Introduction
Primitive neuroectodermal tumor (PNET) is a rare
malignancy that is a member of the family of ‘small round-cell
tumors’ and is often classified as central nervous system PNET or
peripheral PNET, depending on its site of origin (1). Although PNET has been reported in the
brain, ovary and liver (2), renal
(r)PNET is rare, with only 120 cases of rPNET reported in the
medical literature since its discovery in 1975 (3). rPNET usually affects young adults and
features a rapid clinical progression and significant mortality due
to late diagnosis, early metastasis and advanced stage at
presentation (4). The overall
survival of rPNET at the advanced stage is only 15 months, compared
with 60 months in patients with localized tumors (5). In spite of common treatments, including
surgical excision, chemotherapy and radiotherapy, providing a
survival benefit for patients with rPNET at the localized stage,
the prognosis at the advanced stage remains poor (1,6).
Therefore, novel therapeutic approaches to prolong survival are
required, particularly for patients at the advanced stage.
Immunotherapy is a novel treatment for various types of tumor,
including renal cell carcinoma (RCC), and has achieved encouraging
results (7). Numerous studies have
demonstrated the anti-tumor properties of cytokine-induced killer
(CIK) cells, such as enhanced cytotoxic activity and resistance to
Fas-mediated apoptosis (8). To the
best of our knowledge, no previous study has reported on CIK cell
immunotherapy for patients with rPNET. Between December 2004 and
January 2013, eight patients with rPNET at an advanced stage were
treated at Lingnan Hospital (branch of The Third Affiliated
Hospital) and the Cancer Center of Sun Yat-sen University
(Guangzhou, China), of which one case was administered CIK cells,
with the aim of improving the long-term survival after having
obtained informed consent. The present study describes the
pathological and clinical features, as well as the treatment
outcomes, of these cases, in addition to a review of the literature
on rPNET.
Patients and methods
Patients
Between December 2004 and January 2013, eight cases
of rPNET at the advanced stage were treated at two institutions
[Lingnan Hospital (branch of The Third Affiliated Hospital) and the
Cancer Center of Sun Yat-sen University, Guangzhou]. The cohort
comprised five males and three females with a median age of 34
years (range, 17–45 years) at presentation. All of the patients
complained of a palpable abdominal mass; furthermore five cases had
abdominal pain and one case had edema in the lower limbs. Patient
evaluation included history, physical examination, complete blood
count, renal and liver function tests, chest X-ray, computed
tomography (CT) scan of the abdomen and radionuclide renography. On
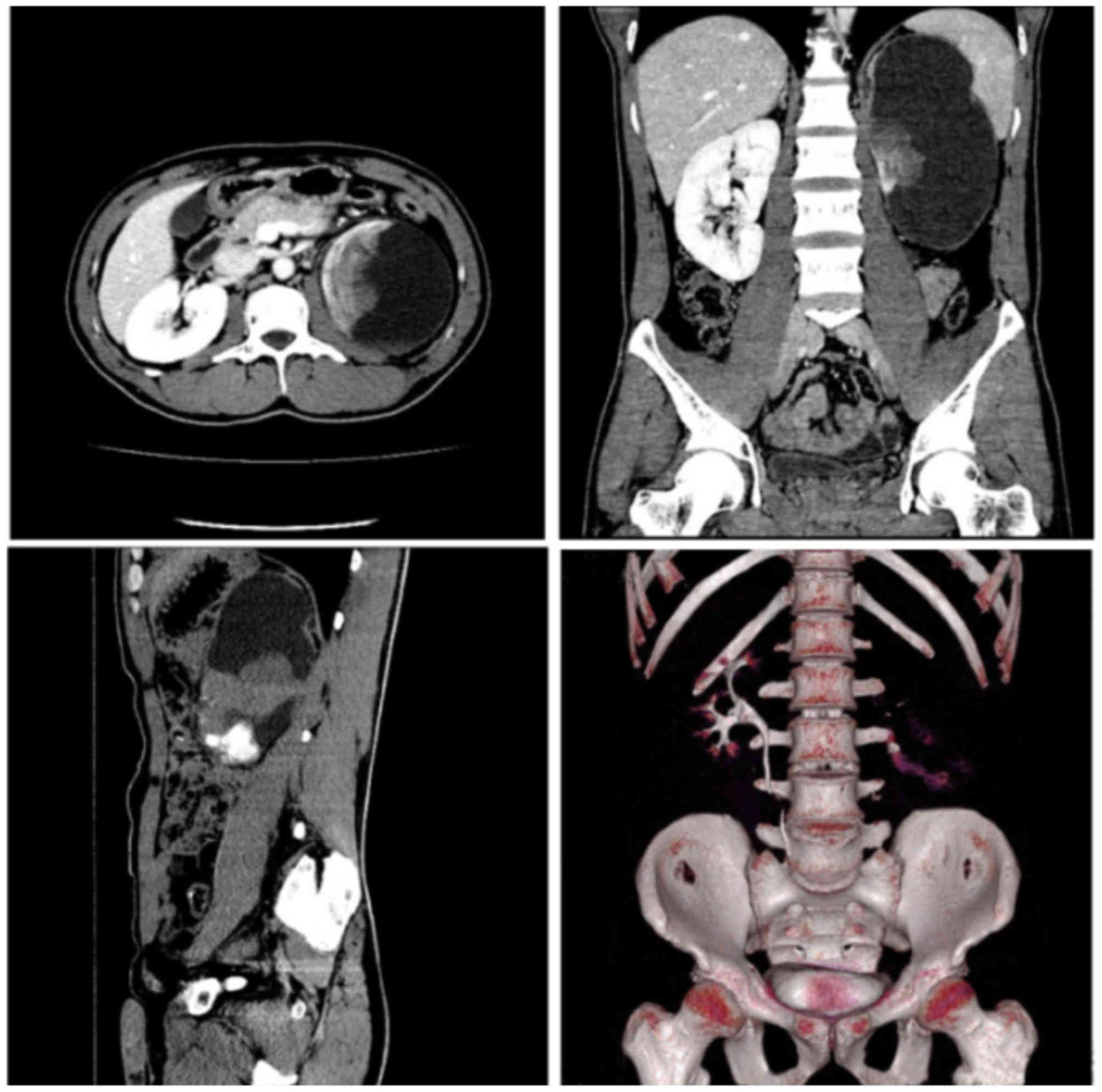
the CT scan with heterogeneous contrast enhancement, the tumors
ranged from 4–22 cm in size (median, 11 cm) and few showed
calcified areas (Fig. 1). Seven
cases received radical surgery, while the remaining case was only
subjected to needle biopsy of the tumor due to tumor invasion of
the inferior vena cava. Five cases received adjuvant chemotherapy,
while three cases received none. Surgical treatment consisted of
radical nephrectomy, as well as resection of parts of regional
organs and lymph nodes as required.
CIK cell preparation
CIK cells were prepared as described in a previous
study (9). In brief, peripheral
blood mononuclear cells (PBMCs) were collected from the patient and
cultured in medium containing 50 ng/ml anti-CD3 antibody, 100 U/ml
recombinant human interleukin (IL)-1α and 1,000 U/ml interferon
(IFN)-γ at 37°C in an atmosphere containing 5% CO2 for
24 h. Subsequently, 300 U/ml recombinant human IL-2 was added to
the media. At day 14, CIK cells were harvested and analyzed for
phenotype and cytotoxicity. All cells, reagents and materials were
free of bacteria, mycoplasma or fungal contamination. The measured
endotoxin levels were <5 endotoxin units.
Statistical analysis
Overall survival was estimated using the
Kaplan-Meier method, and calculated from the date of diagnosis to
the date of death from the disease or the last follow-up. Survival
estimates were calculated by the Kaplan-Meier method using SPSS 19
software (IBM SPSS, Armonk, NY, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Cases
All cases were confirmed to have advanced-stage
disease based on clinical imaging and pathological studies, five
(62.5%) of which had lymph node metastasis, one (12.5%) had
pancreatic and adrenal gland invasion, one (12.5%) had pancreatic
and spleen invasion, and the remaining one (12.5%) had invasion of
the inferior vena cava according to the CT scan. The
clinicopathological features of the eight cases are presented in
Table I.
 | Table I.Clinical and follow-up data of the
cases of the present study. |
Table I.
Clinical and follow-up data of the
cases of the present study.
| Case no. | Age (years) | Sex | Size (cm) | Side | Presentation | Surgery | Chemotherapy | Other therapy | Distant
metastasis | Outcome |
|---|
| 1 | 36 | F | 22 | Left | Palpable abdominal
mass, abdominal pain | Radical nephrectomy +
adrenalectomy + splenectomy + partial pancreatectomy | CTX, DDP | None | No | DOD at 48 months |
| 2 | 36 | M | 10 | Left | Palpable abdominal
mass, abdominal pain | Radical
nephrectomy | CTX, 6-MP, DDP, ADM,
CBP | None | Brain, lung | DOD at 36 months |
| 3 | 17 | M | 6 | Right | Palpable abdominal
mass, edema of lower limbs | Needle biopsy | CTX, VCR, IFO, VP16,
ADM, CBP, MEL | None | Lung | DOD at 7 months |
| 4 | 32 | F | 18 | Left | Palpable abdominal
mass | Radical
nephrectomy | None | None | No | DOD at 12 months |
| 5 | 22 | F | 9 | Left | Palpable abdominal
mass, abdominal pain | Radical
nephrectomy | CTX, VP16, IFO, VCR,
THP | None | No | DOD at 40 months |
| 6 | 21 | M | 4 | Left | Palpable abdominal
mass | Radical
nephrectomy | None | None | No | DOD at 10 months |
| 7 | 45 | M | 12 | Left | Palpable abdominal
mass, abdominal pain | Radical
nephrectomy | None | None | No | DOD at 6 months |
| 8 | 37 | M | 20 | Left | Palpable abdominal
mass, abdominal pain, abdominal pain | Radical nephrectomy +
adrenalectomy + splenectomy + partial pancreatectomy | IFO, VP16, CBP, THP,
ADM, NVB, mesna, DDP | CIK | Lung | DOD at 20 months |
Surgery
Four cases received radical nephrectomy, one case
was subjected to radical nephrectomy and splenectomy, and two cases
received radical nephrectomy combined with splenectomy and partial
pancreatectomy. Lymph node dissection was also performed during the
surgery. The tumor was inoperable in the patient with invasion of
the inferior vena cava according to the CT scan, and only a needle
biopsy was performed to confirm the diagnosis. There were no major
intra-operative and post-operative complications, or post-operative
mortality.
Histopathology
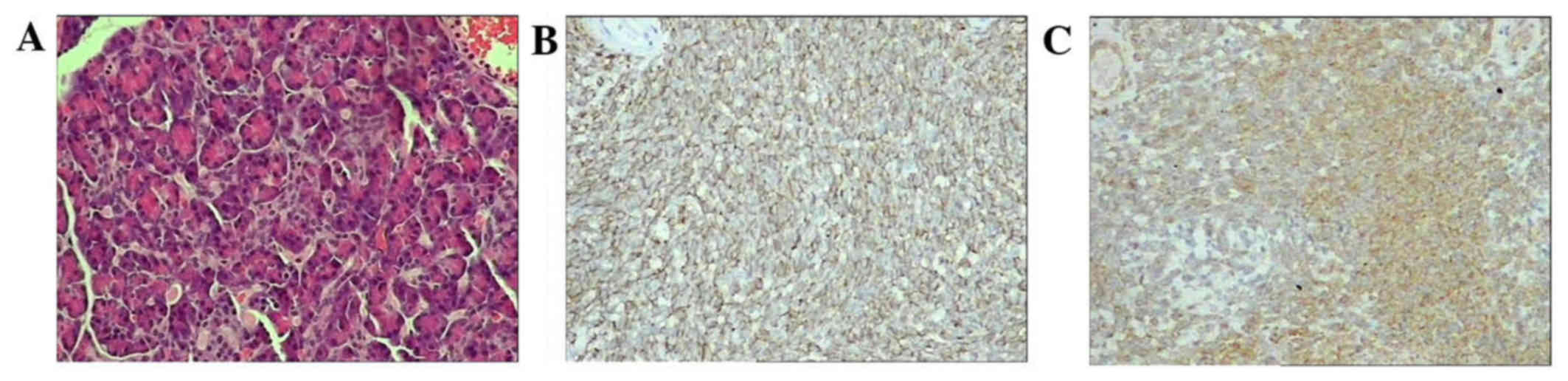
Pathological review and immunohistochemistry (IHC)
were performed to confirm the diagnosis. Histology revealed small
round cells with a high nuclear-to-cytoplasmic ratio with vaguely
defined cytoplasmic borders in the tumor. Homer-Wright rosette
formation was identified in the tumors of three patients. IHC
revealed positivity for CD99, vimentin and neuron-specific enolase
(NSE) in six cases (cases 1, 3, 4, 5, 6 and 7; 75%), Wilms' tumor
protein (WT-1) in two cases (cases 6 and 7; 25%) and desmin in one
case (case 5; 12.5%); furthermore, five cases (62.5%) were negative
for cytokeratin (CK) and one (12.5%) was focal positive (Fig. 2). Fluorescent in situ
hybridization (FISH) analysis using a locus-specific EWS/FLI-1
fusion gene dual color break apart rearrangement probe was
performed in one case, revealing a translocation of chromosomes 11
and 22, t(11;22) (q24;q12).
Adjuvant treatment
Of all the cases, five received adjuvant
chemotherapy (Table I).
Chemotherapeutic agents included cyclophosphamide (CTX),
pirarubicin (THP), cisplatinum (DDP), vinorelbine (NVB), purinethol
(6-MP), melphalan (MEL), adriamycin (ADM), carboplatin (CBP),
vincristine (VCR), etoposide (VP-16), ifosfamide (IFO) and
mesna.
As a novel treatment, CIK cell immunotherapy was
offered to all of the patients, while only one patient decided to
receive this immunotherapy with informed consent. This patient
received two courses of carboplatin (500 mg, d1) and docetaxel (80
mg, d2), and two cycles of CIK cell immunotherapy in parallel with
chemotherapy. This patient received a median of 95×108
CIK cells per cycle. No severe side-effects of the CIK cell
immunotherapy were observed. After CIK was administered, the
patient complained of less gastrointestinal adverse events compared
with the patients receiving chemotherapy only.
Follow-up
Follow-up information was available for all of the
patients, which included physical examination, complete blood
count, renal and liver function tests, chest X-ray and CT scan of
the abdomen. Of the eight cases, five had localized recurrence
only, one had localized recurrence and distant metastasis, and two
had distant metastasis only. Distant metastatic sites included the
lung (three cases) and brain (one case). None of the patients
received any salvage therapy, with the exception of alterations in
the chemotherapy regimens and dosage. Follow-up of these patients
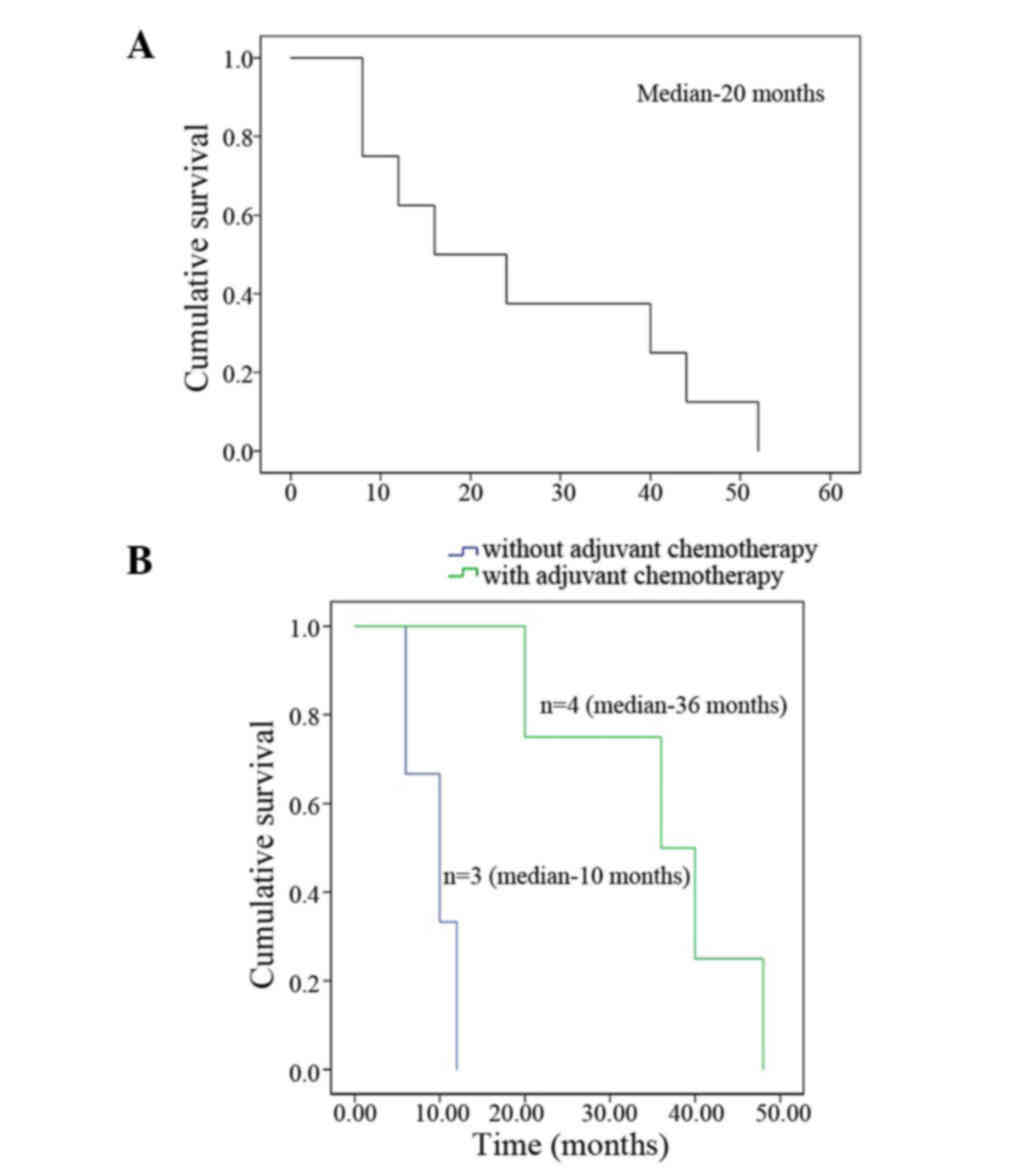
ranged from 6–48 months, with a median of 16 months. Overall median
survival was 20 months with a 3-year survival rate of 25%. Of the
seven cases who had surgery, the overall survival in four patients
who received adjuvant chemotherapy was 36 months, compared with 10
months in the three patients without adjuvant chemotherapy
(Fig. 3). The patient receiving CIK
cell immunotherapy survived for 20 months.
Discussion
Although peripheral PNET may occur in any soft
tissue, it rarely occurs in the genitourinary system (10). Since rPNET was first described in
1975, only 120 confirmed cases have been reported. rPNET frequently
affects young adults (mean age, 26 years) and occurs more commonly
in males (males/females, 1.5:1) (11,12). In
the present study, the patients with advanced-stage rPNET were
relatively young (median age, 34 years; range, 17–45 years), and a
slight male predominance was found (males/females, 1.6:1). Compared
with PNET originating from other sites, rPNET is more aggressive,
with a five-year disease-free survival rate of 45–55% at the
well-confined stage and only 20–30% at the advanced stage. The poor
prognosis of patients with rPNET is due to its non-specific
clinical presentation, tendency to metastasize and advanced stage
at the time of identification in the majority of cases (13,14). The
present study focused on rPNET at the advanced stage, and the cases
presented with non-specific symptoms and clinical signs, such as
palpable abdominal mass (100%), abdominal pain (62.5%) and signs of
metastasis. The overall median survival was 20 months with a
three-year survival rate of 25%, which was markedly lower compared
with the previously reported overall median survival of 40 months
with a 3-year survival rate of 60% in patients who had localized
disease (5). Therefore,
distinguishing rPNET from other kidney malignancies, and detecting
it early, is crucial for the management and prognosis of patients
with rPNET.
Although other small round-cell tumors, including
neuroblastomas (NBs), synovial sarcomas, small cell carcinomas,
lymphomas, Wilms' tumors, and so forth, render the renal tumor
differentiation difficult, rPNET does have specific histological
features, including small uniform round cells with dark nuclei,
ill-defined cytoplasmic borders, and poorly-formed rosette-like
structures (15). In addition, IHC
is important for accurately diagnosing rPNET. Parham et al
(4) reported that it is difficult to
characterize rPNET without IHC. According to IHC, 84–100% of rPNETs
are positive for CD99, a macrophage inhibitory cytokine (MIC-2)
gene product (14,16). In addition, IHC markers, including
vimentin, NSE and S-100, may aid in the diagnosis of rPNET and
differentiation from other tumor types; however, they are not
pathognomonic. Molecular diagnostic markers, such as friend
leukemia integration 1 (FLI-1) and WT-1, were found to be
relatively specific in terms of the diagnosis of rPNET, and a
previous study reported that 60% of rPNETs were positive for FLI-1
expression by IHC (17), indicating
that this marker may aid in the differentiation of rPNET from other
types of renal tumor. Within the cohort of the present study, the
tumors of 37.5% of cases had a specific histological structure
called ‘Homer-Wright rosette’, and IHC analysis revealed that 65%
of cases were positive for CD99, vimentin and NSE. In addition,
positivity for WT-1 was found in two cases (25%) and for desmin in
one case (12.5%); Furthermore, five cases (62.5%) were negative for
CK, while one case (12.5%) was focal positive. WT-1, a common
marker for Wilms' tumor, has been used to rule out the diagnosis of
PNET, while rPNETs may stain positive for WT-1. A cytogenetic study
performed to detect the EWS/FLI-1 fusion gene revealed a
translocation of chromosomes 11 and 22, t(11:22) (q24:q12), in
>90% of rPNET cases (18), while
the use of FISH for the detection of the fusion gene may increase
the specificity of this marker and decrease false-positive test
results. In the present study, FISH detection was performed for one
case, also revealing t(11;22)(q24;q12) translocation.
At present, no definitive guidelines for the
treatment of rPNET are available. For rPNET at the localized stage,
surgical excision is the first choice, providing a greater survival
advantage compared with any other singular treatment (5). However, the prognosis of rPNET at an
advanced stage remains poor in spite of aggressive treatment by
combination therapy, including surgery, chemotherapy and
radiotherapy (1,6). In the present study, seven cases
received radical nephrectomy, of which three cases had surgical
resection of other organs involved. Four cases received adjuvant
chemotherapy and had a significantly better overall survival of 36
months, compared with an overall survival of 10 months in the three
patients without adjuvant chemotherapy. However, severe
gastrointestinal adverse events are usually found during
chemotherapy. In an attempt to improve the survival of high-risk
patients as well as their quality of life, CIK cell immunotherapy
was planned after surgery in combination with the administration of
intensive and multiple large-dose post-operative adjuvant
chemotherapies. In the one patient who consented to the CIK cell
treatment, survival for 20 months was achieved. Of note, the
patient complained of less adverse events associated with
chemotherapy. CIK cells are ex vivo activated lymphocytes
with potent activity against various tumor types and minimal
side-effects. Since CIK cells were first applied for renal cancer
therapy in 1999 (19), the safety
and efficacy of this immunotherapy has been confirmed and the most
frequent adverse event is only mild, transient and easily
controllable (20). The largest
study of autologous CIK cell immunotherapy in metastatic RCC to
date performed by Liu et al (9) found that CIK cell treatment
significantly improved the prognosis of patients with metastatic
RCC, while the prognosis was significantly improved for patients
who received ≥7 cycles of CIK infusions (9). CIK cell therapy is considered to have
synergistic effects with conventional therapies, including
chemotherapy or IL-2/IFN-α biotherapy in patients with RCC. CIK
cells exert cytotoxic activities against solid tumors by
specifically binding to target cells via cell surface adhesion
molecule leukocyte function associated antigen-1 (LFA-1) to form
cellular conjugates (21). Besides
adhesion molecules, CIK cells express activating NK receptors,
including NKG2D and DNAX accessory molecule-1, which leads to
degranulation and activates T-cell receptor-independent tumor cell
recognition and killing (22,23). To
the best of our knowledge, the present study was the first to
report on the clinical application of CIK cell immunotherapy for
the treatment of rPNET. The benefit of CIK cells and the possible
synergy with targeted therapies for rPNET should be assessed in
future studies using larger cohorts.
In conclusion, rPNET is a rare malignancy of the
family of ‘small round-cell tumors’. Patients with rPNET at the
advanced stage have poor prognosis, and aggressive multimodality
treatment, including surgical excision and chemotherapy, is
recommended to manage these tumors. CIK cell immunotherapy may have
the capacity to improve the prognosis and life quality of patients
with rPNET. Further studies are required to validate the benefit of
CIK cells and establish an appropriate immunotherapy protocol.
Acknowledgements
The authors would like to thank all of the patients
enrolled in this study. This work was supported by the Medical
Scientific Research Foundation of Guangdong Province (no. A117),
the Fundamental Research Funds for the Central Universities (no.
16ykjc15) and the Natural Science Foundation of Guangdong Province
(no. 2014A030310158).
References
|
1
|
de Alava E and Gerald WL: Molecular
biology of the Ewing's sarcoma/primitive neuroectodermal tumor
family. J Clin Oncol. 18:204–213. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mani S, Dutta D and De BK: Primitive
neuroectodermal tumor of the liver: A case report. Jpn J Clin
Oncol. 40:258–262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seemayer TA, Thelmo WL, Bolande RP and
Wiglesworth FW: Peripheral neuroectodermal tumors. Perspect Pediatr
Pathol. 2:151–152. 1975.PubMed/NCBI
|
|
4
|
Parham DM, Roloson GJ, Feely M, Green DM,
Bridge JA and Beckwith JB: Primary malignant neuroepithelial tumors
of the kidney: A clinicopathologic analysis of 146 adult and
pediatric cases from the National Wilms' Tumor Study Group
Pathology Center. Am J Surg Pathol. 25:133–146. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thyavihally YB, Tongaonkar HB, Gupta S,
Kurkure PA, Amare P, Muckaden MA and Desai SB: Primitive
neuroectodermal tumor of the kidney: A single institute series of
16 patients. Urology. 71:292–296. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chu WC, Reznikov B, Lee EY, Grant RM,
Cheng FW and Babyn P: Primitive neuroectodermal tumour (PNET) of
the kidney: A rare renal tumour in adolescents with seemingly
characteristic radiological features. Pediatr Radiol. 38:1089–1094.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim JS, Chung IS, Lim SH, Park Y, Park MJ,
Kim JY, Kim YG, Hong JT, Kim Y and Han SB: Preclinical and clinical
studies on cytokine-induced killer cells for the treatment of renal
cell carcinoma. Arch Pharm Res. 37:559–566. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li H, Yu JP, Cao S, Wei F, Zhang P, An XM,
Huang ZT and Ren XB: CD4+CD25+ regulatory T cells decreased the
antitumor activity of cytokine-induced killer (CIK) cells of lung
cancer patients. J Clin Immunol. 27:317–326. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu L, Zhang W, Qi X, Li H, Yu J, Wei S,
Hao X and Ren X: Randomized study of autologous cytokine-induced
killer cell immunotherapy in metastatic renal carcinoma. Clin
Cancer Res. 18:1751–1759. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartholow T and Parwani A: Renal primitive
neuroectodermal tumors. Arch Pathol Lab Med. 136:686–690. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ellinger J, Bastian PJ, Hauser S, Biermann
K and Müller SC: Primitive neuroectodermal tumor: Rare, highly
aggressive differential diagnosis in urologic malignancies.
Urology. 68:257–262. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lam JS, Hensle TW, Delbelenko L,
Granowetter L and Tennenbaum SY: Organ confined primitive
neuroectodermal tumour arising from the kidney. J Pediatr Surg.
38:619–621. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rodríguez-Galindo C, Liu T, Krasin MJ, Wu
J, Billups CA, Daw NC, Spunt SL, Rao BN, Santana VM and Navid F:
Analysis of prognostic factors in ewing sarcoma family of tumors:
Review of St. Jude Children's Research Hospital Studies. Cancer.
110:375–384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ellison DA, Parham DM, Bridge J and
Beckwith JB: Immunohistochemistry of primary malignant
neuroepithelial tumors of the kidney: A potential source of
confusion? A study of 30 cases from the national Wilm's tumor study
pathology center. Hum Pathol. 38:205–211. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wada Y, Yamaguchi T, Kuwahara T, Sugiyama
Y, Kikukawa H and Ueda S: Primitive neuroectodermal tumour of the
kidney with spontaneous regression of pulmonary metastases after
nephrectomy. BJU Int. 91:121–122. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ravindra S and Kini U: Cytomorphology and
morphometry of small round-cell tumors in the region of the kidney.
Diagn Cytopathol. 32:211–216. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Risi E, Iacovelli R, Altavilla A, Alesini
D, Palazzo A, Mosillo C, Trenta P and Cortesi E: Clinical and
pathological features of primary neuroectodermal tumor/Ewing
sarcoma of the kidney. Urology. 82:382–386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kumar R, Gautam U, Srinivasan R, Lal A,
Sharma U, Nijhawan R and Kumar S: Primary Ewing's sarcoma/primitive
neuroectodermal tumor of the kidney: Report of a case diagnosed by
fine needle aspiration cytology and confirmed by
immunocytochemistry and RT-PCR along with review of literature.
Diagn Cytopathol. 40 Suppl 2:E156–E161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schmidt-Wolf IG, Finke S, Trojaneck B,
Denkena A, Lefterova P, Schwella N, Heuft HG, Prange G, Korte M,
Takeya M, et al: Phase I clinical study applying autologous
immunological effector cells transfected with the interleukin-2
gene in patients with metastatic renal cancer, colorectal cancer
and lymphoma. Br J Cancer. 81:1009–1016. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jäkel CE, Hauser S, Rogenhofer S, Müller
SC, Brossart P and Schmidt-Wolf IG: Clinical studies applying
cytokine-induced killer cells for the treatment of renal cell
carcinoma. Clin Dev Immunol. 2012:4732452012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pievani A, Borleri G, Pende D, Moretta L,
Rambaldi A, Golay J and Introna M: Dual-functional capability of
CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and
retains TCR-mediated specific cytotoxicity. Blood. 118:3301–3310.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Girardi M, Oppenheim DE, Steele CR, Lewis
JM, Glusac E, Filler R, Hobby P, Sutton B, Tigelaar RE and Hayday
AC: Regulation of cutaneous malignancy by gammadelta T cells.
Science. 294:605–609. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karimi M, Cao TM, Baker JA, Verneris MR,
Soares L and Negrin RS: Silencing human NKG2D, DAP10 and DAP12
reduces cytotoxicity of activated CD8+ T cells and NK cells. J
Immunol. 175:7819–7828. 2005. View Article : Google Scholar : PubMed/NCBI
|