Introduction
Synovial sarcoma is a clinically rare neoplasm that
accounts for ~10% of all malignant soft tissue tumors. Synovial
sarcoma most commonly arises in the soft tissues of the extremities
in young and middle-aged adults (1).
This tumor may also arise in other parts of the body, but primary
omental synovial sarcoma is extremely rare. To the best of our
knowledge, only 3 cases of primary omental synovial sarcoma (2
cases in women and 1 case in a man) have been documented in the
literature to date, and they were all associated with an
unfavorable clinical course (2–4).
Surgical resection and adjuvant chemotherapy involving
doxorubicin-ifosfamide are the preferred first-line treatments for
synovial sarcoma, whereas pazopanib (a tyrosine kinase inhibitor)
and trabectedin (an anticancer alkaloid agent) (5,6) have
recently been recommended as treatments for patients with advanced
disease, although the optimal therapeutic strategy for primary
omental synovial sarcoma has not been clearly determined due to the
rarity and severity of the disease.
Large complex abdominal masses that are incidentally
discovered in postmenopausal women are often initially suspected to
be pelvic malignancies. The lack of characteristic symptoms and
specific imaging characteristics may lead to the preoperative
misdiagnosis of omental synovial sarcoma as a gynecological
malignancy, particularly ovarian cancer. A specific chromosomal
aberration, t(X;18)(p11.2;q11.2), and its product, the SYT-SSX
fusion protein, are identified in >90% of cases of synovial
sarcoma (7), but the fusion gene may
only be detected postoperatively based on detailed pathological and
immunohistochemical examinations of the tumor.
We herein report a rare case of aggressive primary
omental synovial sarcoma mimicking ovarian cancer that was treated
with multiple surgical resection procedures and adjuvant
chemotherapy involving doxorubicin-ifosfamide, pazopanib and
trabectedin.
Case report
A 51-year-old multigravida woman who experienced
abdominal distension for 4 months was referred to the Department of
Obstetrics and Gynecology of the Hashimoto Municipal Hospital
(Hashimoto, Japan) in April 2014. The patient had no history of
lower abdominal or pelvic discomfort, pelvic surgery, or other
relevant medical conditions. Transvaginal and transabdominal
ultrasound examinations revealed a large solid abdominal mass,
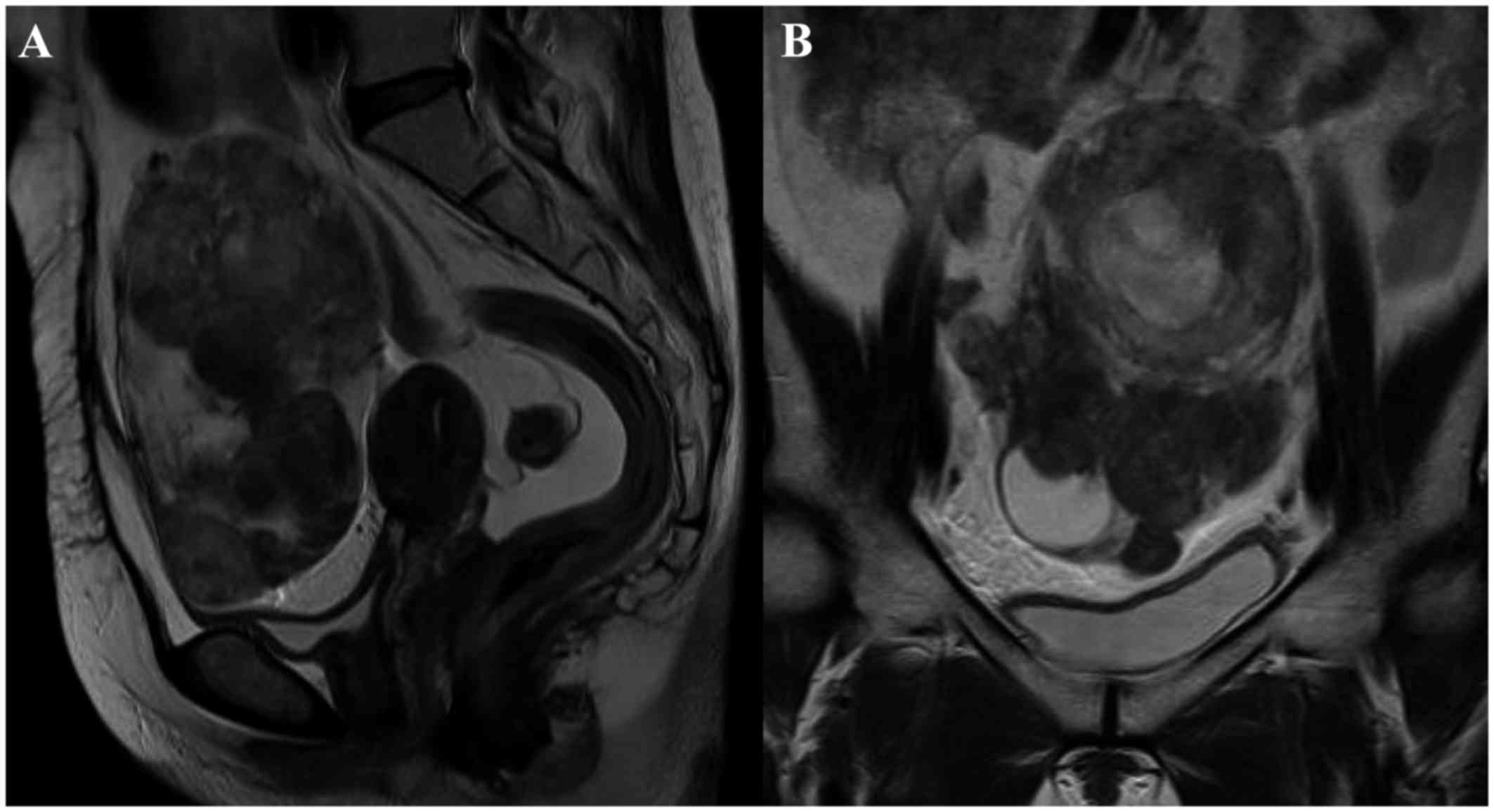
which exhibited iso-echogenicity. Magnetic resonance imaging (MRI)
revealed a large heterogeneous mass with an irregular component
occupying the lower abdominal cavity, with an intact uterus. Based
on these radiological findings, the mass was suspected to be an
ovarian malignancy (Fig. 1). The
results of a laboratory analysis of the patient's peripheral blood
revealed normal tumor marker levels [cancer antigen (CA)125, CA19-9
and carcinoembryonic antigen] and an elevated lactate dehydrogenase
level (291 IU/l). Total abdominal hysterectomy and bilateral
salpingo-oophorectomy were planned. Intraoperative examination
revealed a solid mass arising from the lower omentum. The resected
mass was 12.5×10.0×7.2 cm in size and weighed 502 g. The
consistency of the mass was elastic-hard and partially soft, and
its cut surface was whitish-yellow and contained a cystic and
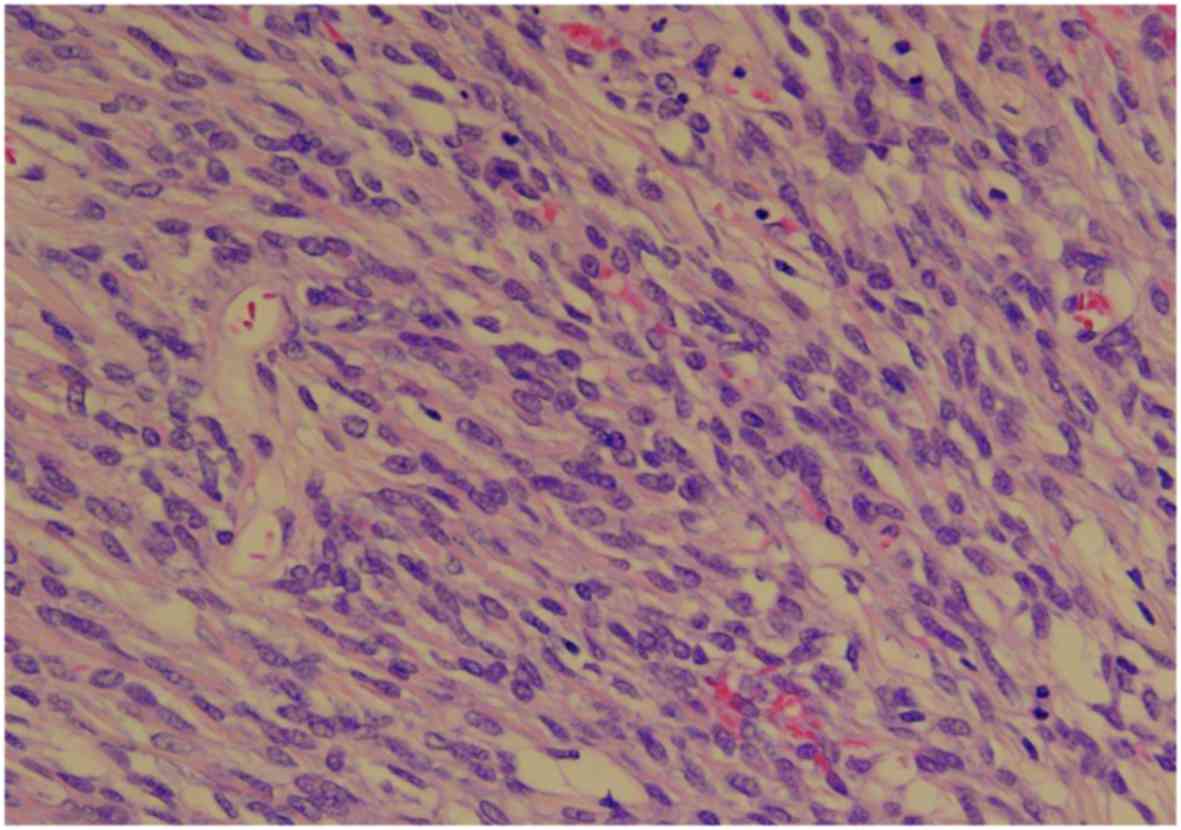
necrotic component measuring 2 cm in diameter. Microscopically, the
tumor included a well-circumscribed region composed of
hyaline-fibrous and mucinous-myxomatous components. The
hyaline-fibrous region exhibited diffuse proliferation of
spindle-shaped and small round tumor cells, with capillaries
arranged in a spider's web-like pattern (Fig. 2). The mucinous-myxomatous region was
composed of highly cellular tissue, which displayed a high
nuclear:cytoplasmic ratio and mitotic figures. Immunohistochemistry
revealed positive staining of the tumor cells for B-cell lymphoma
(Bcl)-2, and partially positive staining for CD99 and epithelial
membrane antigen (EMA). The tumor cells were negative for S100,
α-smooth muscle actin (SMA), desmin, CD34, CD31, cytokeratin (CK)7
and CK AE1/3. The Ki-67 labeling index was 40–60%.
Immunohistochemistry helped to exclude leiomyosarcoma, malignant
schwannoma, gastrointestinal stromal tumor and solitary fibrous
tumor. Although a qualitative analysis of mRNA expression based on
a reverse transcription polymerase chain reaction (RT-PCR) assay
was performed using an RNA sample extracted from formalin-fixed
paraffin-embedded tumor tissue, the SYT-SSX fusion gene transcript
was not detected. The diagnosis of primary omental poorly
differentiated synovial sarcoma was confirmed based on the
pathological findings. The patient's postoperative course was
uneventful, and she was discharged from the hospital on
postoperative day 7. The patient was treated with a combination of
doxorubicin and ifosfamide as adjuvant chemotherapy. Despite normal
findings on physical examination, 7 months after the operation a
metastatic tumor was detected in the liver by positron emission
tomography-computed tomography (PET-CT), and laparoscopic partial
liver resection was performed, followed by adjuvant pazopanib.
Twelve months after the operation, another metastatic tumor was
detected in the liver by PET-CT, and the patient was again treated
with laparoscopic partial liver resection followed by adjuvant
pazopanib. At 14 months after the first operation, PET-CT revealed
multiple recurrent metastatic tumors in the liver, lungs and
pleura. Although the patient was treated with partial lung
resection, aggressive metastatic tumors were detected in the liver,
lungs and abdominal cavity at 2 months after the first operation.
Due to the difficulty of surgical resection, the patient was
treated with trabectedin, and she remained alive with stable
disease at 24 months after the first operation. The patient
provided written informed consent regarding the publication of the
case details and associated images.
Discussion
Synovial sarcoma is a rare soft tissue malignant
tumor. Despite its name, synovial sarcoma does not necessarily
arise from synovial or soft tissue and has been reported to develop
at other sites, such as the kidneys, lungs and pleura. Synovial
sarcoma most commonly develops in the extremities, particularly in
the knee, but primary intra-abdominal synovial sarcoma has rarely
been reported, with only 8 cases published in the literature to
date (2).
Only 3 cases of primary omental synovial sarcoma
have been reported in the English literature since 1945, according
to a search through the PubMed and MEDLINE databases (2–4): 2 of
the cases involved women, aged 16 and 37 years (2,3), and the
remaining case involved a man, aged 66 years (4). Systemic symptoms, such as abdominal
pain, fullness, tenesmus and vomiting, occurred in these cases, and
the tumors ranged in size from 9.5 to 20 cm. None of these cases
were diagnosed as omental synovial sarcoma preoperatively based on
radiological examinations; instead, they were diagnosed
postoperatively based on detailed pathological and
immunohistochemical examinations of the tumor.
The diagnosis of primary omental synovial sarcoma is
difficult in female patients, as surgery is required for a
definitive diagnosis. In a previous study, the results of imaging
analysis were non-specific (8): On
MRI, T1-weighted imaging (WI) revealed areas of isointensity or
hyperintensity relative to muscle in the tumor masses, and T2WI
frequently showed heterogeneous signal intensity, including areas
of triple signal intensity involving regions of high signal
intensity that appeared fluid, isointense, or hyperintense relative
to fat, and hypointense relative to fibrous tissue. In a case
involving a postmenopausal female, a malignant tumor (ovarian
cancer) was initially suspected due to the detection of a large
abdominal mass and signs of necrosis.
Histologically, poorly differentiated synovial
sarcoma is formed from sheets of undifferentiated round cells with
hyperchromatic nuclei, which exhibit frequent mitoses, whereas
biphasic synovial sarcoma is composed of a combination of
epithelial and spindle cell components, and monophasic synovial
sarcoma is diagnosed when only an epithelial or a spindle cell
component is observed (1).
Immunohistochemical studies may confirm the pathological diagnosis.
Poorly differentiated synovial sarcoma is usually positive for
Bcl-2 and CD99, and focally positive for EMA, but it is negative
for S-100, α-SMA, desmin, CD34 and CD31. Poorly differentiated
synovial sarcoma differs from biphasic or monophasic synovial
sarcoma in that it is also negative for CK7 and CK AE1/3, which
emphasizes its poorly differentiated status and the non-epithelial
nature of the tumor cells. The SYT-SSX fusion gene, which is
produced by the chromosomal translocation t(X;18)(p11.2;q11.2), is
found in ~90% of synovial sarcomas during fluorescence in
situ hybridization or RT-PCR analysis of the gene transcript
(7). Although previous studies have
detected the SYT-SSX fusion gene in omental synovial sarcoma
(2,3), its true frequency in omental poorly
differentiated synovial sarcoma remains unknown. Even if molecular
testing for the SYT-SSX fusion gene produces a negative result, the
diagnosis of synovial sarcoma may be confirmed based on
immunohistochemical findings. While the product of the SYT-SSX
fusion gene was not detected in the present case,
immunohistochemistry detected positivity for Bcl-2, focal
positivity for CD99 and EMA, and negativity for S100, α-SMA,
desmin, CD34, CD31, CK7 and CK AE1/3. Thus, a final diagnosis of
poorly differentiated omental synovial sarcoma was made.
The most appropriate clinical management strategy
for primary omental synovial sarcoma may not be clear, as our
experience with such cases is limited. According to the 3 previous
reports on this tumor (2–4), omental synovial sarcoma appears to have
a poor prognosis, although synovial sarcomas characteristically
progress slowly. The primary treatment for this disease is surgery,
but in all 3 reported cases of omental synovial sarcoma, multiple
aggressive metastases developed following complete resection.
Despite receiving combined treatment with surgery and chemotherapy,
the patients succumbed to the disease within 2 months, 13 months
and 3 years, respectively. Patients who present with local
recurrence, distant metastasis, incomplete resection, positive
margins, lymph node involvement, or large bulky tumors, should
undergo adjuvant chemotherapy involving doxorubicin-ifosfamide
(5,6). Some molecular-targeted drugs, including
pazopanib, a selective multi-targeted receptor tyrosine kinase
inhibitor, and trabectedin, an anticancer alkaloid agent, have been
recommended as second-line chemotherapies in cases of poorly
differentiated synovial sarcoma in which combined treatment with
doxorubicin and ifosfamide fails to control metastasis, as they
block tumor growth and inhibit angiogenesis (5,6). In the
present case, the tumor progressed rapidly, with multiple
metastases and locally recurrent lesions developing during the
short follow-up period, which were treated with aggressive surgical
resection and adjuvant chemotherapy. The rapid tumor progression
observed in this case was consistent with the previous 3 reports.
Although treatment with pazopanib and trabectedin was
well-tolerated, it was ultimately unsuccessful. To improve the
treatment of omental synovial sarcoma, further data regarding its
natural history, diagnosis and treatment are required.
In summary, we encountered a rare case of primary
omental synovial sarcoma, in which aggressive metastasis developed
and proved difficult to control with a combination of surgical and
chemotherapeutic interventions, including molecular-targeted drugs.
Gynecologists should be aware of the aggressive metastatic
potential of this rare tumor and the importance of an intensive
diagnostic approach. Furthermore, a new therapeutic strategy for
primary omental synovial sarcoma is required to effectively control
this disease.
References
|
1
|
Fisher C, de Bruijn DRH and van Geurts
Kesse A: Synovial sarcomaWHO pathology and genetics: tumours of
soft tissue and bone. Fletcher CDM, Unni KK and Mertens F: IARC
Press; Lyon, France: pp. 200–204. 2002
|
|
2
|
Indranil G and Divya M: Synovial sarcoma
of the omentum: A rare entity. Indian J Cancer. 52:166–167. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hemmings C and Fisher C: Primary omental
synovial sarcoma: A case with cytogenetic confirmation. Pathology.
36:208–211. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Travaglini G, Biagetti S, Alfonsi S,
Bearzi I and Marmorale C: Primary intra-abdominal synovial sarcoma:
A case report. Ann Ital Chir. Sep 3–2013.(Epub ahead of print).
|
|
5
|
van der Graaf WT, Blay JY, Chawla SP, Kim
DW, Bui-Nguyen B, Casali PG, Schöffski P, Aglietta M, Staddon AP,
Beppu Y, et al: EORTC Soft Tissue and Bone Sarcoma Group; PALETTE
study group: Pazopanib for metastatic soft-tissue sarcoma
(PALETTE): A randomised, double-blind, placebo-controlled phase 3
trial. Lancet. 379:1879–1886. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sanfilippo R, Dileo P, Blay JY,
Constantinidou A, Le Cesne A, Benson C, Vizzini L, Contu M, Baldi
GG, Dei Tos AP, et al: Trabectedin in advanced synovial sarcomas: A
multicenter retrospective study from four European institutions and
the Italian Rare Cancer Network. Anticancer Drugs. 26:678–681.
2015.PubMed/NCBI
|
|
7
|
Aubry MC, Bridge JA, Wickert R and
Tazelaar HD: Primary monophasic synovial sarcoma of the pleura:
Five cases confirmed by the presence of SYT-SSX fusion transcript.
Am J Surg Pathol. 25:776–781. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang C, Mao H, Tan J, Ji Y, Sun F, Dou W,
Wang H, Wang H and Gao J: Synovial sarcoma: Magnetic resonance and
computed tomography imaging features and differential diagnostic
considerations. Oncol Lett. 9:661–666. 2015.PubMed/NCBI
|