Introduction
A neuroendocrine tumor is defined as an epithelial
neoplasm that exhibits neuroendocrine differentiation when analyzed
by conventional histological, immunohistochemical, ultrastructural
and biological evaluations (1).
Neuroendocrine tumors of the ampulla of Vater are extremely rare,
accounting for <2% of all ampullary malignancies (2). Therefore, the natural history of this
disease entity remains to be established, and its clinical behavior
is unknown. Cushing's syndrome, secondary to ectopic
adrenocorticotropic hormone (ACTH) secretion, was first described
in 1928 and subsequently reported in association with various
neuroendocrine tumors (3). The most
common primary site of these tumors is the lung, followed by the
thymus, pancreas and thyroid (4).
Cushing's syndrome arising from the ampulla of Vater has not, to
the best of our knowledge, been reported previously. The present
study reported the first case of ectopic ACTH secretion from a
neuroendocrine tumor of the ampulla of Vater. Written informed
consent was obtained from the patient's family.
Case report
A 69-year-old female presented with general fatigue
and edema of the face and legs lasting for 2 months. The patient
had not taken any hormones or drugs, nor had she undergone surgery.
Physical examination revealed moon face and leg edema. Other
physical examination parameters were normal, including blood
pressure. Laboratory parameters revealed hypokalemia, with a serum
potassium level of 2.5 mmol/l, and slight hyperglycemia, with a
fasting blood glucose level of 154 mg/dl. The level of HbA1c was
elevated to 6.5% (normal range, <6%). Further investigations
revealed hypercortisolemia and high levels of ACTH and cortisol
(98.2 µg/dl and 567.1 pg/ml, respectively). Plasma cortisol levels
at 8 a.m. following overnight 1 and 8 mg dexamethasone suppression
tests were 100.4 and 111.0 µg/dl, respectively. In view of the high
ACTH levels in conjunction with elevated cortisol levels that
failed to be suppressed by dexamethasone, the present study
diagnosed ectopic ACTH-producing disease. Tumor marker
determination revealed increased levels of carcinoembryonic
antigen, carbohydrate antigen 19–9 and neuronal-specific enolase
(Table I).
 | Table I.Laboratory data. |
Table I.
Laboratory data.
| Variable | Value | Reference range |
|---|
| Potassium | 2.5 | 3.5–4.5 mmol/l |
| Blood glucose | 154 | 70–109 mg/dl |
| HbA1c | 6.5 | <6.0% |
| Plasma cortisol | 98.2 | 4.5–21.1 µg/dl |
| ACTH | 567.1 | 7.4–55.7 pg/ml |
| Plasma cortisol after
1 mg DEX test | 100.4 | ≤3.0 µg/dl |
| Plasma cortisol after
8 mg DEX test | 111.0 | ≤1.0 µg/dl |
| CEA | 12.1 | <5.0 ng/ml |
| CA19–9 | 101.9 | <37.0 U/ml |
| NSE | 27.8 | <10.0 ng/ml |
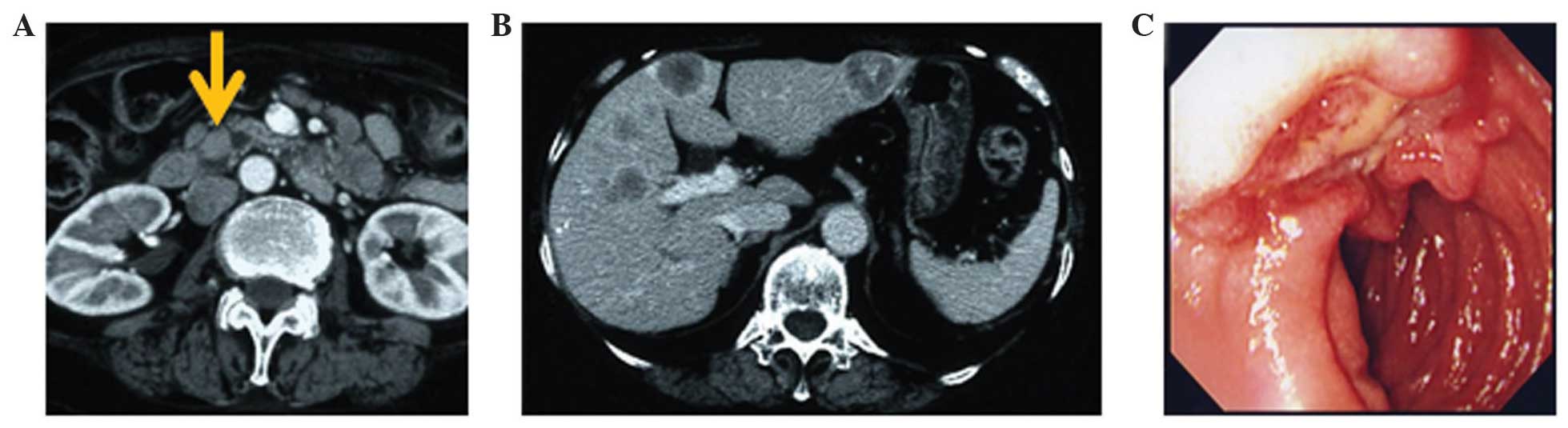
The ultrasonography findings were consistent with
multiple liver tumors. Contrast-enhanced computed tomography (CT)
revealed periampullary growth (15 mm), dilated common bile duct and
main pancreatic duct, masses in both lobes of the liver and
hyperplasia of the adrenal glands (Fig.
1A and B). Magnetic resonance cholangiopancreatography revealed
a soft tissue signal shadow located in the ampulla region,
associated with dilation of both the common bile duct and main
pancreatic duct. An 18F-flurodeoxyglucose positron
emission tomography-CT scan demonstrated an intense uptake of the
radiotracer corresponding to the ampulla of Vater, multiple liver
deposits and both adrenal glands, however not in any other sites.
Head CT revealed no tumor.
The upper gastrointestinal endoscopy revealed a
macroscopic ulceration-like lesion at the duodenal papilla
(Fig. 1C). Biopsies were obtained
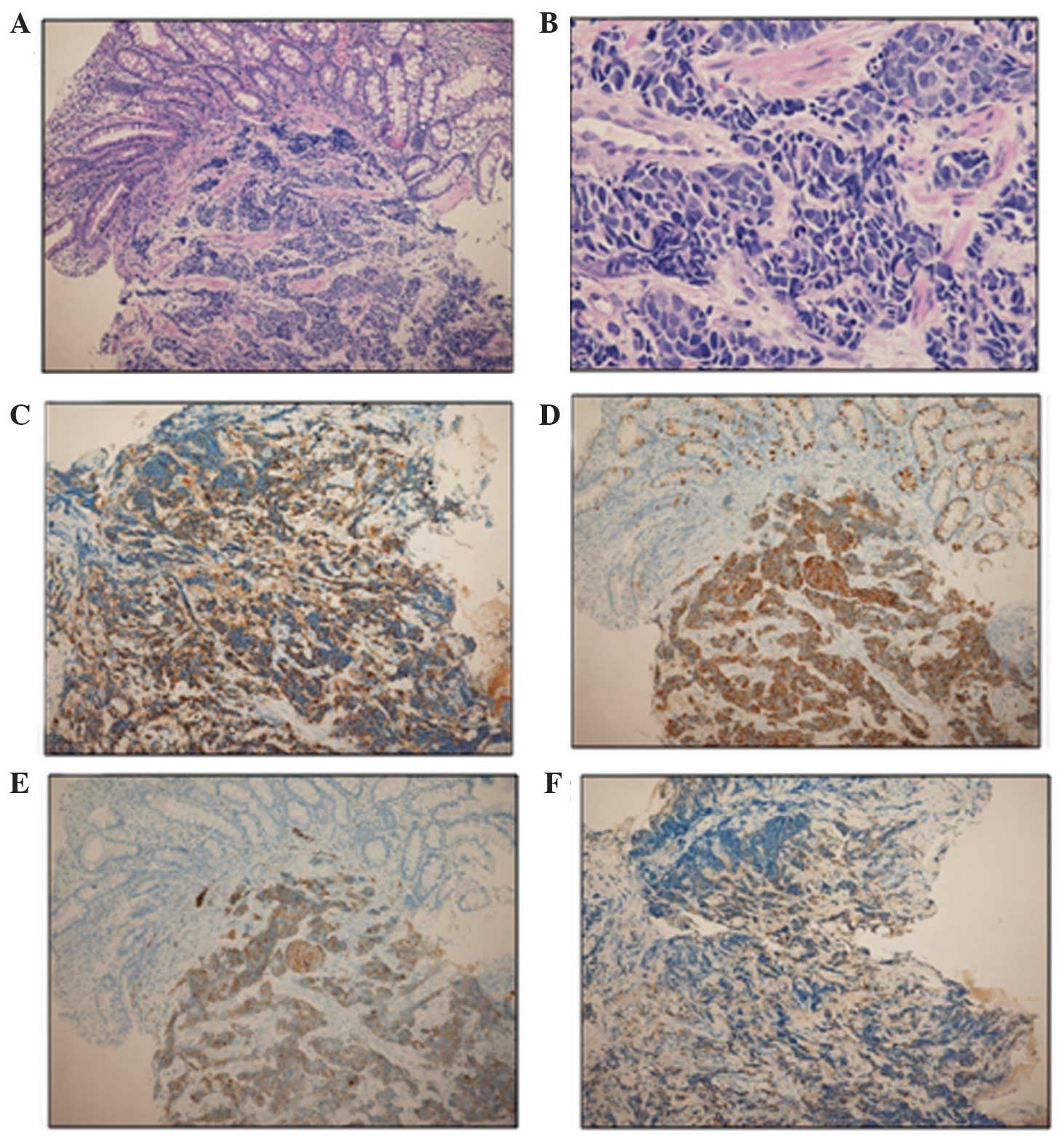
from the ampullary mass, which was histologically confirmed as a
poorly differentiated neuroendocrine tumor. Immunohistochemical
staining was positive for chromogranin A, synaptophysin and cluster
of differentiation (CD)56 (Fig. 2).
The mitotic rate was 24/10 high-power fields (HPFs) and the Ki-67
index was 80%. A neuroendocrine tumor G3 or neuroendocrine
carcinoma was diagnosed, according to the World Health Organization
classification. In addition, immunohistochemical staining for ACTH
was positive, indicating that the tumor produced ACTH. Therefore,
the present study finally diagnosed a functional and metastatic
neuroendocrine carcinoma of the ampulla of Vater with ectopic ACTH
production causing Cushing's syndrome. Chemotherapy with irinotecan
(60 mg/m2, days 1, 8 and 15) and cisplatin (60
mg/m2, day 1) on a 28-day cycle were administered, and
also administered mitotane (3 g/day) pre-chemotherapy to control
the hypercortisolemic state. However, the hematotoxicity worsened
to grade 4 after day 1 administration of the first cycle, and
therefore, chemotherapy had to be discontinued. Nevertheless, 1
month later, the patient had a partial response and exhibited
improvement on imaging findings and reduced levels of ACTH,
cortisol and tumor markers. However, the tumor subsequently regrew
and the patient succumbed to liver failure caused by multiple
hepatic metastases 4 months after the diagnosis.
Discussion
Although neuroendocrine tumors, initially assessed
as carcinoid tumors, were first described >100 years ago, these
tumors still raise many issues regarding their classification,
prognosis and optimum therapeutic approach (5). According to the World Health
Organization classification in 2010, neuroendocrine neoplasms in
the digestive system are categorized as neuroendocrine tumors (NET)
G1 (carcinoid, mitotic count of <2/10 HPF and/or Ki67 index
≤2%), NET G2 (mitotic count of 2–20/10 HPF and/or Ki67 index of
3–20%) and NET G3 (neuroendocrine carcinoma, mitotic count of
>20/10 HPF and/or Ki67 index >20%) (6). Neuroendocrine tumors rarely arise at the
ampulla of Vater, accounting for only 0.05% of all neuroendocrine
tumor types. Its clinical presentation consists of jaundice
(53.1%), abdominal pain (24.6%), pancreatitis (6.0%) and weight
loss (3.6%) (7). Almost all
neuroendocrine tumors of the ampulla are non-functional tumors and
<3% of patients with these tumors have hormonal hypersecretion
syndrome (8). However, no report of
ectopic ACTH secretion associated with neuroendocrine carcinoma of
the ampulla of Vater has been published.
Cushing's syndrome, due to ectopic ACTH secretion,
occurs in around 5–10% of all cases of ACTH-dependent
hypercortisolism (9). These
inappropriately high levels of ACTH are secreted by various types
of tumor, including neuroendocrine tumors, islet cell tumors, small
cell lung carcinomas and medullary thyroid cancers. Clinical
features of ectopic ACTH syndrome depend on the source of
production and rate of ACTH synthesis. Well-defined changes in
Cushingoid body habitus are noticed in slow-growing tumors, whereas
rapidly growing tumors, including that observed in the present
patient, typically produce a profound and sudden onset of symptoms.
Hypokalemia occurs in 80% of reported cases, including the present
patient, and several reports showed that it is more severe compared
with that in Cushing's disease (10).
The more intensive hypokalemia in ectopic ACTH syndrome can be
explained by the mineralocorticoid effect of cortisol, the level of
which tends to be higher in this syndrome compared with in
Cushing's disease (11).
The mortality and morbidity associated with
Cushing's syndrome is associated with the production of excess
cortisol. Hypercortisolism results in multiple medical problems,
including hypertension, obesity, osteoporosis, fractures, impaired
wound healing, infectious diseases, glucose intolerance and
psychosis. A previous study indicated that lowering cortisol levels
prior to attempting curative treatments (surgery or chemotherapy)
may reduce the mortality and morbidity associated with Cushing's
syndrome and, in particular, reduce the rates of opportunistic
infections (12). Therefore, adrenal
blocking agents, including metyrapone or mitotane, may be used to
treat hypercortisolemia. In certain patients with hormonal
hypersecretion, extensive debulking surgery must be considered even
in the presence of distant metastasis (13). However, the present patient had
multiple large liver metastatic deposits and therefore, surgery was
not deemed an option.
The treatment for neuroendocrine tumors of the
ampulla of Vater remains controversial since they are rare tumors
with unpredictable biological behavior and prognosis (13). Pancreaticoduodenectomy or local
excision has been used for these tumors of all sizes with no
distant spread (14,15). Although generally more indolent
compared with carcinomas, once they progress beyond surgical
resectability, they are essentially incurable. Therefore, systemic
treatment options for the management of advanced disease have
expanded substantially in recent years. Not only distant
metastasis, but also tumor grade are important prognostic factors
for survival in patients with neuroendocrine tumors of the ampulla
of Vater (14). Low-grade tumors show
5- and 10-year survival rates of 80 and 71%, respectively, whereas
high-grade neuroendocrine tumors have dismal 5- and 10-year
survival rates (15%) (7). Due to the
paucity of reported cases of neuroendocrine carcinoma, no
standardized chemotherapy exists for this tumor type.
Chemotherapeutic regimens, including cisplatin, irinotecan,
etoposide, doxorubicin and vincristine have been reported.
Large-scale retrospective analyses of advanced gastrointestinal and
pancreatic neuroendocrine carcinoma by a Japanese group
demonstrated that irinotecan plus cisplatin and etoposide plus
cisplatin are the most commonly used regimens (16).
In conclusion, this is the first report, to the best
of our knowledge, describing neuroendocrine carcinoma of the
ampulla of Vater responsible for ectopic production and secretion
of ACTH and resulting in Cushing's syndrome. Further data and
therapies, particularly effective adjuvant chemotherapies, are
urgently required to improve the survival rates of patients with
neuroendocrine tumors of the ampulla of Vater, a rare but
aggressive tumor.
References
|
1
|
Selvakumar E, Rajendran S, Balachandar TG,
Kannan DG, Jeswanth S, Ravichandran P and Surendran R:
Neuroendocrine carcinoma of the ampulla of Vater: A
clinicopathologic evaluation. Hepatobiliary Pancreat Dis Int.
7:422–425. 2008.PubMed/NCBI
|
|
2
|
Ricci JL: Carcinoid of the ampulla of
Vater. Local resection or pancreaticoduodenectomy. Cancer.
71:686–690. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Riggs BL Jr and Sprague RG: Association of
Cushing's syndrome and neoplastic disease: Observations in 232
cases of Cushing's syndrome and review of the literature. Arch
Intern Med. 108:841–849. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jex RK, van Heerden JA, Carpenter PC and
Grant CS: Ectopic ACTH syndrome. Diagnostic and therapeutic
aspects. Am J Surg. 149:276–282. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oberg K: Neuroendocrine tumors (NETs):
Historical overview and epidemiology. Tumori. 96:797–801.
2010.PubMed/NCBI
|
|
6
|
Rindi G, Petrone G and Inzani F: The 2010
WHO classification of digestive neuroendocrine neoplasms: A
critical appraisal four years after its introduction. Endocr
Pathol. 25:186–192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Albores-Saavedra J, Hart A, Chablé-Montero
F and Henson DE: Carcinoids and high-grade neuroendocrine
carcinomas of the ampulla of vater: A comparative analysis of 139
cases from the surveillance, epidemiology, and end results
program-a population based study. Arch Pathol Lab Med.
134:1692–1696. 2010.PubMed/NCBI
|
|
8
|
Solcia E, Fiocca R, Rindi G, Villani L,
Luinetti O, Burrell M, Bosi F and Silini E: Endocrine tumors of the
small and large intestine. Pathol Res Pract. 191:366–372. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wajchenberg BL, Mendonca BB, Liberman B,
Pereira MA, Carneiro PC, Wakamatsu A and Kirschner MA: Ectopic
adrenocorticotropic hormone syndrome. Endocr Rev. 15:752–787. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stewart PM, Walker BR, Holder G,
O'Halloran D and Shackleton CH: 11 beta-Hydroxysteroid
dehydrogenase activity in Cushing's syndrome: Explaining the
mineralocorticoid excess state of the ectopic adrenocorticotropin
syndrome. J Clin Endocrinol Metab. 80:3617–3620. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arteaga E, Fardella C, Campusano C,
Cárdenas I and Martinez P: Persistent hypokalemia after successful
adrenalectomy in a patient with Cushing's syndrome due to ectopic
ACTH secretion: Possible role of 11beta-hydroxysteroid
dehydrogenase inhibition. J Endocrinol Invest. 22:857–859. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ejaz S, Vassilopoulou-Sellin R, Busaidy
NL, Hu MI, Waguespack SG, Jimenez C, Ying AK, Cabanillas M, Abbara
M and Habra MA: Cushing syndrome secondary to ectopic
adrenocorticotropic hormone secretion: The university of texas MD
anderson cancer center experience. Cancer. 117:4381–4389. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klimstra DS, Modlin IR, Coppola D, Lloyd
RV and Suster S: The pathologic classification of neuroendocrine
tumors: A review of nomenclature, grading, and staging systems.
Pancreas. 39:707–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jayant M, Punia R, Kaushik R, Sharma R,
Sachdev A, Nadkarni NK and Attri A: Neuroendocrine tumors of the
ampulla of vater: Presentation, pathology and prognosis. JOP.
13:263–267. 2012.PubMed/NCBI
|
|
15
|
Odabasi M, Yildiz KM, Cengiz E, Hasan AH,
Gunay E, Ozkan E, Aktekin A, Kaya B and Muftuoglu TM: Treatment of
ampullary neuroendocrine tumor by endoscopic snare papillectomy. Am
J Case Rep. 14:439–443. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamaguchi T, Machida N, Morizane C, Kasuga
A, Takahashi H, Sudo K, Nishina T, Tobimatsu K, Ishido K, Furuse J,
et al: Multicenter retrospective analysis of systemic chemotherapy
for advanced neuroendocrine carcinoma of the digestive system.
Cancer Sci. 105:1176–1181. 2014. View Article : Google Scholar : PubMed/NCBI
|