Introduction
Langerhans cell histiocytosis (LCH) is a rare
disease of unknown etiology characterized by mixed cellular
infiltration. Although the etiology of LCH has not been fully
elucidated, the gold standard for the diagnosis of LCH is the
presence of Birbeck granules, and positivity for CD1a, S-100 and/or
CD45 on pathological examination (1,2). LCH may
occur at any age, although it is more common in children, and has
various clinical manifestations, depending on the type and number
of systems or organs involved throughout the body (3).
Specific acute symptoms may include local pain,
weight loss, fatigue, fever, skin rash and neurological changes,
while the bone and lung are the most commonly involved organs
(4). It has been reported that the
proportion of LCH with lung involvement in adults is higher
compared with that in children (4).
Furthermore, the disease is self-limited in the majority of
pediatric patients, which is not the case in adults. A variety of
factors have been implicated in the prognosis of LCH, such as
patient age and extent of the disease. Several therapeutic
approaches may be considered, including surgery, radiotherapy and
chemotherapy; however, there is currently no standard treatment for
LCH patients with multisystem involvement (MS-LCH).
Case report
A 38-year-old male patient with a 2-year history of
left leg pain, involving numbness extending from the left thigh to
the knee, was referred to Nanjing Chest Hospital, Medical School of
Southeast University. The pain and numbness in the left thigh were
aggravated by exertion, such as walking or running, and they
improved by rest, although they did not completely resolve. The
patient had no history of tobacco or alcohol consumption, trauma or
any other significant medical conditions. On physical examination,
the patient appeared to be in a good overall condition.
Neurological examination revealed normal motor and sensory
function, symmetric reflexes, with no evidence of clonus,
fasciculations or ataxia. Other findings on physical examination
were unremarkable. On admission, the immunoglobulin (Ig) laboratory
tests were as follows: IgG, 15.1 g/l (normal range, 7–16 g/l); IgA,
3.87 g/l (normal range, 0.7–4.0 g/l); and IgM, 1.16 g/l (normal
range, 0.4–2.3 g/l). The erythrocyte sedimentation rate (normal
range, 0–20 mm/h) and high-sensitivity C-reactive protein levels
(0–1.0 mg/l) were mildly elevated (31 mm/h and 44.5 mg/l,
respectively). Blood cell count, urinalysis, serum chemistry,
electrolytes and tumor markers were within normal ranges. pulmonary
function is frequently normal at presentation.
The images obtained by a multidetector computed
tomography (CT) device were reconstructed in a variety of planes
and interpreted in soft tissue, bone or lung windows. Pulmonary and
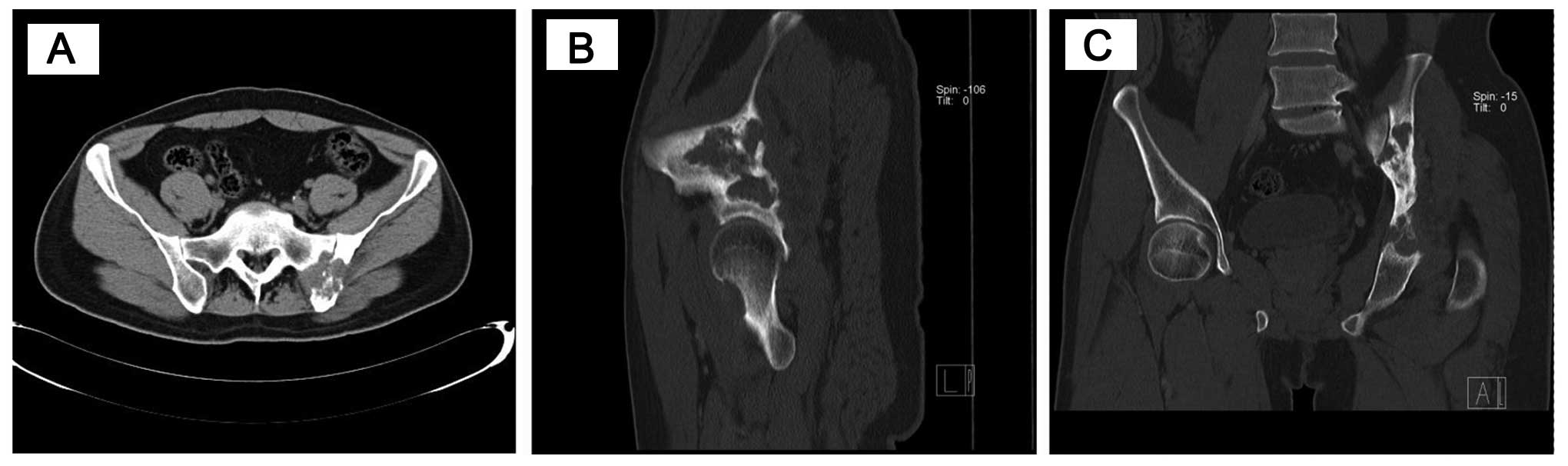
pelvic diagnostic CT examination revealed the distribution of the
disease: The corpus of the sacroiliac joint, sacrum, acetabulum and
femoral head were occupied by a destructive soft tissue mass
(Fig. 1).
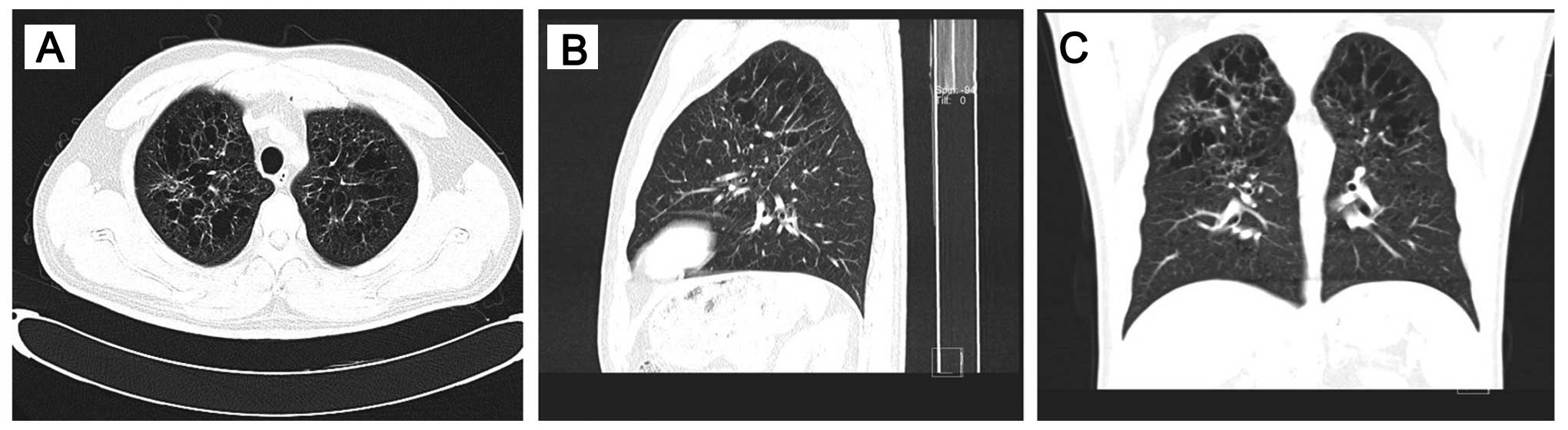
In addition, high-resolution ct (HRCT) of the chest
revealed bilateral, diffuse infiltration of the lungs by numerous
cysts, mainly distributed in the upper and middle lung fields,
while the surrounding pulmonary parenchyma was normal (Fig. 2). A CT-guided percutaneous needle
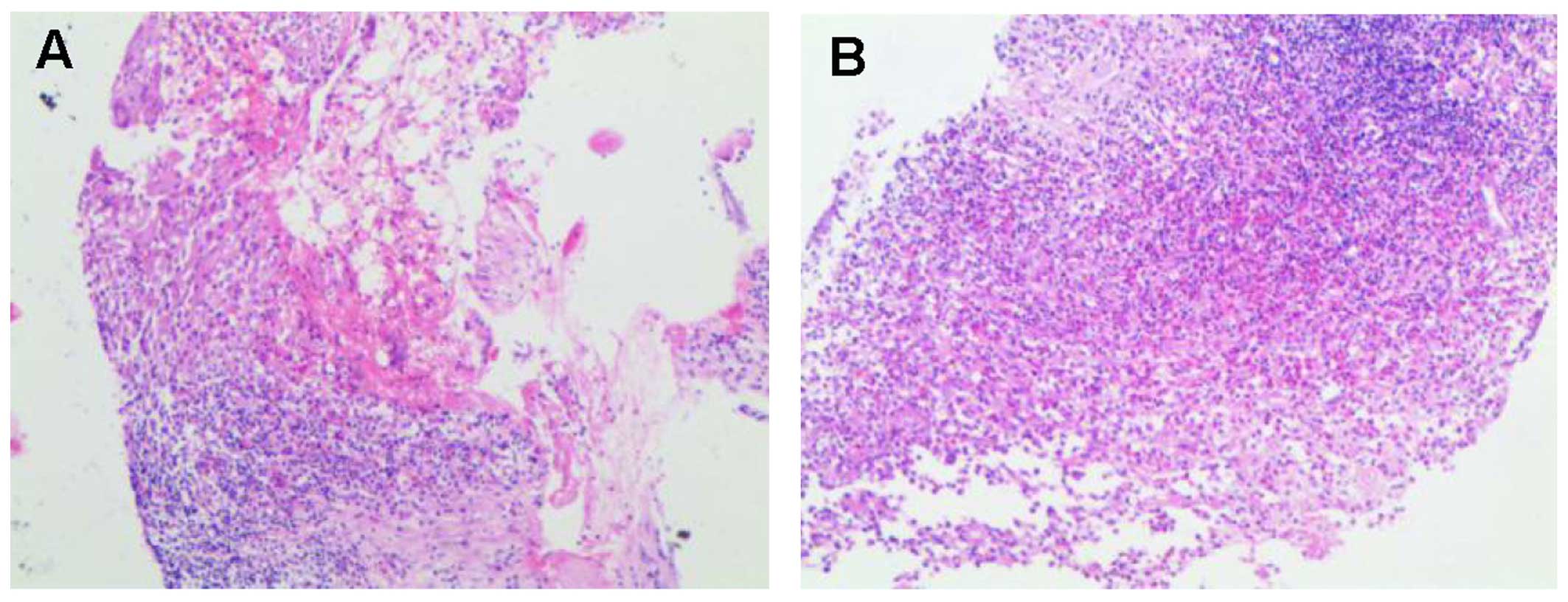
biopsy was performed and the results confirmed the diagnosis of
LCH. Histopathologically, a heterogeneous admixture of inflammatory
cells, including numerous eosinophils, rare multinucleated giant
cells, mononuclear cells and foam cells, was observed (Fig. 3). The histopathological examination
revealed proliferation of histiocytes with an infiltration by
eosinophils. Immunohistochemically, the histiocytes were S-100+,
CD163+, CD1a+, CD68+ (partially), vimentin+, Ki-67+ (~15%) and
langerin+. Although the patient was diagnosed with LCH, the
aggressive appearance of a destructive lesion on CT images made it
necessary to conduct a biopsy in order to establish a definitive
diagnosis. there was no evidence of involvement of any other organ
on magnetic resonance imaging (MRI) of the head and abdomen.
Corticosteroid treatment was administered to the patient. No
further treatment was recommended, such as combination
chemotherapy, surgery or radiotherapy. Corticosteroid therapy was
continued for a few months. There has been no evidence of disease
progression during follow-up.
Written informed consent was obtained from the
patient regarding the publication of this case report and any
accompanying images.
Discussion
LCH, formely referred to as histiocytosis X, is
diagnosed according to clearly established criteria. LCH is a rare,
complex and poorly understood disorder that is associated with a
variety of clinical manifestations. LCH is characterized by the
abnormal proliferation of bone marrow-derived Langerhans cells and
may be divided into three types: Eosinophilic granuloma,
Hand-Schuller-Christian disease and Lettere-Siwe disease (5,6).
Histopathological examination is crucial for the diagnosis of LCH,
and immunoreactivity for S-100 and CD1a, as well as the presence of
Birbeck granules on electron microscopy examination, provide
helpful information (7–9), as does the presence of CD45,
CD56-positive cells (10,11).
LCH is generally considered to be a pediatric
disease and more frequently occurs in males (5,6,12). Adults are rarely diagnosed with LCH,
with an estimated annual incidence of 1–2 cases per million
individuals (6). However, the
patient in the present case was aged 38 years. The clinical
presentation of LCH is highly variable. In addition, a
stratification system depending on the extent and localization of
the disease at the time of evaluation was adopted by the LCH study
group, which classified the disease into single-system LCH (SL-LCH)
and MS-LCH (13); the latter may be
further subdivided into low- and high-risk MS-LCH (14,15). We
herein report a rare adult case of LCH presenting with lung and
bone involvement, which was classified as high-risk MS-LCH. LCH
accounts for <1% of tumor-like bone lesions (16). Adult LCH patients are often afflicted
by chronic pain, such as back pain, and fatigue of undetermined
etiology. The pain may localize to specific sites, despite the lack
of an identifiable pathology. The most frequent sites are the skull
(26%) and jaw (9%), followed in decreasing order of frequency by
the long tubular bones, pelvis, ribs and the spine (17–19);
rarely, the lesion may be located in the pelvis (20).
When a bone lesion is encountered, LCH may not be
considered in the differential diagnosis, as this disease is rare
and may manifest as a heterogeneous spectrum of lesions, ranging
from a single bone lesion to MS-LCH. CT and MRI are effective
approaches to delineating the extent of osseous destruction and
soft tissue involvement. In adults with LCH confirmed by biopsy, a
chest CT is necessary to assess the extent of the disease at
presentation, along with laboratory evaluation. However, a bone
biopsy is required for the diagnosis of LCH in certain cases, in
order to rule out other types of lesions, such as hypoadrenalism,
metastatic carcinoma, lymphoma/myeloma, osteomyelitis,
osteoblastoma, aneurysmal bone cyst and Ewing's sarcoma (21).
In the majority of the cases, LCH in adults
manifests as a widespread, multisystem disease, with the lung being
the most common site of extraosseous involvement. The high
proportion of cases with pulmonary involvement is the most
conspicuous difference between adult and pediatric LCH (4). Although ≤20–30% of adult LCH patients
have isolated pulmonary lesions, the occurrence of LCH in the
pelvis, either as a primary isolated manifestation of the disease
or as part of systemic disease, is rare. In adults, lung
involvement in LHC often coexists with involvement of other
tissues/organs, is poorly recognized and frequently overlooked.
Common symptoms of pulmonary LCH (PLCH) include cough, dyspnea and
chest pain, and they are crucial for diagnosis. However, ~25% of
patients with PLCH are asymptomatic and the disease is diagnosed
due to incidentally discovered radiographic abnormalities (22), as was the case in our patient.
Although the clinical manifestations and imaging
findings of PLCH are different in each stage of the disease, HRCT
was reported to be important in the diagnosis of PLCH (23), and familiarity with characteristic
imaging findings is required for accurate and timely diagnosis. In
addition, imaging methods (particularly HRCT) play a fundamental
role in diagnosing and assessing the stage of LCH, ensuring
administration of appropriate treatment and patient monitoring. In
the early stages of the disease, the typical manifestation of PLCH
on radiological examination is diffuse bilateral and symmetric
ill-defined small or cavitated nodules (both of which may resolve),
ranging from 2 to 10 mm in diameter and predominantly located in
the middle or upper pulmonary fields, with sparing the costophrenic
angles. However, other diseases may present with similar findings
in the lungs, and the differential diagnosis must include
sarcoidosis, silicosis, metastases, miliary tuberculosis,
bronchiectasis and emphysema. Furthermore, the imaging presentation
of PLCH may vary at clinical onset. If the lesions persist or
progress, classic imaging findings typically include small
centrilobular nodules or thick- and thin-walled cysts of varying
sizes, with mid- to upper-lung distribution. Although LCH occurs in
patients below 24 years of age (24), however, the cavitation and the
development of cysts is diagnosed at a later stage of the disease
progression.
The treatment and prognosis, as well as the clinical
course of LCH, remain to be fully determined, particularly in adult
patients (25). Unlike LCH in
children, due to the current lack of standard therapeutic regimens
for adult LCH, age, extent of the disease and dysfunction of vital
organs are considered as the major factors affecting the selection
of the optimal treatment. Lung involvement is almost always
associated with smoking (21,26);
however, the present case indicated that cigarette smoking may not
always be associated with the pathogenesis of PLCH. It was reported
that LCH may be successfully treated by controlling the patient's
smoking habits (27); however,
smoking cessation is not always effective (28). There are several suggested potential
mechanisms through which smoking may promote PLCH in certain
individuals, although the association between cigarette smoking and
progression or regression of adult LCH has not been fully
elucidated. Particularly in non-smokers, the underlying mechanisms
of LCH remain to be determined. novel therapies for LCH are
urgently required, as smoking cessation only showed limited
efficacy. Other treatments, such as corticosteroids, have been
reported in adult LCH. The corticosteroid regimen used in the
present case was selected based on case series reports of
successful treatment (28,29); however, the efficacy of such
treatment in non-smoking adults has not been determined. In the
present case, corticosteroids were used due to their efficacy in
adults with MS-LCH, as LCH lesions do not progress following
corticosteroid therapy, although cystic disease may be
irreversible. In the classic presentation of isolated pulmonary LCH
in non-smoking adults, the condition may resolve with
corticosteroids alone. If the treatment effect is not significant,
chemotherapy with cladribine may be required (30,31).
However, although different regimens have exhibited some efficacy
in the treatment of adult MS-LCH (32), no specific intervention has shown any
benefit with regard to patient survival (33), with rather low complete or partial
remission rates (34). The mortality
rate of patients with LCH receiving no treatment is high, and the
survival probability at 5 years post-diagnosis of patients with
MS-LCH is mildly inferior to that of patients with SL-LCH (4).
As the mechanisms underlying LCH remain unclear,
further studies with a large sample size are required to avoid
misdiagnosis in clinical practice.
LCH is a rare disease with a variable clinical
presentation. Systematic analysis of clinical presentation, imaging
findings and immunohistochemistry is necessary to accurately
diagnose LCH. Bone involvement is common in adults with LCH and may
be indicative of SL-LCH or MS-LCH. chronic pain may be the first or
sole manifestation of the disease. As the lungs are the most common
site of extraosseous involvement, LCH should be considered in
smokers as well as in non-smokers. Imaging, particularly HRCT, is
key to the diagnosis and is useful for detecting lung injury at
each stage of pulmonary involvement in LCH, from small
centrilobular nodules to diffuse cysts and fibrosis. accurate
diagnosis is crucial for improving prognosis. In the present case,
corticosteroids were used for the treatment of LCH, achieving a
positive outcome.
References
|
1
|
Herwig MC, Wojno T, Zhang Q and
Grossniklaus HE: Langerhans cell histiocytosis of the orbit: Five
clinicopathologic cases and review of the literature. Surv
Ophthalmol. 58:330–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kasper EM, Aguirre-Padilla DH, Alter RY
and Anderson M: Histiocytosis X: Characteristics, behavior and
treatments as illustrated in a case series. Surg Neurol Int.
2:572011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Blesa JM Gasent, Candel V Alberola, Vercet
C Solano, Canales J Laforga, Semler C, Pérez Antolí MR and
Rodríguez-Galindo C: Langerhans cell histiocytosis. Clin Transl
Oncol. 10:688–696. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aricò M, Girschikofsky M, Généreau T,
Klersy C, McClain K, Grois N, Emile JF, Lukina E, De Juli E and
Danesino C: Langerhans cell histiocytosis in adults. Report from
the International Registry of the Histiocyte Society. Eur J Cancer.
39:2341–2348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grana N: Langerhans cell histiocytosis.
Cancer Control. 21:328–334. 2014.PubMed/NCBI
|
|
6
|
El Demellawy D, Young JL, De Nanassy J,
Chernetsova E and Nasr A: Langerhans cell histiocytosis: A
comprehensive review. Pathology. 47:294–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pileri SA, Grogan TM, Harris NL, Banks P,
Campo E, Chan JK, Favera RD, Delsol G, De Wolf-Peeters C, Falini B,
et al: Tumours of histiocytes and accessory dendritic cells: An
immunohistochemical approach to classification from the
International lymphoma study group based on 61 cases.
Histopathology. 41:1–29. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herwig MC, Wojno T, Zhang Q and
Grossniklaus HE: Langerhans cell histiocytosis of the orbit: Five
clinicopathologic cases and review of the literature. Surv
Ophthalmol. 58:330–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Histiocytosis syndromes in children, .
Writing Group of the histiocyte society. Lancet. 1:208–209.
1987.PubMed/NCBI
|
|
10
|
Kawase T, Hamazaki M, Ogura M, Kawase Y,
Murayama T, Mori Y, Nagai H, Tateno M, Oyama T, Kamiya Y, et al:
CD56/NCAM-positive langerhans cell sarcoma: A clinicopathologic
study of 4 cases. Int J Hematol. 81:323–329. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kasper EM, Aguirre-Padilla DH, Alter RY
and Anderson M: Histiocytosis X: Characteristics, behavior and
treatments as illustrated in a case series. Surg Neurol Int.
2:572011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Allen CE, Ladisch S and McClain KL: How i
treat Langerhans cell histiocytosis. Blood. 126:26–35. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abla O, Egeler RM and Weitzman S:
Langerhans cell histiocytosis: Current concepts and treatments.
Cancer Treat Rev. 36:354–359. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Allen CE and McClain KL: Langerhans cell
histiocytosis: A review of past, current and future therapies.
Drugs Today (Barc). 43:627–643. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lau EG, Stepenaskie S, Moran R, Quinn R,
Matthew P and Smidt AC: ‘Blueberry muffin’ rash and large right
thigh mass: A unique presentation of Langerhans cell histiocytosis.
Dermatol Online J. 19:185682013.PubMed/NCBI
|
|
16
|
Wells PO: The button sequestrum of
eosinophilic granuloma of the skull. Radiology. 67:746–747. 1956.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stockschlaeder M and Sucker C: Adult
Langerhans cell histiocytosis. Eur J Haematol. 76:363–368. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kilpatrick SE, Wenger DE, Gilchrist GS,
Shives TC, Wollan PC and Unni KK: Langerhans' cell histiocytosis
(histiocytosis X) of bone. A clinicopathologic analysis of 263
pediatric and adult cases. Cancer. 76:2471–2484. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bertram C, Madert J and Eggers C:
Eosinophilic granuloma of the cervical spine. Spine (Phila Pa
1976). 27:1408–1413. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi S, Liu Y, Fu T, Li X and Zhao S:
Multifocal Langerhans cell histiocytosis in an adult with a
pathological fracture of the mandible and spontaneous malunion: A
case report. Oncol Lett. 8:1075–1079. 2014.PubMed/NCBI
|
|
21
|
Haupt R, Minkov M, Astigarraga I, Schäfer
E, Nanduri V, Jubran R, Egeler RM, Janka G, Micic D,
Rodriguez-Galindo C, et al: Langerhans cell histiocytosis (LCH):
guidelines for diagnosis, clinical work-up and treatment for
patients till the age of 18 years. Pediatr Blood Cancer.
60:175–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Satter EK and High WA: Langerhans cell
histiocytosis: A review of the current recommendations of the
Histiocyte Society. Pediatr Dermatol. 25:291–295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tazi A, Soler P and Hance AJ: Adult
pulmonary Langerhans' cell histiocytosis. Thorax. 55:405–416. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martin I, Ballester M, Ruiz Y, Llatjós R,
Alarza F and Molina M: Presentation of pulmonary Langerhans cell
histiocytosis before the development of lung cysts. Respirol Case
Rep. 1:34–35. 2013.PubMed/NCBI
|
|
25
|
Arico M: Langerhans cell histiocytosis in
adults: More questions than answers? Eur J Cancer. 40:1467–1473.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tazi A, Soler P and Hance AJ: Adult
pulmonary Langerhans' cell histiocytosis. Thorax. 55:405–416. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suri HS, Yi ES, Nowakowski GS and Vassallo
R: Pulmonary langerhans cell histiocytosis. Orphanet J Rare Dis.
7:162012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nquyen K and Tazi A: Langerhans cell
histiocytosis in adults. Rev Prat. 56:1863–1871. 2006.(In French).
PubMed/NCBI
|
|
29
|
Tazi A: Adult pulmonary Langerhans' cell
histiocytosis. Eur Respir J. 27:1272–1285. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lorillon G, Bergeron A, Detourmignies L,
Jouneau S, Wallaert B, Frija J and Tazi A: Cladribine is effective
against cystic pulmonary Langerhans cell histiocytosis. Am J Respir
Crit Care Med. 186:930–932. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grobost V, Khouatra C, Lazor R, Cordier JF
and Cottin V: Effectiveness of cladribine therapy in patients with
pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis.
9:1912014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Minkov M, Grois N, Heitger A, Pötschger U,
Westermeier T and Gadner H: Treatment of multisystem Langerhans
cell histiocytosis. Results of the DAL-HX 83 and DAL-HX 90 studies.
DAL-HX Study Group. Klin Padiatr. 212:139–144. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vassallo R, Ryu JH, Schroeder DR, Decker
PA and Limper AH: Clinical outcomes of pulmonary Langerhans'-cell
histiocytosis in adults. N Engl J Med. 346:484–490. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baumgartner I, Von Hochstetter A, Baumert
B, Luetolf U and Follath F: Langerhans'-cell histiocytosis in
adults. Med Pediatr Oncol. 28:9–14. 1997. View Article : Google Scholar : PubMed/NCBI
|