. 3).
On histopathological examination, lymph node
invasion was observed by semicircular tumor cells with large
semicircular nuclei, and a ‘two-cell pattern’ (i.e., large tumor
cells and small mature lymphocytes) was evident. The nucleus of the
tumor cells contained 1–3 nucleoli (Fig.
4). On immunostaining, the tumor cells stained positive for
C-kit and Oct3, but negative for Wilms' tumor-1 and α-inhibin; 60%
of the tumor cells stained positive for Ki-67 (Fig. 5). The final diagnosis was
dysgerminoma. In addition, normal follicles were found in the tumor
tissue (Fig. 6); thus, the
dysgerminoma was hypothesized to have developed from an ectopic
ovary.
The patient's postoperative course was uneventful.
There was no solitary kidney, multiple complications, or residual
tumor, so she received 6 cycles of etoposide as postoperative
treatment and has remained recurrence-free in the 3 years since the
surgery.
The patient and her family provided their consent
regarding the publication of the case details.
Discussion
Ectopic ovary describes the presence of ovarian
tissue outside of a normal ovary. An ectopic ovary is extremely
rare, with an incidence of 1/93,000 (3). Depending on their location, ectopic
ovaries may be classified as ‘accessory’, which are adjacent or
connected to a normal ovary, or ‘supernumerary’, which indicates a
separate location. In 1959, Wharton et al reported 2 cases
of supernumerary ovaries and 1 case of an accessory ovary (7). Since that time, <40 cases of
supernumerary ovaries alone have been reported (8), with several reported cases of both
supernumerary and accessory ovaries. Lachman et al contended
that the principal causes of these conditions were i) ovarian
tissue that was detached during pelvic surgery and implanted
elsewhere, ii) ovarian tissue that was detached due to pelvic
inflammatory disease and implanted elsewhere, or iii) abnormally
located ovarian tissue due to an embryological anomaly (9). The rationale for this distinction is as
follows: Of the reported cases of an ectopic ovary due to ovarian
tissue that was detached during pelvic surgery or pelvic
inflammatory disease and implanted elsewhere, ~50% involved
salpingitis or a history of pelvic surgery or intra-abdominal
surgery. The same literature described cases of an ectopic ovary
due to developmental anomalies at the fetal stage. Primordial germ
cells migrate from the posterior wall of the yolk sac into the
hindgut epithelium and then to the urogenital ridge, where they
differentiate into ovarian tissue. When an anomaly occurs during
this process of cell migration, a congenital ectopic ovary is
formed. Ectopic ovaries may be found at varying sites, including
the perimetrium, intestinal tract, mesentery, retroperitoneal space
and greater omentum (8). In
addition, diagnosing an ectopic ovary prior to surgery is usually
difficult. There are case reports where an ectopic ovary was
suspected prior to surgery, when follicle stimulation with
clomiphene citrate resulted in follicle development within a tumor
(10). In reported cases of an
ectopic ovary where a tumor has formed, that tumor is most often a
mature teratoma, followed by serous or mucinous adenoma (8). In some cases of an ectopic ovary, a
Brenner tumor (4), Wilms' tumor
(5), or epithelial ovarian cancer
(6) have been reported. To the best
of our knowledge, there are currently no case reports of a
dysgerminoma developing in an ectopic ovary, as in the present
case.
It was previously reported that genitourinary or
genetic anomalies are identified in ~11–36% of the cases of an
ectopic ovary (5,9). In the aforementioned case of epithelial
ovarian cancer, the patient had Mayer-Rokitansky-Kuster-Hauser
syndrome. There are also case reports of an ectopic ovary and a
unicorn uterus (10,11). In the aforementioned case of a Wilms'
tumor, the patient had both congenital renal agenesis and an
ectopic ovary. The patient in our case was diagnosed with WAGR
syndrome and had a history of abdominal surgery for Wilms' tumor.
The possibility that the surgery was responsible for separating
ovarian tissue from an ovary is unlikely. We hypothesize that the
ectopic ovary developed as part of the WAGR syndrome.
The ultimate diagnosis of an ectopic ovary is based
on the presence of ovarian cortex, follicles, granulosa cells, or
theca cells (i.e., tissue specific to the ovaries) in a resected
tumor. In the present case, an initial histopathological
examination led to the diagnosis of dysgerminoma, although its
primary origin was not known. A more detailed examination revealed
the presence of follicles in the tumor. If a tumor is large, the
possibility that that tumor contains ovarian tissue may be
overlooked. In cases of an abdominal mass of unknown origin, the
possibility of a tumor developing from an ectopic ovary must be
taken into consideration, and a detailed histological examination
must be performed.
In conclusion, we herein present an extremely rare
case of dysgerminoma developing in an ectopic ovary in a patient
with WAGR syndrome. Identifying the presence of an ectopic ovary
prior to surgery is difficult, but there are reports of a
malignancy developing in patients with that condition. If this
condition is suspected, or a retroperitoneal tumor of unknown
origin is found in the abdomen, the possibility of an ectopic ovary
must be kept in mind, and a detailed histological examination must
be performed.
Acknowledgements
The present study was supported by a Grant-in-Aid
for Cancer Research from the Ministry of Education, Culture,
Sports, Science and Technology (Tokyo, Japan) (grant no. 20591935
to Dr Y. Yokoyama).
References
|
1
|
Riccardi VM, Sujansky E, Smith AC and
Francke U: Chromosomal imbalance in the Aniridia-Wilms' tumor
association: 11p interstitial deletion. Pediatrics. 61:604–610.
1978.PubMed/NCBI
|
|
2
|
Yi T, Weng J, Siwko S, Luo J, Li D and Liu
M: LGR4/GPR48 inactivation leads to aniridia-genitourinary
anomalies-mental retardation syndrome defects. J Biol Chem.
289:8767–8780. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Watkins BP and Kothari SN: True ectopic
ovary: A case and review. Arch Gynecol Obstet. 269:145–146. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heller DS, Harpaz N and Breakstone B:
Neoplasm arising in ectopic ovaries: A case of brenner tumor in an
accessory ovary. Int J of Gynecol Pathol. 9:185–189. 1990.
View Article : Google Scholar
|
|
5
|
Kini H, Baliga PB and Pai KG:
Supernumerary ovary associated with Wilms' tumor. Pediatr Surg Int.
13:67–68. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bae HS, Ryu MJ, Kim IS, Kim SH and Song
JY: Cancer of the supernumerary ovary in
Mayer-Rokitansky-Küster-Hauser Syndrome: A case report. Oncol Lett.
5:598–600. 2013.PubMed/NCBI
|
|
7
|
Wharton LR: Two cases of supernumerary
ovary and one of accessory ovary with an analysis of previously
reported cases. Am J Obstet Gynacol. 78:1101–1109. 1959.
|
|
8
|
El-Gohary Y, Pagkratis S, Lee T and
Scriven RJ: Supernumerary ovary presenting as a paraduodenal
duplication cyst. J Pediatr Surg Case Rep. 3:316–319. 2015.
View Article : Google Scholar
|
|
9
|
Lachman MF and Berman MM: The ectopic
ovary. A case report and review of the literature. Arch Pathol Lab
Med. 115:233–235. 1991.PubMed/NCBI
|
|
10
|
Uyar I, Gulhan I, Sipahi M, Hanhan HM and
Ozeren M: Ectopic ovary confirmed by ovarian stimulation in a case
of unicornuate uterus. Fertil Steril. 96:e122–e124. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ombelet W, Verswijvel G and de Jonge E:
Ectopic ovary and unicornuate uterus. N Engl J Med. 348:667–668.
2003. View Article : Google Scholar : PubMed/NCBI
|