Oxidative stress (OS) refers to the cellular
environment conditions that result from an imbalance between the
generation of reactive oxygen species (ROS) and the response of the
antioxidant defense systems (1). ROS
are short-lived highly reactive molecules and serve a critical role
in the progression of OS. ROS were identified as free radicals for
the first time in 1954 by Gerschman (2). They are metabolites produced during
normal cellular processes, which serve important roles in
activities such as promoting health and longevity (3) and antimicrobial phagocytosis by cells
of the innate immune system (4,5). The
over-generation of ROS without an adequate response from the innate
antioxidant system to maintain the homeostasis eventually leads to
OS. ROS serve a dual role in tumorigenicity, particularly in
hematologic malignancies. ROS can induce the activation of cell
death processes, including apoptosis, which provides a mechanism
for cancer treatment (6); however,
it can also facilitate carcinogenesis by protecting the cell from
apoptosis and promoting cell survival, inducing proliferation
(7), migration (8), metastasis (9) and drug-resistance (10,11). It
has been reported that OS is involved in the development of a
number of hematologic malignancies, including acute myeloid
leukemia (AML), chronic myeloid leukemia (CML), myelodysplastic
syndrome (MDS) and acute lymphoblastic leukemia (ALL) (12–16).
Numerous methods including the use of chemotherapeutic agents and
radiation are reported to generate ROS or other free radicals in
patients undergoing cancer therapy.
The present review focused on exploring the role of
OS in leukemogenesis and determining the association between ROS
and chemotherapy, as well as highlighting the importance of
antioxidant application in leukemia treatment. Improving current
understanding of the underlying mechanisms of OS generation in
leukemogenesis will facilitate significant progress in developing
novel therapeutic measures for various types of leukemia.
OS is a biochemical condition that occurs when
intracellular antioxidants are unable to neutralize pro-oxidants,
such as ROS. Mitochondria are the primary sites for oxidative
phosphorylation, which produces massive highly reactive and
unstable oxygen, thus oxidizing a large number of molecules to form
ROS (17). ROS are generated
intracellularly within various compartments and through multiple
mechanisms (Table I).
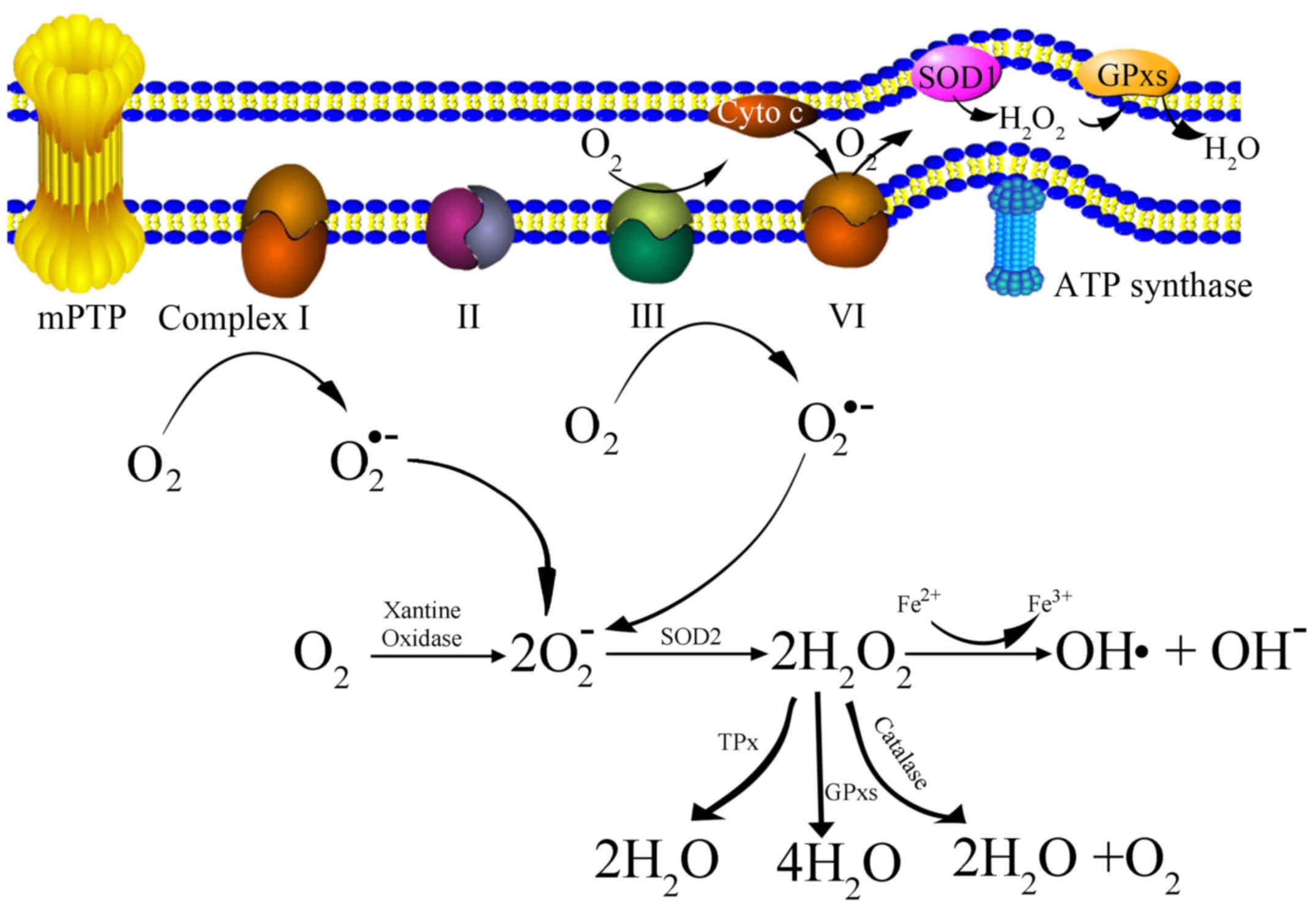
Mitochondria-derived ROS consist of singlet oxygen (O2),
superoxide anions (O2•-), hydrogen peroxide
(H2O2), nitric oxide (NO•), hydroxyl radicals
(OH•) and hydroxyl ions (OH-). The generation of
mitochondria-derived ROS is presented as a schematic in Fig. 1. Initially, oxygen is catalyzed to
transform into a superoxide anion by xanthine oxidase (XO)
(17,18), or by mitochondrial respiratory chain
complexes I (NADH dehydrogenase) and III (bc1 complex) either in
the matrix or in the intermembrane space (19). Subsequently, the superoxide anion is
converted to H2O2 by superoxide dismutase
(SOD). H2O2 can be detoxified to
H2O and O2 with glutathione peroxidase,
catalase (CAT) or thioredoxin peroxidase (TPx) (20). It can also be transformed into an OH•
and an OH-via the Fenton reaction (21).
OS causes cell injury predominantly via the
following three basic pathways: Lipid peroxidation of membranes;
oxidative modification of proteins; and DNA damage (17). Lipid peroxidation affects cell
membranes and other lipid-containing structure via a process known
as the ‘chain reaction of lipid peroxidation’. The critical
intermediate products of this reaction are hydroperoxides (LOOHs),
which can disturb the membrane structure and endanger cells
(22,23). It has been reported that the direct
secondary products of lipid peroxidation are aldehydes,
malondialdehyde (MDA) and 4-hydroxynonenal/4-hydroxy-2-nonenal
(HNE) (24). These products are
considered to be the markers of OS, and their unique property of a
no-charge structure allows them to easily permeate through
membranes and into the cytosol, thus causing far-reaching and
damaging effects inside and outside the cells, rendering them
superior to ROS (25,26). There is evidence that HNE and MDA can
cause protein or nucleic acid damage by modifying the amino acid
residues to form stable adducts or covalent adducts with nucleic
acids and membrane lipids (27,28).
Oxidative modification of proteins is another
pathway by which OS causes cell damage, and thus serves a critical
role in aging and cancer (29). MDA
and HNE can react with and covalently modify numerous proteins,
including amyloid-β peptide, collapsing response mediator protein-2
(CRMP2) and heat shock protein 70 (HSP70) (17,27,28).
HNE- and MDA-protein adducts, including alpha-enolase (ENO1),
phosphoglycerate kinase 1 (PGK1), triosephosphate isomerase (TPI)
and pyruvate kinase (PK), are reported to be involved in cellular
senescence and cancer (30–33). Besides MDA and HNE, ROS-mediated
protein oxidation also can be measured via the concentration of
carbonyl groups, advanced oxidation protein products (AOPPS),
advanced glycation end products (AGE) and S-nitrosylated proteins,
which are considered to be novel markers for OS due to their long
half-life and their ease of detection (34).
With respect to oxidative DNA damage, ROS and
products of lipid peroxidation can have an effect on genomic and
mitochondrial DNA, leading to various types of DNA damage (35,36). The
replication of damaged DNA prior to repair results in DNA mutations
and genomic instability, subsequently leading to a variety of
disorders and tumorigenesis. The molecule 8-oxoGuanine (8-OHG) and
its nucleoside form 8-OHdG are considered to be indicators of
oxidative DNA damage in vivo and in vitro (37,38). The
presence of 8-OHG in the DNA caused a G-T and a C-A transversion,
as 8-OHG allows the incorporation of cytosine and adenine
nucleotides opposite the lesion during DNA replication (39,40).
Numerous studies have reported that 8-OHG/8-OHdG is involved in
carcinogenesis and altered level of them demonstrated an
association with pathogenesis of aging associated disease and
cancer (41–43). For example, Ames and colleagues have
found the age-dependent accumulation of 8-OHdG in DNA from various
aged rat organs (44) and increased
levels of 8-OHdG and OH8Gua were shown in senescent human diploid
fibroblast (45). Mitochondrial
dysfunction and the lack of protective mechanisms mean that
mitochondrial DNA can be more easily and extensively exposed to ROS
than nuclear DNA, which can result in irreversible DNA damage. In
general, ROS and other OS-products attack cells through a variety
of intricate pathways. The lipid peroxidation of membranes, the
oxidative modification of proteins and DNA damage are the major
known mechanisms for oxidative cell damage. Improved understanding
the molecular mechanisms associated with OS will assist in the
development of novel and reliable treatments, as well as preventive
measures, for various types of cancer, particularly for
leukemia.
Leukemia develops when hematopoietic stem cells
(HSC) lose the capacity to differentiate normally into mature blood
cells at various stages during maturation and differentiation
(46). Hypoxia has emerged as a key
regulator of stem cell biology and maintains HSC quiescence with a
condition of metabolic dormancy based on anaerobic glycolysis,
which causes low production of ROS and high antioxidant defense
(47,48). While hematopoietic cell
differentiation is accompanied by changes in oxidative metabolism,
including a decrease in anaerobic glycolysis and an increase in
oxidative phosphorylation, thus producing high levels of ROS
(49–51). Furthermore, evidences have indicated
that leukemia stem cells (LSC) are more dependent on oxidative
respiration and are more sensitive to OS, compared with normal HSCs
(16). Although OS has been linked
to the etiology and development of leukemia, numerous
chemotherapeutic drugs exert their biological effects via the
induction of OS in affected cells. Thus OS serves a dual role in
leukemogenesis. ROS have a pathogenic role in various leukemia
models, including CML, MDS and AML (14,52).
First, BCR-ABL induces ROS production, which then contributes to
malignant transformation, cell growth, resistance to apoptosis and
increased DNA damage (53–55). Second, FLT3-ITD mutants induce
increased production of ROS, which are responsible for increased
DNA double-strand breaks and repair errors (56). Third, activated mutant Ras (N-Ras or
H-Ras) induces the production of superoxide and
H2O2 in human CD34+ cells through
the stimulation of NOX-1 (NADPH oxidase 1) activity; this effect
promotes the growth factor-independent proliferation of these cells
(57).
Conversely, ROS and lipid peroxidation by-products
are reported to be involved in mitochondria-derived apoptosis and
the induction of cell death (6). It
has been reported that ROS or lipid peroxidation by-products
primarily react to cardiolipin molecules in the inner mitochondrial
membrane (IMM), which disturbs the cytochrome c-cardiolipin
interaction and promotes the release of cytochrome c into
the cytoplasm, finally resulting in caspase activation and causing
cell death (58,59). It has also been demonstrated that HNE
reacts with the surrounding molecules near the site of its
formation, thereby stimulating chain-reactions of
mitochondria-derived apoptosis (60). A recent study explored the molecular
mechanisms responsible for the leukemogenesis effect of MLL-AF9 and
revealed an essential role of MEIS1 (61). MEIS1 expression in these leukemia
types limits the extent of OS and responses for leukemia cell
survival, while MEIS1 knockdown in MLL-AF9 leukemic cells induces
ROS production and the inhibition of leukemic cell growth.
Furthermore, a prior study published by our group demonstrated that
increased intracellular ROS levels are important for the induction
of cell death and the downregulation of BCR-ABL (62).
Furthermore, ROS participate in numerous cell growth
pathways by interfering with the regulation of certain genes and
signal transduction pathways, including tumor protein p53 mutation,
activator protein-1 (AP-1) activation, vascular endothelial growth
factor (VEGF) or rat sarcoma/mitogen activated protein kinase
(Ras/MAPK), nuclear factor κ-light-chain-enhancer of activated B
cells (NF-κB) signal pathway and the phosphatidylinositide
3-kinase/protein kinase B (PI3K/AKT) pathway (63). Ras/MAPK cascades consisting of
mitogen-activated protein kinase (ERK1/2), c-Jun N-terminal kinase
(JNK), p38 and 14-3-3β binds to big mitogen-activated protein
kinase 1 (BMK1/ERK5) pathways (64)
are involved in cytokines and growth factors signaling
transmission. The latter, including tumor necrosis factor (TNF)-α,
interferon gamma (IFN-γ), epidermal growth factor (EGF) and
platelet-derived growth factor (PDGF), bind to their receptors
under extracellular or intracellular stimuli and subsequently
activate a series of MAP kinases (MAPKKK, MAPKK, MAPK). The
activated MAPKs phosphorylate various substrate proteins, resulting
in the regulation of various cellular activities (65–67).
Each of aforementioned processes may be a target of ROS regulation.
For example, it has been demonstrated that ROS activates the
receptors of EGF and PDGF without corresponding ligands, thus
stimulating Ras and activating the ERK pathway (68,69).
Furthermore, in certain cells, treatment with
H2O2 leads to the phosphorylation and
activation of phospholipase C-γ (PLC-γ), and results in the
generation of inositol trisphosphate (IP3) and diacylglycerol
(DAG). The increase of the IP3 and DAG induces the release of
calcium from intracellular stores, and activates numerous forms of
Protein Kinase C (PKC), leading to the activation of Ras and Raf
and the initiation of ERK signaling (70,71). Akt
is a serine/threonine kinase, recruited to the cell membrane by
PI3K and activated via phosphorylation. The end result of PI3K/Akt
pathway activation is the stimulation of growth pathways and the
inhibition of apoptosis, or vice versa. ROS not only
activate PI3K directly to amplify its downstream signaling, but
also concurrently inactivate its negative regulator PTEN (72). For example, it is reported that ROS
can induce the phosphorylation of PTEN via casein kinase II, thus
urging it to enter the proteolytic degradation pathway (73). ROS influence the NK-κB pathway mainly
through inhibiting IκBα phosphorylation and degradation, thus
activating the NK-κB pathway. In addition, IKK is the primary
target for ROS through S-glutathionylation of the IKKβ on cysteine
179, resulting in the inhibition of IKKβ activity (74,75).
The current therapy for leukemia primarily consists
of high-dose cytotoxic chemotherapy with or without allogeneic stem
cell transplantation. However, chemotherapeutic treatments are
often accompanied by elevated ROS levels, and cause
drug-intolerance or resistance correspondingly (75). The underlying mechanisms may be
closely associated with the aforementioned ROS-mediated signaling
pathway. Chemotherapy impairs the mitotic and metabolic process of
cancer cells, involving various signal transmission abnormalities
or sub-cellular organ damage, thus causing excess ROS production.
Angsutararux et al (75)
studied doxorubicin (DOX)-induced cardiotoxicity, and proposed that
DOX is particularly harmful to the heart due to its exceptional
effects on mitochondria, which are the home of ROS. Petrola et
al (76) performed a clinical
trial to evaluate OS through detecting the levels of MDA and
nitrite in patients with CML undergoing treatment with 1st and 2nd
generation TKIs. The results indicated that TKIs caused
significantly high concentration of ROS in patients CML who were
undergoing these treatments, and that oxidative damage markers
could indicate resistance to TKIs. Furthermore, it has been
demonstrated that anthracyclines, including DOX, a type of
important component of current cancer treatment, generate high
levels of ROS and cause severe chemotherapy-associated
cardiotoxicity (77–79). Therefore, combinations of
antioxidants and chemotherapeutic agents perhaps have promising
synergistic effects (80). The role
of OS in DOX-induced cardiotoxicity can be attenuated in a
transgenic mouse model containing high levels of cardiac
metallothionein, a potent antioxidant (81). Nakayama et al (82) conducted a systematic review of
published clinical trials to examine the effects of dietary
antioxidants taken concurrently with chemotherapy or radiation
therapy. The results indicated that glutathione (GSH), vitamin E
and N-acetysteine (NAC) were the most frequently used antioxidant
supplements in combination with chemotherapy or radiation therapy
for kinds of cancer treatments, including leukemia. GSH combined
with cisplatin (CDDP)-based chemotherapy accord for 88% of all the
experiments (23/26), and adding GSH to CDDP-based chemotherapy
could improve the antitumor response against solid tumors and
hematological malignancies; some also revealed a neuroprotective
effect. Another study reported a trend of longer clinical PFS and
OS in patients with CML when they were treated with vitamin A in
combination with standard chemotherapy, although this trend was not
statistically significant (83).
However, there are conflicting opinions regarding
the administration of antioxidants during cancer therapy. Certain
researchers suppose that it may reduce the effectiveness of
chemotherapies, which are based on increasing oxidative stress. For
example, Hewish et al (84)
revealed that cytarabine was toxic to MLH1 and MLH2 deficient tumor
cells, but this cytotoxicity was reduced by antioxidants. In
general, the combination of antioxidants and chemotherapy is a
promising strategy for cancer treatment, a number of other studies
have argued their antagonistic effects. Further studies on the use
of this specific combined therapy are required, and further
synergistic effects must be investigated and elucidated.
Leukemia is a type of hematological neoplasm
characterized by the abnormal proliferation and circulation of
immature clonal hematopoietic cells in the blood or bone marrow
(85). OS has been implicated in
leukemogenesis and serves an important role in cell proliferation
and cell signaling regulation. Abnormalities in the
oxidative-antioxidative balance have been observed in numerous
cases of leukemia neoplasm, including ALL, B-CLL and MM.
Indeed, leukemia cells produce higher concentrations
of ROS than non-leukemic cells83. OS has beneficial and
deleterious effects on leukemogenesis. On the one hand, it promotes
leukemia progression through activating oncogenes, including Ras
and VEGF, and the NF-κB signal transduction pathway. Conversely,
mitochondria-derived apoptosis can olso be induced by OS and causes
cell death. It is difficult to separate the oncogenic properties
from the tumor suppressive activity. Therefore, an improved
understanding of the association between OS and leukemogenesis will
provide more insight for leukemia treatments. Chemotherapy is a
commonly used strategy for leukemia treatment. However, the current
cytotoxic drugs available for use in standard leukemia therapy are
often accompanied by elevated ROS production, and cause
drug-intolerance or resistance. Thus, targeting ROS levels during
chemotherapy could constitute a novel approach for various types of
leukemia, particularly for those of refractory and relapsed
hematological neoplasms. Indeed, studies have demonstrated that
antioxidant treatments combined with chemotherapy are effective in
leukemia therapy, but their concurrent negative effects have also
been recorded. Therefore, further studies are required to explore
the synergistic effects, long-term effects and consequences of
using these combination therapies. Potentially, targeted OS therapy
in combination with chemotherapy or other strategies may become a
clinically useful therapeutic approach for various types of
hematological diseases in the near future.
The present study was supported by the National
Natural Science Foundation of China (grant no. 81670178, 81370645),
the Hangzhou Science and Technology Bureau (grant no. 20140633B06)
and the Special Scientific Construction Research Funds of National
Chinese Medicine Clinical Research Center, SATCM (grant no.
JDZX2015113).
The authors declare that they have no competing
interests.
|
1
|
Imbesi S, Musolino C, Allegra A, Saija A,
Morabito F, Calapai G and Gangemi S: Oxidative stress in
oncohematologic diseases: An update. Expert Rev Hematol. 6:317–325.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gerschman R, Gilbert D, Nye SW, Dwyer P
and Fenn WO: Oxygen poisoning and X-irradiation: A mechanism in
common. 1954. Nutrition. 17:1622001.PubMed/NCBI
|
|
3
|
Asthana J, Yadav AK, Pant A, Pandey S,
Gupta MM and Pandey R: Specioside ameliorates oxidative stress and
promotes longevity in Caenorhabditis elegans. Comp Biochem Physiol
C Toxicol Pharmacol. 169:25–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Valko M, Leibfritz D, Moncol J, Cronin MT,
Mazur M and Telser J: Free radicals and antioxidants in normal
physiological functions and human disease. Int J Biochem Cell Biol.
39:44–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weyemi U, Caillou B, Talbot M,
Ameziane-El-Hassani R, Lacroix L, Lagent-Chevallier O, Al Ghuzlan
A, Roos D, Bidart JM, Virion A, et al: Intracellular expression of
reactive oxygen species-generating NADPH oxidase NOX4 in normal and
cancer thyroid tissues. Endocr Relat Cancer. 17:27–37. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Khoshtabiat L, Mahdavi M, Dehghan G and
Rashidi MR: Oxidative stress-induced apoptosis in chronic
myelogenous leukemia K562 cells by an active compound from the
dithio-carbamate family. Asian Pac J Cancer Prev. 17:4267–4273.
2016.PubMed/NCBI
|
|
7
|
Cheng D, Zhao L, Xu Y, Ou R, Li G, Yang H
and Li W: K-Ras promotes the non-small lung cancer cells survival
by cooperating with sirtuin 1 and p27 under ROS stimulation. Tumour
Biol. 36:7221–7232. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weyemi U, Lagente-Chevallier O, Boufraqech
M, Prenois F, Courtin F, Caillou B, Talbot M, Dardalhon M, Al
Ghuzlan A, Bidart JM, et al: ROS-generating NADPH oxidase NOX4 is a
critical mediator in oncogenic H-Ras-induced DNA damage and
subsequent senescence. Oncogene. 31:1117–1129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu D, Shen Z, Liu J, Chen J, Liu Y, Hu C,
Li Z and Li Y: The ROS-mediated activation of STAT-3/VEGF signaling
is involved in the 27-hydroxycholesterol-induced angiogenesis in
human breast cancer cells. Toxicol Lett. 264:79–86. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lina M, Hongfei M, Yunxin X, Dai W and
Xilin Z: The mechanism of ROS regulation of antibiotic resistance
and antimicrobial lethality. Yi Chuan. 38:902–909. 2016.PubMed/NCBI
|
|
11
|
Das DS, Ray A, Das A, Song Y, Tian Z,
Oronsky B, Richardson P, Scicinski J, Chauhan D and Anderson KC: A
novel hypoxia-selective epigenetic agent RRx-001 triggers apoptosis
and overcomes drug resistance in multiple myeloma cells. Leukemia.
30:2187–2197. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Udensi UK and Tchounwou PB: Dual effect of
oxidative stress on leukemia cancer induction and treatment. J Exp
Clin Cancer Res. 33:1062014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Battisti V, Maders LD, Bagatini MD, Santos
KF, Spanevello RM, Maldonado PA, Brulé AO, Araújo Mdo C, Schetinger
MR and Morsch VM: Measurement of oxidative stress and antioxidant
status in acute lymphoblastic leukemia patients. Clin Biochem.
41:511–518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chung YJ, Robert C, Gough SM, Rassool FV
and Aplan PD: Oxidative stress leads to increased mutation
frequency in a murine model of myelodysplastic syndrome. Leuk Res.
38:95–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pawlowska E and Blasiak J: DNA repair-a
double-edged sword in the genomic stability of cancer cells-the
case of chronic myeloid leukemia. Int J Mol Sci. 16:27535–27549.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Testa U, Labbaye C, Castelli G and Pelosi
E: Oxidative stress and hypoxia in normal and leukemic stem cells.
Exp Hematol. 44:540–560. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kudryavtseva AV, Krasnov GS, Dmitriev AA,
Alekseev BY, Kardymon OL, Sadritdinova AF, Fedorova MS, Pokrovsky
AV, Melnikova NV, Kaprin AD, et al: Mitochondrial dysfunction and
oxidative stress in aging and cancer. Oncotarget. 7:44879–44905.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sanganahalli BG, Joshi PG and Joshi NB:
Xanthine oxidase, nitric oxide synthase and phospholipase A(2)
produce reactive oxygen species via mitochondria. Brain Res.
1037:200–203. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aprioku JS: Pharmacology of free radicals
and the impact of reactive oxygen species on the testis. J Reprod
Infertil. 14:158–172. 2013.PubMed/NCBI
|
|
20
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao K, Zeng Q, Bai J, Li J, Xia L, Chen S
and Zhou B: Enhanced organic pollutants degradation and electricity
production simultaneously via strengthening the radicals reaction
in a novel Fenton-photocatalytic fuel cell system. Water Res.
108:293–300. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ayala A, Muñoz MF and Argüelles S: Lipid
peroxidation: Production, metabolism, and signaling mechanisms of
malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev.
2014:3604382014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yin H, Xu L and Porter NA: Free radical
lipid peroxidation: Mechanisms and analysis. Chem Rev.
111:5944–5972. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Breitzig M, Bhimineni C, Lockey R and
Kolliputi N: 4-Hydroxy-2-nonenal: A critical target in oxidative
stress? Am J Physiol Cell Physiol. 311:C537–C543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Negre-Salvayre A, Coatrieux C, Ingueneau C
and Salvayre R: Advanced lipid peroxidation end products in
oxidative damage to proteins. Potential role in diseases and
therapeutic prospects for the inhibitors. Br J Pharmacol. 153:6–20.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Negre-Salvayre A, Auge N, Ayala V, Basaga
H, Boada J, Brenke R, Chapple S, Cohen G, Feher J, Grune T, et al:
Pathological aspects of lipid peroxidation. Free Radic Res.
44:1125–1171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang G, Wang J, Fan X, Ansari GA and Khan
MF: Protein adducts of malondialdehyde and 4-hydroxynonenal
contribute to trichloroethene-mediated autoimmunity via activating
Th17 cells: Dose- and time-response studies in female MRL+/+ mice.
Toxicology. 292:113–122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Domingues RM, Domingues P, Melo T,
Pérez-Sala D, Reis A and Spickett CM: Lipoxidation adducts with
peptides and proteins: Deleterious modifications or signaling
mechanisms? J Proteomics. 92:110–131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stadtman ER and Berlett BS: Reactive
oxygen-mediated protein oxidation in aging and disease. Drug Metab
Rev. 30:225–243. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sharma NK, Sethy NK and Bhargava K:
Comparative proteome analysis reveals differential regulation of
glycolytic and antioxidant enzymes in cortex and hippocampus
exposed to short-term hypobaric hypoxia. J Proteomics. 79:277–298.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen XL, Zhou L, Yang J, Shen FK, Zhao SP
and Wang YL: Hepatocellular carcinoma-associated protein markers
investigated by MALDI-TOF MS. Mol Med Rep. 3:589–596. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu H, Zhu W, Qin J, Chen M, Gong L, Li L,
Liu X, Tao Y, Yin H, Zhou H, et al: Acetylation of PGK1 promotes
liver cancer cell proliferation and tumorigenesis. Hepatology.
65:515–528. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ahmad SS, Glatzle J, Bajaeifer K, Bühler
S, Lehmann T, Königsrainer I, Vollmer JP, Sipos B, Ahmad SS,
Northoff H, et al: Phosphoglycerate kinase 1 as a promoter of
metastasis in colon cancer. Int J Oncol. 43:586–590. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Singh RK, Tripathi AK, Tripathi P, Singh
S, Singh R and Ahmad R: Studies on biomarkers for oxidative stress
in patients with chronic myeloid leukemia. Hematol Oncol Stem Cell
Ther. 2:285–288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rahal ON, Fatfat M, Hankache C, Osman B,
Khalife H, Machaca K and Muhtasib HG: Chk1 and DNA-PK mediate
TPEN-induced DNA damage in a ROS dependent manner in human colon
cancer cells. Cancer Biol Ther. 17:1139–1148. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen CY, Yen CY, Wang HR, Yang HP, Tang
JY, Huang HW, Hsu SH and Chang HW: Tenuifolide B from cinnamomum
tenuifolium stem selectively inhibits proliferation of oral cancer
cells via apoptosis, ROS generation, mitochondrial depolarization,
and DNA damage. Toxins (Basel). 8:pii: E3192016. View Article : Google Scholar
|
|
37
|
Kikuchi A, Takeda A, Onodera H, Kimpara T,
Hisanaga K, Sato N, Nunomura A, Castellani RJ, Perry G, Smith MA
and Itoyama Y: Systemic increase of oxidative nucleic acid damage
in Parkinson's disease and multiple system atrophy. Neurobiol Dis.
9:244–248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weidner AM, Bradley MA, Beckett TL,
Niedowicz DM, Dowling AL, Matveev SV, LeVine H III, Lovell MA and
Murphy MP: RNA oxidation adducts 8-OHG and 8-OHA change with Aβ42
levels in late-stage Alzheimer's disease. PLoS One. 6:e249302011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shibutani S, Takeshita M and Grollman AP:
Insertion of specific bases during DNA synthesis past the
oxidation-damaged base 8-oxodG. Nature. 349:431–434. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sunaga N, Kohno T, Shinmura K, Saitoh T,
Matsuda T, Saito R and Yokota J: OGG1 protein suppresses
G:C->T:A mutation in a shuttle vector containing
8-hydroxyguanine in human cells. Carcinogenesis. 22:1355–1362.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lovell MA, Soman S and Bradley MA:
Oxidatively modified nucleic acids in preclinical Alzheimer's
disease (PCAD) brain. Mech Ageing Dev. 132:443–448. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Valavanidis A, Vlachogianni T and Fiotakis
C: 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of
oxidative stress and carcinogenesis. J Environ Sci Health C Environ
Carcinog Ecotoxicol Rev. 27:120–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Moskalev AA, Shaposhnikov MV, Plyusnina
EN, Zhavoronkov A, Budovsky A, Yanai H and Fraifeld VE: The role of
DNA damage and repair in aging through the prism of Koch-like
criteria. Ageing Res Rev. 12:661–684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fraga CG, Shigenaga MK, Park JW, Degan P
and Ames BN: Oxidative damage to DNA during aging:
8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc Natl
Acad Sci USA. 87:pp. 4533–4537. 1990; View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen Q, Fischer A, Reagan JD, Yan LJ and
Ames BN: Oxidative DNA damage and senescence of human diploid
fibroblast cells. Proc Natl Acad Sci USA. 92:pp. 4337–4341. 1995;
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vyas P and Jacobsen SE: Clever leukemic
stem cells branch out. Cell stem cell. 8:242–244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nombela-Arrieta C and Silberstein LE: The
science behind the hypoxic niche of hematopoietic stem and
progenitors. Hematology Am Soc Hematol Educ Program. 2014:542–547.
2014.PubMed/NCBI
|
|
48
|
Bigarella CL, Liang R and Ghaffari S: Stem
cells and the impact of ROS signaling. Development. 141:4206–4218.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rönn RE, Guibentif C, Saxena S and Woods
NB: Reactive oxygen species impair the function of CD90+
hematopoietic progenitors generated from human pluripotent stem
cells. Stem cells. 35:197–206. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cao Y, Fang Y, Cai J, Li X, Xu F, Yuan N,
Zhang S and Wang J: ROS functions as an upstream trigger for
autophagy to drive hematopoietic stem cell differentiation.
Hematology. 21:613–618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kaur A, Jankowska K, Pilgrim C, Fraser ST
and New EJ: Studies of hematopoietic cell differentiation with a
ratiometric and reversible sensor of mitochondrial reactive oxygen
species. Antioxid Redox Signal. 24:667–679. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hole PS, Darley RL and Tonks A: Do
reactive oxygen species play a role in myeloid leukemias? Blood.
117:5816–5826. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sattler M, Verma S, Shrikhande G, Byrne
CH, Pride YB, Winkler T, Greenfield EA, Salgia R and Griffin JD:
The BCR/ABL tyrosine kinase induces production of reactive oxygen
species in hematopoietic cells. J Biol Chem. 275:24273–24278. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kim JH, Chu SC, Gramlich JL, Pride YB,
Babendreier E, Chauhan D, Salgia R, Podar K, Griffin JD and Sattler
M: Activation of the PI3K/mTOR pathway by BCR-ABL contributes to
increased production of reactive oxygen species. Blood.
105:1717–1723. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Koptyra M, Falinski R, Nowicki MO,
Stoklosa T, Majsterek I, Nieborowska-Skorska M, Blasiak J and
Skorski T: BCR/ABL kinase induces self-mutagenesis via reactive
oxygen species to encode imatinib resistance. Blood. 108:319–327.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sallmyr A, Fan J, Datta K, Kim KT, Grosu
D, Shapiro P, Small D and Rassool F: Internal tandem duplication of
FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and
misrepair: Implications for poor prognosis in AML. Blood.
111:3173–3182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hole PS, Pearn L, Tonks AJ, James PE,
Burnett AK, Darley RL and Tonks A: Ras-induced reactive oxygen
species promote growth factor-independent proliferation in human
CD34+ hematopoietic progenitor cells. Blood. 115:1238–1246. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kagan VE, Tyurin VA, Jiang J, Tyurina YY,
Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V,
et al: Cytochrome c acts as a cardiolipin oxygenase required for
release of proapoptotic factors. Nat Chem Biol. 1:223–232. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu Z, Lin H, Ye S, Liu QY, Meng Z, Zhang
CM, Xia Y, Margoliash E, Rao Z and Liu XJ: Remarkably high
activities of testicular cytochrome c in destroying reactive oxygen
species and in triggering apoptosis. Proc Natl Acad Sci USA.
103:pp. 8965–8970. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhong H and Yin H: Role of lipid
peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing
on mitochondria. Redox Biol. 4:193–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Roychoudhury J, Clark JP, Gracia-Maldonado
G, Unnisa Z, Wunderlich M, Link KA, Dasgupta N, Aronow B, Huang G,
Mulloy JC and Kumar AR: MEIS1 regulates an HLF-oxidative stress
axis in MLL-fusion gene leukemia. Blood. 125:2544–2552. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou W, Zhu W, Ma L, Xiao F and Qian W:
Proteasome inhibitor MG-132 enhances histone deacetylase inhibitor
SAHA-induced cell death of chronic myeloid leukemia cells by an
ROS-mediated mechanism and downregulation of the Bcr-Abl fusion
protein. Oncol Lett. 10:2899–2904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu
Y and Dong W: ROS and ROS-mediated cellular signaling. Oxid Med
Cell Longev. 2016:43509652016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Junttila MR, Li SP and Westermarck J:
Phosphatase-mediated crosstalk between MAPK signaling pathways in
the regulation of cell survival. FASEB J. 22:954–965. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pimienta G and Pascual J: Canonical and
alternative MAPK signaling. Cell cycle. 6:2628–2632. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen Z, Gibson TB, Robinson F, Silvestro
L, Pearson G, Xu B, Wright A, Vanderbilt C and Cobb MH: MAP
kinases. Chem Rev. 101:2449–2476. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
León-Buitimea A, Rodríguez-Fragoso L,
Lauer FT, Bowles H, Thompson TA and Burchiel SW: Ethanol-induced
oxidative stress is associated with EGF receptor phosphorylation in
MCF-10A cells overexpressing CYP2E1. Toxicol Lett. 209:161–165.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lei H and Kazlauskas A: Growth factors
outside of the platelet-derived growth factor (PDGF) family employ
reactive oxygen species/Src family kinases to activate PDGF
receptor alpha and thereby promote proliferation and survival of
cells. J Biol Chem. 284:6329–6336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Franklin RA, Atherfold PA and McCubrey JA:
Calcium-induced ERK activation in human T lymphocytes occurs via
p56(Lck) and CaM-kinase. Mol Immunol. 37:675–683. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Dann SG, Golas J, Miranda M, Shi C, Wu J,
Jin G, Rosfjord E, Upeslacis E and Klippel A: p120 catenin is a key
effector of a Ras-PKCε oncogenic signaling axis. Oncogene.
33:1385–1394. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Leslie NR and Downes CP: PTEN: The down
side of PI 3-kinase signalling. Cell Signal. 14:285–295. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lee SR, Yang KS, Kwon J, Lee C, Jeong W
and Rhee SG: Reversible inactivation of the tumor suppressor PTEN
by H2O2. J Biol Chem. 277:20336–20342. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Reynaert NL, van der Vliet A, Guala AS,
McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS,
Matthews DE, et al: Dynamic redox control of NF-kappaB through
glutaredoxin-regulated S-glutathionylation of inhibitory kappaB
kinase beta. Proc Natl Acad Sci USA. 103:pp. 13086–13091. 2006;
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Angsutararux P, Luanpitpong S and
Issaragrisil S: Chemotherapy-induced cardiotoxicity: Overview of
the roles of oxidative stress. Oxid Med Cell Longev.
2015:7956022015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Petrola MJ, de Castro AJ, Pitombeira MH,
Barbosa MC, Quixadá AT, Duarte FB and Gonçalves RP: Serum
concentrations of nitrite and malondialdehyde as markers of
oxidative stress in chronic myeloid leukemia patients treated with
tyrosine kinase inhibitors. Rev Bras Hematol Hemoter. 34:352–355.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cheuk DK, Sieswerda E, van Dalen EC,
Postma A and Kremer LC: Medical interventions for treating
anthracycline-induced symptomatic and asymptomatic cardiotoxicity
during and after treatment for childhood cancer. Cochrane Database
Syst Rev: CD008011. 2016. View Article : Google Scholar
|
|
78
|
Bartlett JJ, Trivedi PC, Yeung P,
Kienesberger PC and Pulinilkunnil T: Doxorubicin impairs
cardiomyocyte viability by suppressing transcription factor EB
expression and disrupting autophagy. Biochem J. 473:3769–3789.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Piegari E, Russo R, Cappetta D, Esposito
G, Urbanek K, Dell'Aversana C, Altucci L, Berrino L, Rossi F and De
Angelis A: MicroRNA-34a regulates doxorubicin-induced
cardiotoxicity in rat. Oncotarget. 7:62312–62326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sabnis HS, Bradley HL, Tripathi S, Yu WM,
Tse W, Qu CK and Bunting KD: Synergistic cell death in FLT3-ITD
positive acute myeloid leukemia by combined treatment with
metformin and 6-benzylthioinosine. Leuk Res. 50:132–140. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sun X, Zhou Z and Kang YJ: Attenuation of
doxorubicin chronic toxicity in metallothionein-overexpressing
transgenic mouse heart. Cancer Res. 61:3382–3387. 2001.PubMed/NCBI
|
|
82
|
Nakayama A, Alladin KP, Igbokwe O and
White JD: Systematic review: Generating evidence-based guidelines
on the concurrent use of dietary antioxidants and chemotherapy or
radiotherapy. Cancer Invest. 29:655–667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Meyskens FL Jr, Kopecky KJ, Appelbaum FR,
Balcerzak SP, Samlowski W and Hynes H: Effects of vitamin A on
survival in patients with chronic myelogenous leukemia: A SWOG
randomized trial. Leuk Res. 19:605–612. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Hewish M, Martin SA, Elliott R, Cunningham
D, Lord CJ and Ashworth A: Cytosine-based nucleoside analogs are
selectively lethal to DNA mismatch repair-deficient tumour cells by
enhancing levels of intracellular oxidative stress. Br J Cancer.
108:983–992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kennedy JA and Barabé F: Investigating
human leukemogenesis: From cell lines to in vivo models of human
leukemia. Leukemia. 22:2029–2040. 2008. View Article : Google Scholar : PubMed/NCBI
|