Introduction
Autoimmune myelofibrosis (AIMF) is a benign disease
that was first described in 1994 as a distinct clinicopathological
entity associated with diffuse bone marrow fibrosis and autoimmune
phenomena (1). AIMF is mainly
encountered in patients with well-established autoimmune diseases,
such as systemic lupus erythematosus (SLE). In patients without
well-established autoimmune diseases, AIMF is defined as primary,
characteristically follows a benign course and responds well to
treatment with steroids and/or other immunosuppressive agents
(1). Immune thrombocytopenia (ITP)
is also an autoimmune disorder characterized by
antiplatelet-antibody-mediated thrombocytopenia in the absence of
other causes of thrombocytopenia (2). Bone marrow fibrosis has also been
reported in a proportion of ITP patients treated with
thrombopoietin receptor agonists (3). Although low-grade bone marrow fibrosis
[reticulin; grade 0–1 on the European Consensus scale (4)] has also been observed in newly
diagnosed ITP patients, but fibrosis of grade >1 in such
patients is very rare (5). We herein
report a rare case of a patient with severe thrombocytopenia
exhibiting grade 2 bone marrow fibrosis, who was diagnosed with
primary AIMF initially misdiagnosed as ITP.
Case report
A 52-year-old female patient with a 5-year history
of type 2 diabetes mellitus treated with voglibose presented to a
local hospital with extensive petechiae on her legs. The patient
was diagnosed with thrombocytopenia and referred to the Department
of Hematology, Eiju General Hospital (Tokyo, Japan). A laboratory
evaluation revealed pancytopenia with mild leukopenia and anemia,
in addition to marked thrombocytopenia. The white blood cell count
was 3.1×109/l (neutrophils: 48%, monocytes: 6% and
lymphocytes: 44%), there were no atypical cells on the blood smear,
the hemoglobin level was 109 g/l and the platelet count
was 4×109/l. The serum lactate dehydrogenase,
immunoglobulin (Ig)G, platelet-associated IgG and rheumatoid factor
levels were elevated to 251 U/l (normal, <223 U/l), 28.47 g/l
(normal, <17 g/l), 438 ng/107 cells (normal, <46
ng/107 cells), and 70 IU/ml (normal, 15 IU/ml),
respectively. In addition, the reticulated platelet count, plasma
thrombopoietin level and plasma transforming growth factor β
(TGF-β) level were significantly elevated to 13% (normal, <5%),
3.4 fmol/ml (normal range, 0.48±0.28 fmol/ml), and 23.7 ng/ml
(normal, <3.24 ng/ml), respectively. The antinuclear antibody
titer was within normal limits and no anti-GPIIb/IIIa
antibody-producing B cells were detected in the serum. Tests for
mutations in the Janus kinase-2 (JAK-2), thrombopoietin receptor
(MPL) and calreticulin (CALR) genes were all negative. Bone marrow
aspiration was attempted at the sternum and the left and right
posterior inferior iliac spines; however, only a dry tap was
obtained each time. Bone marrow biopsy from the left posterior
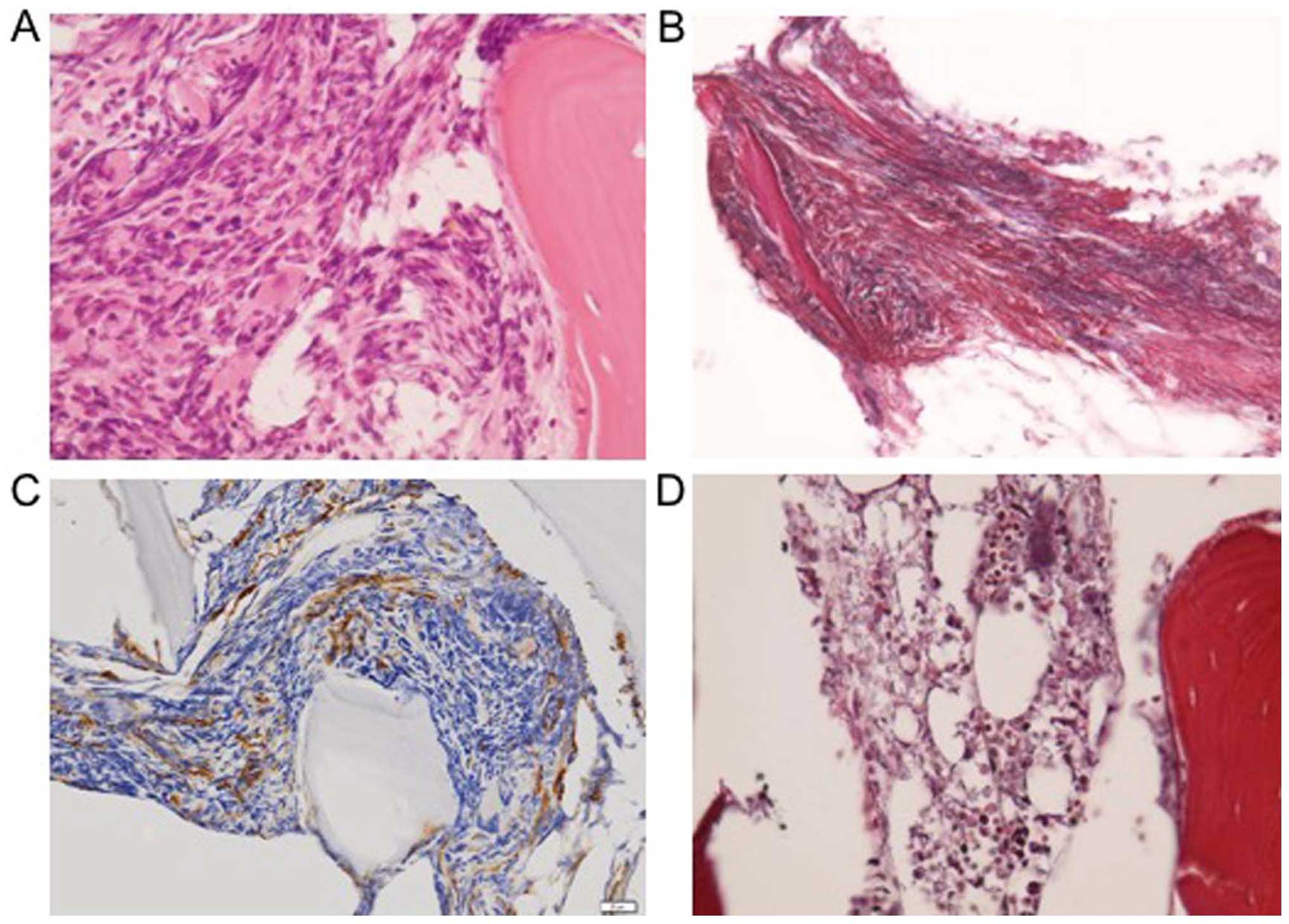
inferior iliac spine revealed a hypercellular bone marrow (Fig. 1A) with high-grade fibrosis (grade 2
on the European Consensus scale; Fig.
1B) and an increase in the number of megakaryocytes (Fig. 1C). Abdominal computed tomography (CT)
showed no splenomegaly or lymphadenopathy. On the basis of these
results, the patient was diagnosed with ITP complicated by bone
marrow fibrosis. After the patient received intravenous IgG
treatment (400 mg/kg for 5 days) from day 2 after admission, the
platelet count increased to 38×109/l on day 8, slowly
decreasing again thereafter to 3×109/l on day 24. On the
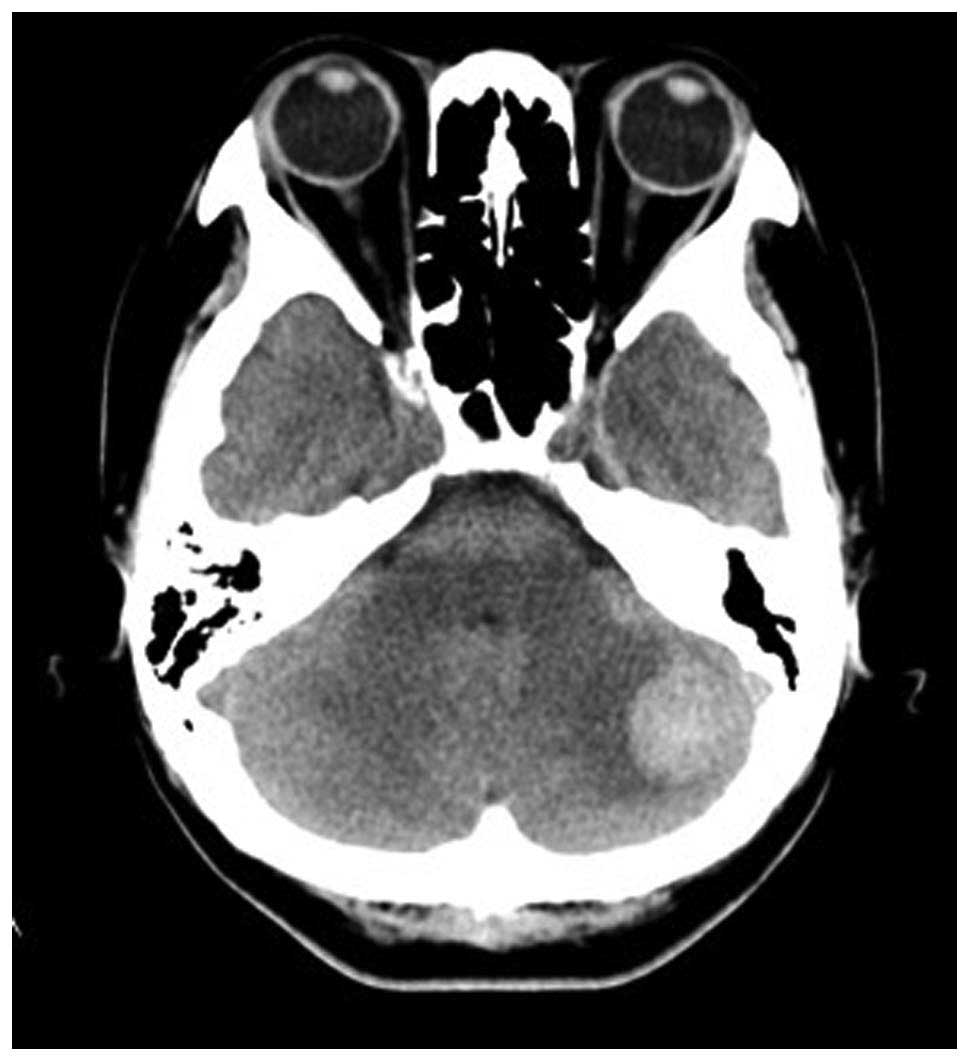
same day (day 24), the patient complained of headache and
dizziness, and was diagnosed with left cerebellar hemorrhage on
head CT (Fig. 2). Intravenous IgG
treatment was repeated at the same doses, followed by oral
administration of prednisolone at 1 mg/kg per day. Thereafter, the
platelet count promptly increased to normal within 7 days, and the
patient was discharged on day 66 after admission. The dose of
prednisolone was slowly tapered, and the treatment was discontinued
at 1 year from the diagnosis, while the patient was followed up on
an outpatient basis. A bone marrow aspirate from the left posterior
inferior iliac spine was repeated after complete withdrawal of the
prednisolone and revealed no abnormalities, and a repeat bone
marrow biopsy at 1 year from the diagnosis revealed no evidence of
bone marrow fibrosis (Fig. 1D). To
date, the patient has been followed up for 2 years without evidence
of disease. The patient provided written informed consent regarding
the publication of the case details.
Discussion
Myelofibrosis, encountered in association with
various benign and malignant disorders, is characterized by
reticulin fibrosis of the bone marrow with resultant myelophthisis.
Primary idiopathic myelofibrosis is well-known as a clonal
Philadelphia chromosome-negative myeloproliferative neoplasm
characterized by extramedullary hematopoiesis and marrow fibrosis,
with a proportion of the patients harboring mutations of the JAK-2,
MPL or CALR genes (6). Secondary
myelofibrosis may occur in various neoplastic disorders, such as
chronic myeloid leukemia, acute megakaryoblastic leukemia,
malignant lymphoma, myelodysplastic syndrome and bone marrow
metastasis (7). In addition to these
malignant causes, AIMF has also been described in association with
well-established autoimmune disorders, particularly SLE (1). Myelofibrosis encountered in cases
without a well-established autoimmune disorder or malignant disease
is defined as primary AIMF (8). The
diagnosis of primary AIMF is based on the presence of reticulin
fibrosis and lymphocyte infiltration of the bone marrow, presence
of autoantibodies (e.g., anti-nuclear antibody and rheumatoid
factor) in the serum, and absence of clustered or atypical
megakaryocytes, myeloid or erythroid dysplasia, eosinophilia or
basophilia, splenomegaly, or any other disorder known to cause
myelofibrosis (8). Although there
was a mild increase of the rheumatoid factor titer in the serum of
our patient, there were no symptoms, such as arthralgia, suggestive
of any of the well-established autoimmune disorders. On the basis
of the abovementioned criteria, the patient was diagnosed with
primary AIMF.
It was previously reported that primary AIMF shows a
favorable response to corticosteroid therapy (1). In our case, prednisolone was initiated
at 1 mg/kg/day and tapered over a 3-month period, achieving
complete normalization of the blood counts. Furthermore, a
follow-up bone marrow biopsy, performed after the steroid
treatment, revealed complete resolution of the bone marrow
fibrosis. The efficacy of the steroid treatment in our case was
consistent with previous reports (1,8).
It has been suggested that certain cytokines, such
as the fibrogenic cytokine TGF-β, the angiogenic cytokine basic
fibroblast growth factor, and tissue inhibitors of
metalloproteinase, may be involved in the development of bone
marrow fibrosis (9). Furthermore,
megakaryocytes and activated monocytes are considered to be
important sources of TGF-β, which induces fibroblast proliferation
(10). Our patient exhibited an
increase of the TGF-β concentration in the serum and of the
megakaryocyte counts in biopsy samples at diagnosis, with
normalization of the serum TGF-β (data not shown) after steroid
treatment. These observations support the hypothesis that TGF-β
generated by megakaryocytes is the most likely cause of marrow
fibrosis in AIMF.
Due to the severe thrombocytopenia in our patient,
the diagnosis of ITP was initially considered on admission. An
increased number of anti-GPIIb/IIIa antibody-producing B cells and
an elevated percentage of reticulated platelets in the peripheral
blood, as well as a normal or mildly increased plasma
thrombopoietin level in ITP cases have been previously reported,
which are recognized as useful adjuncts in the diagnosis of ITP
(11). However, our patient did not
exhibit any increase in the number of anti-GPIIb/IIIa
antibody-producing B cells in the blood, although an elevation in
the percentage of reticulated platelets and mild increase of the
plasma thrombopoietin level were observed. A mild increase in the
marrow reticulin content (grade 0–1 on the European Consensus
scale) has also been reported in untreated adult ITP patients, as
compared to the general adult population (5). Furthermore, bone marrow fibrosis has
been reported in some ITP patients treated with thrombopoietin
receptor agonists (3). However,
marrow reticulin fibrosis of grade >1 is very rare in primary
ITP. Moreover, our patient also exhibited a mildly elevated serum
rheumatoid factor titer, mild leukopenia and anemia, all of which
are well known to occur in AIMF patients (8), in addition to the severe
thrombocytopenia. Thus, the clinical characteristics in our patient
were more consistent with the diagnosis of primary AIMF rather than
ITP.
In conclusion, we herein present a rare case of a
female patient who was diagnosed with primary AIMF characterized by
severe thrombocytopenia, which was similar to the findings in ITP.
The disease symptoms, including pancytopenia and marrow fibrosis,
responded well to glucocorticoid therapy, with complete remission.
To evaluate the pathological mechanisms underlying this disease, a
larger number of cases must be accumulated in the future.
Acknowledgements
The authors would like to thank Dr Masaya Akatsuka
and Dr Masayuki Shimoda for providing the histopathological
findings and images, and Dr Hiroshi Yamaguchi for determining the
mutation status of the JAK-2, MPL and CALR genes.
References
|
1
|
Pullarkat V, Bass RD, Gong JZ, Feinstein
DI and Brynes RK: Primary autoimmune myelofibrosis: Definition of a
distinct clinicopathologic syndrome. Am J Hematol. 72:8–12. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McKenzie CG, Guo L, Freedman J and Semple
JW: Cellular immune dysfunction in immune thrombocytopenia (ITP).
Br J Haematol. 163:10–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brynes RK, Orazi A, Theodore D, Burgess P,
Bailey CK, Thein MM and Bakshi KK: Evaluation of bone marrow
reticulin in patients with chronic immune thrombocytopenia treated
with eltrombopag: Data from the EXTEND study. Am J Hematol.
90:598–601. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thiele J, Kvasnicka HM, Facchetti F,
Franco V, van der Walt J and Orazi A: European consensus on grading
bone marrow fibrosis and assessment of cellularity. Haematologica.
90:1128–1132. 2005.PubMed/NCBI
|
|
5
|
Rizvi H, Butler T, Calaminici M, Doobaree
IU, Nandigam RC, Bennett D, Provan D and Newland AC: United Kingdom
immune thrombocytopenia registry: Retrospective evaluation of bone
marrow fibrosis in adult patients with primary immune
thrombocytopenia and correlation with clinical findings. Br J
Haematol. 169:590–594. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tefferi A: Myeloproliferative neoplasms: A
decade of discoveries and treatment advances. Am J Hematol.
91:50–58. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gianelli U, Vener C, Bossi A, Cortinovis
I, Iurlo A, Fracchiolla NS, Savi F, Moro A, Grifoni F, De Philippis
C, et al: The European Consensus on grading of bone marrow fibrosis
allows a better prognostication of patients with primary
myelofibrosis. Mod Pathol. 25:1193–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vergara-Lluri ME, Piatek CI, Pullarkat V,
Siddiqi IN, O'Connell C, Feinstein DI and Brynes RK: Autoimmune
myelofibrosis: An update on morphologic features in 29 cases and
review of the literature. Hum Pathol. 45:2183–2191. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tefferi A: Pathogenesis of myelofibrosis
with myeloid metaplasia. J Clin Oncol. 23:8520–8530. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuter DJ, Bain B, Mufti G, Bagg A and
Hasserjian RP: Bone marrow fibrosis: Pathophysiology and clinical
significance of increased bone marrow stromal fibres. Br J
Haematol. 139:351–362. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kuwana M, Okazaki Y, Satoh T, Asahi A,
Kajihara M and Ikeda Y: Initial laboratory findings useful for
predicting the diagnosis of idiopathic thrombocytopenic purpura. Am
J Med. 118:1026–1033. 2005. View Article : Google Scholar : PubMed/NCBI
|